

ABSTRACT
BBC3 (BCL2 binding component 3) is a known apoptosis inducer; however, its role in microglial survival remains poorly understood. In addition to the classical transcription factor TRP53, Mir143 is involved in BBC3 expression at the post-transcriptional level. Here, we identify unique roles of Mir143-BBC3 in mediating microglial survival via the regulation of the interplay between apoptosis and autophagy. Autophagy inhibition accelerated methamphetamine-induced apoptosis, whereas autophagy induction attenuated the decrease in microglial survival. Moreover, anti-Mir143-dependent BBC3 upregulation reversed the methamphetamine-induced decrease in microglial survival via the regulation of apoptosis and autophagy. The in vivo relevance of these findings was confirmed in mouse models, which demonstrated that the microinjection of anti-Mir143 into the hippocampus ameliorated the methamphetamine-induced decrease in microglia as well as that observed in heterozygous Mir143+/− mice. These findings provide new insight regarding the specific contributions of Mir143-BBC3 to microglial survival in the context of drug abuse.
KEYWORDS: apoptosis, autophagy, BBC3, methamphetamine, microglia, Mir143
Introduction
Methamphetamine abuse is a major social and health concern. Methamphetamine is a popular addictive pharmacological psychostimulant of the central nervous system (CNS), and its use is associated with multiple neuropsychiatric adverse reactions as well as neurotoxicity to the dopaminergic and serotonergic systems of the brain.1,2 Methamphetamine-induced neurotoxicity is associated with microglial activation that is thought to participate in either pro-toxic or protective mechanisms in the brain. Microglia, CNS cells of the myeloid lineage, play an important role in immune surveillance and compromised microglial function is associated with various neurological pathologies.3,4
Accumulating evidence suggests that microglia might also be activated during the process of methamphetamine-induced toxicity in animals.5-9 A previous study indicated that chronic methamphetamine abuse is also associated with microglial activation in the brains of human methamphetamine addicts.10 Activated microglia eventually undergo apoptosis by a process known as activation-induced cell death (AICD). Activated microglia secrete not onlyneurotrophic factors, thereby prolonging neuronal survival, but also cytotoxic mediators, such as nitric oxide (NO), and cytokines, such as TNF and IL1B, that induce inflammation and neurotoxicity. Cytotoxic factors such as NO and pro-inflammatory cytokines that are secreted from activated microglia have been shown to mediate AICD through the regulation of apoptotic proteins, including caspases and the BCL2 family of proteins.11 Although methamphetamine is known to induce microglial activation, whether methamphetamine induces AICD in microglia and the molecular mechanisms in this process remains elusive.
BBC3/PUMA (BCL2 binding component 3) is an essential apoptosis inducer. BBC3 is one of the most common apoptosis inducers among the BCL2-homology 3 (BH3)-only subgroup of BCL2 family members.12,13 BBC3 is transcriptionally activated by a wide range of apoptotic stimuli and transduces proximal death signals to the mitochondria. BBC3 directly binds to all 5 known anti-apoptotic BCL2 family members with high affinities through its BH3 domain. The binding of BBC3 to the BCL2-like proteins results in the displacement of BAX-BAK1 and the activation of these proteins via the formation of multimeric pore-like structures on the mitochondrial outer membrane, leading to mitochondrial dysfunction and caspase activation. BBC3 is implicated in many pathological and physiological processes, including cancer, tissue injury, neurodegenerative diseases, immune response and bacterial or viral infections.14 Although the function of BBC3 in apoptosis has been extensively illustrated in different tissues, whether BBC3 is involved in methamphetamine-induced microglial apoptosis remains poorly understood.
BBC3 was initially identified as a transcriptional target of TP53 and as a mediator of DNA damage-induced apoptosis;15,16 however, the regulation of Bbc3 expression by noncoding RNAs in microglia has not yet been explored. Computational algorithms such as TargetScan were employed to identify the microRNAs (miRs) that target evolutionarily conserved sequences in the Bbc3 gene. Mir143, which is conserved among vertebrates, was predicted to target Bbc3. miRs are small, evolutionarily conserved noncoding RNAs that are derived from substantially larger primary transcripts. Recent studies have demonstrated that miRs expressed by immune cells can target proteins that regulate inflammation and consequently affect the magnitude of the immune response.17 miRs are becoming increasingly recognized as important regulators of cell functions, including glial activation.17-23 Mir143 has a well-established role in the differentiation and proliferation of bovine intramuscular preadipocytes.24 Moreover, Mir143 decreases cell growth by targeting SDC1 (syndecan1) in melanoma25 as well as in colon tumor cells.26 Jordan et al. demonstrated that obesity-induced Mir143 overexpression (OE) inhibits insulin-stimulated AKT1/Akt activation and impairs glucose metabolism.27 However, the role of Mir143 in mediating microglial survival remains unexplored.
Our study revealed an unexpected function of BBC3 that was regulated by Mir143 at the post-transcriptional level via the regulation of the interplay between autophagy and apoptosis, which contradicts its known role as an essential apoptosis inducer. These findings provide the first evidence that the Mir143-BBC3 axis mediates a regulatory pathway critical for the regulation of microglial survival. Specific blockage of Mir143 could be a potential therapeutic target for treating decreased microglial survival in the context of drug abuse and other neurodegenerative diseases.
Results
Paradoxical role of BBC3 in reduced microglial survival induced by methamphetamine
Although BBC3 has been intensively studied for many years, its role in the viability of microglia treated with methamphetamine has remained elusive. As an initial screen to better understand how methamphetamine affects microglial survival, we examined the effect of methamphetamine on microglial survival in various brain regions in mice. As shown in Fig. S1A, unlike in the striatum and cortex, administration of methamphetamine significantly decreased the number of microglial cells in the hippocampus, as evidenced by the finding that the expression of the microglial marker-AIF1/Iba-1 (allograft inflammatory factor 1) in the hippocampus was significantly decreased compared with the saline control group. Therefore, the hippocampus was chosen as the region of interest for our in vivo study. Because BBC3 functions as an essential apoptosis inducer, we hypothesized that BBC3 deficiency might attenuate methamphetamine-induced cell death. As shown in Fig. 1A and B, methamphetamine administration significantly decreased the number of AIF1-positive cells in the hippocampus. Surprisingly, BBC3 deficiency further decreased the number of microglia in the presence of methamphetamine instead of reversing the methamphetamine-induced decrease in microglia cell number, contradicting the known role of BBC3 as an apoptosis inducer (Fig. 1A and B).
Figure 1.
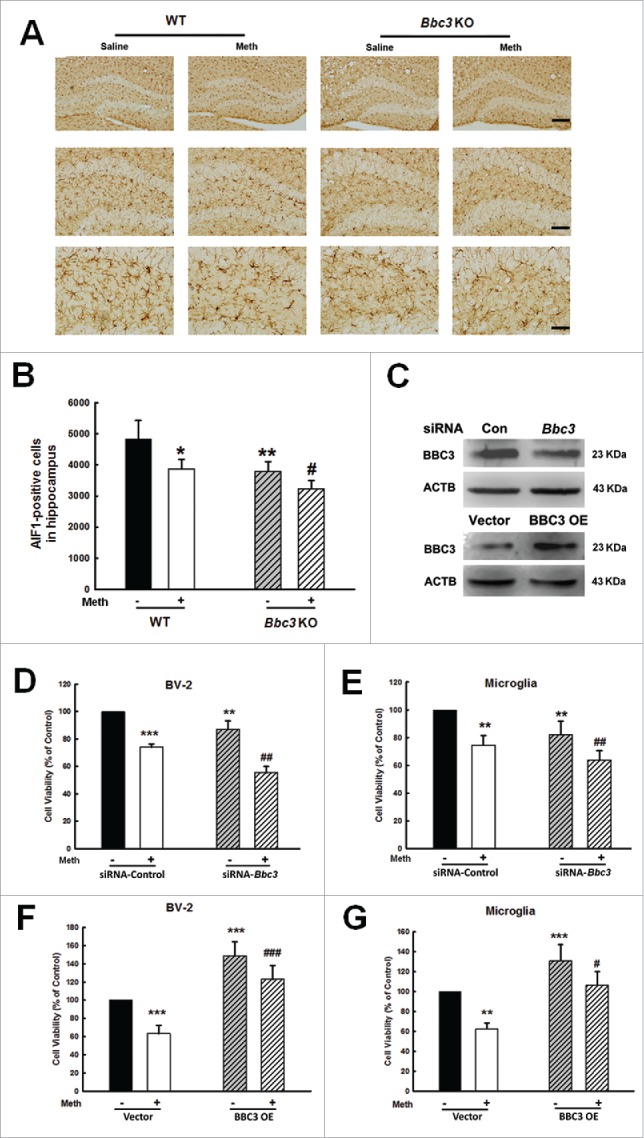
Paradoxical role of BBC3 in the methamphetamine-induced decrease in microglial survival. (A) The effect of methamphetamine on the survival of microglia in the hippocampus of WT and Bbc3 KO mice. WT and Bbc3 KO mice were treated with methamphetamine (intraperitoneal, 30 mg/kg) for 48 h, followed by AIF1 immunostaining. Representative image of AIF1 immunostaining in the hippocampus of mice injected with saline/methamphetamine. Scale bars: 200 μm (upper panel), 100 μm (middle panel), and 50 μm (lower panel). (B) Stereology of AIF1-positive microglia in the hippocampus. N = 6 animals/group. *, p < 0.05, **, p < 0.01 vs. the saline-treated group; #, p < 0.05 vs. the methamphetamine-treated group using one-way ANOVA followed by the Holm-Sidak test. (C) Bbc3 siRNA lentivirus was transduced to decrease BBC3 expression (upper panel), and the cells were transduced with BBC3 OE lentivirus to increase BBC3 expression (lower panel). (D and E) Transduction of cells with Bbc3 siRNA lentivirus failed to ameliorate the decreased microglial survival as assessed by CCK8 assay in both BV-2 cells (D) and primary mouse microglia (E). (F and G) Transduction of cells with a BBC3 OE lentivirus ameliorated the decreased microglial survival as assessed by CCK8 in both BV-2 cells (F) and primary mouse microglia (G). All data are given as the mean ± SD of 3 independent experiments. **, p < 0.01 and ***, p < 0.001 vs. the siRNA-control or vector group; #, p < 0.05, ##, p < 0.01 and ###, p < 0.001 vs. the methamphetamine-treated group using one-way ANOVA followed by the Holm-Sidak test. Meth, methamphetamine.
