

Abstract
Purpose
To identify pathogenic variations in carbohydrate sulfotransferase 6 (CHST6) and transforming growth factor, beta-induced (TGFBI) genes in Turkish patients with corneal dystrophy (CD).
Methods
In this study, patients with macular corneal dystrophy (MCD; n = 18), granular corneal dystrophy type 1 (GCD1; n = 12), and lattice corneal dystrophy type 1 (LCD1; n = 4), as well as 50 healthy controls, were subjected to clinical and genetic examinations. The level of antigenic keratan sulfate (AgKS) in the serum samples of patients with MCD was determined with enzyme-linked immunosorbent assay (ELISA) to immunophenotypically subtype the patients as MCD type I and MCD type II. DNA was isolated from venous blood samples from the patients and controls. Variations were analyzed with DNA sequencing in the coding region of CHST6 in patients with MCD and exons 4 and 12 in TGFBI in patients with LCD1 and GCD1. Clinical characteristics and the detected variations were evaluated to determine any existing genotype–phenotype correlations.
Results
The previously reported R555W mutation in TGFBI was detected in 12 patients with GCD1, and the R124C mutation in TGFBI was detected in four patients with LCD1. Serum AgKS levels indicated that 12 patients with MCD were in subgroup I, and five patients with MCD were in subgroup II. No genetic variation was detected in the coding region of CHST6 for three patients with MCD type II. In other patients with MCD, three previously reported missense variations (c. 1A>T, c.738C>G, and c.631 C>T), three novel missense variations (c.164 T>C, c.526 G>A, c. 610 C>T), and two novel frameshift variations (c.894_895 insG and c. 462_463 delGC) were detected. These variations did not exist in the control chromosomes, 1000 Genomes, and dbSNP.
Conclusions
This is the first molecular analysis of TGFBI and CHST6 in Turkish patients with different types of CD. We detected previously reported, well-known hot spot mutations in TGFBI in the patients with GCD1 and LCD1. Eight likely pathogenic variations in CHST6, five of them novel, were reported in patients with MCD, which enlarges the mutational spectrum of MCD.
Introduction
Corneal dystrophies (CDs) are a group of hereditary disorders that affect one or several layers of the cornea and are usually bilateral, symmetric, and progressive. Affected patients generally suffer from recurrent erosions and/or progressive visual deterioration due to the increasing corneal opacity. Keratoplasty is still the most common treatment method when CD leads to important visual impairment [1]. Although CDs are relatively common in Turkey due to the high rate of consanguinity [2] and are among the most common indications of keratoplasty in Turkey [3], until now no comprehensive study has investigated genotype–phenotype properties of CDs.
During the past decade, important advances have been made in determining the genetic basis of CDs. According to the updated classification based on this increasing knowledge, CDs are divided into four groups: epithelial and subepithelial dystrophies, epithelial-stromal transforming growth factor, beta-induced (TGFBI; Gene ID: 7045, OMIM 601692) dystrophies, stromal dystrophies, and endothelial dystrophies [4]. The causative genes have been mapped, and the specific mutations are known for several types of CDs [5,6].
The carbohydrate sulfotransferase-6 (CHST6; Gene ID: 4166, OMIM 605294) gene, on chromosome 16q22, encodes enzyme N-acetylglucosamine-6-sulfotransferase (GlcNAc6ST) [5,7]. Mutations in CHST6 cause the deposition of low or unsulfated keratan sulfate (KS) in the corneal stroma [8] and result in autosomal recessively inherited macular corneal dystrophy (MCD, OMIM 217800). MCD causes bilateral, progressive corneal clouding and irregular corneal opacities [5]. MCD is the most common stromal CD in Iceland where the gene pool is small [9]. MCD is also common in countries such as Saudi Arabia and Turkey due to the high rate of consanguinity [2,10]. There are three subtypes of MCD (I, IA, and II) based on the absence or presence of antigenic keratan sulfate (AgKS) in the serum and corneal tissue [11]. Since Akama et al. identified CHST6 as a candidate gene for MCD in 2000, more than 150 likely pathogenic variations of this gene have been identified in patients with MCD from various populations [5,12]. CHST6 has been analyzed in many populations, and each study expanded the mutational spectrum of this gene. However, to date, no genetic analysis has been performed on Turkish patients with MCD.
TGFBI, located on chromosome 5q31, consists of 17 exons and encodes an extracellular matrix protein known as keratoepithelin (TGFBIp) [13]. Keratoepithelin is expressed in many tissues, including the corneal epithelium, but mutations in this gene lead to the production of abnormal keratoepithelin, which cannot be metabolized and causes progressive deposits detected only in the cornea [14,15]. TGFBI was identified as the causative gene of lattice corneal dystrophy type I (LCD1), variant lattice dystrophies, granular corneal dystrophy type I (GCD1), granular corneal dystrophy type II (GCD2), Thiel–Behnke corneal dystrophy (TBCD), and Reis–Bücklers corneal dystrophy (RBCD), which were recently classified in one group as epithelial-stromal TGFBI dystrophies [4]. More than 40 mutations in TGFBI that cause epithelial-stromal CDs have been identified thus far in patients from different countries [6,16]. There are two mutational hot spots, corresponding to arginine residues at positions 124 and 555 (located in exons 4 and 12 of TGFBI, respectively). Although there is an apparent genotype–phenotype correlation in this group of CDs, studies from different populations were useful to expand the mutational spectrum of this gene, to strengthen existing correlations, and to detect unique phenotypic variants. Only two studies have investigated Turkish patients, including two large GCD1 families that carry the p.R555W mutation [17,18]. In the present study, we conducted a clinical evaluation and a molecular genetic analysis of CHST6 and TGFBI in 34 Turkish probands with MCD, GCD1, and LCD1.
Methods
This study was approved by the Institutional Review Board of the Ankara Keçiören Training and Research Hospital, Turkey and adhered to the ARVO statement on human subjects. The study was performed according to the principles of the Declaration of Helsinki.
Patient selection and clinical evaluation
Thirty-four unrelated patients with CD, followed in the Department of Ophthalmology of Gazi Medical Faculty, were included in the study. 19 were male and 15 were female. The age of all participants ranged from 18 to 63 years old. A corneal specialist (F.A.) examined the patients and diagnosed 18 patients with MCD, 12 patients with GCD1, and four patients with LCD1 based on their typical clinical corneal features. Fifty unrelated healthy control individuals were also included in the study. Informed consent was obtained from all participants for clinical and molecular genetic studies. All subjects and controls underwent a comprehensive ocular examination, including best-corrected visual acuity (BCVA), intraocular pressure (IOP), slit-lamp examination, fundus examination, and measurement of central corneal thickness (CCT). Age, gender, ethnicity, family history in three generations, known consanguinity in families, age at clinical diagnosis, and previous therapies were recorded.
Determination of serum AgKS
The serum AgKS levels were determined in 18 patients with MCD and 50 controls with enzyme-linked immunosorbent assay (ELISA) using the Human Keratan Sulfate (KS) ELISA Kit (East Bio Pharm, Hangzhou, China) as previously described [19]. Serum AgKS levels that were 10 ng/ml or below were interpreted as MCD types I/IA and levels of 100 ng/ml or above as normal or MCD type II [20].
