

Abstract
Purpose
To identify genetic mutations in three families with early onset high myopia (eoHM) limited to female members.
Methods
Genomic DNA was collected from participating members of families XF1, XF2, and XF3. Genome-wide linkage scans were performed on the largest family (XF1). Whole exome sequencing was performed on seven samples, including five samples (four affected and one unaffected) from family XF1, as well as the two probands from family XF2 and XF3. Variants were analyzed with multistep bioinformatics analyses. Sanger-dideoxy sequencing was used to verify candidate variations in families and controls.
Results
The genome-wide linkage scans performed on family XF1 detected a candidate locus on chromosome Xp11.1-Xq13.3 with a maximum logarithm of the odds (LOD) score of 2.48 and 3.01 for markers DXS991 and DXS986, respectively. Parallel whole exome sequencing identified a novel c.893C>A (p.Ala298Asp) mutation in ARR3 located on Xq13.1 in family XF1, which was shared by all four affected individuals but not the unaffected individual. Two other novel mutations in ARR3, c.298C>T (p.Arg100*) and c.239T>C (p.Leu80Pro), were detected in families XF2 and XF3, respectively. These mutations were predicted to be damaging and were not present in the normal controls and existing databases. All three mutations cosegregated with eoHM in each of the three families, in which all heterozygous female members are affected whereas all hemizygous male family members are not affected. Transmission of the mutations and eoHM in the three families demonstrates an unusual pattern of X-linked female-limited inheritance.
Conclusions
These data suggest that heterozygous mutations in ARR3 might be responsible for X-linked female-limited eoHM in the three families, a pattern contrary to the standard X-linked recessive trait. To our knowledge, eoHM is the first human disease associated with mutations in ARR3 and the second X-linked female-limited disease identified thus far. Identification of ARR3 associated with X-linked female-limited trait provides not only additional evidence of this unusual hereditary pattern but also an additional model for investigating the molecular mechanism responsible for female-limited phenotypes.
Introduction
Some genetic characteristics are more commonly seen in men while others are seen more commonly in women. Many such characteristics are determined by genes located on the X or Y chromosome, that is, sex-linked traits. However, sex-limited traits may be present in men or women alone despite the same genotype in male and female individuals, such as breast cancer and prostate cancer [1,2]. Sex-limited traits outside sex-specific organs are rare [3-7], such as male-limited precocious puberty associated with mutations in the LHCGR gene (OMIM 152790) [4] and female-limited epilepsy and cognitive impairment associated with mutations in PCDH19 (OMIM 300460) [5-7].
Degenerative changes in the retina associated with high myopia have become one of the most common causes of irreversible blindness [8-11]. Mendelian and complex modes of inheritance have been suggested for high myopia [12-21]. Early onset high myopia (eoHM) [22], with minimum influence of the environment and different clinical characteristics, is a unique resource for the identification of genes responsible for high myopia [23-26]. During a genetic study on high myopia, we examined three large families with eoHM limited to female family members but without any affected male family members. Transmission of eoHM in the families demonstrates an unusual pattern of inheritance, which could hardly be explained by traditional X-linked traits or by sex-limited traits in sex-specific organs. Based on a genome-wide linkage scan and whole exome sequencing, novel mutations in ARR3 (Gene ID: 407; OMIM 301770) are responsible for X-linked female-limited eoHM in the three families, the first human disease associated with ARR3 and the second X-linked female-limited disease identified thus far.
Methods
Three large families with eoHM limited to female family members and with no affected male family members were examined during a genetic study on eoHM (Figure 1, Figure 2, and Figure 3). Written informed consent in accordance with the tenets of the Declaration of Helsinki was obtained from the participants or their guardians. This study was approved by the institutional review board of the Zhongshan Ophthalmic Center. Venous blood for genomic DNA preparation was collected from 30 (15 affected; Figure 1), 12 (10 affected; Figure 2), and eight (four affected; Figure 3) individuals in the three families, respectively. All affected female family members had significant nearsightedness in early childhood and demonstrated typical tigroid fundus changes commonly seen in early onset high myopia (Figure 4). Refractive errors were measured with retinoscopy after mydriasis. eoHM was defined as axial length greater than 26.00 mm or spherical refraction in each meridian equal to or greater than −6.00 diopter in both eyes developed before the age of 7 years, with the exclusion of other known ocular or related systemic diseases.
Figure 1.

Family XF1 demonstrating haplotypes around ARR3 and mutation segregation with eoHM. Filled circles represent female family members affected with early onset high myopia (eoHM). V:12 is the proband. The novel c.893C>A (p.Ala298Asp) mutation in ARR3 was present in 15 female patients examined in this family but not in unaffected female family members. Two male family members with mutations (III:1 and III:3) and one obligate male carrier (II:3) did not have eoHM. M: mutation; +: Normal allele.