Activated microglia eventually undergo apoptosis by a process termed AICD; however, whether methamphetamine causes AICD of microglia remains largely unknown. Therefore, we first examined the effect of methamphetamine on cell viability. BV-2 cells were exposed to different concentrations of methamphetamine (15 μM, 150 μM and 1.5 mM), and cell viability was assessed. The rationale for choosing these concentrations was based on the concentration of methamphetamine in the postmortem brains of chronic abusers.28,29 As shown in Fig. S2A, 1.5 mM methamphetamine decreased BV-2 cell viability as determined by Cell Counting Kit-8 (CCK8) assay. Therefore, this concentration of methamphetamine was chosen for all further studies. To address whether methamphetamine-induced cell death is due to mitochondria-dependent mechanisms, the cytosolic and mitochondrial CYCS (cytochrome c, somatic) levels were determined. As shown in Fig. S2B and C, methamphetamine treatment of BV-2 cells increased the CYCS level in the cytosol (Fig. S2B) and decreased the CYCS level in the mitochondrial fraction (Fig. S2C). We next explored whether methamphetamine-induced cell death also involved alterations in mitochondrial membrane potential. Cells were exposed to methamphetamine for different periods (6, 12, and 24 h), and then mitochondrial membrane depolarization was assessed using the JC-1 probe, which is a fluorescent lipophilic cationic dye that accumulates in mitochondria in proportion to the membrane potential that normally exists across the inner mitochondrial membrane. As shown in Fig. S2D and E, methamphetamine treatment increased the ratio of JC-1 aggregate: JC-1 monomer as determined using both a microscope (Fig. S2D) and a plate reader (Fig. S2E). Taken together, these data indicated that methamphetamine-induced cell death is related to mitochondria.
Studies have shown that nitric oxide (NO) is involved in LPS-induced AICD.30,31 Therefore, we next investigated the effect of methamphetamine on the expression of NOS2/iNOS (nitric oxide synthase 2, inducible). As shown in Fig. S3A, methamphetamine significantly increased NOS2 expression, with a peak response at 1 h. Pretreatment of cells with the NOS2 inhibitor-nitro-L-arginine methyl ester (L-NAME) attenuated methamphetamine-induced cell death (Fig. S3B). Consistent with this in vitro finding, administration of another NOS2 inhibitor, aminoguanidine (75 mg/kg), for 5 consecutive d followed by the administration of methamphetamine attenuated the methamphetamine-induced decrease in microglia as determined by Western blot for AIF1 expression in the hippocampus (Fig. S3C and D).
Next, we dissected the role of BBC3 in methamphetamine-induced microglial survival. Lentiviral vector-transduced Bbc3 siRNA in BV-2 cells successfully decreased BBC3 expression as shown in Fig. 1C (upper panel). Methamphetamine significantly decreased the survival of BV-2 cells (Fig. 1D) as well as in primary mouse microglia (Fig. 1E), whereas Bbc3 knockdown using siRNA significantly decreased cell viability and aggravated the methamphetamine-induced decrease in microglial survival. To further confirm the role of BBC3 in microglial survival, cells were transduced with BBC3 OE lentivirus. As shown in Fig. 1F and G, transduction of cells with BBC3 OE lentivirus successfully increased BBC3 expression (Fig. 1C, lower panel) and significantly attenuated the methamphetamine-induced decrease in cell survival in both BV-2 cells (Fig. 1F) and primary mouse microglia (Fig. 1G).
To further confirm our findings, we next examined the role of BBC3 in cell viability using trypan blue dye exclusion assay, as shown in Fig. S4. Methamphetamine treatment increased the number of trypan blue-positive cells; BBC3 knockdown using siRNA further increased the number of trypan blue-positive cells. However, BBC3 OE significantly inhibited the increase in trypan blue-positive cells compared with the methamphetamine-treated group.
Methamphetamine-mediated upregulation of BBC3 in microglia at the transcriptional level
BBC3 plays a seemingly paradoxical role in microglia in in vivo and in vitro studies; however, very little is known regarding the detailed mechanisms underlying BBC3 expression in methamphetamine-induced AICD of microglia. Therefore, we examined the effect of methamphetamine on BBC3 expression. BV-2 cells were exposed to methamphetamine for various periods. Compared with the control group, Bbc3 mRNA was significantly upregulated as determined by real-time PCR (Fig. 2A and B). Having determined that methamphetamine increased BBC3 expression at the mRNA level, we next confirmed the increase in BBC3 expression by Western blot analysis. Exposure to methamphetamine resulted in a biphasic response with an early but significant and transient increase in BBC3 (Fig. 2C, 6 h) followed by later significant repression of BBC3 (Fig. 2C, 24 h). To further confirm the effect of methamphetamine on BBC3 expression, primary mouse microglia were treated with methamphetamine (Fig. 2D). Consistent with the finding in BV-2 cells, methamphetamine treatment induced sustained expression of Bbc3 at the mRNA level but dynamic BBC3 expression at the protein level (Fig. 2B and D). The methamphetamine-mediated regulation of BBC3 expression was further validated by immunostaining (Fig. 2E). Taken together, these data clearly demonstrated that methamphetamine mediated the induction of BBC3 expression in microglia.
Figure 2.
