

Abstract
Small molecule aggregates are considered nuisance compounds in drug discovery, but their unusual properties as colloids could be exploited to form stable vehicles to preserve protein activity. We investigated the co-aggregation of seven molecules chosen because they had been previously intensely studied as colloidal aggregators, co-formulating them with bis-azo dyes. The co-formulation reduced colloid sizes to <100 nm, and improved uniformity of the particle size distribution. The new colloid formulations are more stable than previous aggregator particles. Specifically, co-aggregation of Congo Red with sorafenib, tetraiodophenolphthalein (TIPT) or vemurafenib produced particles that are stable in solutions of high ionic strength and high protein concentrations. Like traditional, single compound colloidal aggregates, the stabilized colloids adsorbed and inhibited enzymes like β-lactamase, malate dehydrogenase and trypsin. Unlike traditional aggregates, the co-formulated colloid-protein particles could be centrifuged and re-suspended multiple times, and from re-suspended particles, active trypsin could be released up to 72 hours after adsorption. Unexpectedly, the stable colloidal formulations can sequester, stabilize, and isolate enzymes by spin-down, resuspension and release.
Keywords: Colloids, aggregation, drug delivery, protein adsorption
INTRODUCTION
Though primarily viewed as a nuisance in early drug discovery1, colloidal aggregation marks a fundamental mechanism for the self-assembly of organic compounds in aqueous media2, creating a kinetically stable species with properties far different from either soluble monomers or precipitants. Whereas we and others have focused on methods to rapidly detect and eliminate these aggregates from early drug discovery3–6, it is possible to wonder if the colloids might have interesting, optimizable properties on their own. Among the most interesting is their ability to adsorb protein and sequester it from solution. In principle one could imagine several uses for such protein-loaded colloids; however, they would first have to be stabilized with a uniform size distribution and be more robust during use in order to be treated as reagents.
Though colloidal aggregates of small molecules are stable enough to cause massive artifacts in high-throughput screening and other discovery techniques6–8, they are not typically stable enough to be treated as reagents. Most colloidal particles are polydisperse in size, with particles of even a single molecule often varying widely in size as visualized by transmission electron microscopy.3,4,9,10 Mechanistic insights into thermodynamic driving forces and association kinetics have provided key details regarding self-assembly and behavior in aqueous solutions3,4,11–14, but utilizing the unique physiochemical properties has proven difficult. Due to the high-energy state of this amorphous phase, typical colloids are especially unstable over long time periods, often growing in size as individual particles fuse, leading to crystallization and precipitation. Whereas colloidal aggregates may be readily centrifuged out of solution—a primary method of controlling for their effects on membrane proteins15—once pelleted, they are not easily re-suspended in aqueous media. Co-formulation using additives, which are primarily comprised of surfactants and polymers,16–18 have been explored to increase stability of metastable colloidal particles for both oral19 and perenteral20–22 drug formulations. However, most colloidal aggregates of organic molecules are polydisperse, relatively unstable, resist useful concentration by centrifugation, as the pelleted particles cannot be resuspended in colloidal form, thus limiting manipulation by processes typical of useful reagents. We wondered, however, if these properties might be optimized.
Here we investigate a strategy to stabilize colloidal aggregates of drug molecules with known dye aggregators, seeking colloids that are optimized for stability, size homogeneity, protein loading, and ability to be treated as a reagents. Specifically, several mole ratios of supramolecular assembling bis-azo dyes, Congo Red and Evans Blue, were incorporated into formulations of aggregating anti-cancer drugs fulvestrant, lapatinib, nilotinib, sorafenib, vemurafenib, and the well-studied aggregator, tetraiodophenolphthalein (TIPT). The properties and stability in aqueous media of such co-formulated colloids were monitored using dynamic light scattering (DLS) and transmission electron microscopy (TEM), seeking stabilized and homogenous colloidal formulations that can be loaded with protein, and used to transfer and release such proteins from one environment to another, over several hours and up to several days.
RESULTS
Dye Effects on Colloidal Aggregation
Seven known aggregators (chlorotrianisene, fulvestrant, nilotinib, lapatinib, sorafenib, tetraiodophenolphthalein [TIPT] and vemurafenib), each with colloidal aggregation concentrations (CAC) values < 5 μM,3,10 were tested with and without the addition of one of two bis-azo dyes: Congo Red (CR) and Evans Blue (EB). We hypothesized that these hydrophobic aromatic compounds, which have been shown previously to aggregate themselves,3,4,10,23 could help control the size and stability of colloids in solution through supramolecular organization24 of the aromatic multiply-charged dye within the colloidal structure that reduces repulsion between the dye-incorporated colloid and aqueous interface. The colloid forming drugs were co-aggregated with CR and EB in PBS buffer at a concentration of 50 μM, with an equal amount of either dye added to the organic phase prior to addition of buffer – i.e. the formal ‘organic-load’ is 100 μM in all formulations.25 DLS was then used to ascertain the properties of colloidal formulations and to measure their hydrodynamic diameter, polydispersity, and scattering (Table 1).
Table 1.
Colloids formulated with one of Congo Red (CR) or Evans Blue (EB) are smaller and generally more monodisperse than those formulated without CR or EB, as determined by dynamic light scattering (DLS).
| Formulation* | Diameter (nm) | Polydispersity | Scattering (cnts/sec × 106) |
|---|---|---|---|
| CR | ND | ND | 0.4 ± 0.01 |
| EB | ND | ND | 1 ± 0.2 |
| Chlorotrianisene | 185 ± 15 | 0.09 ± 0.03 | 499 ± 33 |
| Chlorotrianisene:CR | 68 ± 1 | 0.08 ± 0.01 | 52 ± 3 |
| Chlorotrianisene:EB | 76 ± 3 | 0.17 ± 0.02 | 61 ± 1 |
| Fulvestrant | 1170± 59 | 0.12 ± 0.01 | 143 ± 7 |
| Fulvestrant:CR | 73 ± 4 | 0.22 ± 0.01 | 45 ± 6 |
| Fulvestrant:EB | 77 ± 1 | 0.22 ± 0.01 | 34 ± 2 |
| Nilotinib | 1868± 228 | 0.04 ± 0.06 | 97 ± 6 |
| Nilotinib:CR | 46 ± 2 | 0.16 ± 0 | 14 ± 2 |
| Nilotinib:EB | 89 ± 10 | multimodal | 7 ± 0.4 |
| Lapatinib | 490 ± 16 | 0.13 ± 0.03 | 440 ± 25 |
| Lapatinib:CR | 38 ± 5 | 0.17 ± 0.06 | 16 ± 3 |
| Lapatinib:EB | 248 ± 27 | 0.23 ± 0.01 | 46 ± 4 |
| Sorafenib | 983 ± 27 | 0.21 ± 0.03 | 128 ± 10 |
| Sorafenib:CR | 34 ± 2 | 0.17 ± 0.04 | 9 ± 1 |
| Sorafenib:EB | 41± 1 | 0.12 ± 0.01 | 10 ± 0.4 |
| TIPT | 106 ± 10 | 0.17 ± 0.02 | 99 ± 0.5 |
| TIPT:CR | 41 ± 1 | 0.14 ± 0 | 121 ± 2 |
| TIPT:EB | 54 ± 4 | 0.13 ± 0.02 | 50 ± 10 |
| Vemurafenib | 1037 ±769 | 0.12 ± 0.11 | 91 ± 5 |
| Vemurafenib:CR | 40 ± 0 | 0.16 ± 0.03 | 12 ± 0.4 |
| Vemurafenib:EB | 107 ± 5 | 0.18 ± 0.02 | 110 ± 5 |
50 μM in PBS, 1:1 molar ratio with either CR or EB, 1% DMSO, n=3 independent colloid formulations, mean ± SD
In the absence of azo dye, four of the drugs aggregated to colloids with diameters of approximately 1 μm, and the fifth drug (lapatinib) aggregated to colloids of approximately 0.5 μm. Most had polydisperse size distributions. This is not unexpected given that the concentration is well above the CAC for these compounds. Only two compounds, chlorotrianisene and TIPT, had well-defined colloids immediately after formation, but even these rapidly increased in size within an hour (Supplementary Figure 1).
