

Summary
The phenolic compound salicylic acid (SA) is a key signalling molecule regulating local and systemic plant defense responses, mainly against biotrophs. Many microbial organisms, including pathogens, share the ability to degrade SA. However, the mechanism by which they perceive SA is unknown. Here we show that Ustilago maydis, the causal agent of corn smut disease, employs a so far uncharacterized SA sensing mechanism. We identified and characterized the novel SA sensing regulator, Rss1, a binuclear zinc cluster protein with dual functions as putative SA receptor and transcriptional activator regulating genes important for SA and tryptophan degradation. Rss1 represents a major component in the identified SA sensing pathway during the fungus’ saprophytic stage. However, Rss1 does not have a detectable impact on virulence. The data presented in this work indicate that alternative or redundant sensing cascades exist that regulate the expression of SA‐responsive genes in U. maydis during its pathogenic development.
Introduction
Salicylic acid (SA, 2‐hydroxybenzoic acid) belongs to the class of phenolic compounds and is composed of an aromatic ring with a carboxyl‐ and hydroxyl group. Its biosynthesis is shared between organisms of diverse kingdoms of life ranging from bacteria to plants (Serino et al., 1995; Kerbarh et al., 2005; Dempsey et al., 2011). In bacteria, SA serves as building block for siderophore biosynthesis or as siderophore itself (Visca et al., 1993). In plants, it was identified as important hormone regulating diverse processes, including seed germination, cell growth, respiration, and most prominently defense responses to biotrophic pathogens (Vlot et al., 2009). To limit the spread of biotrophic pathogens that rely on the living host for proliferation (Glazebrook, 2005), SA regulates a complex network of diverse signalling components. Upon pathogen attack, this network activates defense responses which can culminate in hypersensitive cell death at the site of infection (Vlot et al., 2009). Moreover, SA is involved in systemic acquired resistance in distal, non‐infected parts of the plant resulting in broad‐spectrum resistance to diverse pathogens and an immune memory that primes the plant for secondary infections (Fu and Dong, 2013).
One model pathogen for the study of biotrophic interactions between filamentous fungi and their hosts is the smut fungus Ustilago maydis. The basidiomycete overcomes SA‐regulated defense barriers of its host Zea mays, one of the most important crop plants, and successfully proliferates inside the plant (Doehlemann et al., 2008). After recognition during the early phase of infection, U. maydis suppresses the immune responses of its host with the help of small secreted molecules, termed effectors, and establishes a biotrophic interaction (Kamper et al., 2006; Doehlemann et al., 2008; Djamei et al., 2011; Hemetsberger et al., 2012; Mueller et al., 2013). The biotrophic interaction ultimately leads to tumor formation, where fungal proliferation and differentiation of black‐pigmented fungal spores occurs (Christensen, 1963). Not surprisingly, U. maydis as well as other (hemi‐) biotrophic plant pathogens employ diverse strategies to interfere with host SA signalling and production in order to suppress defense responses (Tanaka et al., 2015). For example, they can limit SA biosynthesis through the elimination of SA precursors, like chorismate or isochorismate (Djamei et al., 2011; Liu et al., 2014), or they manipulate the proteasome‐mediated turnover rate of SA signalling components (Ustun et al., 2013). Moreover, we recently showed that U. maydis degrades SA by means of the salicylate hydroxylase Shy1, demonstrating the capability of a biotrophic pathogen to eliminate SA (Rabe et al., 2013). Shy1 is essential for the utilization of SA as carbon source in axenic culture and the shy1 gene is induced during plant colonization. However, since the deletion of shy1 did not affect virulence, the role of SA‐degradation in the pathogenic development of U. maydis remains elusive. Besides Shy1, two additional proteins were predicted to be salicylate hydroxylases but did not show enzymatic activity. Respective genes are upregulated during pathogenic development and show an increase in transcript levels upon SA treatment. The specific induction of a set of genes in the presence of SA indicates that U. maydis is able to sense SA by an as yet unknown mechanism. Shy1 is thought to be part of a negative feedback loop, indirectly regulating SA‐responsive gene expression. By degrading SA, Shy1 reduces the amount of inducer, which subsequently leads to the downregulation of SA‐responsive genes (Rabe et al., 2013).
Studies of model plants like Arabidopsis and tobacco have provided insights into SA perception and signalling in plants, and several SA receptors and binding proteins have been discovered in the recent years (Seyfferth and Tsuda, 2014). Although SA binding is well studied, divergent models propose different bona fide SA receptors important for regulating pathogen defense. Wu and colleagues (2012) showed that upon SA binding the transcriptional co‐activator NPR1 changes its conformation and activates defense‐related genes. Fu et al. (2012) failed to show SA binding by NPR1 and propose NPR3 and NPR4 as bona fide SA receptors. Both proteins bind SA and, depending on the cellular SA concentration, target NPR1 for proteasomal degradation. Moreover, H2O2 scavengers and methyl salicylate esterases are capable of binding SA with high affinities resulting in inhibition of their enzymatic activity (Chen and Klessig, 1991; Durner and Klessig, 1995; Kumar and Klessig, 2003; Forouhar et al., 2005).
Since many filamentous fungi have evolved ways to eliminate SA (Sze and Dagley, 1984; Penn and Daniel, 2013; Rabe et al., 2013; Ambrose et al., 2015; Martins et al., 2015), SA sensing should also be wide spread among these organisms. However, nothing is known about fungal SA perception and signalling. Here we show that the biotrophic fungus U. maydis perceives SA via the response factor Rss1, a putative binuclear zinc cluster protein. The protein constitutes a major component of SA sensing and regulates genes involved in a shared pathway for the metabolism of SA and tryptophan. However, although Rss1 is essential for the activation of SA‐responsive genes in the saprophytic phase of U. maydis, we provide evidence that additional cues and pathways exist that regulate these genes during plant colonization.
Results
Isolation of UMAG_05966 as an important factor for SA sensing
We have previously shown that U. maydis senses SA resulting in the transcriptional induction of SA‐responsive genes. Among these are the salicylate hydroxylase encoding gene shy1, essential for SA degradation, as well as the SA induced gene srg1 (SA‐responsive gene 1; UMAG_05967), coding for a protein of so far unknown function (Rabe et al., 2013). To identify factors involved in the SA sensing pathway we conducted a genetic screen by making use of the strong srg1 promoter. The haploid solopathogenic strain SG200 was transformed with a construct harbouring mCherry‐3xHA under control of the srg1 promoter and subjected to UV mutagenesis to retrieve mutants impaired in SA perception and signalling (Fig. 1A). Since SG200Psrg1mCherry‐3xHA exhibited strong and specific mCherry fluorescence in the presence of SA (Fig. 1B), loss of fluorescence was used as a readout after UV mutagenesis. If mutations led to the disruption of the SA sensing pathway, the respective strains should be unable to activate the srg1 promoter and to produce mCherry‐3xHA upon SA‐treatment. Seven of 86,400 tested colonies showed the expected phenotype and did not exhibit mCherry fluorescence (Supporting Information Fig. 1A). Since induction of shy1 is essential for SA degradation under axenic growth conditions, a disrupted SA sensing pathway might lead to reduced shy1 gene expression and impaired growth on medium with SA as sole carbon source. In line with this assumption, all identified mutants displayed severe growth attenuations on salicylate minimal medium (Fig. 1C). To identify the mutations underlying the phenotype, each mutant strain was transformed with an U. maydis cosmid library (Weinzierl, 2001) and screened for rescue of the mutation through restored mCherry fluorescence. Four candidate genes (UMAG_10865, UMAG_05964, UMAG_05965, UMAG_05966) located at the end of chromosome 20 were identified as putatively involved in SA sensing. Complementation analysis with autonomously replicating plasmids harbouring the individual genes revealed that UMAG_05966 was able to rescue the SA sensing phenotype of all mutants. UMAG_05966 complemented mutant strains were able to grow on minimal medium with salicylate as sole carbon source (Fig. 1C) and showed restored mCherry fluorescence (Supporting Information Fig. 1B). Since UMAG_05966 is located on an autonomously replicating plasmid, which is present in the cell in multiple copies (Tsukuda et al., 1988), its overexpression might have rescued other mutations underlying the mutant phenotype. To exclude this, Next Generation Illumina Sequencing (NGS) and single nucleotide polymorphism (SNP) analysis were performed. Although we found several unique mutations for each mutant in different genomic regions (average number of mutations per genome = 12 ± 9; Supporting Information Table 1), only the chromosomal region encompassing UMAG_05966 harboured mutations or deletions in all sequenced strains: UMAG_05966 of UV1 and UV6 contained non‐synonymous mutations while all other mutants lost more than 45 kb from the end of chromosome 20 including UMAG_05966 (Supporting Information Fig. 2). This makes it unlikely, that additional mutations led to the observed phenotype. Through targeted gene deletion of UMAG_05966 via homologous recombination we confirmed the observed growth defect on SA minimal medium indicating that the deletion of UMAG_05966 blocks SA perception and/or signalling (Supporting Information Fig. 3). The growth attenuation could be partially complemented by ectopic expression of mCherryHA‐UMAG_05966 under control of the UMAG_05966 promoter and fully rescued by an untagged version of UMAG_05966 (Supporting Information Fig. 3). Based on the NGS and complementation data we concluded that UMAG_05966 is a key player in SA perception and/or signal transduction under the tested conditions, and we designated the protein Rss1 (Required for SA sensing 1).
Figure 1.
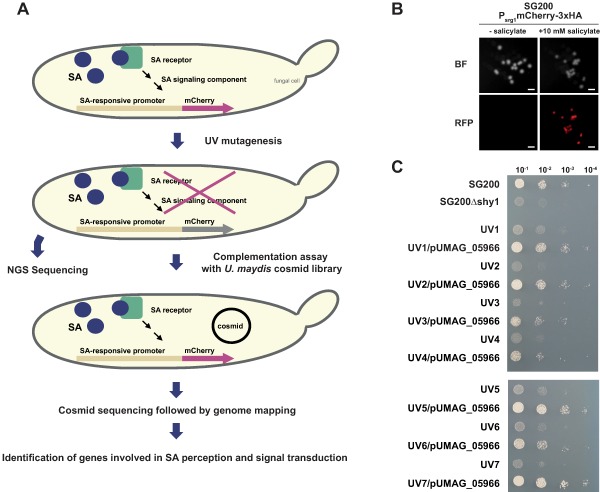
UMAG_05966 (Rss1) represents a major component of SA perception and signalling in U. maydis.