Figure 2.

Family XF2 demonstrating haplotypes around ARR3 and mutation segregation with eoHM. Filled circles represent female family members affected with early onset high myopia (eoHM). III:10 was the proband. The novel c.298C>T (p.Arg100*) mutation in ARR3 was present in all ten female patients examined. M: mutation; +: Normal allele.
Figure 3.

Family XF3 demonstrating haplotypes around ARR3 and mutation segregation with eoHM. Filled circles represent female family members affected with early onset high myopia (eoHM). IV:1 is the proband. The novel c.239T>C (p.Leu80Pro) mutation in ARR3 was present in all four female patients examined. One male family member with the mutation (V:1) did not have eoHM. Except the mutation, genotyping information for microsatellite markers around ARR3 was not available for V:2 because she was recently added. M: mutation; +: Normal allele.
Figure 4.
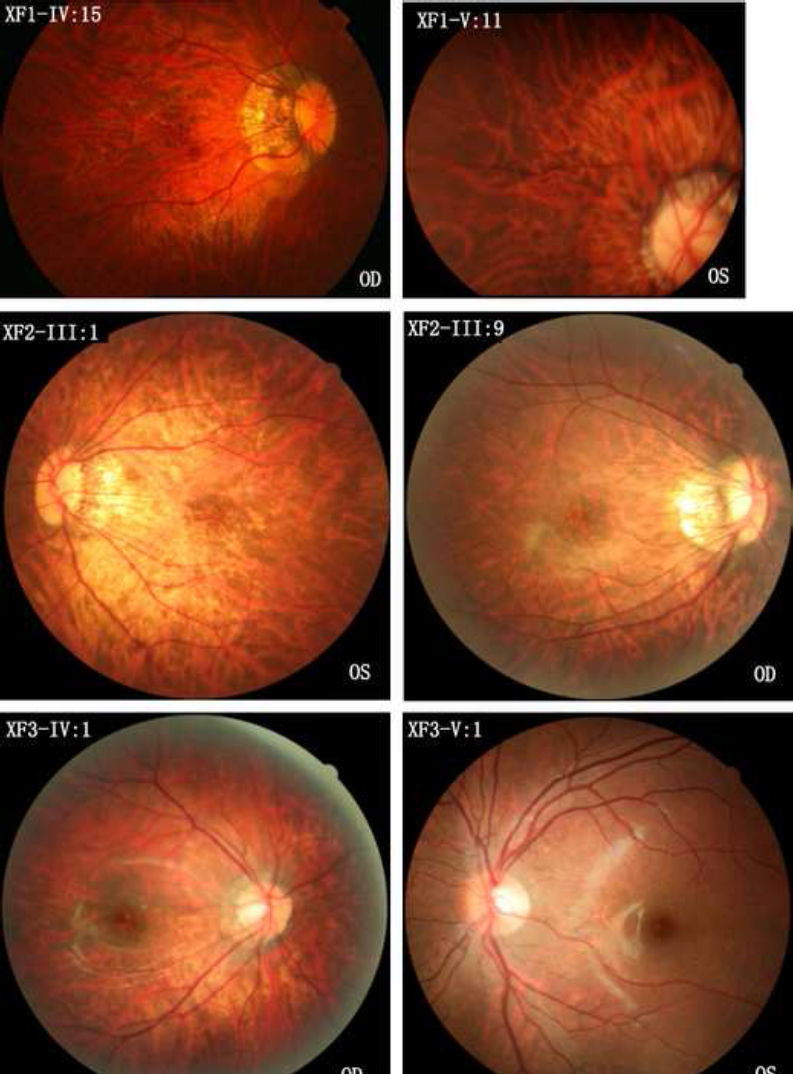
Fundus photographs of female patients with eoHM and different heterozygous mutations in ARR3 and an unaffected male family member with a hemizygous mutation in ARR3. The top two photographs are from family members IV:15 and V:11 of family XF1, respectively. Both have the heterozygous c.893C>A mutation in ARR3. The middle two photographs are from family members III:1 and III:9 of family XF2, respectively. Both have the heterozygous c.298C>T mutation in ARR3. The lower two photographs are from family members IV:1 and V:1 of family XF3, respectively, in which the female patient (IV:1) has the heterozygous c.239T>C mutation in ARR3 and has early onset high myopia (eoHM), but the male family member (V:1) has the hemizygous mutation in ARR3 without eoHM. All five female patients (XF1-IV:15, XF1-V:11, XF2-III:1, XF2-III:9, and XF3-IV:1) with heterozygous mutations in ARR3 demonstrated a temporal crescent of the optic nerve head and tigroid appearance of the posterior retina. However, XF3-V:1 is a 6-year-old boy with a hemizygous mutation in ARR3 who did not have eoHM. OD: right eye. OS: left eye.