Gratifyingly, co-aggregation with CR or EB resulted in generally homogeneous colloids (polydispersity < 0.2) with mean hydrodynamic diameters less than 100 nm (Table 1). Admittedly, such a profound reduction in size and improvement in polydispersity was surprising, especially when compared to ‘bare’ colloids containing the parent compound alone. As a comparison, CR and EB prepared alone at 50 μM resulted in no discernable particles, with scattering intensities falling approximately an order of magnitude below that of all other compounds; however, this may be due, in part, to the optical properties of the dyes interfering with the DLS measurement. Other data that we have collected suggest that CR and EB do in fact form colloids at these concentrations as they inhibit counter-screening enzymes for colloid formation (e.g. AmpC–β-lactamase) in a detergent-dependent manner.26 Still, these initial experiments suggested that both CR and EB could alter the properties of the colloids to produce much smaller particles when co-formulated at concentrations well above the CACs for these compounds.
Drug-based colloids can undergo rapid aggregation/precipitation within minutes to hours after formation, which limits their potential use for delivery. Consistent with this, co-formulated drug colloids with either dye with chlorotrianisene, fulvestrant, lapatinib and nilotinib double in size after about 1 h (Figure 1), while lapatinib:EB precipitated within 30 minutes. Still, dye co-formulations at least partially inhibit the growth rate of these colloids, as they remain far more stable in high salt buffer compared to the parent compounds alone. More encouragingly still, sorafenib:CR, sorafenib:EB, vemurafinib:CR and TIPT:CR maintained their sizes in solution up to 24 h post-formation (Figure 2a, Supplementary Figure 2). For example, sorafenib:EB undergoes little change in diameter (Figure 2a, blue line), and sorafenib:CR undergoes no detectable change at all (Figure 2a, red line). Moreover, sorafenib:CR, TIPT:CR and vemurafenib:CR formed stable particles, each with hydrodynamic diameters below 50 nm, readily characterized by TEM (Figure 2b–c), with clear populations of spherical particles visible across all fields of view.
Figure 1.
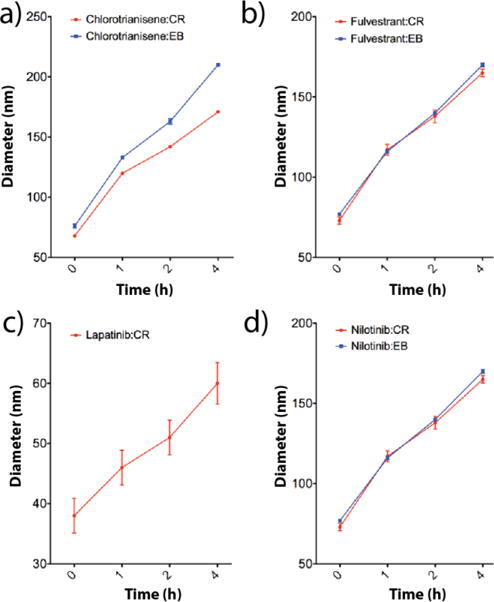
Colloidal formulations containing 50 uM drug and 50 uM of either CR or EB all increase in diameter after 4 h, as measured by DLS: a) chlorotrianisene:CR and chlorotrianisene:EB, b) fulvestrant:CR and fulvestrant:EB, c) lapatinib:CR and d) nilotinib:CR and nilotinib:EB (n=3 independent colloid formulations, mean± SD). Lapatinib:EB is not included as this solution precipitated within 1 h.
Figure 2.
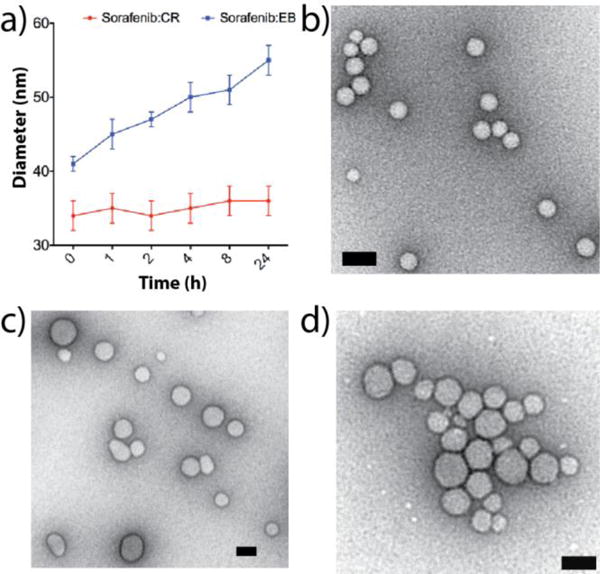
a) Sorafenib:CR (red line) and sorafenib:EB (50 μM per compound, PBS) show little change in hydrodynamic diameters over a 24 h incubation period (n = 3 independent colloid formulations, mean ± SD). b–d) TEM images for b) sorafenib:CR, c) TIPT:CR, and d) vemurafenib:CR reveal homogeneous populations of particles (scale bars 50 nm).