A. Schematic representation of the UV mutagenesis screen to identify proteins involved in SA sensing. SG200Psrg1mCherry‐3xHA, expressing mCherry‐3xHA under control of the SA‐responsive srg1 promoter, was mutagenized with 20 mJ UV. Cells were screened for loss of fluorescence, i. e. for inability to activate the srg1 promoter. Candidate mutants were subjected to NGS whole genome sequencing and complementation analysis employing an autonomously replicating U. maydis cosmid library (Weinzierl, 2001). Re‐isolated cosmids from rescue mutants were sequenced and mapped to the U. maydis genome. Results were compared to NGS sequencing data.
B. Epifluorescence stereomicroscopy was performed with SG200Psrg1mCherry‐3xHA colonies on glucose‐containing YNB‐N medium without (left panel) and with 10 mM sodium salicylate (right panel). Fluorescence could only be detected for SG200Psrg1mCherry‐3xHA spotted on salicylate‐containing medium, indicating a specific and strong activation of the chosen SA‐responsive promoter. Scale bars: 1 mm.
C. UV mutant strains with loss of fluorescence were spotted on YNB‐N + 10 mM salicylate and exhibited severe growth retardation similar to SG200Δshy1. Growth retardation was rescued after introduction of pUMAG_05966 expressing UMAG_05966 under the endogenous UMAG_05966 promoter.
Rss1 likely has a dual function as transcriptional activator and putative SA receptor
rss1 is located in the U. maydis genome on chromosome 20, upstream of the SA‐responsive gene srg1. Both genes share the same promoter region and they are divergently transcribed (A. Czedik‐Eysenberg, J. Bindics, unpublished tiling array data). rss1 encodes an 866 amino acid (aa) long protein that harbours domains often found in ligand binding binuclear zinc cluster transcription factors (Fig. 2; MacPherson et al., 2006; Shelest, 2008): a putative N‐terminal Zn(II)2Cys6‐DNA binding domain (aa 37–82), a predicted PEST motif (aa 244‐257) known to promote proteasomal degradation, a putative fungal transcription factor region (aa 281‐542), a coiled coil region considered to be important for dimerization (aa 678–705), and a predicted monopartite nuclear localization signal (NLS) at the N‐ and a bipartite NLS at the C‐terminus (aa 46–52 and 729–751; Nucpred = 0.95). The consensus sequence CX2CX6CX5‐9CX2CX6‐8C of the DNA binding domain, which is characteristic for binuclear zinc cluster proteins (MacPherson et al., 2006), is highly conserved in Rss1 (Fig. 2).
Figure 2.
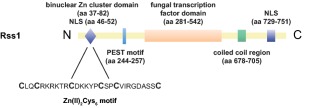
Rss1 harbours domains of binuclear zinc cluster transcription factors. Rss1 (UMAG_05966) contains domains and sequences known to be present in binuclear zinc cluster transcription factors including predicted Nuclear Localization Signal (NLS) sequences, a Zn(II)2Cys6‐DNA binding domain, a putative fungal transcription factor domain, a predicted PEST motif for proteasomal turnover and a coiled‐coil domain important for dimerization. Predicted domains are indicated.
In line with its predicted function as a transcription factor and its nuclear localization signals, mCherryHA‐Rss1 exclusively localizes to nuclei of U. maydis cells grown in YNB‐N supplemented with 10 mM SA (Fig. 3A). Differences in localization in SA‐treated and untreated cells could not be observed (data not shown).
Figure 3.

Rss1 localizes to U. maydis nuclei and responds to SA as a transcriptional activator in a heterologous system.
A. mCherryHA‐Rss1 localizes to the nuclei of U. maydis yeast‐like cells. Localization of mCherryHA‐Rss1 in cells of axenically grown CL13Δrss1‐mCherryHA‐rss1 culture was assessed by confocal laser scanning microscopy. Cells were stained with DAPI to visualize nuclei. Scale bars: 10 μm.
B. Gal4‐BD‐Rss1 activates reporter gene expression in S. cerevisiae in the presence of salicylate and shows a dose‐dependent response to SA. AH109 expressing Gal4‐BD (AH109‐BD; negative control), Gal4‐BD‐rss11‐216 (AH109‐BD‐Rss11‐216; positive control), or Gal4‐BD‐rss1 (AH109‐BD‐Rss1) was spotted in serial dilutions on SD‐Trp, SD‐Trp‐Ade‐His, SD‐Trp‐Ade‐His + 100 µM sodium salicylate and SD‐Trp‐Ade‐His + 1 mM sodium salicylate. Under stringent conditions growth of AH109‐BD‐Rss1 was only detectable with addition of salicylate.
Since bioinformatic predictions as well as the confirmed nuclear localization supported a function as transcription factor, we tested whether Rss1 acts as transcriptional activator. To this end, the DNA binding domain of the Gal4 transcription factor of Saccharomyces cerevisiae (Gal4‐BD) was fused to the N‐terminus of Rss1. The respective construct was introduced into S. cerevisiae AH109 harbouring auxotrophy markers for histidine and adenine biosynthesis that are under control of Gal4‐responsive promoters. Due to recruitment of the fusion protein to the Gal4‐responsive promoters by Gal4‐BD, the capability of Rss1 to act as transcriptional activator could be monitored by the induction of reporter gene expression. Transcriptional induction eventually leads to growth on high stringency medium. The Gal4‐BD‐Rss1 producing strain AH109‐BD‐Rss1 did not grow on high stringency medium (Fig. 3B). However, the addition of 100 µM or 1 mM salicylate restored growth of AH109‐BD‐Rss1 in a dose‐dependent manner (Fig. 3B). We excluded that the exclusively SA‐dependent induction represents an artifact of the medium by including AH109‐BD, expressing Gal4‐BD, as negative control and a strain producing a truncated autoactive mutant version of Rss1, Rss11‐216, fused to Gal4‐BD as positive control.
Since we could not find any Rss1 orthologs in S. cerevisiae by BlastP analysis and the strain used in this study was unable to utilize SA as carbon source (Supporting Information Fig. 4), it is unlikely that an orthologous SA sensing cascade in yeast activates Rss1 post‐translationally. Therefore, we reasoned that Rss1 might not only represent a transcription factor but at the same time a receptor for SA. To test SA binding, we tried to produce Rss1 with N‐terminal affinity tag fusions either in U. maydis or heterologously in Escherichia coli. However, since all tested production methods resulted in protein aggregation, no, or highly unstable soluble protein (Supporting Information Table 2), we could not directly determine SA binding by Rss1 and its binding affinity.
Dimerization of binuclear zinc cluster proteins is often necessary to bind DNA and to regulate transcription (MacPherson et al., 2006). By employing the Yeast two‐hybrid system we could show that Rss1 forms homodimers (Supporting Information Fig. 5). In addition, the presence of the binuclear zinc cluster domain suggests, that Rss1 directly recognizes and binds a certain motif present in its target promoters, e.g. in the srg1 promoter region. However, since the production of biologically active Rss1 protein failed, we could not assess DNA binding and identify specific Rss1‐binding motifs.
rss1 is transcriptionally induced during pathogenic development but does not have a detectable impact on virulence
Since SA‐responsive genes, like shy1 and srg1, are transcriptionally induced during pathogenic development of U. maydis, we reasoned that their regulator might also be present during those developmental stages. To test whether rss1 is expressed, we monitored rss1 transcript levels at different infection stages of the solopathogenic strain SG200 by quantitative real time PCR. rss1 expression was induced 35‐fold in the early stage of infection compared to axenic culture, decreased slightly at two days post infection (dpi), but increased again at four dpi. Transcript levels stayed elevated up to 43‐fold until sporogenesis occurred at twelve dpi (Fig. 4A). Since genes involved in pathogenic development are often induced upon plant infection (Kamper et al., 2006), the expression profile of rss1 suggested that the protein might play a role in virulence. To investigate whether SA sensing by Rss1 is important for the pathogenic development of the fungus, virulence assays with CL13Δrss1 and the progenitor strain CL13 were performed. The haploid solopathogenic strain CL13 was chosen for this experiment, since modest virulence defects can be better assessed with this strain than with SG200 (Di Stasio et al., 2009; Djamei et al., 2011). However, rss1 deletion strains showed no significant alteration in virulence in seedling infections (Fig. 4B). This indicates either that SA perception and signalling are not relevant for virulence under standard laboratory conditions or that Rss1‐mediated SA sensing is part of a complex network where the lack of one pathway can be compensated by others.
Figure 4.
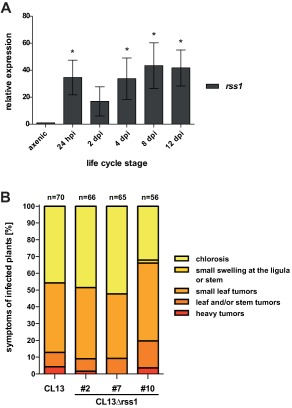
rss1 is transcriptionally induced during the pathogenic development of U. maydis but has no impact on virulence.
A. Transcript levels of rss1 were determined by quantitative real time PCR after RNA isolation and cDNA synthesis from SG200 axenic culture and from different developmental stages. Constitutively expressed peptidyl‐prolyl isomerase (ppi) was used for normalization. Transcript levels were compared to those in axenic culture and levels in axenic culture were set to 1.0. Error bars depict standard deviation calculated from three independent biological replicates using infected areas from 12 plants each (n = 3). Significance was calculated with unpaired t test comparing expression values with those of axenic culture, * P ≤ 0.05.
