

ABSTRACT
Metastasis requires tumor cells to overcome a series of challenges to successfully travel to and colonize new microenvironments. As an adaptive (or maladaptive) response to stress, macroautophagy/autophagy has garnered increasing interest with respect to cancer metastasis, supported by clinical observations of increased autophagic flux in distant metastases relative to primary tumors. Recently, we identified a new role for autophagy in tumor cell motility through the turnover of focal adhesions, large multi-protein structures that link extracellular matrix-bound integrins to the cytoskeleton. The disassembly of focal adhesions at the cell rear is critical to forward movement and successful migration/invasion. We demonstrated that the focal adhesion protein PXN (paxillin), which serves as a crucial scaffolding and signal integrator, binds directly to LC3B through a conserved LC3-interacting region (LIR) motif to stimulate focal adhesion disassembly and metastasis and that this interaction is further promoted by oncogenic SRC.
KEYWORDS: autophagy, cell motility, focal adhesion, metastasis, paxillin, SRC
We identified a specific requirement for autophagy in cell motility during metastasis that is independent of effects on proliferation or viability using an orthotopic murine model of metastatic mammary cancer. Unlike in many other models, the inhibition of autophagy has no effect on primary tumor growth, possibly due to the highly aggressive nature of the tumor model employed. However, autophagy-deficient cells are unable to metastasize from the primary tumor due to the inability to disassemble focal adhesions as the result of increased levels of PXN. Reducing PXN levels restores both focal adhesion morphology and motility, demonstrating that the motility defects in autophagy-deficient cells are due to the inability to degrade PXN and thus properly regulate focal adhesion dynamics.
The interaction of cargo destined for autophagic degradation with LC3/Atg8-related molecules at the elongating phagophore is mediated through LIR motifs characterized by the conserved sequence [W/F/Y]-xx-[L/I/V]. We have added to the ever-growing list of LIR-containing proteins with the identification of an evolutionarily conserved LIR motif at residues 40–43 (YQEI) in the N-terminal region of PXN. In vitro binding assays demonstrated a direct interaction between LC3B and PXN, and the PXN LIR motif is required for LC3B-PXN colocalization and co-immunoprecipitation in metastatic cells. Contrary to other reports, we observed no requirement for the SQSTM1/p62 or NBR1 cargo receptors in PXN degradation.
Given the utility of autophagy for degrading large macromolecular structures and PXN's role as a scaffolding protein within focal adhesions, our work suggests that PXN acts as an autophagy cargo receptor for focal adhesions, forming a bridge between phagophore-bound processed LC3B and focal adhesion proteins. Indeed, other groups have also observed elongating phagophores at focal adhesions. Alternatively, autophagy may siphon PXN from focal adhesions to promote focal adhesion disassembly, although ongoing work in our lab suggests that PXN localization at focal adhesions is required for interaction with LC3B, possibly because of additional direct or receptor-mediated interactions between phagophores and other focal adhesion proteins, including autophagy-regulated proteins such as SRC. Whether autophagic degradation of PXN represents a unique method of regulation in metastatic cells or a general requirement in cell motility remains to be determined.
Interactions of LIR-containing proteins with LC3 can be regulated by post-translational modifications, including phosphorylation both around (e.g., in the cases of OPTN and BNIP3) and within (e.g., in the case of FUNDC1) the LIR motif. The +1 residue of the PXN LIR motif, Y40, is an experimentally validated but previously uncharacterized target of SRC, a known modulator of PXN and focal adhesion turnover. We demonstrated that oncogenic SRC strongly promotes the PXN-LC3B interaction in a manner dependent on Y40 but independent of canonical SRC phosphorylation at Y31 and Y118. In conjunction with observations that the interaction is dependent on SRC kinase activity, these data suggest that phosphorylation of the LIR Y40 residue promotes the PXN-LC3B interaction, in contrast to negative SRC regulation of the FUNDC1-LC3 interaction. Interestingly, we observed that a Y40E phosphomimetic PXN mutant is highly unstable in cells, consistent with increased autophagic turnover of PXN phosphorylated on Y40. However, we cannot exclude the possibility that the SRC effect on focal adhesion disassembly involves the phosphorylation of other sites or proteins. Most intriguingly, the ability of oncogenic SRC to promote migration/invasion was completely abrogated in the absence of intact autophagy, which represents the first suggestion that autophagy may be critical to SRC-mediated motility (Fig. 1).