Mutational analysis
After informed consent was obtained, peripheral blood samples (5 ml) were obtained from each subject for DNA isolation and molecular analysis. DNA was extracted using the spin column–based nucleic acid purification method (MN Macherey-Nagel, Düren, Germany). The exon–intron boundaries in the neighbourhood of the coding region and the coding region of CHST6, as well as exons 4 and 12 of TGFBI, were amplified with PCR using the newly designed primers listed in Table 1. Each PCR was performed in a 50 μl reaction mixture containing genomic DNA (100 ng), primers (20 pmol/µl each), MgCl2 (25 mM), deoxynucleoside triphosphate (dNTPs; 2.5 mM), PCR buffer (Fermentas, Burlington, Canada), and Taq polymerase (0.5 IU/µl; Fermentas). The PCR protocol was as follows: initial denaturation at 94 °C for 5 min; 35 cycles of 94 °C for 30 s, 56 °C for 30 s, and 72 °C for 45 s; and final extension at 72 °C for 3 min. PCR products were purified using Wizard® SV Gel and PCR Clean-Up System kits (Promega, Madison, WI). Bidirectional sequencing was performed by using BigDye Terminator Mix, version 3.1 (Applied Biosystems Inc., Foster City, CA) and analyzed on an ABI-3100 genetic analyzer (Applied Biosystems). The chromatograms were analyzed with the ChromasPro software version 1.7.7 (Technelysium, South Brisbane, Australia). The pathogenicity of novel missense variations was evaluated with the SIFT and PolyPhen-2 software programs. Additionally, conservation of the involved amino acids among several sulfotransferases of human and mouse origins was evaluated using Clustal Omega. Human Genome Variation Society (HGVS) nomenclature was used for the description of all detected variations.
Table 1. Primer list of CHST6 and TGFBI genes.
| CHST6 | Sequences of primers (5′-3′) | Tm(°C) | PCR product |
|---|---|---|---|
|
Primer 1 |
F: CTCGGGTCTGGTGGTAGAATCT
R: TTGAAGAAGCGCACCTCCTTG |
60.9
61.1 |
658 bp |
|
Primer 2 |
F: CTCTTCCAGTGGGCCGTGAG
R: TTGAGCGCATTCCTGGACGAA |
62,5
62,6 |
626 bp |
|
Primer 3 |
F: GCAGAAATCCGTGCGCTCTAC
R: AGAGAAAGAAACGTGCAGTCCTT |
61,9
60,4 |
505 bp |
|
TGFBI |
Sequences of primers (5′-3′) |
Tm(°C) |
PCR product |
|
Exon 4 |
F: TCGTCCTCTCCACCTGTAGA
R: AACATGTTCTCAGCCCTCGT |
59,0
59,3 |
548 bp |
| Exon 12 | F: AACCAAGGTGTGTGCATTCC R: TTTAGTCCCGCCCACTCTTT | 59,0 59,0 | 415 bp |
Results
Clinical findings
Thirty-four Turkish patients with CD were included in the study. Patients were unrelated as we questioned their family history in three generations. Eighteen of the patients were clinically diagnosed with MCD (Figure 1A), 12 with GCD1 (Figure 1B), and four with LCD1 (Figure 1C). Demographic characteristics, family history, known consanguinity, clinical findings, and previous therapeutic procedures, as well as the genetic analysis results for TGFBI and CHST6, are summarized in Table 2 and Table 3, respectively. In all patients, the eyes were affected bilaterally. The CCT of seven patients with MCD without previous surgical procedures was measured with ultrasonic pachymetry, and the mean values were 456.4±23.6 µm (435–497 µm) in the right eye and 458.4±31.2 µm (430–520 µm) in the left eye.
Figure 1.

AS photographs of patients. A: Macular corneal dystrophy patient (patient 9). B: Patient with granular corneal dystrophy type 1 (patient 5). C: Patient with lattice corneal dystrophy type 1 (patient 2).
Table 2. Clinical findings of Turkish LCD1 and GCD1 patients carrying TGFBI variations.
| Patient no | Age/Sex | Diagnosis | Family history | Consanguinity | Age at diagnosis | BCVA (OD/OS) | Treatment | Variation |
|---|---|---|---|---|---|---|---|---|
| 1 |
28/F |
LCD1 |
+ |
- |
18 |
0.8/0.7 |
- |
R124C/- |
| 2 |
23/F |
LCD1 |
- |
- |
18 |
0.2/0.4 |
- |
R124C/- |
| 3 |
45/M |
LCD1 |
+ |
- |
24 |
0.05/0.4 |
OD ALK |
R124C/- |
| 4 |
40/M |
LCD1 |
- |
- |
20 |
0.4/0.5 |
OD-OS PTK |
R124C/- |
| 5 |
36/F |
GCD1 |
+ |
+ |
15 |
0.05/0.05 |
OD PPK |
R555W/- |
| 6 |
35/M |
GCD1 |
+ |
+ |
20 |
0.05/0.3 |
- |
R555W/- |
| 7 |
18/F |
GCD1 |
+ |
- |
10 |
0.9/0.8 |
- |
R555W/- |
| 8 |
53/M |
GCD1 |
+ |
- |
30 |
0.4/0.4 |
- |
R555W/- |
| 9 |
27/M |
GCD1 |
+ |
- |
20 |
0.05/0.05 |
OD-OS ALK |
R555W/R555W |
| 10 |
42/F |
GCD1 |
+ |
+ |
18 |
0.3/0.4 |
- |
R555W/- |
| 11 |
26/M |
GCD1 |
- |
- |
18 |
0.5/0.3 |
OS PTK |
R555W/- |
| 12 |
54/F |
GCD1 |
+ |
- |
30 |
0.6/0.5 |
- |
R555W/- |
| 13 |
40/M |
GCD1 |
+ |
- |
20 |
0.1/0.2 |
OD ALK |
R555W/- |
| 14 |
43/M |
GCD1 |
+ |
+ |
15 |
0.05/0.05 |
OD-OS PPK |
R555W/- |
| 15 |
35/F |
GCD1 |
- |
- |
20 |
0.4/0.3 |
- |
R555W/- |
| 16 | 33/F | GCD1 | + | - | 18 | 0.8/0.7 | - | R555W/-. |
OD: Right eye; OS: Left eye; ALK: Anterior lamellar keratoplasty; PPK: Partial penetrating keratoplasty, PTK: Phototheraupetic keratectomy, NA: Nonavailable
Table 3. Clinical findings of Turkish MCD patients carrying CHST6 gene variations.