Effects of Concentration and Drug:Dye Ratio on Colloid Size/Stability
Encouraged by the substantial effect CR and EB had on both particle size and overall stability in PBS, we endeavored to push concentration well above the CAC for each of the best behaved drug aggregators (sorafenib, TIPT, vemurafenib) and attempted to optimize the drug:dye ratio. Figure 3 shows scattering data obtained for each of the four co-aggregate solutions at a parent compound concentration of 500 μM and varied dye concentration. A common trend among the drug colloid formers is that a 500:1 molar ratio (drug:dye) is insufficient to alter colloidal size, with precipitation occurring almost immediately in all cases. For CR containing colloids (Figure 3a–c) the same can be said of the 50:1 ratio. By 25:1 a significant shift in scattering properties was observed, with hydrodynamic diameters dropping to < 100 nm and remaining unchanged for up to 24 h. For example, the mean diameter of sorafenib:CR at 500 μM and a drug:dye ratio of 25:1 after 24 h incubation is 80 ± 8 nm with good monodispersity (Figure 3a, brown bars), extending solubility well beyond the parent compound itself (Figure 3a, brown bars), extending solubility well beyond the parent compound itself. Importantly, both the mean diameter and polydispersity undergo little change within the 24 h incubation window for all drug:dye ratios starting from 25:1. A similar trend is observed for TIPT:CR and vemurafenib:CR (Figure 3b and c, respectively), where particle sizes are mostly stable at the ratio of 25:1 and remain stable as the amount of CR is increased within the formulation. The stability of the co-formulated colloids at 500 μM is striking, as all three of sorafenib, TIPT and vemurafenib on their own transition from colloid formation to outright precipitation in buffer well-below this concentration (TIPT, for instance, will precipitate on its own at around 50 μM). TEM confirmed the spherical nature of these highly concentrated colloids (Supplementary Figure 3) and zeta potential measurements show large negative surface potentials (sorafenib:CR −42 ± 4 mV, TIPT:CR −41 ± 1 mV and vemurafenib:CR −34 ± 4 mV), suggesting that CR is on the surface. By screening molar ratios between dye and small molecule, and increasing concentration, we found stable colloid formation well beyond the solubility limits for these known aggregating compounds.
Figure 3.
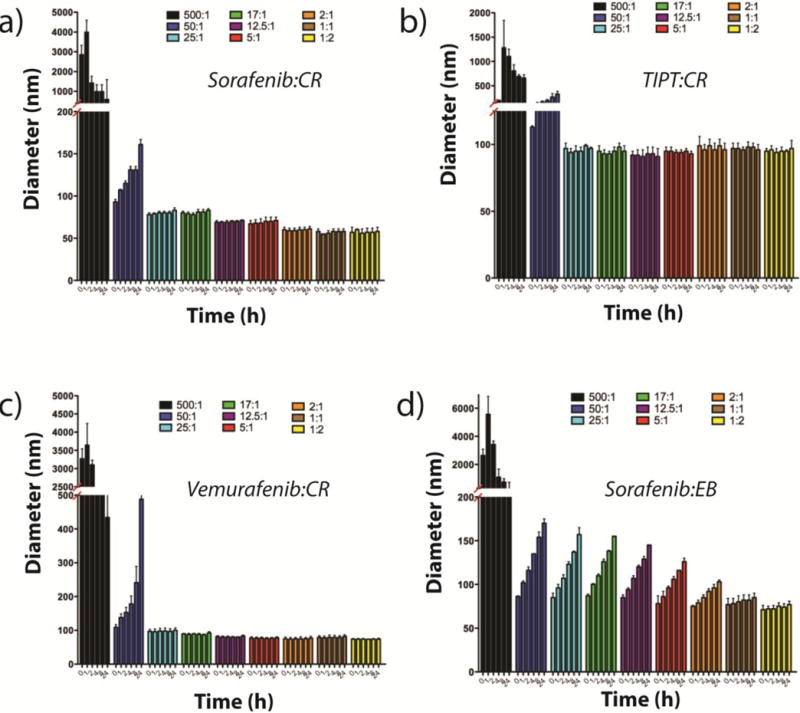
DLS measurements taken over a 24 h incubation period for a) sorafenib:CR, b) TIPT:CR, c) sorafenib:CR, and d) sorafenib:EB reveal changes in particle diameters based on the concentration of dye used. For all CR formulated colloids (a–c), excellent sizing and stability are achieved starting at a drug:dye ratio of 25:1 (drug:dye = 500 μM:X, where X = 1, 10, 20, 30, 40, 100, 250, 500 and 1000 μM; n = 3 independent colloid formulations, mean ± SD).
Co-formulated Aggregates Adsorb and Inhibit Proteins
Though the inclusion of CR improved colloid stability, the ability of these stabilized colloids to adsorb proteins was uncertain. Previous work has established that pre-incubation of colloids with protein (i.e. BSA) leads to adsorption and inhibition of most proteins.3,4,11,26–28 To test the ability of these new colloidal formulations to adsorb enzyme, their ability to inhibit the model enzyme AmpC β-lactamase (AmpC) was investigated. Consistent with their ability to adsorb protein, both sorafenib:CR and sorafenib:EB inhibit AmpC activity substantially (99% and 89%, respectively at 100 μM of both drug and dye, and 2 nM enzyme) (Figure 4a). For standard, single compound colloids, such enzyme inhibition can be reversed by pre-incubation of the colloids with a high-concentration of a second protein, inactive in the assay, such as bovine serum albumin (BSA), which is thought to prophylactically coat the colloid, preventing adsorption of AmpC, or any other enzyme. Consistent with that expectation, and the overall ability of the colloids to adsorb protein, pre-incubation of colloids with 0.16 mg/mL BSA substantially reduced their ability to inhibit AmpC, while pre-incubation with 0.32 mg/mL BSA eliminated enzyme inhibition entirely (Figure 4a). DLS confirmed that BSA addition did not substantially alter colloidal size (Figure 4b), suggesting that it was not directly disrupting the colloids. The scattering data in Figure 4b also demonstrates that these formulations are not affected by the change in formulation media from PBS to KPi. The colloids co-formulated with CR and EB thus retain their ability to adsorb proteins; they are far more stable in solution then colloids comprised of the parent compound alone.
Figure 4.
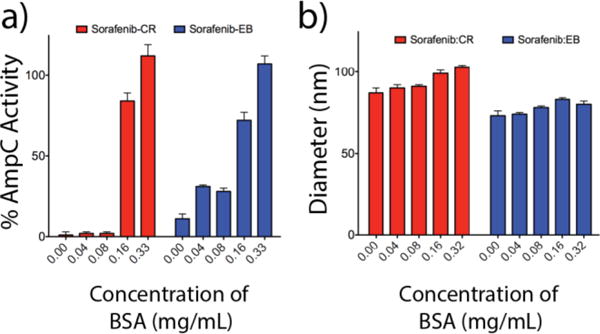
Effect of BSA on enzyme activity. a) Inhibition data for sorafenib:CR and sorafenib:EB (1:1, 100 μM, KPi) with varying concentrations of BSA obtained using the enzyme AmpC β-lactamase (n=3, mean ± SD). Full enzyme activity is recovered upon dilution into 0.32 mg/mL BSA for both formulations. b) Corresponding hydrodynamic diameters for each of the enzyme inhibition data points reveal particle stability despite the adsorption of protein.