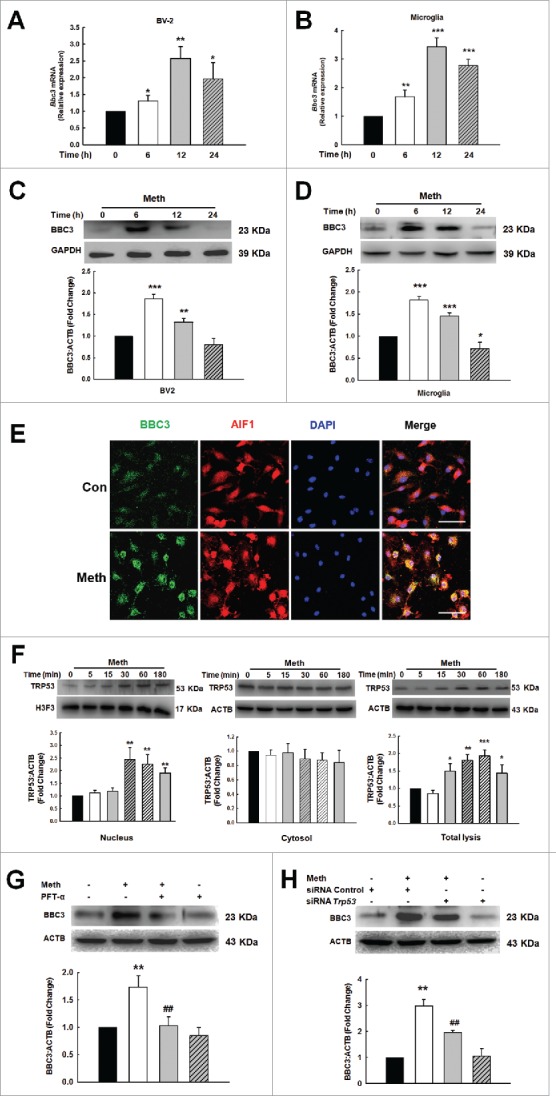
Methamphetamine mediated upregulation of BBC3 in microglia. (A and B) Methamphetamine increased Bbc3 expression at the mRNA level in BV-2 cells (A) and in primary mouse microglia (B) as determined by real-time PCR. (C and D) Methamphetamine induced a biphasic response of BBC3 expression in BV-2 cells (C) and in primary mouse microglia (D) as determined by Western blot analysis. Cells were treated with methamphetamine (1.5 mM) for 6, 12 and 24 h, followed by collection of RNA and protein for assessment of BBC3 expression. All data are presented as the mean ± SD of 3 independent experiments. *, p < 0.05, **, p < 0.01 and ***, p < 0.001 vs. the control group using one-way ANOVA followed by the Holm-Sidak test. (E) Representative image of BBC3 immunofluorescence staining in primary mouse microglia. Cells were treated with methamphetamine (1.5 mM) for 6 h. Red, AIF1; green, BBC3; blue, DAPI. Scale bar: 50 μm. (F) Methamphetamine increased TRP53 expression and induced TRP53 translocation into the nuclei of BV-2 cells. Cells were treated with methamphetamine for 5, 15, 30, 60 and 180 min followed by isolation of nuclear and cytosolic fractions as well as collection of the total cell lysates for the detection of TRP53 expression using Western blot analysis. All data are presented as the mean ± SD of 3 independent experiments. *, p < 0.05, **, p < 0.01 and ***, p < 0.001 vs. the control group using one-way ANOVA followed by the Holm-Sidak test. (G and H) Pretreatment of primary mouse microglia with the TRP53 inhibitor pifithrin-α (PFT-α; 10 μM) for 1 h (G) or Trp53 siRNA (H), followed by treatment with methamphetamine for 6 h. Inhibition of the TRP53 pathway significantly decreased the BBC3 expression induced by methamphetamine. All data are presented as the mean ± SD of 3 independent experiments. **, p < 0.01 vs. the control group; ##, p < 0.01 vs. the methamphetamine-treated group using one-way ANOVA followed by the Holm-Sidak test. Meth, methamphetamine.
While methamphetamine induced BBC3 expression, Bbc3 mRNA and protein expression patterns were inconsistent. This inconsistency may be due to the co-regulation of BBC3 expression at the transcriptional and post-transcriptional levels. BBC3 was initially identified as a transcriptional target of TP53 (TRP53 in mice) and a mediator of DNA damage-induced apoptosis; our study also indicated that methamphetamine treatment triggered the translocation of TRP53 into the nucleus (Fig. 2F, left panel) in a time-dependent manner without significant effect on the level of TRP53 in the cytosolic fraction (Fig. 2F, middle panel). Interestingly, methamphetamine treatment increased the expression of TRP53 in the total cell lysates (Fig. 2F, right panel). Moreover, blockage of the TRP53 pathway using a TRP53 inhibitor and Trp53-specific siRNA significantly decreased BBC3 expression in primary mouse microglia (Fig. 2G and H). As expected, Trp53 siRNA successfully decreased TRP53 expression in primary mouse microglia (Fig. S5). Therefore, the transcription factor TRP53 was involved in methamphetamine-induced BBC3.
Mir143 regulates BBC3 expression in microglia at the post-transcriptional level
Intriguingly, methamphetamine treatment induced a transient increase in the BBC3 protein levels before it decreased to a level comparable to that in the control group, despite a sustained increase in Bbc3 expression at the mRNA level (Fig. 2A to D). This result strongly suggested that an additional mechanism exists that negatively regulates BBC3 protein expression likely at the post-transcriptional level. Therefore, we further examined the detailed mechanisms of this process. We determined that BBC3 was regulated by the transcription factor TRP53 in microglia cells; however, whether BBC3 expression is regulated by noncoding RNAs has not previously been explored in microglia. The next step was to examine the miR that targets BBC3 using the TargetScan algorithm. Computational algorithms such as TargetScan were employed, and Mir143, which is conserved among vertebrates, was predicted to target Bbc3.
As shown in Fig. 3A, Bbc3 has a conserved Mir143 binding site within its 3′-untranslated region (UTR) in most species that was identified as the putative target of Mir143. Intriguingly, cotransfecting a Mir143-overexpressing vector and the pmiR-GLO plasmid with the Bbc3 wild type 3′-UTR resulted in the downregulation of luciferase activity, and this effect was reversed in H293 cells transfected with a mutated Bbc3 3′-UTR (Fig. 3B). The next step was to determine whether methamphetamine mediated its effects via the induction of Mir143 and to assess the kinetics of the methamphetamine response. Methamphetamine treatment of primary mouse microglia induced a biphasic response resulting in early but significant and transient repression of Mir143 (Fig. 3C; 6h) that was followed by later but sustained and significant upregulation of Mir143 (Fig. 3C; 24 h). Interestingly and as expected, methamphetamine-induced modulation of Mir143 correlated inversely with BBC3 expression (Fig. 2C and D). Methamphetamine-mediated regulation of Mir143 was further validated using fluorescence in situ hybridization (FISH; Fig. 3D). In line with this finding, Mir143 decreased BBC3 expression, whereas anti-Mir143 increased BBC3 expression at both the mRNA (Fig. 3E) and protein (Fig. 3F) levels in primary mouse microglia.
Figure 3.

Mir143 regulates BBC3 expression at the post-transcriptional level in microglia. (A) Putative Mir143 binding sites in the Bbc3 gene. The potential complementary residues are shown in red. (B) Relative luciferase activity of wild type 3′-UTR mutant constructs of Bbc3 cotransfected with the Mir143 OE vector and the pmiR-GLO plasmid. All the data are indicated as the mean ± SD of 3 individual experiments. *, p < 0.05 vs. the Mircontrol co-transfected with WT group using one-way ANOVA followed by the Holm-Sidak test. (C) Effect of methamphetamine on Mir143 expression at the mRNA level in primary mouse microglia as determined by real-time PCR. Cells were incubated with methamphetamine (1.5 mM) for 6, 12 and 24 h, followed by collection of RNA for assay of Mir143 expression. All data are presented as the mean ± SD of 3 independent experiments. *, p < 0.05; **, p < 0.01; and ***, p < 0.001 vs. the control group using one-way ANOVA followed by the Holm-Sidak test. (D) Fluorescence in situ hybridization of mature Mir143 in primary mouse microglia combined with immunostaining for the microglial marker AIF1. Cells were incubated with methamphetamine (1.5 mM) for 24 h. Red, Mir143; green, AIF1; blue, DAPI. Scale bar: 20 µm. (E and F) Primary mouse microglia infected with Mircontrol + Mir143 and anti-Mircontrol + anti-Mir143 lentivirus for 48 h were assessed for BBC3 expression at the mRNA (E) and protein (F) levels. All data are presented as the mean ± SD of 3 individual experiments. **, p < 0 .01 vs. the Mircontrol or anti-Mircontrol group using Student ttest. Meth, methamphetamine.
Role of BBC3 in methamphetamine-induced apoptosis of microglia
Because BBC3 expression was increased in the BV-2 cells in the presence of methamphetamine, we next sought to explore the role of BBC3 in the methamphetamine-mediated apoptosis of BV-2 cells. As shown in Fig. 4A to C, methamphetamine induced the expression of BAX, but failed to affect the levels of BCL2L1/Bcl-xL, cleaved CASP9 (caspase 9) and cleaved CASP3. Moreover, knockdown of Bbc3 using Bbc3-specific siRNA alleviated the expression of methamphetamine-mediated markers of apoptosis (Fig. 4D to F). To corroborate the above findings, terminal deoxynucleotidyl transferase-mediated dUTP nick end-labeling (TUNEL) staining was performed, which demonstrated that 25.8% of cells treated with methamphetamine were TUNEL-positive and that this increase in cell apoptosis was significantly reduced in the cells transduced with Bbc3 siRNA (Fig. 4G and H). To further confirm the role of BBC3 in microglial apoptosis, the effect of methamphetamine on cleaved CASP9 and cleaved CASP3 was examined in primary mouse microglia. As shown in Fig. 5A and B, methamphetamine increased the levels of cleaved CASP9 and cleaved CASP3. Moreover, knockdown of BBC3 expression significantly inhibited the increase in cleaved CASP3 induced by methamphetamine (Fig. 5C). Because BBC3 is a common apoptosis inducer, we next examined the effect of BBC3 efficiency on cell apoptosis in vivo. As shown in Fig. 5D and E, methamphetamine-mediated upregulation of BAX-BCL2L1 and cleaved CASP3 was ameliorated in the hippocampus of Bbc3 knockout (KO) mice compared with WT controls. Taken together, these findings suggested that BBC3 is involved in methamphetamine-mediated apoptosis.