| Patient no | Age/Sex | Family history | Consanguinity | Age at diagnosis | VA(OD/OS) | CCT(OD/OS) | Treatment | KS level (ng/ml) | İmmunotype | Variation |
|---|---|---|---|---|---|---|---|---|---|---|
| 1 |
32/M |
+ |
+ |
18 |
0.05/0.7 |
NA |
OD-OS PPK |
0 |
I |
C246W |
| 2 |
42/M |
+ |
+ |
15 |
0.05/0.05 |
NA |
OD-OS PPK |
125 |
II |
M1L/- |
| 3 |
27/M |
+ |
- |
15 |
0.4/0.3 |
NA |
OD-OS PPK |
0 |
I |
P204S |
| 4 |
33/M |
- |
+ |
10 |
0.05/0.05 |
NA |
OD-OS PPK |
0 |
I |
Q298fs |
| 5 |
21/F |
- |
- |
13 |
0.6/0.5 |
482/475 |
- |
146 |
II |
- |
| 6 |
24/M |
- |
- |
10 |
0.1/0.2 |
452/444 |
OD-OS PPK |
0 |
I |
R155fs/P204S |
| 7 |
55/F |
- |
+ |
20 |
0.05/0.05 |
NA |
OD-OS PPK |
0 |
I |
Q298fs |
| 8 |
63/M |
+ |
+ |
20 |
0.05/0.05 |
NA |
OD-OS PPK |
0 |
I |
V176M |
| 9 |
29/F |
- |
- |
15 |
0.1/0.1 |
NA |
OD-OS PPK |
241 |
II |
V176M/F55S |
| 10 |
27/M |
+ |
+ |
15 |
0.2/0.4 |
445/430 |
- |
0 |
I |
R211W |
| 11 |
55/M |
+ |
+ |
20 |
0.16/0.1 |
NA |
OD-OS PTK |
0 |
I |
Q298fs |
| 12 |
25/M |
+ |
+ |
15 |
0.16/0.2 |
440/462 |
- |
0 |
I |
R211W |
| 13 |
27/F |
- |
- |
18 |
0.2/0.3 |
NA |
OD ALK |
203 |
II |
- |
| 14 |
48/M |
- |
+ |
18 |
0.1/0.2 |
NA |
OD PPK |
NA |
NA |
Q298fs |
| 15 |
37/F |
+ |
+ |
15 |
0.05/0.05 |
435/438 |
OS PPK |
0 |
I |
V176M |
| 16 |
26/F |
+ |
+ |
15 |
0.05/0.2 |
NA |
- |
305 |
II |
- |
| 17 |
18/F |
+ |
+ |
14 |
0.05/0.05 |
497/520 |
OS PTK |
0 |
I |
R211W |
| 18 | 32/M | - | + | 18 | 0.4/0.6 | 444/440 | OD-OS PTK | 0 | I | Q298fs |
OD: Right eye; OS: Left eye; ALK: Anterior lamellar keratoplasty; PPK: Partial penetrating keratoplasty, PTK: Phototheraupetic keratectomy, NA: Nonavailable
Sulfated KS level in patients’ serum
The AgKS level was detected in 17 patients with MCD and 50 control patients. The mean concentration of AgKS was 259.3±66.4 ng/ml (120–384 ng/ml) in the control patients. Twelve patients were diagnosed with MCD type I because the concentration of sulfated KS in their serum was less than 10 ng/ml (Table 3). Five patients were diagnosed with MCD type 2 because the mean AgKS level in their serum was 204±72.7 ng/ml (125–305 ng/ml).
Molecular findings
Exons 4 and 12 in TGFBI were analyzed in 12 patients with GCD1 and four patients with LCD1. In all patients with GCD1, the c.1663 C>T (p. R555W) variation was detected in TGFBI exon 12 (Figure 2A). This variation was heterozygous in 11 patients and homozygous in one patient (Figure 2B). In two patients with GCD1, the c.1620 C>T (p. F540F) variation was detected heterozygously. This synonymous variation was included in the dbSNP database as a polymorphism. In all patients with LCD1, the c.370 T>C (p. R124C) variation was detected heterozygously in exon 4 in TGFBI (Figure 2C). These variations were not detected in 100 control chromosomes.
Figure 2.
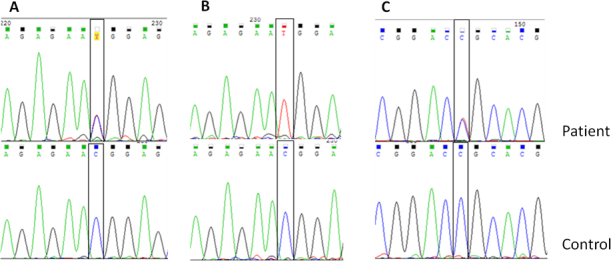
Sequencing chromatograms of variations in TGFBI identified in this study. A: The c.1663 C>T (p. R555W) variation (heterozygote) in granular corneal dystrophy type 1 (GCD1). B: The c.1663 C>T (p. R555W) variation (homozygote) in GCD1. C: The c.370 T>C (p. R124C) variation (heterozygote) in lattice corneal dystrophy type 1 (LCD1).
The coding region and the flanking exon–intron boundaries in exon 4 of CHST6 were analyzed in 18 MCD probands. Eight variations were detected in 18 patients: three previously reported missense variations (c. 1A>T, p. M1L; c.738C>G, p.C246W; and c.631 C>T, p. R211W), three novel missense variations (c.164 T>C, p. F55S; c.526 G>A, p. V176M; and c. 610 C>T, p. P204S), and two novel frameshift variations (c.894_895 insG, p. Q298fs and c. 462_463 delGC, p. R155Afs). The variation c.894_895 insG (p. Q298fs) was the most common variation in CHST6 in the study. No variation was detected in three of the five patients with MCD type II. A homozygous or compound heterozygous variation was detected in the coding region in all patients with MCD type I. Patient 14 who could not be immunophenotyped had a homozygous p. Q298fs variation. All patients with known consanguinity had homozygous variations with the exception of patient 2, who was heterozygous for c.1A>T (p. M1L).
The nucleotide sequences of the three previously reported variations in CHST6 in the patients are shown in Figure 3. Figure 4A shows the c.894_895insG variation in CHST6 that was detected in five unrelated patients. This variation results in a frameshift after codon 298 and early stop codon formation in codon 304. Figure 4B shows the c.462_463 delGC variant, which results in a frameshift after codon 155 (p.R155Afs). This variant was found only in patient 6, who is a compound heterozygote with the other novel variant c. 610 C>T (p. P204S). The c.610 C>T (p. P204S) variant was detected in two patients and leads to a proline to serine substitution (Figure 5A). Another novel missense variation, c. 526 G>A, was homozygously and compound heterozygously detected (Figure 5B,C). This mutation leads to a valine to methionine substitution at codon 176. The last novel missense variation, c.164T>C, was compound heterozygously detected in patient 9. This mutation leads to a phenylalanine to serine substitution at codon 55 (Figure 5D). When the pathogenic effect of the three novel missense variations was evaluated with SIFT and PolyPhen-2 in silico analysis software, the results were “probably damaging” and “not tolerated,” respectively (Table 4). Additionally, all newly detected variations in the CHST6 gene were not included in 100 control chromosomes, 1,000 Genomes, the dbSNP database, Exome Aggregation Consortium (ExAC), and the Human Gene Mutation Database (HGMD). Additionally, amino acid sequence analyses between several sulfotransferases of human and mouse origin revealed that the amino acids substituted in the missense variations detected in the patients were highly conserved residues (Table 5).
Figure 3.
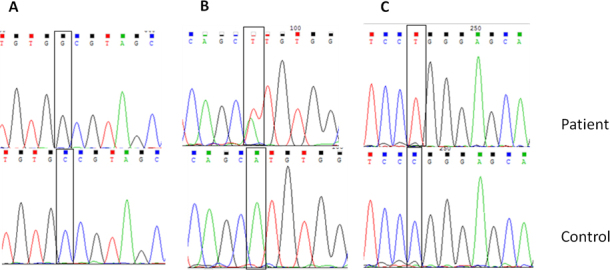
Sequencing chromatograms of previously reported variations in CHST6 identified in this study. A: The c.738 C>G (p.C246W) variation (homozygote) in macular corneal dystrophy (MCD) patient 1. B: The c.1 A>T (p.M1?) variation (heterozygote) in MCD patient 2. C: The c.631 C>T (p.R211W) variation (homozygote) in MCD patient 10.