Previous work had found that the anti-neoplastic sorafenib undergoes a drastic reduction in cytotoxic activity above its CAC in cell culture, even in the presence of 10% FBS (or in ~ 4 mg/mL BSA).10,29 This reflects formation of the colloidal species, which cannot penetrate cells, and act as reservoirs for what would otherwise be active monomer. Disruption of the colloids with Tween-80 restores full drug activity. We used sorafenib as a benchmark aggregator to further examine colloidal stability in the presence of higher protein concentrations more typically found in cell culture experiments (and in vivo). Sorafenib:CR and sorafenib:EB were prepared in PBS at 500 μM (1:1 ratio with dye) and diluted 10-fold into media. Both co-formulated colloids showed reduced cytotoxic activity, compared to colloids comprised of the parent compound alone, when incubated with the breast cancer cell line MDA-MB-231 (Figure 5a). Control cell studies show no cytotoxicity with CR and EB alone (Supplementary Figure 4). This is consistent with the greater stability of the co-formulated colloid compared to colloids of sorafenib alone, and suggests that such stability limits cytotoxicity.
Figure 5.
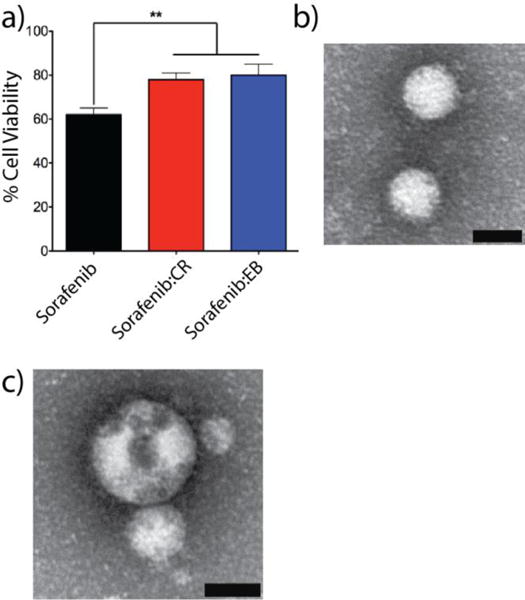
a) In vitro assessment of co-aggregated formulations sorafenib:CR (red bars) and sorafenib:EB (blue bars, 1:1, 50 μM each) with MDA-MB-231 cells reveals significantly less cytotoxicity compared to sorafenib colloids alone (black bars) (n = 3 independent measurements, mean ± SD, ** denotes p < 0.01). Representative TEM fields of view for b) sorafenib:CR and c) sorafenib:EB (1:1, 50 μM each, PBS) obtained after incubation in BSA (4 mg/mL, PBS, 4h, 37 °C). Scale bars are 50 nm.
To further demonstrate stability in high protein milieus, the co-formulated colloids were incubated with BSA (4 mg/mL) at 37 °C followed by imaging with TEM. Typical fields of view reveal the presence of well-distributed colloids for both sorafenib:CR and sorafenib:EB (Figure 5b and c, Supplementary Figure 5) formulations. Taken together, the introduction of high BSA concentrations, typically used for in vitro cell culture experiments, are not enough to disrupt these co-aggregated colloids. Furthermore, dye-formulated colloids interact with biomacromolecules in a similar fashion as the parent colloid forming compounds, but maintain their size under these conditions.
Stabilized Co-Aggregates as Enzyme Carriers
Having established that dye-formulated drug colloids were stable and monodisperse, and still retained the core property of adsorbing and inhibiting protein activity, we considered the stability of the enzymes sequestered on the colloids. We first incubated CR-stabilized colloids with three well-characterized enzymes, AmpC, MDH and trypsin, monitoring activity over 4 hours. Early optimization for the colloid-enzyme inhibition experiments identified that a minimum five-minute incubation was required to effectively adsorb and inhibit the enzymes on the colloids.3,4,11 To investigate stability over time, sorafenib:CR and vemurafenib:CR were incubated with AmpC, MDH and trypsin (all at 2 nM) for 5 min, 1, 2 and 4 hours, after which activity was re-measured following colloidal disruption with 0.1% Triton X-100 (Trx) (Figure 6). Over the course of the experiment, controls of each enzyme alone in solution (no colloid) lost most enzyme activity, dropping to 19% for AmpC (Figure 6a, black bars), 13% for MDH (Figure 6b, black bars) and 18% for trypsin (Figure 6c, black bars) after 4 hours (240 minutes). This loss in enzyme activity is expected for enzymes at these concentrations. Conversely, with sorafenib:CR (blue bars) and vemurafenib:CR (red bars) co-formulated colloids, the three enzymes retain most of their function upon colloid disruption, even after having been adsorbed on the colloid for 4 h (Figure 6). Indeed, for AmpC (Figure 6a) and trypsin (Figure 6c), both enzymes retain nearly 100% activity after 4 h (240 min) on the colloid. In contrast, whereas pure sorafenib or vemurafenib colloids also sequester and inhibit AmpC, trypsin, and MDH10, their suspensions are not stable over any length of time, and once they settle out they cannot be re-suspended in colloidal form. These results highlight the unique capacity of stable colloidal aggregates to act as chaperones for enzymes.
Figure 6.
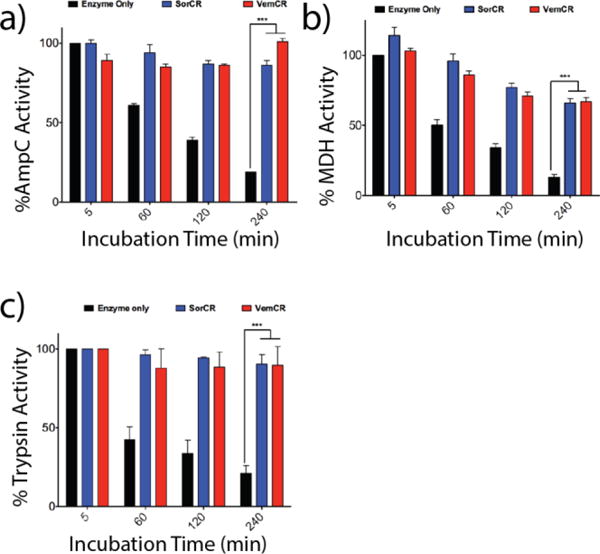
Incubation of enzyme alone (black bars), sorafenib:CR (blue bars) and vemurafenib:CR (red bars) with enzymes (a) AmpC, (b) MDH and (c) trypsin reveal significant differences in activity over a 4 h incubation period (n = 3, mean ± SD ***p < 0.001). For all three enzymes, activity is drastically reduced for the enzyme only samples with time. Adsorption to either colloidal formulation followed by subsequent particle disruption using Triton X-100 (0.1%) restored enzyme activity.