B. Disease symptoms of maize seedlings were determined 12 days post infection with either U. maydis CL13 or three independent isolates of CL13Δrss1 according to Kamper et al. (2006). Disease symptom categories are colour‐coded and depicted on the right. Mean values from two independent infections are shown with the total number of infected plants above each column. No significant differences in disease symptoms were scored.
Rss1‐independent SA sensing pathways exist regulating SA‐responsive genes during pathogenic development
To investigate whether compensatory SA sensing pathways are active during plant infection, we determined the expression of previously identified SA‐responsive genes (Rabe et al., 2013) in SG200Δrss1 and its progenitor SG200 during pathogenic development and in a time course of SA‐treated axenic culture (Fig. 5). During pathogenic development srg1 was significantly upregulated in SG200 compared to axenic culture (P < 0.022). SG200Δrss1 failed to induce srg1 expression to the same degree as SG200 resulting in up to 8‐fold lower transcript levels. However, srg1 expression in SG200Δrss1 was still significantly up‐regulated during pathogenic development (P < 0.018) and srg1 transcript levels increased over time with a peak of 360‐fold at 12 dpi compared to axenic culture (Fig. 5A, left panel). A different expression pattern was observed when measuring srg1 transcript levels in SA‐treated axenic cultures. While srg1 transcript levels were induced up to 4,400‐fold in SG200, no significant induction of srg1 could be detected in the Δrss1 mutant strain. Expression of srg1 in SG200Δrss1 was reduced more than 4,000‐fold compared to SG200 (Fig. 5A, right panel). These findings indicate that, in contrast to axenic culture, Rss1‐independent pathways must exist that activate srg1 expression in planta.
Figure 5.
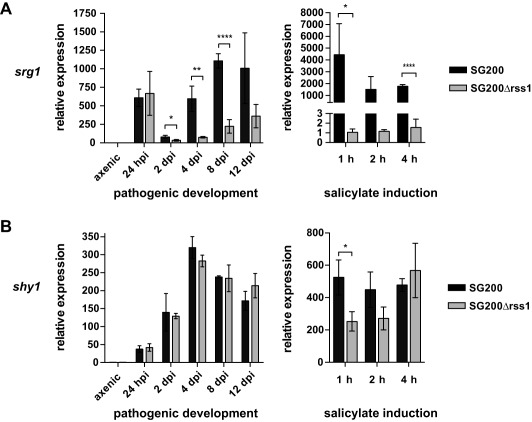
Transcriptional induction of SA‐responsive genes during pathogenic development and during growth in SA supplemented medium does not exclusively depend on Rss1. Transcript levels of the previously identified SA‐responsive genes srg1 (A) and shy1 (B) were quantified in SG200 and SG200Δrss1 by real time PCR. RNA was isolated from the indicated life cycle stages of pathogenic development (‘pathogenic development’, left panel) and from a time course after shift to YNB‐N medium containing 2% glucose and 10 mM salicylate (‘salicylate induction’, right panel). Constitutively expressed peptidyl‐prolyl isomerase transcript levels (ppi) were used for normalization. Transcript levels of the indicated genes were either compared to levels in axenic culture grown in YEPSlight medium (left panel) or grown in YNB‐N medium with 2% glucose (right panel). Expression levels in axenic culture (left panel) or in cultures grown in YNB‐N with glucose (right panel) were set to 1.0. Error bars depict standard deviation calculated from three independent biological replicates (n = 3). Significance was calculated with unpaired t test comparing transcript levels of indicated genes in SG200 with those in SG200Δrss1, * P ≤ 0.05, ** P ≤ 0.01, *** P ≤ 0.001. For transcriptional profiling of different life cycle stages, RNA extracted from twelve infected plants per time point and replicate was used.
Surprisingly, shy1 expression was not significantly altered during pathogenic development of SG200Δrss1 compared with SG200 (Fig. 5B, left panel). Upon salicylate treatment of axenic culture, shy1 transcript levels were initially 2‐fold reduced in the rss1 mutant compared to the progenitor strain but levels increased again over time (Fig. 5B, right panel).
In line with the finding that additional pathways contribute to the regulation of SA‐responsive genes, global transcriptional profiling of SG200 and SG200Δrss1 identified only nine genes that were differentially regulated during plant infection at four dpi (adjusted P‐value < 0.05). Six of those were significantly repressed in SG200Δrss1 while three were induced (Table 1). The majority of them showed only minor differences in expression levels compared to SG200 (fold change < 2.0; Table 1). Among the repressed genes were srg1, the SA marker gene, as well as UMAG_02142 and UMAG_12178, which encode enzymes predicted to be involved in fungal metabolic pathways. UMAG_02142 displays homologies to muconate cycloisomerases, which are known to act in degradation pathways downstream of catechol (Martins et al., 2015). The functional classification system FunCat 2 (Ruepp et al., 2004) predicts that UMAG_12178, the gene with the most severe reduction in transcript levels (Log FC = −2.069), is involved in the metabolism of secondary products derived from tryptophan. Among the induced genes, UMAG_06076 codes for a putative sugar transporter specialized in quinate uptake. In line with the expression data based on quantitative real time PCR, shy1 was not among the differentially regulated genes. Since UMAG_02142 might be involved in the downstream degradation of SA, we assessed the transcriptional profile of this gene by quantitative real time PCR under different conditions: UMAG_02142 was transcriptionally induced upon SA treatment of axenic culture in SG200 and during pathogenic development. Similar to srg1, its transcript levels were significantly reduced in SG200Δrss1 (P < 0.019) and the reduction was more severe in axenic culture one hour after SA‐shift than during biotrophic growth (Supporting Information Fig. 6).
Table 1.
Differentially expressed genes in SG200Δrss1 compared to SG200 at 4 dpi.
| Gene ID | Log FC | P‐value | Adjusted P‐value | Gene prediction |
|---|---|---|---|---|
| UMAG_12178 | −2.069 | 3.23 E −26 | 0.00000 | Related to o‐pyrocatechuate decarboxylase |
| UMAG_02142 | −0.805 | 5.49 E −09 | 0.00002 | Related to muconate cycloisomerase |
| UMAG_11117 | −0.58 | 1.99 E −05 | 0.01930 | Uncharacterized protein |
| UMAG_11020 | −0.551 | 3.09 E −05 | 0.02620 | Uncharacterized protein |
| UMAG_04481 | −0.533 | 4.75 E −05 | 0.03580 | Related to ADH2 ‐ Alcohol dehydrogenase II |
| UMAG_05967 | −0.542 | 6.90 E −05 | 0.04680 | SA‐responsive gene srg1 |
| UMAG_03349 | 0.625 | 1.69 E −05 | 0.01920 | Uncharacterized protein |
| UMAG_06076 | 0.672 | 1.45 E −05 | 0.01920 | Related to quinate transport protein |
| UMAG_05819 | 0.611 | 6.33 E −06 | 0.01070 | Uncharacterized protein |
Rss1 is essential for utilizing tryptophan as a carbon source
Since global transcriptional profiling data indicated that Rss1 does not only regulate genes of the downstream pathway of catechol but might also be involved in the regulation of genes for tryptophan degradation, we assessed whether U. maydis is impaired in growth on tryptophan minimal medium in absence of rss1. Indeed, rss1 deletion mutants showed attenuated growth when tryptophan was provided as sole carbon source (Fig. 6). Similar to the growth attenuation of CL13Δrss1, the deletion of srg1 also resulted in growth retardation on tryptophan minimal medium (Fig. 6). To test whether tryptophan is an inducer of Rss1 activity, we repeated the heterologous yeast‐based transcriptional activation assay with tryptophan. In contrast to medium supplemented with salicylate, AH109‐BD‐Rss1 failed to grow when tryptophan was added (Supporting Information Fig. 7). These results indicate that Rss1 might not perceive tryptophan as a direct signal leading to its activation. The inability of Rss1 to sense tryptophan is also reflected by transcriptional profiling of SA‐responsive genes. Expression levels of the SA‐responsive genes shy1, srg1, and UMAG_02142 were quantified by real time PCR and compared to those in untreated control cells. All tested genes showed significant lower transcript levels upon tryptophan treatment than after addition of salicylate (P < 0.033): shy1 and UMAG_02142 were only 2‐ and 6‐ fold induced upon tryptophan treatment compared to 388‐ and 34‐fold induction upon salicylate treatment (Supporting Information Fig. 8). srg1 showed the highest induction (550‐fold) after the shift to tryptophan‐containing medium. However, the induction was significantly lower than after growth in medium supplemented with salicylate (P = 0.033), which resulted in a relative expression of more than 1,800‐fold (Supporting Information Fig. 8). The significantly weaker induction of SA‐responsive genes upon tryptophan treatment together with the transcriptional activation assay suggests that secondary products derived from the amino acid, and not tryptophan itself, are probably capable of activating the expression of the tested genes.
Figure 6.

CL13Δrss1 and CL13Δsrg1 show growth attenuation on medium with tryptophan as sole carbon source. Growth of CL13 and deletion mutants of the SA‐responsive genes shy1, srg1, and UMAG_03408 as well as CL13Δrss1 was assessed on YNB‐N supplemented with 2% glucose (YNB‐N + Glc), with 10 mM sodium salicylate (YNB‐N + 10 mM salicylate), with 10 mM tryptophan (YNB‐N + 10 mM tryptophan), or without any carbon source (YNB‐N). While shy1 and UMAG_03408 were not required for growth on tryptophan as sole carbon source, deletion of srg1 and rss1 resulted in growth attenuation on the respective medium. Images for ‘YNB‐N + Glc’ plate were acquired three days after spotting, for ‘YNB‐N + 10 mM salicylate’ plate after four days, and for ‘YNB‐N + 10 mM tryptophan’ and ‘YNB’ plates after six days.