A genome-wide linkage scan on family XF1 was performed using panels 1–28 of the ABI PRISM linkage Mapping Set Version 2, which includes 400 markers spaced at intervals of about 10 cM. Genotyping for all participating family members was performed using 5′-fluorescently labeled microsatellite markers, as previously described [27]. Two-point linkage analysis was performed using the MLINK program of the FASTLINK implementation of the LINKAGE program package [28,29]. The eoHM in the family was analyzed as an autosomal dominant trait with incomplete penetrance for panels 1–27 markers or as X-linked inheritance limited to female family members for panel 28 markers.
Whole exome sequencing was performed on genomic DNA from five (four affected and one unaffected) individuals of family XF1, one affected individual of family XF2, and one affected individual of family XF3, using a commercial service from Macrogen, as described in our previous study [25,30]. Variants detected were initially filtered with multistep bioinformatics analyses, as described in our previous study [25,30]. Then, variants shared by four affected individuals but not the unaffected individual in family XF1 were selected. Potential variants in the other two families were also analyzed. Candidate variants were also filtered by comparing with existing databases, including HGMD, EVS, and ExAC. The possible impact of missense changes was predicted by using the SIFT [31] and PolyPhen-2 [32] online tools. Sanger-dideoxy sequencing was used to confirm potential causative variants and to validate their cosegregation in family members, as well as the novelty of the variants by analyzing controls.
Results
Genome-wide linkage scan on family XF1 resulted in logarithm of the odds (LOD) scores higher than 1.5 in only four markers: 1.54 for marker D2S206, 1.67 for D9S285, 2.48 for DXS991, and 3.01 for DXS986, suggesting a candidate locus on Xp11.1-Xq13.3 for eoHM in family XF1 (Figure 1).
Parallel whole exome sequencing identified only one novel candidate variant shared by four affected individuals (IV:4, IV:13, IV:15, and VI:1) but absent in the unaffected female member (VI:3) in family XF1 (Figure 1), that is, the c.893C>A (p.Ala298Asp) mutation in the AAR3 gene located at Xq13.1, a region within the linkage interval. Analyzing whole exome sequencing data of the two probands (III:10 in family XF2 and IV:1 in family XF3) from the other two families identified other novel mutations in ARR3: c.298C>T (p.Arg100*) and c.239T>C (p.Leu80Pro; Figure 2 and Figure 3), respectively. These three mutations were confirmed with Sanger sequencing (Figure 5). Analysis of these mutations in available family members showed complete segregation of heterozygous mutations with affected female family members, in which unaffected female family members did not harbor the mutation (Figure 1, Figure 2, and Figure 3). The most striking phenomenon is that hemizygous male family members were not affected. The p.Ala298Asp mutation was predicted to be damaging with a SIFT score of zero and probably damaging with a PolyPhen2 score of 1.0. The p.Arg100* mutation would result in truncation of most of the 388 residues but is more likely to be a null allele due to nonsense-mediated decay. The p.Leu80Pro mutation was predicted to be possibly damaging with a PolyPhen2 score of 0.523 but tolerated with a SIFT score of 0.07. All three mutations were not present in 192 normal controls (263 X chromosomes) or in existing databases (HGMD, EVS, ExAC, and 1000G). An analysis of the whole exome data of patients from these three families did not identify mutations in genes responsible for other forms of syndromic or nonsyndromic high myopia or in genes associated with other known retinal diseases [23,24].
Figure 5.

Sequence chromatography from Sanger sequencing. Mutations in ARR3 identified with whole exome sequencing were further confirmed with Sanger sequencing and then validated in family members. The sequence with heterozygous mutations detected in female patients with early onset high myopia (eoHM) are shown on the left, whereas the corresponding normal sequences from controls are shown on the right. Arrows indicate the sites with and without mutations in the patients and controls, respectively.
Discussion
In this study, female-limited eoHM was mapped to a novel locus on Xp11.1-Xq13.3 and was associated with novel mutations in ARR3. This association is supported by linkage mapping, identification of novel mutations in ARR3 in three families, segregation of heterozygous mutations with eoHM in the families, absence of the mutations in controls and existing databases, absence of known disease with ARR3, retinal-specific and highly enriched expression of ARR3, and exclusion of other potential mutations in the whole genome. To our knowledge, female-limited eoHM is the first human disease associated with mutations in ARR3 and is the second X-linked female-limited disease identified thus far.