Observing that both the co-formulated colloids and their enzyme complexes had been much stabilized, we next investigated whether they could be purified and then re-used as one might want for a reliable reagent. To do so, we used centrifugation, which previous work has shown could be used to pellet colloid-protein complexes.4,9,28 For normal, single compound colloids, however, the solid pellet that results from centrifugation cannot be re-suspended into a homogenous colloidal population. We first looked at the centrifugation of formulations (500 μM, 25:1) without enzyme addition. After centrifugation, dark red pellets for sorafenib:CR and vemurafinib:CR could be readily re-suspended in KPi buffer with hydrodynamic diameters of 69 ± 2 and 71 ± 5 nm, respectively, very similar to their original diameters prior to centrifugation. This was already a substantial improvement from the irreversibly denatured protein and colloid mixture that results from after pelleting single component drug colloids from the aqueous phase.
We then incubated AmpC with sorafenib:CR colloids, followed by centrifugation. After removal of the supernatant, the pellet was re-suspended to yield a homogeneous colloid-enzyme complex of sorafenib:CR:AmpC with a hydrodynamic diameter of 70 ± 2 nm. No enzyme activity above background was observed upon addition of the β-lactamase substrate CENTA (Figure 7a, green line), indicating that the enzyme remained adsorbed to the colloid and inhibited. After the addition of Triton X-100 (TrX, 0.1%), however, the colloids were disrupted and enzymatic activity was fully restored (Figure 7a, black line). Moreover, the colloid-enzyme complex could be left for 12 h in solution without apparent increase in size (Supplementary Figure 6) or attenuation in activity (Figure 7, blue and red lines). Vemurafenib:CR colloids with adsorbed AmpC behaved essentially identically to those of sorafenib:CR (Supplementary Figure 7), with a hydrodynamic diameter of 72 ± 1 nm, enabled centrifugation, resuspension, and ultimately full restoration of enzyme activity.
Figure 7.
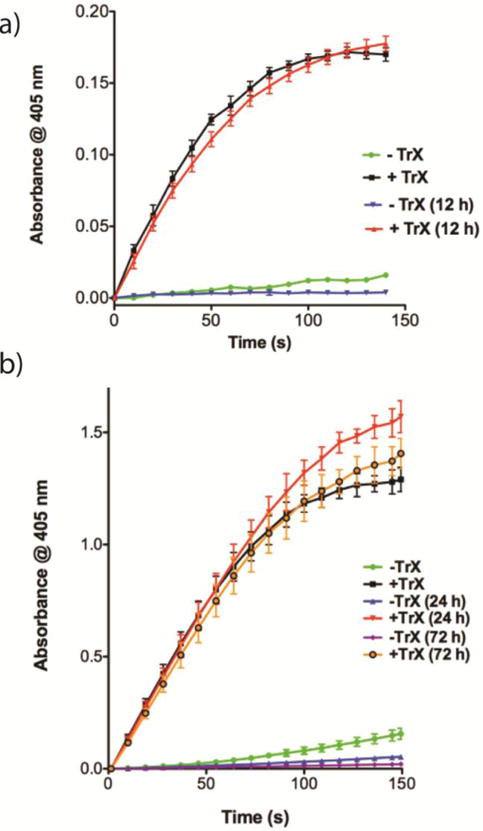
a) Centrifugation after sorafenib:CR (25:1, 500 μM, KPi) incubation with AmpC resulted in a red pellet that could be easily re-suspended in KPi buffer (n = 3, mean ± SD). Addition of β-lactamase substrate CENTA™ substrate led to limited substrate cleavage (green line). Colloid disruption using 0.1% TrX restored activity and resulted in substrate cleavage as indicated by the increase in absorbance at 405 nm (black line). Colloid-enzyme complexes were left for 12 h, after which nearly identical activity was found for solutions without (blue line) and with (red line) TrX addition. b) Studies with trypsin revealed similar trends as outlined above (n = 3, mean ± SD). Activity here was monitored after 0, 24 and 72 h. For both a) and b), activity of the released enzymes between 0 and 12, 24 or 72 h is not statistically significant.
We then undertook the same experiments with trypsin (Figure 7b), which, as a widely used proteomic reagent, has more pragmatic applications than AmpC β-lactamase. For trypsin, we observed identical behaviour to that of AmpC for sorafenib:CR colloids, indeed showing that activity could be sustained out to 72 h incubation, an extraordinary extension of the lifetime of the enzyme which, on its own, loses most of its activity after 4 hours in solution. Though only a preliminary assessment, these results demonstrate the robustness of these formulations and an increased potential for complex formation.
DISCUSSION
Colloidal aggregation of small molecules has been viewed as a nuisance for drug discovery.1,6,27 Colloids have unique properties that differ entirely from soluble monomers or precipitant. It seemed as though there was room to optimize for these special properties, rather than try to rapidly control and eliminate them. To this end, three key observations emerge from this study. First, well-known colloidal aggregating drugs may be co-formulated with bis-azo dyes to form colloids that are much more stable and much more mono-disperse than the colloidal aggregates that have been characterized over the last 15 years. In such co-formulations, the co-colloids are stable in media, more monodisperse, smaller and more stable in size than the same molecules in single molecule colloids. This increase in stability enables the formation of colloids at concentrations up to 500 μM, far above the precipitation point for pure compound in aqueous media. Second, despite this move to stability and homogeneity, the new co-formulated colloids can still adsorb protein, inhibit a range of enzymes, and indeed remain stable in a high-protein milieu, akin to those used in typical cell culture experiments. Third, these colloid-enzyme adducts are stable, the enzymes do not lose activity, as free enzymes do, and they may be spun down, re-suspended, and eventually disrupted to release fully active enzyme.
These results suggest that colloidal aggregates may be optimized as stabilizing agents for enzymes and their subsequent release. Their small size (over 30-fold smaller in radius, and 27,000-fold smaller in volume) vs their parent colloids, their size homogeneity, and their long time stability distinguish them from small molecule aggregates studied to date. Meanwhile, the ability of the colloids to stably sequester and inhibit any protein of interest, pellet and re-suspend it, and then release it fully active, portends applications where active proteins must be separated from their substrates and products, as is, for instance, the case in the proteolysis of target proteins in proteomics experiments.