Anthranilic acid, a possible degradation product of tryptophan, can modulate Rss1 activity
Tryptophan degradation via L‐kynurenine is widespread among eukaryotes and is found in both fungi and animals (Ternes and Schonknecht, 2014). For fungi, it was shown that L‐kynurenine is converted into anthranilate which then is channeled via several intermediates into the 3‐oxoadipate pathway (Rao et al., 1971; Anderson and Dagley, 1981; Martins et al., 2015). SA and anthranilate are structurally similar, differing only in one of their two functional groups. Therefore, we tested whether anthranilate can activate Rss1 by repeating the yeast‐based transcriptional activation assay with anthranilate as a putative inducer. The addition of anthranilate resulted in growth on high stringency medium, suggesting that Rss1 is able to sense this compound and to activate reporter gene expression (Fig. 7).
Figure 7.
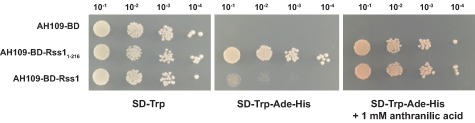
Anthranilate can induce Rss1 activity in yeast. The yeast‐based transcriptional activation assay was performed with anthranilate as a putative inducer. AH109 expressing Gal4‐BD (AH109‐BD; negative control), Gal4‐BD‐rss11‐216 (AH109‐BD‐Rss11‐216; positive control), or Gal4‐BD‐rss1 (AH109‐BD‐Rss1) were spotted in serial dilutions on SD‐Trp (growth control) (A), on SD‐Trp‐Ade‐His (B) and on SD‐Trp‐Ade‐His + 1 mM anthranilic acid (C). Addition of anthranilic acid resulted in an activation of reporter gene expression and growth of AH109‐BD‐Rss1.
SA sensing is conserved among smuts
Since the smuts Sporisorium reilianum and Ustilago hordei share highly conserved orthologs of SA‐responsive genes with U. maydis (Rabe et al., 2013), their capability to respond to SA with an induction of those genes was tested by quantitative real time PCR. To this end, S. reilianum SRZ1 and U. hordei Uh4875‐4 were shifted to YNB‐N medium supplemented with glucose and 10 mM salicylate for one hour. Upon salicylate treatment the shy1 ortholog in S. reilianum, sr_shy1, was only weakly induced, while U. hordei responded to SA with a 150‐fold transcriptional induction of UHOR_shy1. For srg1 orthologs a different expression pattern was observed: sr_srg1 in S. reilianum showed more than 100‐fold higher transcript levels upon SA treatment compared to levels in untreated cells, whereas UHOR_srg1 was not significantly induced in an identical experimental set‐up (Supporting Information Fig. 9). The transcriptional profiles indicate that, although transcript levels of orthologous genes vary dramatically between smuts, SA can trigger the induction of genes in species related to U. maydis.
BlastP analyses revealed highly conserved Rss1 orthologs in S. reilianum (Sr16594), Sporisorium scitamineum (SPSC_06050), and Melanopsichium pennsylvanicum (Bn887_02897; Supporting Information Fig. 10). Rss1 and its orthologs in S. reilianum and S. scitamineum show conserved local synteny and orientation. Although U. hordei shares synteny of the respective region, no rss1 ortholog could be found in the fungal genome. The presence of small blocks of rss1 coding sequence remaining in the syntenic region of U. hordei indicates that the fungus likely once harboured a functional ortholog but might lost it after divergence from U. maydis.
Discussion
In this study, we provide insights into a novel SA sensing and degradation mechanism in U. maydis. We show that a biotrophic fungus is able to sense SA by means of the response factor Rss1. This multifunctional protein, belonging to the family of binuclear zinc cluster proteins, constitutes a major component of an SA sensing mechanism present in U. maydis. It perceives SA and the structural analog anthranilate, presumably by direct binding, and regulates the expression of genes for SA and tryptophan degradation. SA signal transduction by Rss1 represents a novel mechanism that has not been described in other organisms. Rss1 has no homology to known SA sensing and signalling regulators in plants, such as the NPR proteins, thus making an identical mode of action unlikely (Fu et al., 2012; Wu et al., 2012). Also, bioinformatic comparisons with the bacterial SA response factor NahR did not reveal any significant similarity (data not shown) (Schell and Poser, 1989; Schell et al., 1990) suggesting that sensing and activation by Rss1 differs from bacterial systems and may have evolved independently.
Rss1 harbours domains known for binuclear zinc cluster transcription factors (Fig. 2). Binuclear zinc cluster proteins are exclusively found in fungi and they regulate diverse cellular processes, including sugar and amino acid metabolism, nitrogen utilization, respiration, as well as cell cycle regulation (MacPherson et al., 2006; Shelest, 2008). Moreover, they often share functional similarity with transcription factors of the nuclear receptor protein family found in metazoans. Nuclear receptor proteins are able to bind small‐molecule ligands and, as a result of ligand binding, activate the transcription of target genes (Naar and Thakur, 2009). The bioinformatic predictions (Fig. 2) together with the data of the yeast‐based transcriptional activation assay (Fig. 3) suggest that Rss1 might share the ability to act both as signalling sensor and concomitantly as a transcriptional activator. The binuclear zinc cluster proteins Leu3p and Pdr1 of S. cerevisiae are, like many other proteins of this class, composed of an internal domain with low conservation, termed the middle homology region (MHR) (MacPherson et al., 2006). For Pdr1, the MHR domain is important for ligand binding. It was shown that ligand binding leads to a conformational change of Pdr1 which in turn enables the C‐terminal activation domain to interact with a subunit of the Mediator complex and to initiate transcription of target genes (Thakur et al., 2008). Moreover, mutations in this region render the protein constitutively active (Carvajal et al., 1997; Nourani et al., 1997). Similar to Pdr1, deletion and mutational analyses of Rss1 as along with interaction studies might provide further insight into which domains are important for ligand binding and how Rss1 triggers transcriptional activation.
Several binuclear zinc cluster proteins, including the Pdr1 paralog Pdr3, were shown to form positive autoregulatory feedback loops modulating their own expression (Delahodde et al., 1995; Zhang et al., 2001). Since rss1 transcript levels are significantly upregulated during pathogenic development (Fig. 4) and rss1 shares the promoter region with its target gene srg1, it is conceivable that Rss1 also regulates its own expression.
Due to their coiled coil domains binuclear zinc cluster proteins often form homo‐ or heterodimers that recognize response elements with CGG triplets as inverted, everted, or direct repeats (Hellauer et al., 1996; Todd and Andrianopoulos, 1997; MacPherson et al., 2006). In line with this, Rss1 forms a homodimer (Supporting Information Fig. 5) and its promoter as well as those of its putative target genes, like srg1, UMAG_02142 and UMAG_01278, contain several inverted and direct CGG repeats. It will be of future interest to experimentally elucidate the binding motif of the Rss1‐homodimer.
Although Rss1 is an essential factor in SA sensing in the saprophytic phase of the fungus, virulence was not altered when rss1 was deleted (Fig. 4). Since we showed that transcript levels of target genes are more dramatically reduced in axenic culture than during pathogenic development in the rss1 mutant (Fig. 5, Supporting Information Fig. 6), we conclude that alternative signalling pathways must be present during biotrophic growth. However, the identification of pathways that are exclusively active in biotrophy will be technically challenging.
SA growth assays presented in this work show that Rss1 regulates genes involved in SA degradation (Supporting Information Fig. 3). We supported these findings by global transcriptional profiling (Table 1). Among Rss1‐regulated targets are genes that may be involved in the downstream metabolism of SA, such as the putative muconate cycloisomerase encoding gene UMAG_02142. However, the SA sensing and degradation pathway we identified seems to be tightly linked to tryptophan metabolism and Rss1 represents a regulator for the metabolism of both compounds. Since Srg1, although displaying sequence similarity to salicylate hydroxylases, is not involved in the conversion of SA to catechol (Rabe et al., 2013) but is essential for tryptophan degradation (Fig. 6), we assume that the protein converts the structural analog of SA, anthranilate. The intermediate 2,3‐dihydroxybenzoate, which is produced by anthranilate hydroxylases (Rao et al., 1971), could be subsequently converted to catechol by the Rss1 target UMAG_12178. UMAG_12178 is related to o‐pyrocatechuate decarboxylases which facilitate the aforementioned reaction (Rao et al., 1967). Catechol can be further metabolized via the 3‐oxoadipate pathway and TCA cycle (Martins et al., 2015). Moreover, we provide evidence that by reacting to the tryptophan degradation product anthranilate Rss1 could be directly involved in the regulation of this pathway (Fig. 7). In contrast to srg1, the deletion of shy1 had no impact on growth on tryptophan as sole carbon source (Fig. 6). The production of catechol by salicylate hydroxylases (Yamamoto et al., 1965) makes it likely that the Shy1‐mediated SA degradation pathway converges with the tryptophan branch and the shared intermediate catechol enters the 3‐oxoadipate pathway. Although shy1 was not differentially regulated in a rss1 deletion mutant during pathogenic development, transcript levels were significantly reduced in the early phase after shifting cells to SA containing medium (Fig. 5), indicating that Rss1 contributes, albeit only under certain conditions, to the regulation of shy1 and SA degradation. Whether the ability to sense SA and regulate shy1 expression represents a neofunctionalization of Rss1 or is an evolutionary remnant which will disappear over time remains speculative.