The patterns of disease transmission in the three families with eoHM with mutations in ARR3 is highly likely to be X-linked female-limited [33]. Such an unusual pattern of inheritance has been rarely reported, except epilepsy and mental retardation limited to women (EFMR) that is caused by mutations in the PCDH19 gene located at chromosome X [5,6,34], where female family members with heterozygous mutations are affected while hemizygous male family members are spared, a pattern contrary to the standard X-linked recessive trait. In addition, a similar but slight different pattern was also observed in ephrin-B1 (EFNB1, OMIM 300035)-related craniofrontonasal syndrome, where female family members are affected while male family members had no or only mild abnormalities [35,36]. In this unusual pattern of inheritance, it is unclear why heterozygous female family members are affected while hemizygous male family members are not affected. It has been postulated that an alternative pathway may compensate the complete loss of the functional products encoded by the mutant gene. In the heterozygous female family members, however, random inactivation of one X chromosome may create mosaic cells that express either a normal or mutant gene so that these two types of cells may behave differently in cell interaction, migration, connection, metabolism, or even signal transmission [7,35,37-40]. Uncompromised behavior of these two types of cells may be harmful in development or in performing their natural function [6]. This proposed mechanism might also explain the unusual pattern of inheritance seen in families with eoHM with mutations in ARR3. Although diseases with X-linked female-limited inheritance are rare thus far, this mechanism may represent a special molecular pathological mechanism for a new class of diseases. This mechanism may be increasing recognized in other diseases of unknown causes if it can be investigated further, especially in those with developmental anomalies or functional abnormalities of the neurosystems.
ARR3, located at Xq13.1 with 17 coding exons, encodes cone arrestin with retina-specific and retina-enriched expression [41-43]. ARR3 has been speculated to play a role in as-yet undefined retina-specific signal transduction [44,45]. To date, mutations in ARR3 have not been associated with any human disease. Based on a study of Arr4 (the ortholog of human ARR3) knockout mice [46], 2-month-old Arr4-null mice had diminished visual acuity and contrast sensitivity but higher b-wave amplitudes, while 7-month-old Arr4-null mice had significantly reduced a-wave amplitudes compared with normal controls. The older Arr4-null mice had reduced cone numbers and cone opsin expression with normal thickness of the outer nuclear layer, suggesting a model of age-related cone dystrophy [46]. These data from Arr4-null mice suggest the involvement of cone arrestin in the structural and functional circuit of cones but do not explain why eoHM was present in heterozygous female family members but not in hemizygous male family members. Phenotypic differences in mice and humans have been observed in other genes, such as a null mutation in LOXL3 (OMIM 607163) that causes embryonic lethality in mice [15] but eoHM in human beings [26]. In addition, mutations in genes with encoded products that participate in the cone signal pathway have previously been reported in patients with eoHM, such as those in OPN1LW (OMIM 300822) [21,24] and NYX (OMIM 300278) [47,48]. Nevertheless, our results suggest that ARR3 plays important role in cone-related function. Further study of ARR3 in suitable knockout animal models, such as the rat or even the monkey, may help to elucidate the underlying molecular mechanism related to female-limited expression of mutations in X-linked genes.
Acknowledgments
The authors are grateful to the families for their participation. This study was supported by grants from National Natural Science Foundation of China (U1201221 and 31371276), Natural Science Foundation of Guangdong Province (S2013030012978), and the Fundamental Research Funds of the State Key Laboratory of Ophthalmology.
References
- 1.Maxwell KN, Domchek SM. Cancer treatment according to BRCA1 and BRCA2 mutations. Nat Rev Clin Oncol. 2012;9:520–8. doi: 10.1038/nrclinonc.2012.123. https://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&list_uids=22825375&dopt=Abstract [DOI] [PubMed] [Google Scholar]