Certain caveats bear mentioning. Whereas there is precedence in materials science for stabilizing colloids by co-formulation, the origins for the large effects of even small amounts of Congo Red and Evans Blue on the drug colloids is unclear. The similarities and differences between the co-formulated colloids and their parent drug colloids have yet to be fully determined. For instance, while the co-formulated colloids act in many ways like their single compound analogs in sequestering and inhibiting proteins, their capacity to load these proteins has only begun to be explored, as has the exact mechanism by which they do so. Finally, we have focused on well-studied colloidal aggregators here, which are mostly (but not exclusively) drugs. For uses as enzyme sequestering reagents, non-drug aggregators may be preferred to the drugs investigated here, though we expect the principles and empirical behavior would be largely the same, as supported by what we observed for tetraiodophenolphthalein.
CONCLUSIONS
A key observation from this study is that colloidal aggregates of small organic molecules may be optimized for stability, longevity, and size homogeneity. The dye co-formulated colloids are stable in high protein milieus, including in cell culture. This is compelling because drug colloids offer highly concentrated particles of bio-active molecules—in the case of the colloids here, they are tightly concentrated spheres of cytotoxic anti-neoplastics. Just as interesting, the stabilized colloids retain their ability to sequester and inhibit proteins and enzymes. Intriguingly, and very different from previously studied colloids, the co-formulated colloids act almost as protein chaperones that can be handled in a more robust manner. Advancing our molecular understanding of these amorphous colloidal phases and respective protein binding properties will make it possible to further explore their application in biological studies and medicine.
MATERIALS AND METHODS
Materials
Cell culture grade DMSO (Cat No D2650), Congo Red (Cat No C6277), Dulbecco’s phosphate buffered saline (DPBS, D8537), Evans Blue (Cat No E2129), β-nicotinamide adenine dinucleotide (NADH, Cat No N4505), oxaloacetate (Cat No O4126) and RPMI 1640 cell culture media (Cat No R8758) were purchased from Sigma-Aldrich. Fulvestrant (Cat No S1191) was purchased from Selleckchem. Lapatinib (HY-50898), nilotinib (Cat No HY-10159), sorafenib (Cat No HY-10201) and vemurafenib (Cat No HY-12057) were purchased from MedChem Express. 3′,3′′,5′,5′′-Tetraiodophenolphthalein (TIPT, Cat No sc-216639) was purchased from Santa Cruz Biotechnology. AmpC β-lactamase was purified from Escherichia coli as previously described.30 Malate dehydrogenase (MDH, Cat No 442610) and CENTA (Cat No 219475) were purchased from EMD Millipore. Trypsin (Cat No T0303) and Suc-Ala-Ala-Pro-Arg-pNA (Cat No L1720) were purchased from Sigma-Aldrich and BACHEM respectively. Cell line MDA-MB-231 (Cat No HTB-26) was purchased from ATCC. Triton X-100 (Cat No 21568-2500) was purchased from Acros. Presto Blue cell viability reagent (Cat No A13262) was purchased from Invitrogen. 96-well plates (Cat No 655096) for DLS were purchased from Greiner Bio-One.
Colloidal Formulations
Stock solutions of all drugs and dyes were first prepared at 100 mM in DMSO. For initial experiments, stock solutions were diluted to 10 mM. Colloid forming compounds (1 μL) were combined with either DMSO (1 μL) or dye (1 μL). Solutions were mixed briefly and DPBS (198 μL) added rapidly to form colloids. Final concentration of each component was 50 μM, with an overall 1% DMSO (v/v). For higher concentration screening experiments, the 100 mM stock solutions of colloid forming compounds (1 μL) were combined with an appropriate amount of DMSO or dye to yield a total volume of 3 μL. Correspondingly, 100 mM stock solutions of CR and EB were diluted accordingly, such that combination 2 μL would generate the desired final ratios of drug:dye after rapid addition of DPBS (197 μL). Final % DMSO was 1.5 for these experiments. For all sizing/stability experiments buffers were filtered (0.1 μm) prior to colloid preparation.
Cell Culture and Proliferation Assays
MDA-MB-231 cells were maintained (< 10 passages) in a tissue culture incubator (37 °C, 5% CO2, 95% humidified) in plastic culture flasks in RPMI 1640. Growth media was supplemented with 10% FBS, 10 U/mL penicillin, and 10 μg/mL streptomycin. Cells were grown to be ~70% confluent and then passaged at a 1:4 ratio (cells:media) according to the manufacturer’s protocol. For proliferation assays with sorafenib, cells were seeded at 10,000 cells/well and allowed to adhere overnight. Co-aggregate formulations were prepared as described above in DPBS, with the concentration of each component at 500 μM for CR, EB, sorafenib:CR and sorafenib:CR. These solutions were then diluted 10-fold in RPMI/10% FBS to a final concentration of 50 μM and 8% FBS (0.15% DMSO). Colloids comprised solely of sorafenib were prepared directly from the 100 mM stock solution (1.5 μL) and addition of 998.5 μL DPBS/RPMI/8% FBS. All drug formulations and control media were incubated with cells for 4 h, after which solutions were removed and full media (RPMI/10% FBS) was added. Three different flasks of cells were used for each biological replicate, with a technical replicate of 3 within each plate for the various formulations tested. After an additional 20 h growth period, proliferation was determined using Presto Blue according to the manufacturer’s protocol. All experiments were performed in triplicate. Relative proliferation is defined as (fluorescence of treated cells/fluorescence if untreated cell) × 100%.
Dynamic Light Scattering and Zeta Potential
Colloid hydrodynamic diameters, polydispersity and scattering intensities were determined using a DynaPro Plate Reader II (Wyatt Technology) with a 60 mW laser operating at 830 nm and detector angle of 158°. All formulations were prepared in triplicate, with measurements being performed in a 96-well format (200 μL sample/well) at 25 °C. All data were acquired using the Dynamics software (Wyatt Technology). For each sample, 15 acquisitions were acquired, with the individual averages indicated by the software. Laser power was automatically adjusted for each experiment, necessitating the use of normalized scattering intensity (cnts/sec) for data interpretation.
Zeta potential values were determined using a Malvern Zetasizer Nano ZS, equipped with a 4 mW, 633 nm laser, with all measurements carried out at 25 °C. All colloidal samples were prepared at 500 μM (25:1 ratio of drug to dye, 1 mL total volume) in 0.1 μm filtered 10 mM NaCl (pH ~ 7). Zeta potentials were measured using folded capillary cells (Malvern, DTS 1060). The average of three individual samples, with ~20 runs per sample, are reported.