It might not be coincidental that genes involved in tryptophan metabolism are induced during biotrophy. Many phytopathogenic fungi are able to metabolize amino acids as carbon and nitrogen sources (Solomon et al., 2003). Among these are obligate biotrophic fungi, like Blumeria graminis and Uromyces fabae, which employ a set of transporters and permeases during infection that facilitate amino acid uptake (Struck, 2015). U. maydis also induces genes coding for amino acid transporters during the early phase of infection and it harbours all enzymes important for amino acid degradation, including those for tryptophan (McCann and Snetselaar, 2008; Lanver et al., 2014). Although genes important for amino acid degradation are repressed during appressoria formation (Lanver et al., 2014), it is likely that uptake and metabolism of amino acids contributes to growth of U. maydis during later stages. In line with those findings, Horst and colleagues (2010) demonstrated that, due to metabolic reprogramming upon U. maydis infection, plant‐derived amino acids are relocated to tumor tissue. Therefore, the upregulation of the tryptophan pathway we identified could contribute to feeding during these developmental stages. This might represent a survival strategy of biotrophic pathogens; to rely not only on few nitrogen and carbon sources but to employ a multitude of pathways to obtain nutrients that are abundant in the host and that are of diverse chemical origin, like hexoses, tryptophan, or even SA. Although it was shown that the uptake of sucrose and hexose plays an important role in nutrition during biotrophy (Wahl et al., 2010; Schuler et al., 2015), the ability to use alternative carbon sources, might ensure that in the absence of the favorable nutrient source the fungus retains its biosynthetic capacity and is able to complete its life cycle.
The transcriptional profiling data of shy1 in the rss1 deletion mutant (Fig. 5) indicated that additional factors must be involved in the regulation of Shy1‐dependent SA degradation. Tryptophan and SA degradation might not be entirely co‐regulated and induction might depend on different additional stimuli. This could imply that SA sensing and degradation might have a biological function unrelated to the tryptophan pathway. Therefore, it is tempting to speculate whether U. maydis employs SA sensing not only to detect a suitable carbon source in planta but also to sense the defense status of the host, to adapt to its environment, and to modulate its own machinery to ensure a successful infection. In this scenario, SA degradation via Shy1 would be an effective mechanism for eliminating SA and resetting Rss1 and other regulators for SA sensing. The lack of an observable virulence phenotype in the rss1 and shy1 deletion mutants can be explained by additional or redundant SA sensing and degradation cascades in planta. The identification of additional SA sensors, which remained undiscovered in this screen that was based on a single promoter and axenic conditions, and their target genes will provide further insights into why U. maydis is able to sense one of the major defense hormones of its host.
Since SA sensing could be a versatile survival strategy of pathogens related to U. maydis both as saprophytes as well as in their hosts, we tested whether SA sensing per se is conserved among smuts. We showed that S. reilianum and U. hordei respond to SA with the induction of orthologs of SA‐responsive marker genes (Supporting Information Fig. 9). However, differences in transcriptional responses along with the finding that U. hordei likely lost the rss1 ortholog while retaining the capability to activate UHOR_shy1, point towards Rss1‐independent sensing pathways. In line with these findings, we could not identify Rss1‐orthologs by BlastP in SA degrading fungal species including the saprophytes Trichosporon cutaneum, the symbiont Epichloe festucae, the hemi‐biotroph Fusarium graminearum as well as the necrotroph Sclerotinia sclerotiorum, suggesting that Rss1‐independent SA‐sensing mechanisms in fungi might exist (Sze and Dagley, 1984; Qi et al., 2012; Penn and Daniel, 2013; Ambrose et al., 2015).
Several phytohormone nanosensors have been developed enabling the quantification of hormone levels in the cell (Brunoud et al., 2012; Jones et al., 2014; Waadt et al., 2014). Although high‐affinity SA‐binding proteins were identified (dissociation constants of K d = 90 nM for SABP2 and K d = 45 nM for NPR4) (Du and Klessig, 1997; Fu et al., 2012), no SA nanosensor has been established to date. Once the affinity between Rss1 and SA is determined, Rss1 might fill the gap of a missing SA nanosensor and could be employed to assess SA levels inside eukaryotic cells in a quantitative way. The yeast‐based transcriptional activation assay provided already evidence that SA can be detected in a different heterologous eukaryotic system (Fig. 3). Since Rss1 functions as transcriptional activator, quantification of SA could be coupled to a reporter system such as GUS or fluorescence markers. In a further step, it would need to be evaluated whether Rss1 could be used to generate a FRET‐based nanosensor, similar to those established for Auxin and ABA (Brunoud et al., 2012; Jones et al., 2014; Waadt et al., 2014).
With the identification of Rss1 we were not only able to shed light on SA sensing via a multifunctional protein acting as putative receptor and transcriptional activator but also provide the foundation for the generation of valuable tools to assess and monitor cellular SA levels in the future.
Experimental procedures
Plasmids, strains and culture conditions
Plasmids were generated according to standard molecular cloning procedures described in Sambrook et al. (1989). Primers, plasmids, and cloning strategies employed in this study are compiled in Supporting Information Tables 3 and 4.
U. maydis strains used in this study are listed in Supporting Information Table 5. They were generated by gene replacement via homologous recombination with PCR‐generated constructs (Kamper, 2004) or by insertion of p123 derivatives into the ip locus (Loubradou et al., 2001). Gene deletions and insertions were verified by PCR and/or Southern analysis. To assess SA‐responsiveness of S. reilianum and U. hordei, the strains SRZ1 (Schirawski et al., 2005b) and Uh4875‐4 (Linning et al., 2004) were used. For U. maydis pathogenicity assays three independent mutants were tested in replicates for virulence on 7‐day‐old maize seedlings of the variety Early Golden Bantam (Olds Seeds, Madison, USA). Disease symptoms were scored 12 days post infection following described protocols (Kamper et al., 2006). Statistical analysis was performed using the R statistical environment (R Core Team, 2011)
UV mutagenesis and cosmid complementation
U. maydis SG200Psrg1mCherry‐3xHA was grown to an exponential phase and adjusted to OD600nm = 1 with H20dd. 15 ml of the cell suspension, diluted 1:103 with H20dd, were transferred into a petri dish (diameter: 90 mm). To lower surface tension 1 µl 10% Tween 20 was added. UV mutagenesis with a survival rate of 40–50% was achieved by irradiating the cell suspension with 20 mJ using a UV crosslinker (UV Stratalinker 2400; Stratagene, La Jolla, CA, USA). The mutagenized cell suspension was plated on YNB‐N medium supplemented with 2% glucose and 10 mM salicylate. Single colonies were screened for loss of mCherry fluorescence with a widefield stereomicroscope for brightfield and fluorescence (Lumar; Zeiss, Jena, Germany) four days after plating. Images were acquired with the SPOT Pursuit‐XS Monochrome camera controlled by the SPOT Basic Image Capture Software (SPOT Imaging, Sterling Heights, MI, USA). Colonies of cells displaying the expected phenotype were used for inoculation of liquid YEPSlight medium (Schirawski et al., 2005a) and tested again for loss of fluorescence after spotting in serial dilution on YNB‐N medium (Rabe et al., 2013) supplemented with 2% glucose and 10 mM salicylate.
For complementation analysis, SA sensing mutants were transformed with an U. maydis cosmid library (Weinzierl, 2001). Colonies were replica plated on YNB‐N plates containing 2% glucose, 10 mM salicylate, and 200 µg ml−1 Hygromycin B and screened for rescue of mCherry fluorescence by fluorescence stereomicroscopy. Rescue mutants were grown in YEPSlight supplemented with 200 µg ml−1 Hygromycin B to retain the autonomously replicating cosmids. Cosmids were re‐isolated by employing the genomic DNA isolation protocol according to Hoffman and Winston (1987) and amplified in E. coli. The complementing U. maydis fragment, which is inserted in the cosmid, was sequenced with primers 5′ pScos seq and 3′ pScos seq. Sequenced reads were mapped to the U. maydis genome by using CLC Main Workbench (Qiagen, Hilden, Germany) to narrow down regions that harbour genes for SA sensing.
U. maydis growth assay on minimal media
Growth assays on salicylate and tryptophan minimal medium were performed as described in Rabe et al. (2013). In brief, U. maydis strains were grown in YEPSlight until OD600nm = 0.8 was reached. Cells were washed three times with H20dd and resuspended in H20dd to OD600nm = 1. Adjusted cultures were spotted in serial dilutions on YNB‐N minimal medium, pH 7.0, supplemented with either 10 mM sodium salicylate or 10 mM L‐tryptophan.
Yeast‐based assays
AH109 (Clontech/Takara Bio, Saint‐Germain‐en‐Laye, France) was transformed with pGBKT7 or pGADT7 derivatives described in Supporting Information Table 4 using standard protocols (Clontech/Takara Bio, Saint‐Germain‐en‐Laye, France). Strains were grown in selective medium, adjusted to OD600nm = 1 and spotted on indicated media in serial dilutions. Yeast strains used in this study are compiled in Supporting Information Table 6.
To test for transcriptional activation by Rss1, AH109 producing Gal4‐BD‐Rss1 was spotted on selective dropout media plates lacking tryptophan, adenine, and histidine (SD‐Trp‐Ade‐His) and SD‐Trp‐Ade‐His plates supplemented with either 100 µM or 1 mM sodium salicylate, 1 mM L‐tryptophan, or 1 mM anthranilic acid. SD‐Trp was used as growth control plate.
For homodimerization tests of Rss1, in‐frame fusions of Rss1 with Gal4 binding and activation domain, respectively, were produced in yeast. AH109 and Y187 from the Matchmaker™ GAL4 Two‐Hybrid System (Clontech/Takara Bio, Saint‐Germain‐en‐Laye, France) were transformed with either pGADT7 or pGBKT7 derivative containing the respective constructs and mated according to the manufacturer's protocol. Diploids harbouring both plasmids were selected on SD‐Trp‐Leu and homodimerization was assessed by testing growth on high stringency medium (SD‐Trp‐Leu‐Ade‐His).
Confocal laser scanning microscopy
For colocalization experiments U. maydis cells were grown in YNB‐N medium with 10 mM sodium salicylate for 5 h (OD600nm = 0.8). Cells were harvested by centrifugation (2400 g, 5 min) and fixed by addition of 2% formaldehyde in 1x PBS. Samples were incubated for 5 min, centrifuged at 2400 g for 5 min, and washed once with 1x PBS. Afterwards the pellet was resuspended in DAPI (4′,6‐Diamidine‐2′‐phenylindole dihydrochloride) solution (0.5 µg ml−1) and incubated for 10 min. After an additional washing step, samples were subjected to confocal microscopy. Colocalization of mCherry‐HA‐Rss1 and DAPI stained nuclei was microscopically assessed by employing an LSM780 Axio Observer confocal laser‐scanning microscope (Zeiss, Jena, Germany) with the following settings: mCherry: Laser DPSS 15 mW, excitation 561 nm, detection 578‐648 nm; DAPI: Laser Diode 25 mW, excitation 405 nm, detection 415–510 nm.