Figure 4.
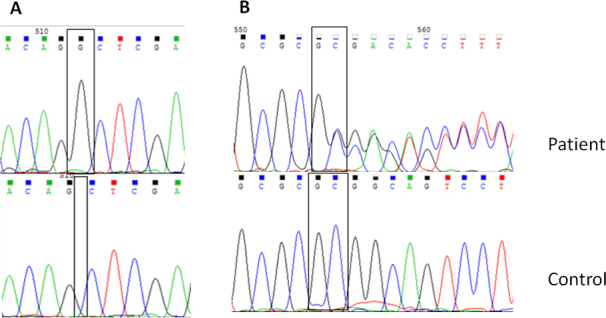
Sequencing chromatograms of novel frameshift variations in CHST6 detected in this study. A: The c.894_895 insG (p.Q298fs) variation (homozygote) in macular corneal dystrophy (MCD) patient 4. B: The c.462_463delGC (p.R155Afs) variation (heterozygote) in MCD patient 6.
Figure 5.
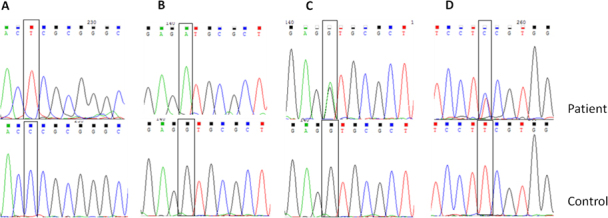
Sequencing chromatograms of novel missense variations in CHST6 detected in this study. A: The c.610 C>T (p.P204S) variation (homozygote) in macular corneal dystrophy (MCD) patient 3. B: The c.526 G>A (p.V176M) variation (homozygote) in MCD patient 8. C: The c.526 G>A (p.V176M) variation (heterozygote) in MCD patient 9. D: The c.164 T>C (p.F55S) variation (heterozygote) in MCD patient 9.
Table 4. Variations of CHST6 identified in Turkish MCD patients.
| Nucleotide change | Amino acid change | Variation type | Immunotype | Patient no | Polyphen | SIFT | HGMD No | ||
|---|---|---|---|---|---|---|---|---|---|
|
c.738 C>G |
p.C246W |
missense |
I |
1(1:homozygote) |
1
(probably damaging) |
0
(not tolerated) |
CM065078 |
||
|
c.1 A>T |
p. M1L |
First codon loss |
II |
1(2:heterozygote) |
MD |
MD |
CM0650699 |
||
|
c.894_895 insG |
p. Q298fs |
frameshift |
I |
5(4,7,11,14,18: homozygote) |
MD |
MD |
Novel |
||
|
c.610 C>T |
p. P204S |
missense |
I |
3(3: homozygote; 6: compound heterozygote |
1
(probably damaging) |
0,01
(not tolerated) |
Novel |
||
|
c.462_463 del GC |
p. R155Afs |
frameshift |
I |
1(6:compound heterozgote) |
MD |
MD |
Novel |
||
|
c.526 G>A |
p. V176M |
missense |
I,II |
2(8,15:homozygote; 9:compound heterozygote) |
1
(probably damaging) |
0
(not tolerated) |
Novel |
||
|
c. 631 C>T |
p. R211W |
missense |
I |
3(10,12,17: homozygote) |
1
(probably damaging) |
0
(not tolerated) |
CM002586 |
||
| c.164 T>C | p. F55S | missense | I | 1(9:compund heterozygote) | 0,968 (probably damaging) | 0 (not tolerated) | Novel | ||
Table 5. Multiple sequence alignments of sulfotransferases showing conservation of amino acid residues mutated in Turkish MCD patients.
| Sulfotransferase | F55V | V176M | P204S | R211W | C246W |
|---|---|---|---|---|---|
|
CHST6 (human) |
SFVG |
EVRF |
DPRA |
SREQ |
VCRS |
|
CHST5 (human) |
SFLG |
EVRF |
DPRA |
SREA |
VCRS |
|
CHST4 (human) |
SFVG |
EVRF |
DPRA |
SRER |
ICQS |
|
CHST3 (human) |
SFVG |
AVRI |
DPRA |
SR– |
NCES |
|
CHST2 (human) |
SFFG |
GVRF |
DPRA |
SRIR |
ICNS |
|
CHST5 (mouse) |
SFVG |
EVRF |
DPRA |
SREQ |
VCRS |
|
CHST4(mouse) |
SFVQ |
EVRF |
DPRA |
SREH |
ICKS |
|
CHST3(mouse) |
SFFG |
AVRI |
DPRA |
SR-I |
NCES |
| CHST2(mouse) | SFVG | GVRV | DPRA | SRIR | ICNS |
Discussion
In this study, we describe variations in CHST6 and TGFBI in 34 unrelated Turkish patients with CD. We detected five novel and three previously reported variations in CHST6 in patients with MCD and two previously reported variations in TGFBI in patients with GCD1 and LCD1.
TGFBI
Since Munier et al. reported mutations in TGFBI in 61 indexed patients with CD (30 LCD, 12 GCD, seven ACD, eight RBCD, and four TBCD) in 2002, this gene has been investigated in autosomal dominant epithelial-stromal CDs in numerous populations [6]. TGFBI encodes keratoepithelin (transforming growth factor beta-induced protein, TGFBIp) that was described as an extracellular matrix protein [13]. Two mutation hot spots correspond to keratoepithelin arginine residues at positions 124 and 555, where 50% of mutations were detected [21]. Five genotype–phenotype correlations were detected in these studies: R555Q in TBCD, R555W in GCD1, R124C in LCDI, R124H in ACD, and R124L in RBCD [6]. In addition to these defined correlations, molecular studies in a growing number of patients revealed mutational and phenotypic heterogeneity. To date, more than 60 mutations have been reported in TGFBI [6,16,22-24]. We detected the R555W mutation in patients with GCD1 in agreement with previous reports from Turkey [17,18] and from other populations [6,25-31]. The R555W mutation was heterozygous in 11 patients with GCD1 but homozygous in patient 9 who had known consanguinity in the family. His BCVA was severely affected although he was 27 years old and had undergone early keratoplasty surgery in both eyes. The phenotype of this patient was more severe than that of patients who carry the R555W mutation heterozygously. This condition was reported in the literature for patients with GCD and ACD and explained by semidominancy: Patients who are homozygous for ACD (R124H) or GCD (R555W) mutations have been reported, and these patients were more severely affected than heterozygotes [32-34]. Furthermore, the clinical severity of the disease was different in patients with GCD although they carried the same variation in one allele and had similar ages. Patient 12 was a 54-year-old woman. Her BCVA was above 0.4 in both eyes, and she had no previous surgery. However, patient 14 was a 43-year-old man. His BCVA was below 0.1 in both eyes, and he had undergone keratoplasty surgery in both eyes. Phenotypical differences in patients with the same mutation have been reported in the literature even between members of the same family [25]. This condition could be explained by two mechanisms: There could be other genetic variations in TGFBI that we did not analyze or there could be other genetic and/or environmental factors that affect protein–protein interactions [35,36] or protein degradation mechanisms [37,38], which should be evaluated to reveal the exact mechanism of corneal deposit formation in TGFBI-associated CDs.