- 2.Attard G, Parker C, Eeles RA, Schroder F, Tomlins SA, Tannock I, Drake CG, de Bono JS. Prostate cancer. Lancet. 2016;387:70–82. doi: 10.1016/S0140-6736(14)61947-4. https://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&list_uids=26074382&dopt=Abstract [DOI] [PubMed] [Google Scholar]
- 3.Ryan SG, Chance PF, Zou CH, Spinner NB, Golden JA, Smietana S. Epilepsy and mental retardation limited to females: an X-linked dominant disorder with male sparing. Nat Genet. 1997;17:92–5. doi: 10.1038/ng0997-92. https://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&list_uids=9288105&dopt=Abstract [DOI] [PubMed] [Google Scholar]
- 4.Shenker A, Laue L, Kosugi S, Merendino JJ, Jr, Minegishi T, Cutler GB., Jr A constitutively activating mutation of the luteinizing hormone receptor in familial male precocious puberty. Nature. 1993;365:652–4. doi: 10.1038/365652a0. https://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&list_uids=7692306&dopt=Abstract [DOI] [PubMed] [Google Scholar]
- 5.Dibbens LM, Tarpey PS, Hynes K, Bayly MA, Scheffer IE, Smith R, Bomar J, Sutton E, Vandeleur L, Shoubridge C, Edkins S, Turner SJ, Stevens C, O’Meara S, Tofts C, Barthorpe S, Buck G, Cole J, Halliday K, Jones D, Lee R, Madison M, Mironenko T, Varian J, West S, Widaa S, Wray P, Teague J, Dicks E, Butler A, Menzies A, Jenkinson A, Shepherd R, Gusella JF, Afawi Z, Mazarib A, Neufeld MY, Kivity S, Lev D, Lerman-Sagie T, Korczyn AD, Derry CP, Sutherland GR, Friend K, Shaw M, Corbett M, Kim HG, Geschwind DH, Thomas P, Haan E, Ryan S, McKee S, Berkovic SF, Futreal PA, Stratton MR, Mulley JC, Gecz J. X-linked protocadherin 19 mutations cause female-limited epilepsy and cognitive impairment. Nat Genet. 2008;40:776–81. doi: 10.1038/ng.149. https://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&list_uids=18469813&dopt=Abstract [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Duszyc K, Terczynska I, Hoffman-Zacharska D. Epilepsy and mental retardation restricted to females: X-linked epileptic infantile encephalopathy of unusual inheritance. J Appl Genet. 2015;56:49–56. doi: 10.1007/s13353-014-0243-8. https://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&list_uids=25204757&dopt=Abstract [DOI] [PubMed] [Google Scholar]
- 7.Depienne C, Bouteiller D, Keren B, Cheuret E, Poirier K, Trouillard O, Benyahia B, Quelin C, Carpentier W, Julia S, Afenjar A, Gautier A, Rivier F, Meyer S, Berquin P, Helias M, Py I, Rivera S, Bahi-Buisson N, Gourfinkel-An I, Cazeneuve C, Ruberg M, Brice A, Nabbout R, Leguern E. Sporadic infantile epileptic encephalopathy caused by mutations in PCDH19 resembles Dravet syndrome but mainly affects females. PLoS Genet. 2009;5:e1000381. doi: 10.1371/journal.pgen.1000381. https://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&list_uids=19214208&dopt=Abstract [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Yamada M, Hiratsuka Y, Roberts CB, Pezzullo ML, Yates K, Takano S, Miyake K, Taylor HR. Prevalence of visual impairment in the adult Japanese population by cause and severity and future projections. Ophthalmic Epidemiol. 2010;17:50–7. doi: 10.3109/09286580903450346. https://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&list_uids=20100100&dopt=Abstract [DOI] [PubMed] [Google Scholar]
- 9.Xu L, Wang Y, Wang S, Wang Y, Jonas JB. High myopia and glaucoma susceptibility the Beijing Eye Study. Ophthalmology. 2007;114:216–20. doi: 10.1016/j.ophtha.2006.06.050. https://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&list_uids=17123613&dopt=Abstract [DOI] [PubMed] [Google Scholar]
- 10.Liang YB, Friedman DS, Wong TY, Zhan SY, Sun LP, Wang JJ, Duan XR, Yang XH, Wang FH, Zhou Q, Wang NL. Prevalence and causes of low vision and blindness in a rural chinese adult population: the Handan Eye Study. Ophthalmology. 2008;115:1965–72. doi: 10.1016/j.ophtha.2008.05.030. https://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&list_uids=18684506&dopt=Abstract [DOI] [PubMed] [Google Scholar]
- 11.McCarty CA, Taylor HR. Myopia and vision 2020. Am J Ophthalmol. 2000;129:525–7. doi: 10.1016/s0002-9394(99)00444-4. https://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&list_uids=10764864&dopt=Abstract [DOI] [PubMed] [Google Scholar]
- 12.Feldkamper M, Schaeffel F. Interactions of genes and environment in myopia. Dev Ophthalmol. 2003;37:34–49. doi: 10.1159/000072037. https://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&list_uids=12876828&dopt=Abstract [DOI] [PubMed] [Google Scholar]