Transmission Electron Microscopy
For both 50 and 500 μM colloidal formulations, 5 μL was deposited onto a freshly glow-discharged 400 mesh carbon coated copper TEM grid (Ted Pella, Inc.) and allowed to adhere for 5 min. Excess liquid was removed with filter paper, followed by a quick wash with double-distilled water (5 μL). Particles were then stained with 1% ammonium molybdate (w/v, pH 7, 5 μL) for 30 seconds. Stain was removed and samples imaged using a Hitachi H-7000 microscope operating at 75 kV. Images were captured using an Advanced Microscopy Techniques (AMT) XR-60 CCD camera with typical magnifications between 30000 – 100000×. Images were analyzed using ImageJ 64 software and processed with Photoshop. For stability experiments, sorafenibCR and sorafenib:EB (1:1, 500 μM each, PBS, 1.5% DMSO) were diluted after formation into PBS containing BSA (4 mg/mL). After incubation for 4 h at 37 °C, samples were deposited onto TEM grids and imaged as outlined above.
Enzyme Assays
AmpC and MDH experiments were performed in 50 mM potassium phosphate (KPi, pH 7) buffer and reactions assayed in a total volume of 200 μL using a plate reader. Trypsin experiments were performed using a HP8453a spectrophotometer in kinetic mode using UV-Vis Chemstation software (Agilent Technologies). First order reaction rates were calculated manually using linear regression analysis. In each experiment, three independent measurements were performed.
For BSA-enzyme experiments, sorafenib:CR and sorafenib:EB (each 1:1, respectively) were diluted to 50 μM in KPi buffer (final 0.15% DMSO). BSA was added to each colloidal solution to reach final concentrations of 0, 0.04, 0.08, 0.16 and 0.32 mg/mL, followed by a 5-minute incubation time at room temperature. AmpC β–lactamase (AmpC) was then added (Final concentration = 2 nM) and incubated for an additional 5 minutes. Colloid solutions were then added to the chromogenic substrate CENTA (Final concentration = 200 μM) and the reaction followed by spectrophotometric monitoring at 405 nm using a Tecan plate reader (96-well format) for a total of 120 seconds (10 seconds/reading).
For enzyme protection experiments, sorafenib:CR and vemurafenib:CR (each 25:1, respectively) were diluted to 100 μM in KPi buffer (final 0.30% DMSO). A 1000 μL solution of each colloid with either AmpC (Final concentration = 2 nM), malate dehydrogenase (MDH, Final concentration = 2 nM) or trypsin (Final concentration = 4nM) was prepared. From this colloid-enzyme stock solution, 200 μL were immediately taken out, transferred a new centrifuge tube and allowed to incubate at room temperature for 5, 60, 120 and 240 minutes. Samples were then added to the appropriate reagents to initiate enzymolysis. For AmpC, the reaction was monitored as outlined above using CENTA. For MDH, the reaction was initiated with oxaloacetate (Final concentration = 200 μM) and NADH (Final concentration = 200 μM), which were freshly prepared for each time point, and monitored at 340 nm using a Biotech Plate Reader. For trypsin, the reaction was monitored by cleavage of the Suc-Ala-Ala-Pro-Arg-pNA substrate (Final concentration = 60 μM) at 405 nm using the spectrophotometer. Control experiments were performed for each enzyme in KPi buffer with 0.3% DMSO.
Enzyme-centrifugation experiments were performed using 1000 μL undiluted formulations of sorafenib:CR and vemurafenib:CR (each 25:1, respectively) at 500 μM in KPi buffer (final 1.5% DMSO). AmpC (750 ng) and trypsin (1000ng) was added to colloid solutions, followed by a 30-minute incubation period at room temperature. Samples were then centrifuged (16000× g, 4 °C, 60 min). The supernatant was carefully removed. For enzyme activity experiments, the pellet was rapidly re-suspended in 1000 μL KPi buffer. DLS measurements were first performed on 200 μL of this solution. AmpC and trypsin activity were monitored using CENTA and Suc-Ala-Ala-Pro-Arg-pNA respectively as described above with or without the addition of Triton X-100 (Final concentration = 0.1%).
Graphing and Statistics
All statistical analyses were performed using Graph Pad Prism version 5.00 for Mac (Graph Pad Software, San Diego California USA, www.graphpad.com). Differences among groups were assessed by one-way ANOVA with Bonferroni post hoc correction to identify statistical differences among three or more treatments. Alpha levels were set at 0.05, and a p-value of ≤0.05 was set as the criteria for statistical significance. Data are presented as mean ± standard deviation.
Supplementary Material
Acknowledgments
We thank Mr. S. Doyle and Mr. B. Calvieri from the University of Toronto Microscopy Imaging Laboratory (MIL) for assistance with TEM imaging, Dr. L. Nedyalkova for preparation of AmpC and members of the Shoichet lab for their thoughtful review.
Funding Sources
This work was supported by grants from the National Institutes of Health (GM71630 to BKS and MSS) and the Canadian Cancer Society Research Institute (to MSS). CKM is supported in part by a Natural Sciences and Engineering Research Council (NSERC) postdoctoral fellowship. AG is supported in part by an NSERC graduate scholarship.
ABBREVIATIONS
- API
active pharmaceutical ingredient
- BSA
Bovine serum albumin
- CR
Red
- EB
Evans Blue
- DLS
dynamic light scattering
- SD
standard deviation
- TEM
transmission electron microscopy
- TrX
Triton X-100
Footnotes
Supporting Information. Additional colloid characterization using DLS and TEM, control cytoxicity studies, colloid-enzyme studies. This material is available free of charge via the Internet at http://pubs.acs.org.
Notes
The authors declare no competing financial interest.
Contributor Information
Brian K. Shoichet, Email: bshoichet@gmail.com.
Molly S. Shoichet, Email: molly.shoichet@utoronto.ca.