Quantitative real time PCR and microarray analyses
Quantitative real time PCR was performed as described in Rabe et al. (2013). In brief, RNA from axenic culture or from infected plant material was extracted using the TRIzol method according to the manufacturer's protocol (Thermo Fisher Scientific, Waltham, MA, USA), DNA was removed by DNase I treatment (DNA‐free Kit; Thermo Fisher Scientific, Waltham, MA, USA) and RNA was reverse transcribed using the RevertAid First Strand cDNA Synthesis Kit (Thermo Fisher Scientific, Waltham, MA, USA). Quantitative real time PCR measurements were performed with a Roche LightCycler® 96 system (Roche Diagnostics, Rotkreuz, Switzerland) according to manufacturer's instructions. Relative expression values were calculated with the 2−ΔΔCt method (Livak and Schmittgen, 2001). Graphical outputs and statistical analyses were performed using GraphPad Prism (v6.0; GraphPad Software, La Jolla, CA, USA).
For global transcriptional profiling, maize plants (Early Golden Bantam) were grown in a plant growth chamber and infected with SG200 or SG200Δrss1 as described previously (Doehlemann et al., 2008). Samples were collected in three independently conducted experiments by sampling 12 plants per experiment. To this end, two cm of the third leaf were harvested two cm below the injection point. For RNA isolation, material from the 12 plants was pooled, ground in liquid nitrogen and extracted using TRIzol (Thermo Fisher Scientific, Waltham, MA, USA) according to the manufacturer's protocol. RNA was additionally purified with the RNeasy Kit (Qiagen, Hilden, Germany). Quality and quantity was assessed with the Agilent 2100 Bioanalyzer (Agilent Technologies, Santa Clara, CA, USA). 200 ng purified RNA was subjected to microarray analysis using custom designed Agilent expression microarrays. Chips (8x60K array format) containing U. maydis and Z. mays gene probes were designed with the online design application eArray (Agilent Technologies, Santa Clara, CA, USA). For probe design the U. maydis orfeome (http://www.helmholtz-muenchen.de/en/ibis/institute/groups/fungal-microbial-genomics/resources/mumdb/index.html) and pre‐designed probes from a 4x44K maize gene expression microarray of the eArray AgilentCatalog (Agilent Technologies, Santa Clara, CA, USA) were used. Each Chip included a total of 6,782 sense probes against U. maydis genes, along with 6,782 U. maydis antisense probes and 42,030 probes against maize genes as well as control probes. The subsequent experimental procedure was performed according to Agilent's Two‐Colour Microarray‐Based Gene Expression Analysis protocol using the Low Input Quick Amp Labelling Kit (Agilent Technologies, Santa Clara, CA, USA). For each individual microarray chip, the respective RNA sample, which was labelled with Cyanine 3‐CTP, was hybridized to the chip together with a common reference pool derived from all samples. The reference pool was labelled with Cyanine 5‐CTP. Data normalization and analysis was conducted in the R statistical environment (R Core Team, 2011) using the limma package (Ritchie et al., 2015). Raw expression data for each chip were background normalized by the normexp algorithm. The overall distribution of expression ratios between the green and red channel in each chip was normalized by the loess method. In a subsequent step, the expression data were filtered such that only probes targeting sense transcripts of U. maydis were retained for further analysis. This removed noise between samples caused by the plant side and therefore improved the detection of the relatively weak expression changes in the mutant compared to SG200. The log2 expression ratios for the remaining probes were normalized between arrays by the quantile method. A linear model was used to test for significant expression differences between the SG200 and SG200Δrss1 samples. Since location‐dependent effects on plant growth between the three replicates could be observed in the plant growth chamber, the estimation of a “sampling date” effect was included in the model to subtract background noise between replicates. Differential expression was determined by the limma ebayes function. P‐values were corrected for multiple testing by the Benjamini‐Hochberg method (Benjamini and Hochberg, 1995). Expression data were submitted to GeneExpressionOmnibus (http://www.ncbi.nlm.nih.gov/geo/) under the accession number GSE83576.
Next generation illumina sequencing
To sequence genomes of mutant strains, their genomic DNA was subjected to Next Generation Illumina Sequencing. To this end, genomic DNA was extracted according to the method of Hoffman and Winston (1987) and purified by an additional purification step using the MasterPure Complete DNA and RNA Purification Kit (Epicentre, Madison, WI, USA). DNA concentration was determined by PicoGreen measurements (Thermo Fisher Scientific, Waltham, MA, USA). 1 µg genomic DNA was subsequently sheared in micro AFA tubes using an S220 focused ultra‐sonicator with AFA technology (Covaris, Woburn, MA, USA). Average fragmentation size was assessed by performing a Fragment Analyzer run with the High Sensitivity NGS Fragment Analysis Kit (Advanced Analytical Technologies, Ankeny, IA, USA). Fragmented DNA was subsequently converted into indexed libraries for Next Generation Sequencing using the NEBNext®Ultra™ DNA Library Prep Kit for Illumina®. The library was prepared according to the manufacturer's protocol. Quality control and Illumina 125 bp paired end sequencing on a HiSeq 2500 instrument was carried out by the Next Generation Sequencing Facility (VBCF, Vienna, Austria).
Removal of adapter contamination and trimming of low quality 3′ read ends was performed with BBDUK (BBMap ‐ Bushnell B. ‐ sourceforge.net/projects/bbmap/) and Trimmomatic (v0.33; Bolger et al., 2014), respectively. Paired‐end reads were mapped to the U. maydis reference genome with BWA (v0.7.8; Li and Durbin, 2009), while duplicated reads were removed with Picard (v1.101; http://broadinstitute.github.io/picard). Local realignment around indels and base recalibration were done with GATK (v3.5; DePristo et al., 2011). All format conversions were done with Samtools (v0.1.18; Li et al., 2009). Joint genotyping of all sequenced strains was performed with GATK/UnifiedGenotyper in SNP mode setting the ploidy parameter to 1 for haploid individuals. Variants common to all strains, including the progenitor SG200Psrg1mCherry‐3xHA, were discarded for considering that those mutations with respect to the reference existed before UV mutagenesis. In addition, variant positions with quality ≤ 50 or supported by more than one read with mapping quality zero were filtered out in all strains. At the sample level, variants where the log2 of the ratio between the read depth and the median coverage of the strain was either > 0.5 or < −0.5 were excluded from the analysis. Mapped reads were visualized with IGV (v2.3.57; Robinson et al., 2011; Thorvaldsdottir et al., 2013). Sequencing data were submitted to NCBI under the BioProject accession number PRJNA326324, Study ID SRP076835.
Bioinformatic analyses
Gene and protein sequences of U. maydis, S. reilianum, and U. hordei as well as gene and protein information were obtained from the MIPS Ustilago maydis database (http://www.helmholtz-muenchen.de/en/ibis/institute/groups/fungal-microbial-genomics/resources/mumdb/index.html), the MIPS Sporisorium reilianum database (http://www.helmholtz-muenchen.de/ibis/institute/groups/fungal-microbial-genomics/resources/msrdb/index.html), and the MIPS Ustilago hordei database (http://www.helmholtz-muenchen.de/ibis/institute/groups/fungal-microbial-genomics/resources/muhdb/index.html). The Rss1 protein sequences of S. scitamineum (GeneBank Accession No. CDU25879) and M. pennsylvanicum (GeneBank Accession No. CDI53350) were taken from the ‘National Center of Biotechnology Information’ (NCBI; www.ncbi.nlm.nih.gov/). BlastP (Basic Local Alignment Search Tool) version 2.225 was employed for the identification of potential Rss1 orthologs using standard search parameters (Altschul et al., 1990). Homologous amino acid sequences were compared with CLC Main Workbench (v7.0.2; Qiagen, Hilden, Germany) using progressive alignment algorithms. Protein domains were identified with the Simple Modular Architecture Research Tool ‚SMART’ (Schultz et al., 1998; Letunic et al., 2015) and nuclear localizations were predicted with NucPred (Brameier et al., 2007). PEST motifs were determined with epestfind (v5.0.0; Rice et al., 2000)
Authors contributions
Conceived and designed the experiments: FR and AD. Performed the wet bench experiments: FR, DS, LB, and FN. Performed bioinformatics analyses: ACE and FAR. Wrote the manuscript: FR and AD with input from all co‐authors. Directed the project: AD.
Authors declaration
The Authors declare that there is no conflict of interest in the research.
Supporting information
Supporting Information
Acknowledgements
We would like to thank the GMI/IMBA/IMP service facilities for excellent technical support, the Vienna Biocenter Core Facilities (VBCF) for next generation sequencing, Ulrich Güldener and his team from the Helmholtz Zentrum München for support with array design, and James Matthew Watson as well as Simon Uhse for input on the manuscript. The research leading to these results has received funding from the European Research Council under the European Union's Seventh Framework Programme (FP7/2007‐2013)/ERC grant agreement n° [EUP0012 ‘Effectomics’], the Austrian Science Fund (FWF): [P27429‐B22, P27818‐B22] and the Austrian Academy of Science (OEAW). FR is member of the International Max Planck Research School for Environmental, Cellular and Molecular Microbiology.