Figure 1.
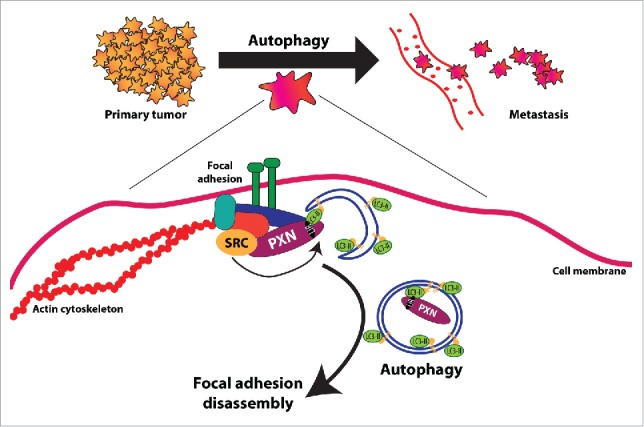
Novel role for autophagy downstream of oncogenic SRC in promoting focal adhesion disassembly and tumor cell motility. Analyses from primary human cancers and data from genetically engineered mouse models link autophagy to tumor progression to metastasis. However, the molecular mechanisms by which autophagy promotes metastasis are relatively unknown. Our work contributes to a greater understanding of how autophagy promotes metastasis by identifying a critical role for autophagy in metastatic breast and melanoma cells in stimulating focal adhesion disassembly and tumor cell motility. This was achieved through autophagic targeting and degradation of PXN, a critical focal adhesion protein that we show interacts directly with LC3B at the phagophore. Significantly in terms of tumorigenesis, this direct protein-protein interaction between PXN and LC3B is strongly enhanced by oncogenic SRC and, indeed, we also showed that functional autophagy is required for SRC-induced migration and invasion by metastatic tumor cells.
SRC is frequently activated or overexpressed in solid tumors. Although not a primary driver of tumorigenesis, SRC has been suggested to play a critical role in bone metastases in breast cancer. Thus, SRC inhibitors such as dasatinib may represent an effective means of halting metastasis of breast and other cancers, and improved SRC inhibitors with increased selectivity and efficacy are currently being developed. Although clinical trials indicate that SRC inhibitor monotherapy may be ineffective in cancer therapy, our work suggests that autophagy inhibitors may potentiate SRC therapy by targeting a specific requirement for autophagy in SRC-regulated focal adhesion turnover. The potential to prevent tumor cell metastasis could prove to be highly useful within the context of ductal carcinoma in situ (DCIS) and other isolated primary tumors.
Our work has focused on the role of autophagy in focal adhesion turnover in metastatic cells (Fig. 1). There is a growing body of work linking autophagy to different stages of the metastatic cascade, including the secretion of pro-migratory factors, epithelial-to-mesenchymal acquisition, and anoikis resistance. With respect to migration, autophagy exhibits reciprocal regulation of the RHO family of GTPases and cytoskeletal dynamics, and may be involved in the intracellular degradation of extracellular matrix components. As in primary tumor growth, the effect of autophagy on motility is likely to differ among cell types, and our work indicates that SRC status may dictate which cells rely on autophagy for turnover of focal adhesions. It will be interesting to determine whether autophagic focal adhesion turnover and the requirement for intact autophagy in SRC-stimulated motility are required during nonpathological wound healing or immune cell migration. Indeed, autophagy is well-positioned to act as a key regulatory node that allows cells to respond to extracellular or intracellular conditions by balancing cell movement to nutrient-replete areas or metastatic niches with motility arrest, and prosurvival autophagy or cellular death. Future work exploring autophagy in the context of physiological and pathological cell migration will likely reveal many more connections between autophagy and cell motility.
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
Funding
Grant support for this work includes NIH RO1 CA 162405 to KFM, T32 GM007281 supported EEM and T32 CA009594 supported MNS.