Figure 4.

Role of BBC3 in methamphetamine-induced cell apoptosis of BV-2 cells. (A to C) Exposure of BV-2 cells to methamphetamine (1.5 mM) for different time periods (3, 6, 12 and 24 h) increased the expression of BAX-BCL2L1 (A), cleaved CASP9:CASP9 (B) and cleaved CASP3:CASP3 (C). All data are presented as the mean ± SD of 3 independent experiments. **, p < 0.01 and ***, p < 0.001 vs. the control group using one-way ANOVA followed by the Holm-Sidak test. (D to F) Methamphetamine-induced expression of BAX-BCL2L1 (D), cleaved CASP9:CASP9 (E) and cleaved CASP3:CASP3 (F) was attenuated by BBC3 knockdown in BV-2 cells using Bbc3 siRNA in BV-2 cells. BV-2 cells were transfected with Bbc3 siRNA for 24 h followed by methamphetamine treatment (1.5 mM) for another 6 h, and the cells were homogenized for detection of cleaved CASP9 and CASP3 expression using Western blotting. (G and H) TUNEL staining (green) in BV-2 cells transfected with Bbc3 siRNA for 24 h and treated with methamphetamine for another 24 h. All data are presented as the mean ± SD of 3 independent experiments. *, p < 0.05 and **, p < 0.01 vs. the control group; #, p < 0.05 and ##, p < 0.01 vs. the methamphetamine-treated group using one-way ANOVA followed by the Holm-Sidak test.
Figure 5.

Role of BBC3 in methamphetamine-induced microglial apoptosis. (A and B) Exposure of primary mouse microglia to methamphetamine (1.5 mM) for different time periods (6, 12 and 24 h) increased the expression of cleaved CASP9 and CASP3. All data are presented as the mean ± SD of 3 independent experiments. *, p < 0.05 and **, p < 0.01 vs. the control group using one-way ANOVA followed by the Holm-Sidak test. (C) Bbc3 siRNA inhibited the methamphetamine-induced increase in cleaved CASP3 expression. Primary mouse microglia were transduced with Bbc3 siRNA for 24 h followed by methamphetamine treatment (1.5 mM) for another 6 h, and the cells were homogenized for detection of cleaved CASP3 expression using Western blotting. All data are presented as the mean ± SD of 3 independent experiments. **, p < 0.01 vs. the control group; #, p < 0.05 vs. the methamphetamine-treated group using one-way ANOVA followed by the Holm-Sidak test. (D and E) Expression of BAX-BCL2L1 (D) and cleaved CASP3:CASP3 (E) in the hippocampus of WT and Bbc3 KO mice as determined by Western blotting. WT and Bbc3 KO mice were treated with methamphetamine (intraperitoneal, 30 mg/kg) for 48 h followed by examination for apoptosis. N = 6 animals/group. *, p < 0.05 and **, p < 0.01 vs. the saline-treated WT group; ##, p < 0 .01 vs. the methamphetamine-treated WT group using one-way ANOVA followed by the Holm-Sidak test. Meth: methamphetamine.
Role of BBC3 in methamphetamine-induced autophagy of microglia
To explain the paradoxical role of BBC3 in microglial survival, we investigated whether BBC3 affected autophagy, a critical regulatory process of cell survival and proliferation.32 Two well-known pathways are involved in autophagy: the phosphoinositide 3-kinase-AKT1-MTOR-RPS6KB signaling pathway and the RAS-RAF1-MAPK1/2 signaling pathway. Both pathways are often activated in numerous types of cancers.33 Several anticancer agents are known to inhibit the phosphoinositide 3-kinase-AKT1-MTOR-RPS6KB signaling pathway and to simultaneously activate the MAPK1/ERK2 pathway, resulting in autophagy induction.34-36 Therefore, we first examined the effect of methamphetamine on these pathways. As shown in Fig. S6, methamphetamine treatment suppressed the phosphorylation of AKT1, MTOR, and RPS6K but increased the phosphorylation of MAPK1. Next, we examined MAP1LC3-II/LC3-II expression in the presence of methamphetamine. As shown in Figure 6A and B, methamphetamine treatment of both BV-2 and primary mouse microglia increased the expression of BECN1/beclin 1 and induced the production of MAP1LC3-II, which is a cleaved MAP1LC3-phosphatidylethanolamine conjugate and a general autophagosomal marker.37 In the context of autophagy, SQSTM1/p62 acts as a receptor protein that links MAP1LC3 with ubiquitin moieties on misfolded proteins. Therefore, autophagy mediates the clearance of SQSTM1 together with ubiquitinated proteins. Consistently, SQSTM1 expression was downregulated by methamphetamine treatment in both BV-2 and primary mouse microglia (Fig. 6A and B).
Figure 6.

Methamphetamine induced microglial autophagy. (A and B) The expression of BECN1, MAP1LC3-II and SQSTM1 in BV-2 cells (A) and primary mouse microglia (B) induced by methamphetamine. Cells were treated with methamphetamine (1.5 mM) for 6, 12 and 24 h. All data are presented as the mean ± SD of 3 independent experiments. *, p < 0.05; **, p < 0.01; and ***, p < 0.001 vs. the control group using one-way ANOVA followed by the Holm-Sidak test. (C) Primary mouse microglia infected with RFP-GFP-MAP1LC3 adenovirus and then treated with 1.5 mM methamphetamine for 24 h. Effect of methamphetamine on RFP- and GFP-MAP1LC3 puncta (left panel). Scale bar: 5 μm. The RFP- and GFP-MAP1LC3 puncta per cell were counted, and the quantification is shown in the right panel. (D) Transmission electron microscopy imaging of cells showing autolysosomes (arrows) and double-membraned autophagosomes (arrowheads) in primary microglia (left panel) and quantification of autolysosomes and autophagosomes (right panel) in primary microglial cells treated with methamphetamine (1.5 mM) for 24 h. Scale bar: 200 nm. Quantification of cell numbers is presented as the mean ± SD of 3 independent experiments. **, p < 0.01 and ***, p < 0.001 vs. the control group; ###, p < 0.01 vs. the methamphetamine-treated group using one-way ANOVA followed by the Holm-Sidak test.
Autophagic flux was further monitored in primary mouse microglia that were transduced with tandem fluorescent-mRFP-GFP-MAP1LC3-adenovirus, a specific marker for autophagosome formation that relies on the different nature of GFP and RFP fluorescence under acidic conditions. GFP fluorescence is sensitive to the acidic condition of the lysosome lumen, while RFP is relatively stable under acidic conditions. Thus, the colocalization of GFP and RFP signals (yellow dots) indicates the phagophores or autophagosomes that have not fused with lysosomes, whereas RFP-only signals (red puncta) stain the autolysosomes. As shown in Fig. 6C, methamphetamine treatment significantly increased the number of yellow dots per cell with a concomitant greater increase in RFP-only MAP1LC3 dots in primary mouse microglia transduced with tandem fluorescent-tagged nRFP-GFP-MAP1LC3. Autophagy is a dynamic process of protein degradation that is characterized by the formation of double-membraned cytoplasmic vesicles.38 Structural analysis via electron microscopy allows the visualization of autophagy with the massive accumulation of autophagic vacuoles (autophagosomes) and autolysosomes in the cytoplasm. As shown in Fig. 6D, methamphetamine treatment resulted in the accumulation of numerous autolysosomes (arrow) and double-membraned autophagic vacuoles (arrowhead) in primary microglia (Fig. S7).