References
- Altschul, S.F. , Gish, W., Miller, W., Myers, E.W., and Lipman, D.J. (1990) Basic local alignment search tool. J Mol Biol 215: 403–410. [DOI] [PubMed] [Google Scholar]
- Ambrose, K.V. , Tian, Z., Wang, Y., Smith, J., Zylstra, G., Huang, B., and Belanger, F.C. (2015) Functional characterization of salicylate hydroxylase from the fungal endophyte Epichloe festucae . Sci Rep 5: 10939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anderson, J.J. , and Dagley, S. (1981) Catabolism of tryptophan, anthranilate, and 2,3‐dihydroxybenzoate in Trichosporon cutaneum . J Bacteriol 146: 291–297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benjamini, Y. , and Hochberg, Y. (1995) Controlling the false discovery rate: a practical and powerful approach to multiple testing. J R Stat Soc B 57: 289–300. [Google Scholar]
- Bolger, A.M. , Lohse, M., and Usadel, B. (2014) Trimmomatic: a flexible trimmer for illumina sequence data. Bioinformatics 30: 2114–2120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brameier, M. , Krings, A., and MacCallum, R.M. (2007) NucPred–predicting nuclear localization of proteins. Bioinformatics 23: 1159–1160. [DOI] [PubMed] [Google Scholar]
- Brunoud, G. , Wells, D.M., Oliva, M., Larrieu, A., Mirabet, V., Burrow, A.H., Beeckman, T., Kepinski, S., et al (2012) A novel sensor to map auxin response and distribution at high spatio‐temporal resolution. Nature 482: 103–106. [DOI] [PubMed] [Google Scholar]
- Carvajal, E. , van den Hazel, H.B., Cybularz‐Kolaczkowska, A., Balzi, E., and Goffeau, A. (1997) Molecular and phenotypic characterization of yeast PDR1 mutants that show hyperactive transcription of various ABC multidrug transporter genes. Mol Gen Genet 256: 406–415. [DOI] [PubMed] [Google Scholar]
- Chen, Z. , and Klessig, D.F. (1991) Identification of a soluble salicylic acid‐binding protein that may function in signal transduction in the plant disease‐resistance response. Proc Natl Acad Sci USA 88: 8179–8183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christensen, J.J. 1963. Corn Smut Caused by Ustilago maydis. St Paul, MN: American Phytopathological Society; Monograph no. 2. [Google Scholar]
- Delahodde, A. , Delaveau, T., and Jacq, C. (1995) Positive autoregulation of the yeast transcription factor Pdr3p, which is involved in control of drug resistance. Mol Cell Biol 15: 4043–4051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dempsey, D.A. , Vlot, A.C., Wildermuth, M.C., and Klessig, D.F. (2011) Salicylic acid biosynthesis and metabolism. Arabidopsis Book 9: e0156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DePristo, M.A. , Banks, E., Poplin, R., Garimella, K.V., Maguire, J.R., Hartl, C., Philippakis, A.A., del Angel, G., et al (2011) A framework for variation discovery and genotyping using next‐generation DNA sequencing data. Nat Genet 43: 491–498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Stasio, M. , Brefort, T., Mendoza‐Mendoza, A., Munch, K., and Kahmann, R. (2009) The dual specificity phosphatase Rok1 negatively regulates mating and pathogenicity in Ustilago maydis . Mol Microbiol 73: 73–88. [DOI] [PubMed] [Google Scholar]
- Djamei, A. , Schipper, K., Rabe, F., Ghosh, A., Vincon, V., Kahnt, J., Osorio, S., Tohge, T., et al (2011) Metabolic priming by a secreted fungal effector. Nature 478: 395–398. [DOI] [PubMed] [Google Scholar]
- Doehlemann, G. , Wahl, R., Horst, R.J., Voll, L.M., Usadel, B., Poree, F., Stitt, M., Pons‐Kuhnemann, J., et al (2008) Reprogramming a maize plant: transcriptional and metabolic changes induced by the fungal biotroph Ustilago maydis . Plant J 56: 181–195. [DOI] [PubMed] [Google Scholar]
- Du, H. , and Klessig, D.F. (1997) Identification of a soluble, high‐affinity salicylic acid‐binding protein in tobacco. Plant Physiol 113: 1319–1327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Durner, J. , and Klessig, D.F. (1995) Inhibition of ascorbate peroxidase by salicylic acid and 2,6‐dichloroisonicotinic acid, two inducers of plant defense responses. Proc Natl Acad Sci USA 92: 11312–11316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Forouhar, F. , Yang, Y., Kumar, D., Chen, Y., Fridman, E., Park, S.W., Chiang, Y., Acton, T.B., et al (2005) Structural and biochemical studies identify tobacco SABP2 as a methyl salicylate esterase and implicate it in plant innate immunity. Proc Natl Acad Sci USA 102: 1773–1778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu, Z.Q. and Dong, X. (2013) Systemic acquired resistance: turning local infection into global defense. Annu Rev Plant Biol 64: 839–863. [DOI] [PubMed] [Google Scholar]
- Fu, Z.Q. , Yan, S., Saleh, A., Wang, W., Ruble, J., Oka, N., Mohan, R., Spoel, S.H., et al (2012) NPR3 and NPR4 are receptors for the immune signal salicylic acid in plants. Nature 486: 228–232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glazebrook, J. (2005) Contrasting mechanisms of defense against biotrophic and necrotrophic pathogens. Annu Rev Phytopathol 43: 205–227. [DOI] [PubMed] [Google Scholar]
- Hellauer, K. , Rochon, M.H., and Turcotte, B. (1996) A novel DNA binding motif for yeast zinc cluster proteins: the Leu3p and Pdr3p transcriptional activators recognize everted repeats. Mol Cell Biol 16: 6096–6102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hemetsberger, C. , Herrberger, C., Zechmann, B., Hillmer, M., and Doehlemann, G. (2012) The Ustilago maydis effector Pep1 suppresses plant immunity by inhibition of host peroxidase activity. PLoS Pathog 8: e1002684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoffman, C.S. , and Winston, F. (1987) A ten‐minute DNA preparation from yeast efficiently releases autonomous plasmids for transformation of Escherichia coli . Gene 57: 267–272. [DOI] [PubMed] [Google Scholar]
- Horst, R.J. , Doehlemann, G., Wahl, R., Hofmann, J., Schmiedl, A., Kahmann, R., Kamper, J., Sonnewald, U., et al (2010) Ustilago maydis infection strongly alters organic nitrogen allocation in maize and stimulates productivity of systemic source leaves. Plant Physiol 152: 293–308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones, A.M. , Danielson, J.A., Manojkumar, S.N., Lanquar, V., Grossmann, G., and Frommer, W.B. (2014) Abscisic acid dynamics in roots detected with genetically encoded FRET sensors. Elife 3: e01741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamper, J. (2004) A PCR‐based system for highly efficient generation of gene replacement mutants in Ustilago maydis . Mol Genet Genomics 271: 103–110. [DOI] [PubMed] [Google Scholar]
- Kamper, J. , Kahmann, R., Bolker, M., Ma, L.J., Brefort, T., Saville, B.J., Banuett, F., Kronstad, J.W., et al (2006) Insights from the genome of the biotrophic fungal plant pathogen Ustilago maydis . Nature 444: 97–101. [DOI] [PubMed] [Google Scholar]
- Kerbarh, O. , Ciulli, A., Howard, N.I., and Abell, C. (2005) Salicylate biosynthesis: overexpression, purification, and characterization of Irp9, a bifunctional salicylate synthase from Yersinia enterocolitica . J Bacteriol 187: 5061–5066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar, D. , and Klessig, D.F. (2003) High‐affinity salicylic acid‐binding protein 2 is required for plant innate immunity and has salicylic acid‐stimulated lipase activity. Proc Natl Acad Sci USA 100: 16101–16106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lanver, D. , Berndt, P., Tollot, M., Naik, V., Vranes, M., Warmann, T., Munch, K., Rossel, N., et al (2014) Plant surface cues prime Ustilago maydis for biotrophic development. PLoS Pathog 10: e1004272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Letunic, I. , Doerks, T., and Bork, P. (2015) SMART: recent updates, new developments and status in 2015. Nucleic Acids Res 43: D257–D260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li, H. , and Durbin, R. (2009) Fast and accurate short read alignment with Burrows‐wheeler transform. Bioinformatics 25: 1754–1760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li, H. , Handsaker, B., Wysoker, A., Fennell, T., Ruan, J., Homer, N., Marth, G., Abecasis, G., et al (2009) The sequence alignment/map format and SAMtools. Bioinformatics 25: 2078–2079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Linning, R. , Lin, D., Lee, N., Abdennadher, M., Gaudet, D., Thomas, P., Mills, D., Kronstad, J.W., et al (2004) Marker‐based cloning of the region containing the UhAvr1 avirulence gene from the basidiomycete barley pathogen Ustilago hordei . Genetics 166: 99–111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu, T. , Song, T., Zhang, X., Yuan, H., Su, L., Li, W., Xu, J., Liu, S., et al (2014) Unconventionally secreted effectors of two filamentous pathogens target plant salicylate biosynthesis. Nat Commun 5: 4686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Livak, K.J. , and Schmittgen, T.D. (2001) Analysis of relative gene expression data using real‐time quantitative PCR and the 2(‐Delta Delta C(T)) method. Methods 25: 402–408. [DOI] [PubMed] [Google Scholar]
- Loubradou, G. , Brachmann, A., Feldbrugge, M., and Kahmann, R. (2001) A homologue of the transcriptional repressor Ssn6p antagonizes cAMP signalling in Ustilago maydis . Mol Microbiol 40: 719–730. [DOI] [PubMed] [Google Scholar]
- MacPherson, S. , Larochelle, M., and Turcotte, B. (2006) A fungal family of transcriptional regulators: the zinc cluster proteins. Microbiol Mol Biol Rev 70: 583–604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martins, T.M. , Hartmann, D.O., Planchon, S., Martins, I., Renaut, J., and Silva Pereira, C. (2015) The old 3‐oxoadipate pathway revisited: new insights in the catabolism of aromatics in the saprophytic fungus Aspergillus nidulans . Fungal Genet Biol 74: 32–44. [DOI] [PubMed] [Google Scholar]
- McCann, M.P. , and Snetselaar, K.M. (2008) A genome‐based analysis of amino acid metabolism in the biotrophic plant pathogen Ustilago maydis . Fungal Genet Biol 45 (Suppl.1): S77–S87. [DOI] [PubMed] [Google Scholar]
- Mueller, A.N. , Ziemann, S., Treitschke, S., Assmann, D., and Doehlemann, G. (2013) Compatibility in the Ustilago maydis‐maize interaction requires inhibition of host cysteine proteases by the fungal effector Pit2. PLoS Pathog 9: e1003177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naar, A.M. , and Thakur, J.K. (2009) Nuclear receptor‐like transcription factors in fungi. Genes Dev 23: 419–432. [DOI] [PubMed] [Google Scholar]
- Nourani, A. , Papajova, D., Delahodde, A., Jacq, C., and Subik, J. (1997) Clustered amino acid substitutions in the yeast transcription regulator Pdr3p increase pleiotropic drug resistance and identify a new central regulatory domain. Mol Gen Genet 256: 397–405. [DOI] [PubMed] [Google Scholar]
- Penn, C.D. , and Daniel, S.L. (2013) Salicylate degradation by the fungal plant pathogen Sclerotinia sclerotiorum . Curr Microbiol 67: 218–225. [DOI] [PubMed] [Google Scholar]
- Qi, P.F. , Johnston, A., Balcerzak, M., Rocheleau, H., Harris, L.J., Long, X.Y., Wei, Y.M., Zheng, Y.L., et al (2012) Effect of salicylic acid on Fusarium graminearum, the major causal agent of fusarium head blight in wheat. Fungal Biol 116: 413–426. [DOI] [PubMed] [Google Scholar]
- R Core Team . (2011). R: A language and environment for statistical computing. R Foundation for Statistical Computing, Vienna, Austria.