References
- 1.Shoichet BK. Screening in a spirit haunted world. Drug discovery today. 2006;11:607–615. doi: 10.1016/j.drudis.2006.05.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Brick MC, Palmer HJ, Whitesides TH. Formation of colloidal dispersions of organic materials in aqueous media by solvent shifting. Langmuir. 2003;19:6367–6380. [Google Scholar]
- 3.McGovern SL, Caselli E, Grigorieff N, Shoichet BK. A common mechanism underlying promiscuous inhibitors from virtual and high-throughput screening. J Med Chem. 2002;45:1712–1722. doi: 10.1021/jm010533y. [DOI] [PubMed] [Google Scholar]
- 4.McGovern SL, Helfand BT, Feng B, Shoichet BK. A specific mechanism of nonspecific inhibition. J Med Chem. 2003;46:4265–4272. doi: 10.1021/jm030266r. [DOI] [PubMed] [Google Scholar]
- 5.Thorne N, Auld DS, Inglese J. Apparent activity in high-throughput screening: origins of compound-dependent assay interference. Curr Opin Chem Biol. 2010;14:315–324. doi: 10.1016/j.cbpa.2010.03.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Coan KE, Ottl J, Klumpp M. Non-stoichiometric inhibition in biochemical high-throughput screening. Expert Opin Drug Discov. 2011;6:405–417. doi: 10.1517/17460441.2011.561309. [DOI] [PubMed] [Google Scholar]
- 7.Dahlin JL, Walters MA. The essential roles of chemistry in high-throughput screening triage. Future Med Chem. 2014;6:1265–1290. doi: 10.4155/fmc.14.60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Feng BY, Simeonov A, Jadhav A, Babaoglu K, Inglese J, Shoichet BK, Austin CP. A high-throughput screen for aggregation-based inhibition in a large compound library. J Med Chem. 2007;50:2385–2390. doi: 10.1021/jm061317y. [DOI] [PubMed] [Google Scholar]
- 9.Doak AK, Wille H, Prusiner SB, Shoichet BK. Colloid formation by drugs in simulated intestinal fluid. J Med Chem. 2010;53:4259–4265. doi: 10.1021/jm100254w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Owen SC, Doak AK, Wassam P, Shoichet MS, Shoichet BK. Colloidal aggregation affects the efficacy of anticancer drugs in cell culture. ACS Chem Biol. 2012;7:1429–1435. doi: 10.1021/cb300189b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Seidler J, McGovern SL, Doman TN, Shoichet BK. Identification and prediction of promiscuous aggregating inhibitors among known drugs. J Med Chem. 2003;46:4477–4486. doi: 10.1021/jm030191r. [DOI] [PubMed] [Google Scholar]
- 12.Irwin JJ, Duan D, Torosyan H, Doak AK, Ziebart KT, Sterling T, Tumanian G, Shoichet BK. An Aggregation Advisor for Ligand Discovery. J Med Chem. 2015 doi: 10.1021/acs.jmedchem.5b01105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ilevbare GA, Taylor LS. Liquid-Liquid Phase Separation in Highly Supersaturated Aqueous Solutions of Poorly Water-Soluble Drugs: Implications for Solubility Enhancing Formulations. Crystal Growth & Design. 2013;13:1497–1509. [Google Scholar]
- 14.Almeida e Sousa L, Reutzel-Edens SM, Stephenson GA, Taylor LS. Assessment of the amorphous “solubility” of a group of diverse drugs using new experimental and theoretical approaches. Mol Pharm. 2015;12:484–495. doi: 10.1021/mp500571m. [DOI] [PubMed] [Google Scholar]
- 15.Sassano MF, Doak AK, Roth BL, Shoichet BK. Colloidal aggregation causes inhibition of G protein-coupled receptors. J Med Chem. 2013;56:2406–2414. doi: 10.1021/jm301749y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ilevbare GA, Liu H, Edgar KJ, Taylor LS. Impact of polymers on crystal growth rate of structurally diverse compounds from aqueous solution. Mol Pharm. 2013;10:2381–2393. doi: 10.1021/mp400029v. [DOI] [PubMed] [Google Scholar]
- 17.Ilevbare GA, Liu HY, Edgar KJ, Taylor LS. Maintaining Supersaturation in Aqueous Drug Solutions: Impact of Different Polymers on Induction Times. Crystal Growth & Design. 2013;13:740–751. [Google Scholar]
- 18.Jackson MJ, Toth SJ, Kestur US, Huang J, Qian F, Hussain MA, Simpson GJ, Taylor LS. Impact of polymers on the precipitation behavior of highly supersaturated aqueous danazol solutions. Mol Pharm. 2014;11:3027–3038. doi: 10.1021/mp500201s. [DOI] [PubMed] [Google Scholar]
- 19.Grohganz H, Priemel PA, Lobmann K, Nielsen LH, Laitinen R, Mullertz A, Van den Mooter G, Rades T. Refining stability and dissolution rate of amorphous drug formulations. Expert opinion on drug delivery. 2014;11:977–989. doi: 10.1517/17425247.2014.911728. [DOI] [PubMed] [Google Scholar]
- 20.D’Addio SM, Prud’homme RK. Controlling drug nanoparticle formation by rapid precipitation. Adv Drug Deliv Rev. 2011;63:417–426. doi: 10.1016/j.addr.2011.04.005. [DOI] [PubMed] [Google Scholar]
- 21.Lepeltier E, Bourgaux C, Couvreur P. Nanoprecipitation and the “Ouzo effect”: Application to drug delivery devices. Adv Drug Deliv Rev. 2014;71:86–97. doi: 10.1016/j.addr.2013.12.009. [DOI] [PubMed] [Google Scholar]
- 22.Fuhrmann K, Gauthier MA, Leroux JC. Targeting of injectable drug nanocrystals. Mol Pharm. 2014;11:1762–1771. doi: 10.1021/mp5001247. [DOI] [PubMed] [Google Scholar]
- 23.Feng BY, Toyama BH, Wille H, Colby DW, Collins SR, May BC, Prusiner SB, Weissman J, Shoichet BK. Small-molecule aggregates inhibit amyloid polymerization. Nat Chem Biol. 2008;4:197–199. doi: 10.1038/nchembio.65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Skowronek M, Roterman I, Konieczny L, Stopa B, Rybarska J, Piekarska B. Why Do Congo Red, Evans Blue, and Trypan Blue Differ in Their Complexation Properties? Journal of Computational Chemistry. 2000;21:656–667. [Google Scholar]
- 25.Feng BY, Shoichet BK. Synergy and antagonism of promiscuous inhibition in multiple-compound mixtures. J Med Chem. 2006;49:2151–2154. doi: 10.1021/jm060029z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Feng BY, Shelat A, Doman TN, Guy RK, Shoichet BK. High-throughput assays for promiscuous inhibitors. Nat Chem Biol. 2005;1:146–148. doi: 10.1038/nchembio718. [DOI] [PubMed] [Google Scholar]
- 27.Feng BY, Shoichet BK. A detergent-based assay for the detection of promiscuous inhibitors. Nature protocols. 2006;1:550–553. doi: 10.1038/nprot.2006.77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Coan KE, Maltby DA, Burlingame AL, Shoichet BK. Promiscuous aggregate-based inhibitors promote enzyme unfolding. J Med Chem. 2009;52:2067–2075. doi: 10.1021/jm801605r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Owen SC, Doak AK, Ganesh AN, Nedyalkova L, McLaughlin CK, Shoichet BK, Shoichet MS. Colloidal drug formulations can explain “bell-shaped” concentration-response curves. ACS Chem Biol. 2014;9:777–784. doi: 10.1021/cb4007584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Weston GS, Blazquez J, Baquero F, Shoichet BK. Structure-based enhancement of boronic acid-based inhibitors of AmpC beta-lactamase. J Med Chem. 1998;41:4577–4586. doi: 10.1021/jm980343w. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.