Because MAP1LC3-II can be increased by either enhanced autophagy initiation or inhibition of lysosomal degradation,39 we next examined the mechanisms underlying the methamphetamine-induced increase in MAP1LC3-II expression. BV-2 cells were pretreated with the lysosome inhibitors bafilomycin A1 (BafA1) and chloroquine (CQ) for 1 h followed by methamphetamine treatment for 24 h. The cells were then collected, and homogenates were prepared for detection of MAP1LC3-II levels. As shown in Fig. 7, methamphetamine increased the level of MAP1LC3-II in the presence of CQ (Fig. 7A and C) or BafA1 (Fig. 7B and D) compared with BV-2 cells (Fig. 7A and B) and primary mouse microglia (Fig. 7C and D) treated with CQ or BafA1 alone, suggesting that methamphetamine increased MAP1LC3-II levels primarily through enhancing autophagy initiation.
Figure 7.
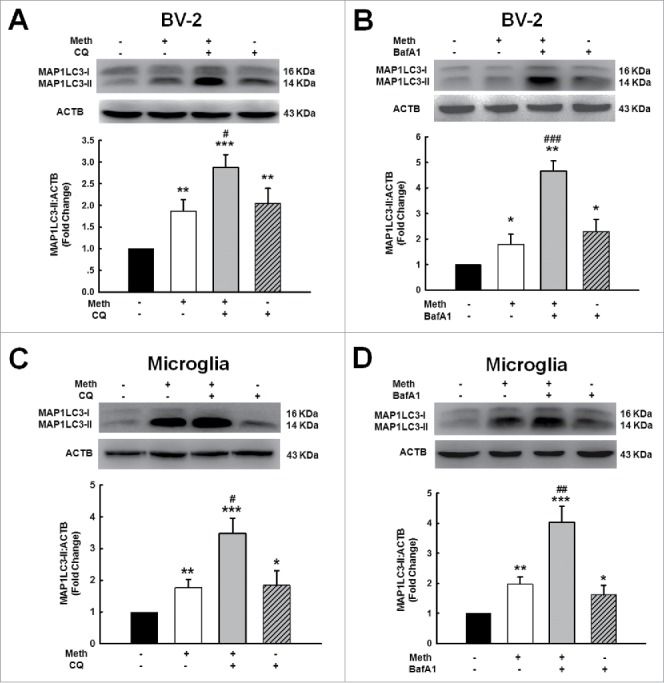
Methamphetamine increased MAP1LC3-II level primarily through enhancing the initiation of autophagy. (A and B) BV-2 cells were pretreated with or without CQ (45 μM) (A) or BafA1 (50 nM) (B) for 1 h followed by methamphetamine (1.5 mM) treatment for 24 h. (C and D) primary mouse microglia were pretreated with or without CQ (45 μM) (C) or BafA1 (50 nM) (D) for 1 h followed by methamphetamine (1.5 mM) treatment for 24 h. All data are presented as the mean ± SD of 3 independent experiments. *, p < 0.05, **, p < 0.01 and ***, p < 0.001 vs. the control group; #, p < 0.05, ##, p < 0.01 and ###, p < 0.01 vs. the CQ- or BafA1-treated group using one-way ANOVA followed by the Holm-Sidak test. Meth, methamphetamine; CQ, chloroquine; BafA1, bafilomycin A1.
The anti-apoptotic protein BCL2 binds with BECN1 to inhibit the autophagic process, and a previous study demonstrated that methamphetamine treatment resulted in disruption of the interaction between BCL2 and BECN1.40 To determine whether such a mechanism was also responsible for methamphetamine-induced autophagy, we investigated the binding status of BCL2 with BECN1 following methamphetamine treatment. Using co-immunoprecipitation assays, the amount of BECN1 in the BCL2-immunoprecipitated protein complex was shown to decrease while the amount of total BCL2 remained unchanged in methamphetamine-treated microglia, as shown in Fig. S8.
Our findings demonstrated that the change in the levels of MAP1LC3-II occurred at 24 h following methamphetamine treatment (Fig. 6), when BBC3 expression had already returned to the basal level (Fig. 2). According to this finding, the role of autophagy as a mechanism of BBC3-mediated cell survival seems to be confusing considering the timing of these events. However, as shown in Fig. 6A, methamphetamine treatment resulted in the increased expression of BECN1 as early as 6 h, which is consistent with the response of BBC3; methamphetamine induced a transient increase in the BBC3 protein level at 6 h followed by reduction to a level comparable to that of the control group (Fig. 2). BECN1 is an adaptor protein that assembles a core complex with phosphatidylinositol 3-kinase to recruit other autophagy-related proteins that orchestrate autophagosome formation and thus plays a central role in autophagy induction.40-42 Therefore, our current finding suggested that the early increase in BBC3 expression could initiate autophagy by increasing the level of BECN1 and that BECN1 expression further initiated the process of autophagy in a dynamic, time-dependent manner. To confirm this possibility, BV-2 cells were transfected with Bbc3 siRNA followed by treatment with methamphetamine. As shown in Fig. S9A, knockdown of BBC3 expression significantly inhibited methamphetamine-induced BECN1 expression. Moreover, we also demonstrated that BECN1 knockdown significantly diminished MAP1LC3-II expression, thus indicating that BECN1 lies upstream of MAP1LC3-II (Fig. S9B). As expected, BECN1 siRNA successfully decreased BECN1 expression as shown in Fig. S9C.
MAP1LC3-II formation was enhanced by the autophagy inducer rapamycin, as shown in Fig. 8A and B. This effect was also confirmed by immunofluorescence staining showing that rapamycin enhanced the number of vacuoles as indicated by endogenous MAP1LC3-II (Fig. 8A and B). Having determined that methamphetamine induced BBC3 expression and autophagy, we next sought to explore the role of BBC3 in methamphetamine-mediated autophagy. As shown in Fig. 8C, transduction of primary mouse microglia with a lentiviral vector carrying Bbc3 siRNA significantly reversed methamphetamine-induced MAP1LC3-II expression. In contrast, BBC3 OE further enhanced the methamphetamine-induced increase in MAP1LC3-II expression (Fig. 8D). Furthermore, the level of MAP1LC3-II was significantly increased in WT mice treated with methamphetamine, and this level was reduced in the Bbc3 KO mice (Fig. 8E). Taken together, these findings suggested that BBC3 plays a critical role in methamphetamine-induced autophagy.
Figure 8.
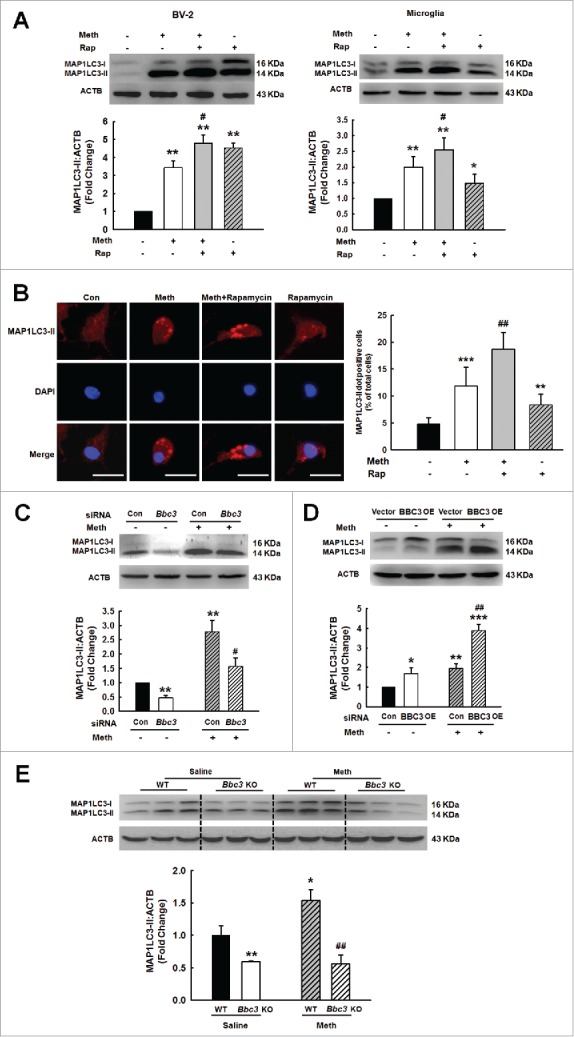
Role of BBC3 in methamphetamine-induced autophagy of microglia. (A) Pretreatment of BV-2 cells (left panel) and primary mouse microglia (right panel) with the autophagy inducer rapamycin (0.1 μM) for 1 h enhanced the methamphetamine-induced increase in MAP1LC3-II expression. (B) Effect of methamphetamine on the formation of MAP1LC3-II puncta. Cells were pretreated with rapamycin for 1 h and then treated with methamphetamine (1.5 mM) for another 24 h. MAP1LC3-II puncta were then analyzed by confocal microscopy. Scale bar: 20 μm. (C) Knockdown of Bbc3 expression by siRNA decreased the methamphetamine-induced expression of MAP1LC3-II in primary mouse microglia. Primary mouse microglia were transfected with Bbc3 siRNA for 24 h followed by methamphetamine treatment (1.5 mM) for another 24 h, and the cells were processed for detection of MAP1LC3-II expression using Western blotting. (D) Transduction of primary mouse microglia with BBC3 OE lentivirus further enhanced the methamphetamine-induced increase in MAP1LC3-II expression in primary mouse microglia. Primary mouse microglia were transduced with BBC3 OE lentivirus for 24 h followed by methamphetamine treatment (1.5 mM) for another 24 h, and the cells were processed for detection of MAP1LC3-II expression using Western blotting. All data are presented as the mean ± SD of 3 independent experiments. *, p < 0.05, **, p < 0.01 and ***, p < 0.001 vs. the control group; #, p < 0.05 and ##, p < 0.01 vs. the methamphetamine-treated group using one-way ANOVA followed by the Holm-Sidak test. (E) Expression of MAP1LC3-II in the hippocampus of WT and Bbc3 KO mice as determined by Western blotting. WT and Bbc3 KO mice were treated with methamphetamine (intraperitoneal, 30 mg/kg) for 48 h, followed by examination for autophagy. N = 6 animals/group. *, p < 0.05 and **, p < 0.01 vs. the saline-treated WT group; ##, p < 0.01 vs. the methamphetamine-treated WT group using one-way ANOVA followed by the Holm-Sidak test. Meth, methamphetamine; Rap, rapamycin.