In patients with LCD1, R124C was the most common mutation in TGFBI, but various mutations were detected in exons 4, 11, and 12 in previous studies [6,24-31,39-41]. In all of the present patients with LCD1, a heterozygous R124C mutation was detected in exon 4 in TGFBI. There was no known consanguinity in the LCD families, and the patients had similar phenotypes, except patient 3 in whom the disease showed asymmetry between two eyes. The asymmetric progression of LCD was reported in the literature [25,34], but further studies are needed to explain this asymmetry by identifying other pathogenic factors that underlie amyloid formation in the cornea. This study was the first report to examine genotype–phenotype relations in Turkish patients with LCD1.
CHST6
In this study, the coding region of CHST6 was analyzed in 18 unrelated patients with MCD. MCD was the most frequent CD type in the study population. MCD is also one of the most common CD observed in patients undergoing cornea transplantation in Saudi Arabia and India due to the high rate of consanguinity as in Turkey [10,42-44].
CHST6 encodes the enzyme N-acetyl-glucosamine-6-O-sulfotransferase (C-GlcNAc6ST), a member of the carbohydrate sulfotransferase family. C-GlcNAc6ST contains a short cytosolic tail at the N-terminal, a single transmembrane span, and a C-terminal domain that contains two sulfate donor [adenosine 30-phosphate-50-phosphosulfonate (PAPS)] binding sites, the catalytic domain, and an area that determines carbohydrate specificity in vivo [7,45,46]. The mutations of these three important sequences were reported to deteriorate the enzyme function and cause the deposition of low or unsulfated KS in the corneal stroma, leading to MCD [47-49].
There are three clinically indistinguishable immunophenotypes of MCD (I, IA, and II) based on the absence or presence of AgKS in the serum and corneal tissue [11]. In the present study population, we detected AgKS levels only in the serum samples of patients with MCD as histopathological cornea material was not available. We detected a variation in the coding region of CHST6 in all patients with type I, but not in three of the five patients with MCD type II. Akama et al. detected deletions or rearrangements of the upstream region of CHST6 in patients with MCD type II and suggested that MCD type II is characterized by the absence of mutations in the coding region in one or two alleles [5]. In the present study, we analyzed only the coding sequence. Thus, it is possible that the variation was located in the promoter or in a non-coding upstream or downstream region of this gene. Additionally, we could not correlate MCD diagnosis with histopathology in these three patients. Thus, these cases without coding region mutations might represent phenocopies. Furthermore, genetic heterogeneity could explain the patients without coding region mutations reported in the present study and in previous studies [50-55]. In one of the patients with type II MCD, we detected only a single heterozygous M1L variation that was previously reported in patients with MCD type I homozygously [56,57]. Nowinska et al. detected this start codon alteration heterozygously in a Polish patient with MCD with an unknown immunophenotype [50]. There could be a second undetected variation outside the coding region of CHST6 in the present patient, or genetic heterogeneity could be another possible explanation in this case. In the fifth patient with MCD type II, we detected two novel heterozygote missense variations. One missense variation, V176M, was also homozygously detected in two patients with MCD type I. Although Akama et al. proposed that MCD types I and II were discernible by the type of CHST6 variation [5], recent reports and the present study suggest that there is no correlation between immunophenotypes and variations in CHST6 [58-60]. The same variation could be associated with all three immunophenotypes as reported from a single family in India [20].
To date, 165 variations in CHST6 in patients with MCD from different populations have been reported in the HGMD. This means strong allelic heterogeneity in CHST6. The identification of eight variations in 15 patients with MCD in the present study also supports allelic heterogeneity in this gene.
The most common variation detected in the patients with MCD was c.894_895 insG (p. Q298fs). This variation was homozygously detected in five unrelated patients with MCD type 1. This insertion variation was not previously reported and resulted in the occurrence of a frameshift after the 298th glutamine residue and premature stop codon formation. This variation was found in ten of 36 alleles in the present study population. This could be explained in two ways. First, this could be a hot spot mutation, but this variation was not detected in other patients from different populations studied until now. Second, this variation could be a founder mutation, but to evaluate a founder effect, a haplotype analysis of the family members of these patients, whom we could not reach, should be performed. The other novel frameshift variation detected in the present study was c.462_463delGC (p.R155Afs). This deletion, detected in patient 6 compound heterozygously, resulted in the occurrence of a frameshift after the 155th arginine residue and premature stop codon formation. According to the recommended standards for the interpretation of sequence of the American College of Medical Genetics and Genomics (ACMG), these two variations belong to the PVS1 null variant class that can often be assumed to disrupt gene function by leading to a complete absence of the gene product by a lack of transcription or nonsense-mediated decay of an altered transcript [61]. Null variants were reported less frequently than missense variants in MCD [4,62]. In French and German families, frameshift mutations correlated with more severe phenotypes and earlier requirement of keratoplasty [51,56]. However, El-Ashry et al. and Sultana et al. observed that there is no consistent phenotypic difference between truncating and missense mutations in their study populations [43,63]. In the present study, we detected two frameshift variations that resulted in premature stop codon formation in six patients of whom four underwent keratoplasty (66%). We detected missense variations in nine patients of whom six underwent keratoplasty (66%). Regarding previous treatments, other phenotypic properties listed in Table 3 were not different between patients carrying missense and frameshift variations in accordance with reports from Egypt and India [43,63].
The second common variation in the present study was c. 631 C>T (p. R211W), which was previously reported in populations from Iceland [5] and Japan [49]. The other previously reported variation homozygously detected in patient 1 was c.738 C>G (p. C246W). This missense variation was reported in populations from North America in 2006 [62].
In the present study, we also detected three novel missense variations in patients with MCD: c.610 C>T (p. P204S) was located in the RX7S sequence for the 3′-PB domain, c. 526 G>A (p. V176M) was located in the sequence between the 5′-PSB domain and the 3′-PB domain called the binding pocket, and c.164 T>C (p. F55S) was located in the 5′-PSB domain. All the novel variations were located in the binding sites or binding pocket that was important for enzyme function [45,46]. In addition, none of these missense variations was detected in the 100 control chromosomes or the ExAC, 1000 Genomes, and dbSNP databases. These three novel missense variations were predicted to be pathogenic by SIFT and PolyPhen-2 software (Table 4). Moreover, the amino acid sequence analysis between several sulfotransferases of human and mouse origin demonstrated that the novel variations substitute well-conserved amino acid residues, which supports the pathogenic effect of these novel missense variations (Table 5). However, functional studies are warranted to determine the exact consequences of these changes for protein functions.
Analysis of clinical parameters between different variations and between different immunophenotypes indicated that most patients in the present study had similar features as reported in various studies in different populations [5,12,22]. As a result, it was not possible to detect consistent genotype and phenotype correlations in patients with MCD.
This study presents some limitations. The patient population was small, and the families of the patients were not methodically evaluated.
In conclusion, this is the first molecular analysis of TGFBI and CHST6 in Turkish patients with CD. Well-known mutations in TGFBI were detected in patients with GCD1 and LCD1, supporting the existence of hot spot mutations in this gene. Moreover, five novel and three previously reported likely pathogenic variations in CHST6 were identified in patients with MCD, which highlights the allelic heterogeneity of this gene.