- 13.Young TL. Molecular genetics of human myopia: an update. Optom Vis Sci. 2009;86:E8–22. doi: 10.1097/OPX.0b013e3181940655. https://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&list_uids=19104467&dopt=Abstract [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Young TL, Ronan SM, Drahozal LA, Wildenberg SC, Alvear AB, Oetting WS, Atwood LD, Wilkin DJ, King RA. Evidence that a locus for familial high myopia maps to chromosome 18p. Am J Hum Genet. 1998;63:109–19. doi: 10.1086/301907. https://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&list_uids=9634508&dopt=Abstract [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zhang Q. Genetics of Refraction and Myopia. Prog Mol Biol Transl Sci. 2015;134:269–79. doi: 10.1016/bs.pmbts.2015.05.007. https://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&list_uids=26310160&dopt=Abstract [DOI] [PubMed] [Google Scholar]
- 16.Fan Q, Barathi VA, Cheng CY, Zhou X, Meguro A, Nakata I, Khor CC, Goh LK, Li YJ, Lim W, Ho CE, Hawthorne F, Zheng Y, Chua D, Inoko H, Yamashiro K, Ohno-Matsui K, Matsuo K, Matsuda F, Vithana E, Seielstad M, Mizuki N, Beuerman RW, Tai ES, Yoshimura N, Aung T, Young TL, Wong TY, Teo YY, Saw SM. Genetic variants on chromosome 1q41 influence ocular axial length and high myopia. PLoS Genet. 2012;8:e1002753. doi: 10.1371/journal.pgen.1002753. https://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&list_uids=22685421&dopt=Abstract [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Meng W, Butterworth J, Bradley DT, Hughes AE, Soler V, Calvas P, Malecaze F. A genome-wide association study provides evidence for association of chromosome 8p23 (MYP10) and 10q21.1 (MYP15) with high myopia in the French Population. Invest Ophthalmol Vis Sci. 2012;53:7983–8. doi: 10.1167/iovs.12-10409. https://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&list_uids=23049088&dopt=Abstract [DOI] [PubMed] [Google Scholar]
- 18.Mordechai S, Gradstein L, Pasanen A, Ofir R, El Amour K, Levy J, Belfair N, Lifshitz T, Joshua S, Narkis G, Elbedour K, Myllyharju J, Birk OS. High myopia caused by a mutation in LEPREL1, encoding prolyl 3-hydroxylase 2. Am J Hum Genet. 2011;89:438–45. doi: 10.1016/j.ajhg.2011.08.003. https://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&list_uids=21885030&dopt=Abstract [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Shi Y, Qu J, Zhang D, Zhao P, Zhang Q, Tam PO, Sun L, Zuo X, Zhou X, Xiao X, Hu J, Li Y, Cai L, Liu X, Lu F, Liao S, Chen B, He F, Gong B, Lin H, Ma S, Cheng J, Zhang J, Chen Y, Zhao F, Yang X, Yang C, Lam DS, Li X, Shi F, Wu Z, Lin Y, Yang J, Li S, Ren Y, Xue A, Fan Y, Li D, Pang CP, Zhang X, Yang Z. Genetic variants at 13q12.12 are associated with high myopia in the Han Chinese population. Am J Hum Genet. 2011;88:805–13. doi: 10.1016/j.ajhg.2011.04.022. https://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&list_uids=21640322&dopt=Abstract [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Aldahmesh MA, Khan AO, Alkuraya H, Adly N, Anazi S, Al-Saleh AA, Mohamed JY, Hijazi H, Prabakaran S, Tacke M, Al-Khrashi A, Hashem M, Reinheckel T, Assiri A, Alkuraya FS. Mutations in LRPAP1 are associated with severe myopia in humans. Am J Hum Genet. 2013;93:313–20. doi: 10.1016/j.ajhg.2013.06.002. https://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&list_uids=23830514&dopt=Abstract [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.McClements M, Davies WI, Michaelides M, Young T, Neitz M, MacLaren RE, Moore AT, Hunt DM. Variations in opsin coding sequences cause x–linked cone dysfunction syndrome with myopia and dichromacy. Invest Ophthalmol Vis Sci. 2013;54:1361–9. doi: 10.1167/iovs.12-11156. https://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&list_uids=23322568&dopt=Abstract [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wang P, Xiao X, Huang L, Guo X, Zhang Q. Cone-rod dysfunction is a sign of early-onset high myopia. Optom Vis Sci. 2013;90:1327–30. doi: 10.1097/OPX.0000000000000072. https://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&list_uids=24100477&dopt=Abstract [DOI] [PubMed] [Google Scholar]
- 23.Sun W, Huang L, Xu Y, Xiao X, Li S, Jia X, Gao B, Wang P, Guo X, Zhang Q. Exome Sequencing on 298 Probands With Early-Onset High Myopia: Approximately One-Fourth Show Potential Pathogenic Mutations in RetNet Genes. Invest Ophthalmol Vis Sci. 2015;56:8365–72. doi: 10.1167/iovs.15-17555. https://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&list_uids=26747767&dopt=Abstract [DOI] [PubMed] [Google Scholar]