Role of autophagy in the methamphetamine-induced decrease in cell viability
The finding that BBC3 is required for microglial survival, as shown in Fig. 1, was unexpected as these data contradicted the common view that BBC3 is an essential apoptosis inducer. Because BBC3 was involved in methamphetamine-induced autophagy and because autophagy is considered to have a prosurvival role under pathological conditions, we next determined the role of autophagy in the survival of microglia treated with methamphetamine. Primary mouse microglia were treated with the autophagy inhibitor 3-methyladenine (3-MA), which is a class III phosphatidylinositol 3-kinase inhibitor that prevents autophagy at an early stage of autophagosome formation. As shown in Fig. 9A, pretreatment of cells with 3-MA enhanced the methamphetamine-induced decrease in cell viability. 3-MA alone had no effect on cell viability. However, pretreatment of cells with the autophagy inducer rapamycin significantly ameliorated the methamphetamine-induced decrease in cell viability (Fig. 9B) suggesting that autophagy exerted a prosurvival role in microglia, whereas the inhibition of autophagy decreased cell viability. In order to confirm the role of autophagy in methamphetamine-induced decreased cell viability of microglia, cells were transduced with siRNA Atg7 (autophagy-related 7) lentivirus. As shown in Fig. 9C and D, knockdown of ATG7 further decreased microglial survival in both BV-2 and primary mouse microglia. As expected, lentiviral vector-transduced Atg7 siRNA significantly decreased ATG7 expression in primary mouse microglia and enhanced BAX-BCL2L1 expression, as shown in Fig. S10. To further confirm the role of autophagy in methamphetamine-induced apoptosis, cells were treated with 3-MA, which enhanced the level of cleaved CASP3 in both BV-2 cells and primary mouse microglia (Fig. 10A and B). Conversely, pretreatment of cells with rapamycin significantly inhibited the increased level of cleaved CASP3 in both BV-2 cells and primary mouse microglia (Fig. 10C and D).
Figure 9.
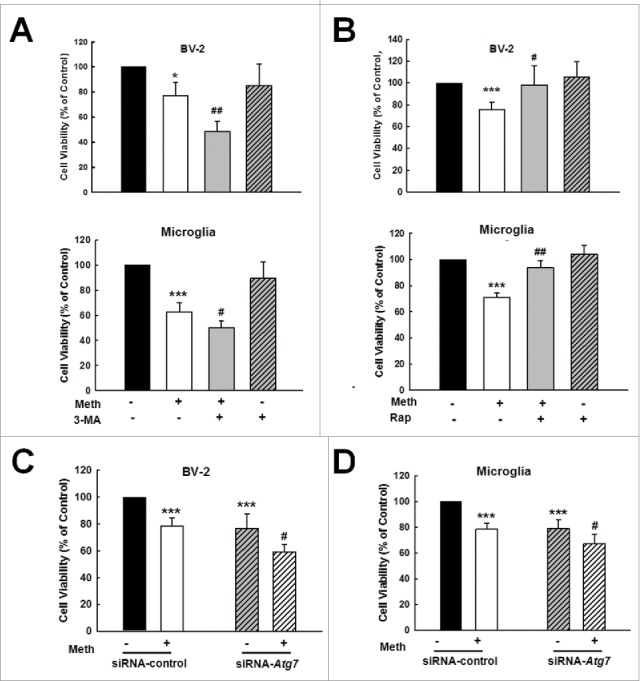
Role of autophagy in methamphetamine-induced microglial survival. (A) Pretreatment of cells with 3-MA augmented the methamphetamine-induced decrease in cell viability in BV-2 cells (upper panel) and in primary mouse microglia (lower panel). Cells were pretreated with 3-MA (1 mM) for 1 h and then treated with methamphetamine (1.5 mM) for 24 h. (B) Pretreatment of cells with the autophagy inducer rapamycin attenuated the methamphetamine-induced decrease in cell viability in BV-2 cells (upper panel) and in primary mouse microglia (lower panel). Cells were pretreated with rapamycin (0.1 μM) for 1 h, and then treated with methamphetamine (1.5 mM) for 24 h. Cells were pretreated with rapamycin (0.1 μM) for 1 h followed by methamphetamine (1.5 mM) treatment for 6 h. (C and D) Transfection of cells with Atg7 siRNA lentivirus decreased cell viability in both BV-2 cells (C) and primary mouse microglia (D) as determined by CCK8 assay. Cells were transduced with Atg7 siRNA lentivirus for 24 h and treated with methamphetamine (1.5 mM) for 24 h. All data are presented as the mean ± SD of 3 independent experiments. *, p < 0.05 and ***, p < 0.001 vs. the control group; #, p < 0.05 and ##, p < 0.01 vs. the methamphetamine-treated group using one-way ANOVA followed by the Holm-Sidak test. Meth, methamphetamine; 3-MA, 3-methyladenine; Rap, rapamycin.
Figure 10.
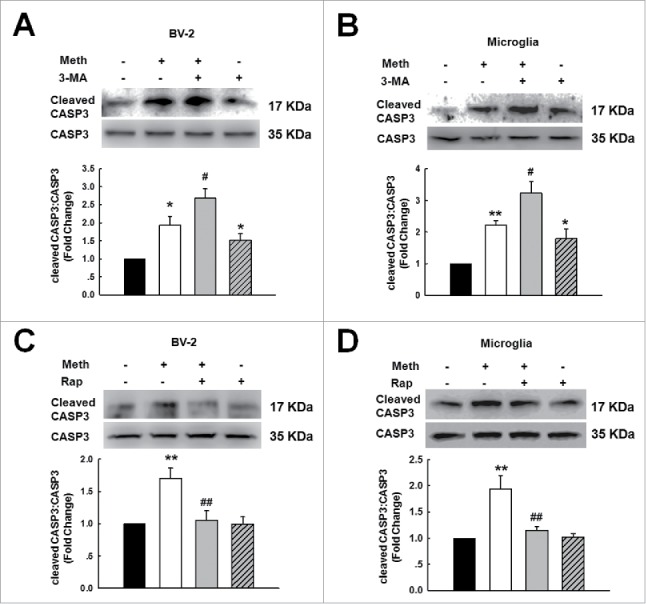
Role of autophagy in methamphetamine-induced microglial apoptosis. (A and B) Pretreatment with 3-MA (1 mM) for 1 h enhanced the level of cleaved CASP3 in both BV-2 cells (A) and primary mouse microglia (B). Cells were pretreated with 3-MA (1 mM) for 1 h followed by methamphetamine (1.5 mM) treatment for 6 h. (C and D) Pretreatment with rapamycin (0.1 μM) for 1 h significantly decreased the level of cleaved CASP3 in both BV-2 cells (C) and primary mouse microglia (D). All data are presented as the mean ± SD of 3 independent experiments. *, p < 0.05, and **, p < 0.01 vs. the control group; #, p < 0.05 and ##, p < 0.01 vs. the methamphetamine-treated group using one-way ANOVA followed by the Holm-Sidak test. Meth, methamphetamine; 3-MA, 3-methyladenine; Rap, rapamycin.