Acknowledgments
This study was supported by Scientific Research Fund of The Scientific and Technological Research Council of Turkey (TUBITAK). (Grant no: 114S126).
References
- 1.Seitz B, Lisch W. Stage-related therapy of corneal dystrophies. Dev Ophthalmol. 2011;48:116–53. doi: 10.1159/000324081. [DOI] [PubMed] [Google Scholar]
- 2.Nurözler AB, Akkaya ZY, Yıldız HE, Onat M, Budak K, Örnek F. Penetrating Keratoplasty Indications and Outcomes. Turkiye Klinikleri J Ophthalmol. 2009;18:85–91. [Google Scholar]
- 3.Tunçbilek E, Koç İ. Consanguineous Marriage in Turkey and its impact on fertility and mortality. Ann Hum Genet. 1994;58:321–9. doi: 10.1111/j.1469-1809.1994.tb00729.x. [DOI] [PubMed] [Google Scholar]
- Weiss JS, Møller HU, Lisch W, Aldave AJ, Seitz B, Bredrup C, Kivela T, Munier FL, Rapuano CJ, Nischal KK, Kim EK, Sutphin J, Busin M, Labbe A, Kenyon KR, Kinoshita S, Lisch W. IC3D classification of the corneal dystrophies- Edition 2. Cornea. 2015;34:117–59. [DOI] [PubMed] [Google Scholar]
- 5.Akama TO, Nishida K, Nakayama J, Watanabe H, Ozaki K, Nakamura T, Dota A, Kawasaki S, Inoue Y, Maeda N, Yamamoto S, Fujiwara T, Thonar EJ, Shimomura Y, Kinoshita S, Tanigami A, Fukuda MN. Macular corneal dystrophy type I and type II are caused by distinct mutations in a new sulphotransferase gene. Nat Genet. 2000;26:237–41. doi: 10.1038/79987. [DOI] [PubMed] [Google Scholar]
- 6.Munier FL, Frueh BE, Othenin-Girard P, Uffer S, Cousin P, Wang MX, Heon E, Black GC, Blasi MA, Balestrazzi E, Lorenz B, Escoto R, Barraquer R, Hoeltzenbein M, Gloor B, Fossarello M, Singh AD, Arsenijevic Y, Zografos L, Schorderet DF. BIGH3 mutation spectrum in corneal dystrophies. Invest Ophthalmol Vis Sci. 2002;43:949–54. [PubMed] [Google Scholar]
- 7.Musselmann K, Hassell JR. Focus on molecules: CHST6 (carbohydrate sulfotransferase 6; corneal N-acetylglucosamine-6-sulfotransferase). Exp Eye Res. 2006;83:707–8. doi: 10.1016/j.exer.2005.11.020. [DOI] [PubMed] [Google Scholar]
- 8.Hassell JR, Newsome DA, Krachmer JH, Rodrigues MM. Macular cornealdystrophy: failure to synthesize a mature keratan sulfate preteoglycan. Proc Natl Acad Sci USA. 1980;77:3705–9. doi: 10.1073/pnas.77.6.3705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Jonasson F, Oshima E, Thonar EJ, Smith CF, Johannsson JH, Klintworth GK. Macular corneal dystrophy in Iceland. A clinical, genealogic, and immunohistochemical study of 28 patients. Ophthalmology. 1996;103:1111–7. doi: 10.1016/s0161-6420(96)30559-9. [DOI] [PubMed] [Google Scholar]
- 10.Klintworth GK, Oshima E. AL-Rajhi A, Al-Saif A, Thonar EJ, Karcioglu ZA. Macular corneal dystrophy in Saudi Arabia: A study of 56 cases and recognition of a new immunophenotype. Am J Ophthalmol. 1997;124:9–18. doi: 10.1016/s0002-9394(14)71637-x. [DOI] [PubMed] [Google Scholar]
- 11.Saito T, Nishida K, Nakayama J, Akama TO, Fukuda MN, Watanabe K, Quantock AJ, Maeda N, Watanabe H, Tano Y. Sulfation patterns of keratan sulfate in different macular corneal dystrophy immunophenotypes using three different probes. Br J Ophthalmol. 2008;92:1434–6. doi: 10.1136/bjo.2008.139527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Park SH, Ahn YJ, Chae H, Kim Y, Kim MS, Kim M. Molecular analysis of the CHST6 gene in Korean patients with macular corneal dystrophy: Identification of three novel mutations. Mol Vis. 2015;21:1201–9. [PMC free article] [PubMed] [Google Scholar]
- 13.Escribano J, Hernando N, Ghosh S, Crabb J, Coca-Prados M. cDNA from human ocular ciliary epithelium homologous to beta ig-h3 is preferentially expressed as an extracellular protein in the corneal epithelium. J Cell Physiol. 1994;160:511–21. doi: 10.1002/jcp.1041600314. [DOI] [PubMed] [Google Scholar]
- 14.El Kochairi I, Letovanec I, Uffer S, Munier FL, Chaubert P, Schorderet DF. Systemic investigation of keratoepithelin deposits in TGFBI/BIGH3-related corneal dystrophy. Mol Vis. 2006;12:461–6. [PubMed] [Google Scholar]
- 15.Skonier J, Neubauer M, Madisen L, Bennett K, Plowman GD, Purchio AF. cDNA cloning and sequence analysis of beta igh3, a novel gene induced in a human adenocarcinoma cell line after treatment with transforming growth factor-beta. DNA Cell Biol. 1992;11:511–22. doi: 10.1089/dna.1992.11.511. [DOI] [PubMed] [Google Scholar]
- 16.Han KE, Choi SI, Kim TI, Maeng YS, Stulting RD, Ji YW, Kim EK. Pathogenesis and treatments of TGFBI corneal dystrophies. Prog Retin Eye. 2016;50:67–88. doi: 10.1016/j.preteyeres.2015.11.002. [DOI] [PubMed] [Google Scholar]
- 17.Kocak-Altintas AG, Kocak-Midillioglu I, Akarsu AN, Duman S. BIGH3 gene analysis in the differential diagnosis of corneal dystrophies. Cornea. 2001;20:64–8. doi: 10.1097/00003226-200101000-00013. [DOI] [PubMed] [Google Scholar]
- 18.Kıratlı H, Irkeç M, Ozgül K, Ogüş A. The phenotype of arg555trp mutation in a large Turkish family with corneal granular dystrophy. Eur J Ophthalmol. 2001;11:333–7. doi: 10.1177/112067210101100403. [DOI] [PubMed] [Google Scholar]
- 19.Thonar EJ, Lenz ME, Klinthworth GK, Caterson B, Pachman LM, Glickman P, Katz R, Huff J, Kuettner KE. Quantification of keratan sulfate in blood as a marker of cartilage catabolism. Arthritis Rheum. 1985;28:1367–76. doi: 10.1002/art.1780281209. [DOI] [PubMed] [Google Scholar]
- 20.Sultana A, Klintworth GK, Thonar EJMA, Vemuganti GK, Kannabiran C. Immunophenotypes of macular corneal dystrophy in India and correlation with mutations in CHST6. Mol Vis. 2009;15:319–25. [PMC free article] [PubMed] [Google Scholar]
- 21.Kannabiran C, Klintworth GK. TGFBI gene mutations in corneal dystrophies. Hum Mutat. 2006;27:615–62. doi: 10.1002/humu.20334. [DOI] [PubMed] [Google Scholar]