- 24.Li J, Gao B, Guan L, Xiao X, Zhang J, Li S, Jiang H, Jia X, Yang J, Guo X, Yin Y, Wang J, Zhang Q. Unique Variants in OPN1LW Cause Both Syndromic and Nonsyndromic X–Linked High Myopia Mapped to MYP1. Invest Ophthalmol Vis Sci. 2015;56:4150–5. doi: 10.1167/iovs.14-16356. https://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&list_uids=26114493&dopt=Abstract [DOI] [PubMed] [Google Scholar]
- 25.Jiang D, Li J, Xiao X, Li S, Jia X, Sun W, Guo X, Zhang Q. Detection of mutations in LRPAP1, CTSH, LEPREL1, ZNF644, SLC39A5, and SCO2 in 298 families with early-onset high myopia by exome sequencing. Invest Ophthalmol Vis Sci. 2014;56:339–45. doi: 10.1167/iovs.14-14850. https://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&list_uids=25525168&dopt=Abstract [DOI] [PubMed] [Google Scholar]
- 26.Li J, Gao B, Xiao X, Li S, Jia X, Sun W, Guo X, Zhang Q. Exome sequencing identified null mutations in LOXL3 associated with early-onset high myopia. Mol Vis. 2016;22:161–7. https://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&list_uids=26957899&dopt=Abstract [PMC free article] [PubMed] [Google Scholar]
- 27.Zhang Q, Zulfiqar F, Xiao X, Amer Riazuddin S, Ayyagari R, Sabar F, Caruso R, Sieving PA, Riazuddin S, Fielding Hejtmancik J. Severe autosomal recessive retinitis pigmentosa maps to chromosome 1p13.3-p21.2 between D1S2896 and D1S457 but outside ABCA4. Hum Genet. 2005;118:356–65. doi: 10.1007/s00439-005-0054-4. https://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&list_uids=16189710&dopt=Abstract [DOI] [PubMed] [Google Scholar]
- 28.Lathrop GM, Lalouel JM. Easy calculations of lod scores and genetic risks on small computers. Am J Hum Genet. 1984;36:460–5. https://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&list_uids=6585139&dopt=Abstract [PMC free article] [PubMed] [Google Scholar]
- 29.Schaffer AA, Gupta SK, Shriram K, Cottingham RW., Jr Avoiding recomputation in linkage analysis. Hum Hered. 1994;44:225–37. doi: 10.1159/000154222. https://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&list_uids=8056435&dopt=Abstract [DOI] [PubMed] [Google Scholar]
- 30.Li J, Jiang D, Xiao X, Li S, Jia X, Sun W, Guo X, Zhang Q. Evaluation of 12 myopia-associated genes in Chinese patients with high myopia. Invest Ophthalmol Vis Sci. 2015;56:722–9. doi: 10.1167/iovs.14-14880. https://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&list_uids=25587058&dopt=Abstract [DOI] [PubMed] [Google Scholar]
- 31.Kumar P, Henikoff S, Ng PC. Predicting the effects of coding non-synonymous variants on protein function using the SIFT algorithm. Nat Protoc. 2009;4:1073–81. doi: 10.1038/nprot.2009.86. https://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&list_uids=19561590&dopt=Abstract [DOI] [PubMed] [Google Scholar]
- 32.Flanagan SE, Patch AM, Ellard S. Using SIFT and PolyPhen to predict loss-of-function and gain-of-function mutations. Genet Test Mol Biomarkers. 2010;14:533–7. doi: 10.1089/gtmb.2010.0036. https://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&list_uids=20642364&dopt=Abstract [DOI] [PubMed] [Google Scholar]
- 33.Scheffer IE, Turner SJ, Dibbens LM, Bayly MA, Friend K, Hodgson B, Burrows L, Shaw M, Wei C, Ullmann R, Ropers HH, Szepetowski P, Haan E, Mazarib A, Afawi Z, Neufeld MY, Andrews PI, Wallace G, Kivity S, Lev D, Lerman-Sagie T, Derry CP, Korczyn AD, Gecz J, Mulley JC, Berkovic SF. Epilepsy and mental retardation limited to females: an under-recognized disorder. Brain. 2008;131:918–27. doi: 10.1093/brain/awm338. https://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&list_uids=18234694&dopt=Abstract [DOI] [PubMed] [Google Scholar]
- 34.Depienne C, LeGuern E. PCDH19-related infantile epileptic encephalopathy: an unusual X-linked inheritance disorder. Hum Mutat. 2012;33:627–34. doi: 10.1002/humu.22029. https://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&list_uids=22267240&dopt=Abstract [DOI] [PubMed] [Google Scholar]
- 35.Wieland I, Jakubiczka S, Muschke P, Cohen M, Thiele H, Gerlach KL, Adams RH, Wieacker P. Mutations of the ephrin-B1 gene cause craniofrontonasal syndrome. Am J Hum Genet. 2004;74:1209–15. doi: 10.1086/421532. https://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&list_uids=15124102&dopt=Abstract [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Twigg SR, Kan R, Babbs C, Bochukova EG, Robertson SP, Wall SA, Morriss-Kay GM, Wilkie AO. Mutations of ephrin-B1 (EFNB1), a marker of tissue boundary formation, cause craniofrontonasal syndrome. Proc Natl Acad Sci USA. 2004;101:8652–7. doi: 10.1073/pnas.0402819101. https://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&list_uids=15166289&dopt=Abstract [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Evers C, Jungwirth MS, Morgenthaler J, Hinderhofer K, Maas B, Janssen JW, Jauch A, Hehr U, Steinbeisser H, Moog U. Craniofrontonasal syndrome in a male due to chromosomal mosaicism involving EFNB1: further insights into a genetic paradox. Clin Genet. 2014;85:347–53. doi: 10.1111/cge.12171. https://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&list_uids=23614707&dopt=Abstract [DOI] [PubMed] [Google Scholar]