Role of Mir143-BBC3 in methamphetamine-induced apoptosis and autophagy in vitro
Having determined that Mir143 regulated BBC3 expression, we next examined the role of Mir143 in microglial survival. Primary mouse microglia were transduced with a lentivirus expressing anti-Mir143, and cell viability was assessed. As shown in Fig. 11A, transduction of cells with anti-Mir143 lentivirus significantly inhibited the methamphetamine-induced decrease in cell survival as determined by CCK8 assay. The role of Mir143 in methamphetamine-induced apoptosis and autophagy in microglia was further examined. Transduction of cells with anti-Mir143 lentivirus significantly enhanced the levels of cleaved CASP3 (Fig. 11B) and MAP1LC3-II (Fig. 11C) in primary mouse microglia. Next, to determine whether anti-Mir143-mediated functional effects depend specifically on the upregulation of BBC3, primary mouse microglia were cotransduced with lentiviral vectors expressing anti-Mir143 and Bbc3 siRNA. Transduction of cells with anti-Mir143 lentivirus enhanced cell viability, and this effect was significantly inhibited in cells cotransduced with lentiviral vectors expressing anti-Mir143 and Bbc3 siRNA (Fig. 11D). Moreover, the transduction of cells with anti-Mir143 lentivirus resulted in enhanced cleaved CASP3 and MAP1LC3-II (Figs. 11E and F), and this effect was inhibited in cells cotransduced with lentiviral vectors expressing anti-Mir143 and Bbc3 siRNA (Figs. 11E and F).
Figure 11.
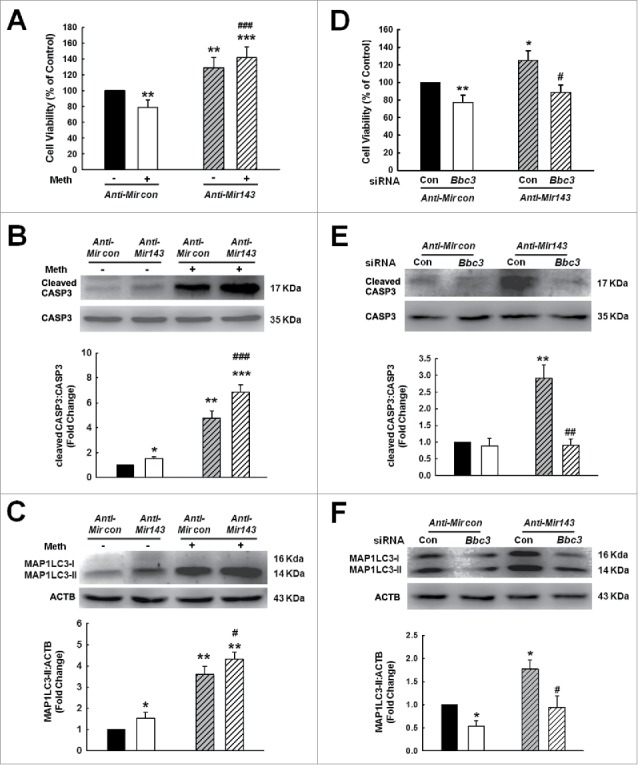
Role of Mir143-BBC3 in methamphetamine-induced apoptosis and autophagy in vitro. (A to C) Transduction of cells with anti-Mir143 attenuated the methamphetamine-induced decrease in cell viability as determined by CCK8 assay (A) and enhanced the level of cleaved CASP3 (B) and MAP1LC3-II (C) as determined by Western blotting. Cells were transduced with anti-Mir143 lentivirus for 24 h and then treated with methamphetamine (1.5 mM) for 24 h to examine the cell viability and MAP1LC3-II expression and for 6 h to evaluate the level of cleaved CASP3. All data are presented as the mean ± SD of 3 independent experiments. *, p < 0.05, **, p < 0.01 and ***, p < 0.001 vs. the anti-Mircontrol group; #, p < 0.05 and ###, p < 0.001 vs. the anti-Mircontrol treated with methamphetamine group using one-way ANOVA followed by the Holm-Sidak test. (D to F) Transduction of cells with the Bbc3 siRNA lentivirus significantly inhibited the anti-Mir143-induced increase in cell viability as determined by CCK8 assay (D) and decreased the levels of cleaved CASP3 (E) and MAP1LC3-II (F) as determined by Western blotting. Cells were transduced with Bbc3 siRNA lentivirus for 24 h, and then were treated with methamphetamine (1.5 mM) for another 24 h to examine the cell viability and MAP1LC3-II expression, and for another 6 h to evaluate the level of cleaved CASP3. All data are presented as the mean ± SD of 3 independent experiments. *, p < 0.05 and **, p < 0.01 vs. the anti-Mircontrol group; #, p < 0.05 and ##, p < 0.01 vs. the anti-Mir143 cotransduced with siRNA-control group using one-way ANOVA followed by the Holm-Sidak test. Meth: methamphetamine.
Reciprocally, transduction of cells with the Mir143 precursor had opposing effects. To further determine whether Mir143-mediated functional effects depend specifically on BBC3 suppression, an expression construct encoding the entire BBC3 coding sequence but lacking the 3′-UTR (BBC3 OE), which yields an mRNA resistant to Mir143-mediated suppression, was generated. The transduction of cells with Mir143 lentivirus decreased cell survival, and this effect was ameliorated in cells cotransduced with BBC3 OE lentivirus in primary mouse microglia (Fig. S11).
Role of Mir143 in methamphetamine-induced microglial death in vivo
Our findings demonstrate that Mir143, which is involved in the regulation of BBC3 expression, plays a critical role in microglial apoptosis and autophagy. Notably, one of the unavoidable caveats of in vitro studies involving either BV-2 or primary microglia is their inherent ability to become activated during culture. Therefore, we sought to validate the role of Mir143 in vivo by microinjecting anti-Mir143 lentivirus into the hippocampus of C57BL/6J mice. C57BL/6J mice were microinjected bilaterally with either the anti-Mir control-RFP lentivirus or the anti-Mir143-RFP lentivirus in the hippocampus and monitored for microglial survival in response to methamphetamine. However, first determining the efficacy of anti-Mir143-RFP lentivirus transduction in vivo was important. As shown in Fig. 12A, RFP expression was largely restricted to the hippocampus, and a certain number of AIF1-positive cells colocalized with RFP. As expected, increased BBC3 expression was observed in the anti-Mir143-injected side compared with that in the side injected with anti-Mir control (Fig. 12B). Two wk following lentivirus injections, the mice were administered methamphetamine (30 mg/kg), and AIF1-positive cells in the hippocampus were assessed. As shown in Fig. 12C and D, compared with the saline control group, anti-Mir control-RFP lentivirus-microinjected mice exhibited a decreased number of AIF1-positive microglia in the presence of methamphetamine, which was significantly attenuated by microinjection of anti-Mir143.
Figure 12.
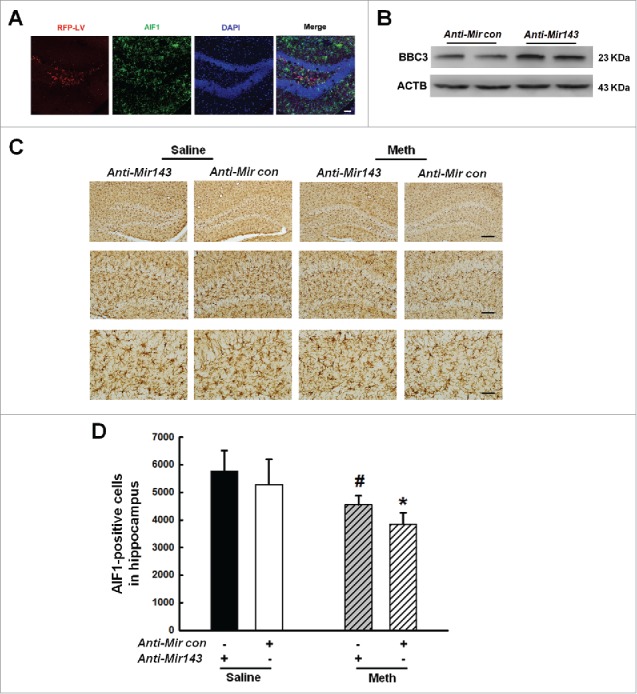
Role of Mir143 in methamphetamine-induced microglial death in vivo. (A) Representative images of C57BL/6J mice microinjected with either anti-Mir control-RFP lentivirus or anti-Mir143-RFP lentivirus (LV) in microglia of the hippocampus. Scale bar: 100 μm. Mice were microinjected bilaterally with anti-Mir-RFP lentivirus (2 μl of 109 viral genomes µl−1) into the hippocampus, and 2 wk after microinjection, mice were sacrificed and examined for RFP and AIF1 expression. (B) Anti-Mir143 lentivirus injection successfully increased BBC3 expression as determined by Western blotting. Two wk after microinjection, mice were sacrificed and examined for BBC3 expression. (C) Effect of methamphetamine on the survival of microglia in the hippocampus of C57BL/6J mice, which were microinjected with either anti-Mircontrol-RFP lentivirus or anti-Mir143-RFP lentivirus. Two wk after microinjection, mice were treated with methamphetamine (intraperitoneal, 30 mg/kg) for another 48 h, followed by AIF1 immunostaining. Representative images of AIF1 immunostaining in the hippocampus of mice injected with saline or methamphetamine. Scale bars: 200 μm (upper panel), 100 μm (middle panel), and 50 μm (lower panel). (D) Stereology of AIF1-positive microglia in the hippocampus. N = 5 animals/group. *, p < 0.05 vs. the anti-Mircontrol mice treated with saline group; #, p < 0.05 vs. the anti-Mircontrol mice treated with methamphetamine-treated group using one-way ANOVA followed by the Holm-Sidak test. Meth, methamphetamine.