- 22.Aldave AJ. The genetics of the corneal dystrophies. Dev Ophthalmol. 2011;48:51–66. doi: 10.1159/000324077. [DOI] [PubMed] [Google Scholar]
- 23.Zenteno JC, Ramirez-Miranda A, Santacruz-Valdes C, Suarez-Sanchez R. Expanding the mutational spectrum in TGFBI-linked corneal dystrophies: identification of a novel and unusual mutation (Val113Ile) in a family with granular dystrophy. Mol Vis. 2006;12:331–5. [PubMed] [Google Scholar]
- 24.Sommer JR, Wong F, Klintworth GK. Phenotypic variations of corneal dystrophies caused by different mutations in the big-h3 gene. Invest Ophthalmol Vis Sci. 1998;39:5–13. ARVO Abstract. [Google Scholar]
- 25.Long Y, Gu YS, Han W, Li XY, Yu P, Qi M. Genotype-phenotype correlations in Chinese patients with TGFBI gene-linked corneal dystrophy. J Zhejiang Univ Sci B. 2011;12:287–92. doi: 10.1631/jzus.B1000154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Pampukha VM, Drozhyna GI, Livshits LA. TGFBI gene mutation analysis in families with hereditary corneal dystrophies from Ukraine. Ophthalmologica. 2004;218:411–4. doi: 10.1159/000080945. [DOI] [PubMed] [Google Scholar]
- 27.Vincent AL, de Karolyi B, Patel DV, Wheeldon CE, McGhee CN. TGFBI mutational analysis in a New Zealand populationof inherited corneal dystrophy patients. Br J Ophthalmol. 2010;94:836–42. doi: 10.1136/bjo.2009.159632. [DOI] [PubMed] [Google Scholar]
- 28.Li Y, Li T, Song XS, Li JZ, Wu QS, Li HY. TGFBI and CHST6 gene analysis in Chinese stromal corneal dystrophies. Int J Ophthalmol. 2012;5:301–6. doi: 10.3980/j.issn.2222-3959.2012.03.10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hou YC, Wang IJ, Hsiao CH, Chen WL, Hu FR. Phenotypegenotype correlations in patients with TGFBI-linked corneal dystrophies in Taiwan. Mol Vis. 2012;18:362–71. [PMC free article] [PubMed] [Google Scholar]
- 30.Gonzalez-Rodriguez J, Ramirez-Miranda A, Hernandez-Da Mota SE, Zenteno JC. TGFBI, CHST6, and GSN gene analysis in Mexican patients with stromal corneal dystrophies. Graefes Arch Clin Exp Ophthalmol. 2014;252:1267–72. doi: 10.1007/s00417-014-2648-9. [DOI] [PubMed] [Google Scholar]
- 31.Cho KJ, Mok JW, Na KS, Rho CR, Byun YS, Hwang HS, Hwang KY, Joo CK. TGFBI gene mutations in a Korean population with corneal dystrophy. Mol Vis. 2012;18:2012–21. [PMC free article] [PubMed] [Google Scholar]
- 32.Okada M, Yamamoto S, Watanabe H, Inoue Y, Tsujikawa M, Maeda N, Shimomura Y, Nishida K, Kinoshita S, Tano Y. Granular corneal dystrophy with homozygous mutations in the kerato-epithelin gene. Am J Ophthalmol. 1998;126:169–76. doi: 10.1016/s0002-9394(98)00075-0. [DOI] [PubMed] [Google Scholar]
- 33.Fujiki K, Hotta Y, Nakayasu K, Kanal A. Homozygotic patient with BIGH3 gene mutation in granular dystrophy. Cornea. 1998;17:288–92. [PubMed] [Google Scholar]
- 34.Romero P, Vogel M, Diaz JM, Romero MP, Herrera L. Anticipation in familial corneal dystrophy type I with R124C mutation in the TGFBI (BIGH3) gene. Mol Vis. 2008;14:829–35. [PMC free article] [PubMed] [Google Scholar]
- 35.Basaiawmoit RV, Oliveira CL, Runager K, Sørensen CS, Behrens MA, Jonsson BH, Kristensen T, Klintworth GK, Enghild JJ, Pedersen JS, Otzen DE. SAXS models of TGFBIp reveal a trimeric structure and show that the overall shape is not affected by the Arg124His mutation. J Mol Biol. 2011;408:503–13. doi: 10.1016/j.jmb.2011.02.052. [DOI] [PubMed] [Google Scholar]
- 36.Kim BY, Olzmann JA, Choi SI, Ahn SY, Kim TI, Cho HS, Suh H, Kim EK. Corneal dystrophy-associated R124H mutation disrupts TGFBI interaction with Periostin and causes mislocalization to the lysosome. J Biol Chem. 2009;284:19580–91. doi: 10.1074/jbc.M109.013607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Zhu M, Yu P, Jiang B, Gu Y. Investigation of the influence of Arg555Trp and Thr538Pro TGFBI mutations on C-terminal cleavage and cell endoplasmic reticulum stress. Mol Vis. 2012;18:1156–64. [PMC free article] [PubMed] [Google Scholar]
- 38.Choi SI, Maeng YS, Kim KS, Kim TI, Kim EK. Autophagy is induced by raptor degradation via the ubiquitin/ proteasome system in granular corneal dystrophy type 2. Biochem Biophys Res Commun. 2014;450:1505–11. doi: 10.1016/j.bbrc.2014.07.035. [DOI] [PubMed] [Google Scholar]
- 39.Chau HM, Ha NT, Cung LX, Thanh TK, Fujiki K, Murakami A, Kanai A. H626R and R124C mutations of the TGFBI (BIGH3) gene caused lattice corneal dystrophy in Vietnamese people. Br J Ophthalmol. 2003;87:686–9. doi: 10.1136/bjo.87.6.686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Zhang T, Yan N, Yu W, Liu Y, Liu G, Wu X, Lian J, Liu X. Molecular genetics of Chinese families with TGFBI corneal dystrophies. Mol Vis. 2011;17:380–7. [PMC free article] [PubMed] [Google Scholar]
- 41.Yamamoto S, Okada M, Tsjuikawa M, Shimomura Y, Nishida K, Inoue Y, Watanabe H, Maeda N, Kurahashi H, Kinoshita S, Nakamura Y, Tano Y. A kerato-epithelin (big-h3) mutation in lattice corneal dystrophy type IIIA. Am J Hum Genet. 1998;62:719–22. doi: 10.1086/301765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Weiss JS, Fifth ARVO/Pfizer Ophthalmics Research Institute Conference Working Group Corneal dystrophies: molecular genetics to therapeutic intervention--Fifth ARVO/Pfizer Ophthalmics Research Institute Conference. Invest Ophthalmol Vis Sci. 2010;51:5391–402. doi: 10.1167/iovs.09-4746. [DOI] [PubMed] [Google Scholar]
- 43.Sultana A, Sridhar MS, Jagannathan A, Balasubramanian D, Kannabiran C, Klintworth GK. Novel mutations of the carbohydrate sulfotransferase-6 (CHST6) gene causing macular corneal dystrophy in India. Mol Vis. 2003;9:730–4. [PubMed] [Google Scholar]