- 38.Twigg SR, Babbs C, van den Elzen ME, Goriely A, Taylor S, McGowan SJ, Giannoulatou E, Lonie L, Ragoussis J, Sadighi Akha E, Knight SJ, Zechi-Ceide RM, Hoogeboom JA, Pober BR, Toriello HV, Wall SA, Rita Passos-Bueno M, Brunner HG, Mathijssen IM, Wilkie AO. Cellular interference in craniofrontonasal syndrome: males mosaic for mutations in the X-linked EFNB1 gene are more severely affected than true hemizygotes. Hum Mol Genet. 2013;22:1654–62. doi: 10.1093/hmg/ddt015. https://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&list_uids=23335590&dopt=Abstract [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Bush JO, Soriano P. Ephrin-B1 forward signaling regulates craniofacial morphogenesis by controlling cell proliferation across Eph-ephrin boundaries. Genes Dev. 2010;24:2068–80. doi: 10.1101/gad.1963210. https://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&list_uids=20844017&dopt=Abstract [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Terracciano A, Trivisano M, Cusmai R, De Palma L, Fusco L, Compagnucci C, Bertini E, Vigevano F, Specchio N. PCDH19-related epilepsy in two mosaic male patients. Epilepsia. 2016;57:e51–5. doi: 10.1111/epi.13295. https://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&list_uids=26765483&dopt=Abstract [DOI] [PubMed] [Google Scholar]
- 41.Sakuma H, Murakami A, Fujimaki T, Inana G. Isolation and characterization of the human X-arrestin gene. Gene. 1998;224:87–95. doi: 10.1016/s0378-1119(98)00510-1. https://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&list_uids=9931451&dopt=Abstract [DOI] [PubMed] [Google Scholar]
- 42.Craft CM, Whitmore DH, Wiechmann AF. Cone arrestin identified by targeting expression of a functional family. J Biol Chem. 1994;269:4613–9. https://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&list_uids=8308033&dopt=Abstract [PubMed] [Google Scholar]
- 43.Sakuma H, Inana G, Murakami A, Higashide T, McLaren MJ. Immunolocalization of X-arrestin in human cone photoreceptors. FEBS Lett. 1996;382:105–10. doi: 10.1016/0014-5793(96)00163-9. https://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&list_uids=8612728&dopt=Abstract [DOI] [PubMed] [Google Scholar]
- 44.Murakami A, Yajima T, Sakuma H, McLaren MJ, Inana G. X-arrestin: a new retinal arrestin mapping to the X chromosome. FEBS Lett. 1993;334:203–9. doi: 10.1016/0014-5793(93)81712-9. https://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&list_uids=8224247&dopt=Abstract [DOI] [PubMed] [Google Scholar]
- 45.Maeda T, Ohguro H, Sohma H, Kuroki Y, Wada H, Okisaka S, Murakami A. Purification and characterization of bovine cone arrestin (cArr). FEBS Lett. 2000;470:336–40. doi: 10.1016/s0014-5793(00)01334-x. https://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&list_uids=10745092&dopt=Abstract [DOI] [PubMed] [Google Scholar]
- 46.Deming JD, Pak JS, Brown BM, Kim MK, Aung MH, Eom YS, Shin JA, Lee EJ, Pardue MT, Craft CM. Visual Cone Arrestin 4 Contributes to Visual Function and Cone Health. Invest Ophthalmol Vis Sci. 2015;56:5407–16. doi: 10.1167/iovs.15-16647. https://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&list_uids=26284544&dopt=Abstract [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Zhang Q, Xiao X, Li S, Jia X, Yang Z, Huang S, Caruso RC, Guan T, Sergeev Y, Guo X, Hejtmancik JF. Mutations in NYX of individuals with high myopia, but without night blindness. Mol Vis. 2007;13:330–6. https://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&list_uids=17392683&dopt=Abstract [PMC free article] [PubMed] [Google Scholar]
- 48.Yip SP, Li CC, Yiu WC, Hung WH, Lam WW, Lai MC, Ng PW, Fung WY, Chu PH, Jiang B, Chan HH, Yap MK. A novel missense mutation in the NYX gene associated with high myopia. Ophthalmic Physiol Opt. 2013;33:346–53. doi: 10.1111/opo.12036. https://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&list_uids=23406521&dopt=Abstract [DOI] [PubMed] [Google Scholar]