- Rabe, F. , Ajami‐Rashidi, Z., Doehlemann, G., Kahmann, R., and Djamei, A. (2013) Degradation of the plant defence hormone salicylic acid by the biotrophic fungus Ustilago maydis . Mol Microbiol 89: 179–188. [DOI] [PubMed] [Google Scholar]
- Rao, P.V. , Moore, K., and Towers, G.H. (1967) O‐pyrocatechiuc acid carboxy‐lyase from Aspergillus niger . Arch Biochem Biophys 122: 466–473. [DOI] [PubMed] [Google Scholar]
- Rao, P.V. , Sreeleela, N.S., Premakumar, R., and Vaidyanathan, C.S. (1971) Regulation of the pathway for the degradation of anthranilate in Aspergillus niger . J Bacteriol 107: 100–105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rice, P. , Longden, I., and Bleasby, A. (2000) EMBOSS: the European molecular biology open software suite. Trends Genet 16: 276–277. [DOI] [PubMed] [Google Scholar]
- Ritchie, M.E. , Phipson, B., Wu, D., Hu, Y., Law, C.W., Shi, W., and Smyth, G.K. (2015) Limma powers differential expression analyses for RNA‐sequencing and microarray studies. Nucleic Acids Res 43: e47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson, J.T. , Thorvaldsdottir, H., Winckler, W., Guttman, M., Lander, E.S., Getz, G., and Mesirov, J.P. (2011) Integrative genomics viewer. Nat Biotechnol 29: 24–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruepp, A. , Zollner, A., Maier, D., Albermann, K., Hani, J., Mokrejs, M., Tetko, I., Guldener, U., et al (2004) The FunCat, a functional annotation scheme for systematic classification of proteins from whole genomes. Nucleic Acids Res 32: 5539–5545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sambrook, J. , Frisch, E.F., and Maniatis, T. (1989) Molecular Cloning: A Laboratory Manual. New York: Cold Spring Harbour Laboratory Press, Cold Spring Harbour. [Google Scholar]
- Schell, M.A. , and Poser, E.F. (1989) Demonstration, characterization, and mutational analysis of NahR protein binding to nah and sal promoters. J Bacteriol 171: 837–846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schell, M.A. , Brown, P.H., and Raju, S. (1990) Use of saturation mutagenesis to localize probable functional domains in the NahR protein, a LysR‐type transcription activator. J Biol Chem 265: 3844–3850. [PubMed] [Google Scholar]
- Schirawski, J. , Bohnert, H.U., Steinberg, G., Snetselaar, K., Adamikowa, L., and Kahmann, R. (2005a) Endoplasmic reticulum glucosidase II is required for pathogenicity of Ustilago maydis . Plant Cell 17: 3532–3543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schirawski, J. , Heinze, B., Wagenknecht, M., and Kahmann, R. (2005b) Mating type loci of Sporisorium reilianum: novel pattern with three a and multiple b specificities. Eukaryot Cell 4: 1317–1327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schuler, D. , Wahl, R., Wippel, K., Vranes, M., Munsterkotter, M., Sauer, N., and Kamper, J. (2015) Hxt1, a monosaccharide transporter and sensor required for virulence of the maize pathogen Ustilago maydis . New Phytol 206: 1086–1100. [DOI] [PubMed] [Google Scholar]
- Schultz, J. , Milpetz, F., Bork, P., and Ponting, C.P. (1998) SMART, a simple modular architecture research tool: identification of signaling domains. Proc Natl Acad Sci USA 95: 5857–5864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Serino, L. , Reimmann, C., Baur, H., Beyeler, M., Visca, P., and Haas, D. (1995) Structural genes for salicylate biosynthesis from chorismate in Pseudomonas aeruginosa . Mol Gen Genet 249: 217–228. [DOI] [PubMed] [Google Scholar]
- Seyfferth, C. , and Tsuda, K. (2014) Salicylic acid signal transduction: the initiation of biosynthesis, perception and transcriptional reprogramming. Front Plant Sci 5: 697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shelest, E. (2008) Transcription factors in fungi. FEMS Microbiol Lett 286: 145–151. [DOI] [PubMed] [Google Scholar]
- Solomon, P.S. , Tan, K.C., and Oliver, R.P. (2003) The nutrient supply of pathogenic fungi; a fertile field for study. Mol Plant Pathol 4: 203–210. [DOI] [PubMed] [Google Scholar]
- Struck, C. (2015) Amino acid uptake in rust fungi. Front Plant Sci 6: 40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sze, I.S. , and Dagley, S. (1984) Properties of salicylate hydroxylase and hydroxyquinol 1,2‐dioxygenase purified from Trichosporon cutaneum . J Bacteriol 159: 353–359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanaka, S. , Han, X., and Kahmann, R. (2015) Microbial effectors target multiple steps in the salicylic acid production and signaling pathway. Front Plant Sci 6: 349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ternes, C.M. , and Schonknecht, G. (2014) Gene transfers shaped the evolution of de novo NAD+ biosynthesis in eukaryotes. Genome Biol Evol 6: 2335–2349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thakur, J.K. , Arthanari, H., Yang, F., Pan, S.J., Fan, X., Breger, J., Frueh, D.P., Gulshan, K., et al (2008) A nuclear receptor‐like pathway regulating multidrug resistance in fungi. Nature 452: 604–609. [DOI] [PubMed] [Google Scholar]
- Thorvaldsdottir, H. , Robinson, J.T., and Mesirov, J.P. (2013) Integrative genomics viewer (IGV): high‐performance genomics data visualization and exploration. Brief Bioinform 14: 178–192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Todd, R.B. , and Andrianopoulos, A. (1997) Evolution of a fungal regulatory gene family: the Zn(II)2Cys6 binuclear cluster DNA binding motif. Fungal Genet Biol 21: 388–405. [DOI] [PubMed] [Google Scholar]
- Tsukuda, T. , Carleton, S., Fotheringham, S., and Holloman, W.K. (1988) Isolation and characterization of an autonomously replicating sequence from Ustilago maydis . Mol Cell Biol 8: 3703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ustun, S. , Bartetzko, V., and Bornke, F. (2013) The Xanthomonas campestris type III effector XopJ targets the host cell proteasome to suppress salicylic‐acid mediated plant defence. PLoS Pathog 9: e1003427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Visca, P. , Ciervo, A., Sanfilippo, V., and Orsi, N. (1993) Iron‐regulated salicylate synthesis by Pseudomonas spp. J Gen Microbiol 139: 1995–2001. [DOI] [PubMed] [Google Scholar]
- Vlot, A.C. , Dempsey, D.A., and Klessig, D.F. (2009) Salicylic acid, a multifaceted hormone to combat disease. Annu Rev Phytopathol 47: 177–206. [DOI] [PubMed] [Google Scholar]
- Waadt, R. , Hitomi, K., Nishimura, N., Hitomi, C., Adams, S.R., Getzoff, E.D., and Schroeder, J.I. (2014) FRET‐based reporters for the direct visualization of abscisic acid concentration changes and distribution in Arabidopsis. Elife 3: e01739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wahl, R. , Wippel, K., Goos, S., Kamper, J., and Sauer, N. (2010) A novel high‐affinity sucrose transporter is required for virulence of the plant pathogen Ustilago maydis . PLoS Biol 8: e1000303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weinzierl, G. (2001) Isolierung und Charakterisierung von Komponenten der b‐vermittelten Regulationskaskade in Ustilago maydis. Philipps‐Universität Marburg, Marburg. [Google Scholar]
- Wu, Y. , Zhang, D., Chu, J.Y., Boyle, P., Wang, Y., Brindle, I.D., De Luca, V., and Despres, C. (2012) The Arabidopsis NPR1 protein is a receptor for the plant defense hormone salicylic acid. Cell Rep 1: 639–647. [DOI] [PubMed] [Google Scholar]
- Yamamoto, S. , Katagiri, M., Maeno, H., and Hayaishi, O. (1965) Salicylate hydroxylase, a monooxygenase requiring flavin adenine dinucleotide. I. Purification and general properties. J Biol Chem 240: 3408–3413. [PubMed] [Google Scholar]
- Zhang, X. , Cui, Z., Miyakawa, T., and Moye‐Rowley, W.S. (2001) Cross‐talk between transcriptional regulators of multidrug resistance in Saccharomyces cerevisiae . J Biol Chem 276: 8812–8819. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting Information