- 44.Warren JF, Aldave AJ, Srinivasan M, Thonar EJ, Kumar AB, Cevallos V, Whitcher JP, Margolis TP. Novel mutations in the CHST6 gene associated with macular corneal dystrophy in southern India. Arch Ophthalmol. 2003;121:1608–12. doi: 10.1001/archopht.121.11.1608. [DOI] [PubMed] [Google Scholar]
- 45.Kakuta Y, Pedersen LG, Pedersen LC, Negishi M. Conserved structural motifs in the sulfotransferase family. Trends Biochem Sci. 1998;23:129–30. doi: 10.1016/s0968-0004(98)01182-7. [DOI] [PubMed] [Google Scholar]
- 46.Ong E, Yeh JC, Ding Y, Hindsgaul O, Pedersen LC, Negishi M, Fukuda M. Structure and function of HNK-1 sulfotransferase: identification of donor and acceptor binding sites by site-directed mutagenesis. J Biol Chem. 1999;274:25608–12. doi: 10.1074/jbc.274.36.25608. [DOI] [PubMed] [Google Scholar]
- 47.Lewis D, Davies Y, Nieduszynski IA, Lawrence F, Quantock AJ, Bonshek R, Fullwood NJ. Ultrastructural localization of sulfated and unsulfated keratan sulfate in normal and macular corneal dystrophy type I. Glycobiology. 2000;10:305–12. doi: 10.1093/glycob/10.3.305. [DOI] [PubMed] [Google Scholar]
- 48.Nakayama J, Nishida K, Hiraoka N, Suzuki M, McAuliffe J, Hindsgaul O, Fukuda M, Fukuda MN. Human corneal GlcNAc 6-sulfotransferase and Mouse intestinal GlcNAc 6-O-sulfotransferase both produce keratan sulfate. J Biol Chem. 2001;276:16271–8. doi: 10.1074/jbc.M009995200. [DOI] [PubMed] [Google Scholar]
- 49.Iıda-Hasegawa N, Furuhata A, Hayatsu H, Murakami A, Fujiki K, Nakayasu K, Kanai A. Mutations in the CHST6 Gene in Patients with Macular Corneal Dystrophy: Immunohistochemical Evidence of Heterogeneity. Invest Ophthalmol Vis Sci. 2003;44:3272–7. doi: 10.1167/iovs.02-0910. [DOI] [PubMed] [Google Scholar]
- 50.Nowinska AK, Wylegala E, Teper S, Wróblewska-Czajka E, Aragona P, Roszkowska AM, Micali A, Pisani A, Puzzolo D. Phenotype and genotype analysis in patients with macular corneal dystrophy. Br J Ophthalmol. 2014;98:1514–21. doi: 10.1136/bjophthalmol-2014-305098. [DOI] [PubMed] [Google Scholar]
- 51.Niel F, Ellies P, Dighiero P, Soria J, Sabbagh C, San C, Renard G, Delpech M, Valleix S. Truncating mutations in the carbohydrate sulfotransferase 6 gene (CHST6) result in macular corneal dystrophy. Invest Ophthalmol Vis Sci. 2003;44:2949–53. doi: 10.1167/iovs.02-0740. [DOI] [PubMed] [Google Scholar]
- 52.Aldave AJ, Yellore VS, Thonar EJ, Udar N, Warren JF, Yoon MK, Cohen EJ, Rapuano CJ, Laibson PR, Margolis TP, Small K. Novel mutations in the carbohydrate sulfotransferase gene (CHST6) in American patients with macular corneal dystrophy. Am J Ophthalmol. 2003;137:465–73. doi: 10.1016/j.ajo.2003.09.036. [DOI] [PubMed] [Google Scholar]
- 53.Sultana A, Sridhar MS, Klintworth GK, Balasubramanian D, Kannabiran C. Allelic heterogeneity of the carbohydrate sulfotransferase-6 gene in patients with macular corneal dystrophy. Clin Genet. 2005;68:454–60. doi: 10.1111/j.1399-0004.2005.00517.x. [DOI] [PubMed] [Google Scholar]
- 54.Birgani SA, Salehi Z, Houshmand M, Mohamadi MJ, Promehr LA, Mozafarzadeh Z. Novel mutations of CHST6 in Iranian patients with macular corneal dystrophy. Mol Vis. 2009;15:373–7. [PMC free article] [PubMed] [Google Scholar]
- 55.Paliwal P, Sharma A, Tandon R, Sharma N, Titiyal JS, Sen S, Vajpayee RB. Molecular genetic analysis of macular corneal dystrophy patients from North India. Ophthalmic Res. 2012;48:28–32. doi: 10.1159/000334911. [DOI] [PubMed] [Google Scholar]
- 56.Gruenauer-Kloevekorn C, Bracutigam S, Heinritz W, Froster UG, Duncker GI. Macular corneal dystrophy: mutational spectrum in German patients, novel mutations and therapeutic options. Graefes Arch Clin Exp Ophthalmol. 2008;246:1441–7. doi: 10.1007/s00417-008-0836-1. [DOI] [PubMed] [Google Scholar]
- 57.Liskova P, Veraitch B, Jirsova K, Filipec M, Neuwirth A, Ebenezer ND, Hysi PG, Hardcastle AJ, Tuft SJ, Bhattacharya SS. Sequencing of the CHST6 gene in Czech macular corneal dystrophy patients supports the evidence of a founder mutation. Br J Ophthalmol. 2008;92:265–7. doi: 10.1136/bjo.2007.125252. [DOI] [PubMed] [Google Scholar]
- 58.Bao W, Smith CF, al-Rajhi A, Chandler JW, Karcioglu ZA, Akama TO, Fukuda MN, Klintworth GK. Novel mutations in the CHST6 gene in Saudi Arabic patients with macular corneal dystrophy. Invest Ophthalmol Vis Sci (Suppl) 2001;42:483–6. [Google Scholar]
- 59.Liu NP, Baldwin J, Lennon F, Stajich JM, Thonar EJ, Pericak-Vance MA, Klintworth GK, Vance JM. Coexistence of macular corneal dystrophy types I and II in a single sibship. Br J Ophthalmol. 1998;82:241–4. doi: 10.1136/bjo.82.3.241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Liu NP, Bao W, Smith CF, Vance JM, Klintworth GK. Different mutations in 5 sulfotransferase 6 (CHST6) gene cause macular corneal dystrophy types I and II in a single sibship. Am J Ophthalmol. 2005;139:1118–20. doi: 10.1016/j.ajo.2004.11.054. [DOI] [PubMed] [Google Scholar]
- 61.Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, Grody WW, Hedge M, Lyon E, Spector E, Voelkerding K, Rehm HL, ACMG Laboratory Quality Assurance Committee Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. 2015;17:405–24. doi: 10.1038/gim.2015.30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Klintworth GK, Smith CF, Bowling BL. CHST6 mutations in North American subjects with macular corneal dystrophy: comprehensive molecular genetic review. Mol Vis. 2006;12:159–76. [PubMed] [Google Scholar]
- 63.El-Ashry MF, El-Aziz MM, Wilkins S, Cheetham ME, Wilkie SE, Hardcastle AJ, Halford S, Bayoumi AY, Ficker LA, Tuft S, Bhattacharya SS, Ebenezer ND. Identification of novel mutations in the carbohydrate sulfotransferase gene (CHST6) causing macular corneal dystrophy. Invest Ophthalmol Vis Sci. 2002;43:377–82. [PubMed] [Google Scholar]