To further confirm the role of Mir143 in methamphetamine-induced decrease in microglial survival, we sought to generate mir143−/- mice. Because we failed to generate homozygous mir143−/- mice from 8 littermates thus far, we used heterozygous Mir143+/− mice to confirm our findings. As shown in Fig. 13A, Mir143 expression was significantly decreased in heterozygous Mir143+/− mice compared with WT mice. Moreover, methamphetamine treatment increased Mir143 expression in the hippocampus of WT mice but not in Mir143+/− mice (Fig. 13B). Consistent with the in vitro findings, methamphetamine treatment increased the mRNA level of Bbc3 (Fig. S12A) but decreased BBC3 expression at the protein level (Fig. S12B) in the WT mice, which further confirming that the decreased expression of BBC3 was regulated at the post-transcriptional level in vivo. However, methamphetamine treatment failed to affect the expression of BBC3 at either the mRNA or protein level in Mir143+/− mice compared with the saline control group (Fig. S12). To confirm the role of Mir143 in methamphetamine-induced microglial survival, we examined microglial survival in Mir143+/− mice. As shown in Fig. 13C and D, methamphetamine administration significantly decreased the number of AIF1-positive cells in the hippocampus of WT mice; however, methamphetamine failed to induce microglial death in Mir143+/− mice. These findings thus underpin the role of Mir143 in regulating the methamphetamine-mediated decrease in microglial survival in vivo.
Figure 13.
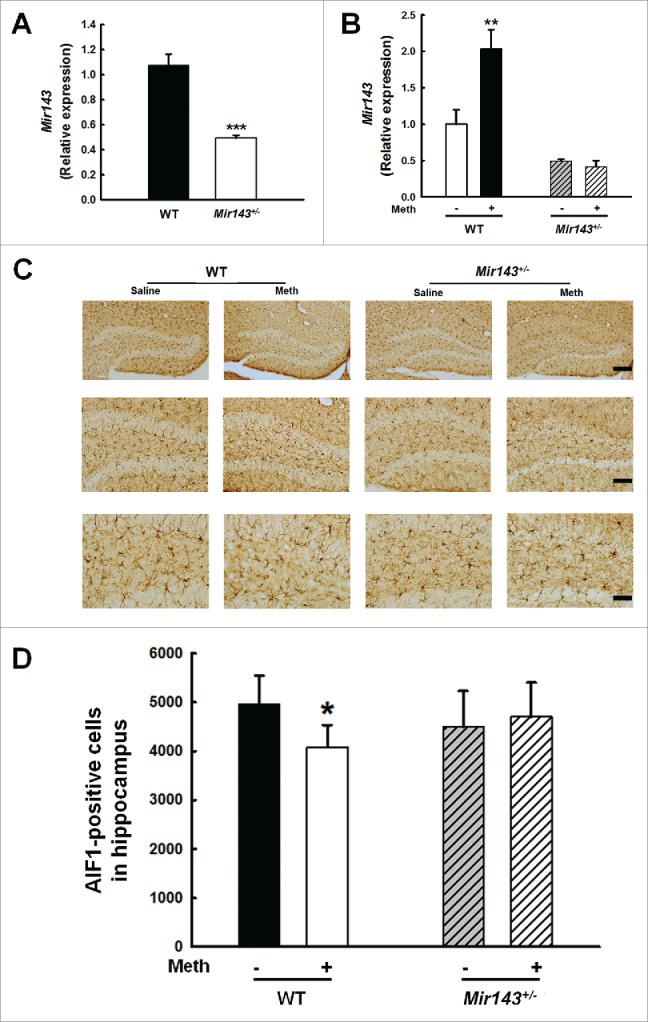
Role of Mir143 in methamphetamine-induced decreased microglial survival in vivo. (A) Mir143 expression was decreased in heterozygous Mir143+/− mice compared with WT mice. WT and Mir143+/− mice were treated with methamphetamine (intraperitoneal, 30 mg/kg) for 48 h. N = 6 animals/group. ***, p < 0.001 vs. the WT group using Student ttest. (B) Administration of methamphetamine significantly increased hippocampal Mir143 expression in WT mice but not in Mir143+/− mice compared with the saline control group. WT and Mir143+/− mice were treated with methamphetamine (intraperitoneal, 30 mg/kg) for 48 h. N = 6 animals/group. **, p < 0.01 vs. the saline-treated WT group using one-way ANOVA followed by the Holm-Sidak test. (C) Effect of methamphetamine on the survival of microglia in the hippocampus of heterozygous Mir143+/− and WT mice. WT and Mir143+/− mice were treated with methamphetamine (intraperitoneal, 30 mg/kg) for 48 h. Representative images of AIF1 immunostaining in the hippocampus of mice injected with saline or methamphetamine. Scale bars: 200 μm (upper panel), 100 μm (middle panel), and 50 μm (lower panel). (D) Stereology of AIF1-positive microglia in the hippocampus. N = 5 animals/group. *, p < 0.05 vs. the saline-treated WT group using one-way ANOVA followed by the Holm-Sidak test. Meth, methamphetamine.
Discussion
Our study provides new insights into the role of Mir143 in the regulation of microglial survival: Mir143 modulates BBC3 expression, which regulates the interplay of apoptosis and autophagy. The modulation of Mir143-BBC3 expression may be a therapeutic intervention for the regulation of microglial survival in the context of drug abuse.
Our finding indicated that methamphetamine increased BBC3 expression with a concomitant increase in apoptosis, thus resulting in decreased cell viability. We anticipated that BBC3 deficiency would attenuate the methamphetamine-induced decrease in microglia survival. However, to our surprise, BBC3 deficiency failed to attenuate the methamphetamine-induced decrease in microglial survival and even exacerbated the decreased cell viability, contradicting the common view that BBC3 is an essential apoptosis inducer.14,15 This finding, along with the previous report that the number of microglia in the retina was significantly decreased in Bbc3 KO mice compared with WT,43 suggests a novel function of BBC3 in microglial survival.
Mechanistically, our study indicated that the transcription factor TRP53 and Mir143 co-coordinately regulated methamphetamine-induced BBC3 expression. This finding is in agreement with a previous study that demonstrated that Mir9-5p and SP1 coregulated target genes that cooperatively activate or block NFKB target gene expression.44 Consistent with previous studies showing that BBC3 is the downstream target of TRP53,16,45,46 our study also demonstrated that the TRP53 pathway was activated in methamphetamine-treated microglia and contributed to BBC3 expression. Indeed, methamphetamine induced an almost 2-fold increase in Bbc3 mRNA levels. Intriguingly, Western blot assay showed that methamphetamine treatment induced a transient increase in BBC3 protein levels followed by reduction to a level comparable to the control group, although the Bbc3 mRNA level significantly increased simultaneously. This result strongly suggested that an additional mechanism negatively regulates BBC3 protein expression likely at the post-transcriptional level. Therefore, we further examined the detailed mechanisms in this process and found that Mir143 regulated BBC3 protein expression. Although mounting evidence has demonstrated the critical role of Mir143 in cancer cells24-26 and in cell metabolism,27 studies in microglia in the CNS are lacking. To our knowledge, our study is the first to indicate that Mir143 regulates BBC3 expression, revealing a new mechanism for BBC3 expression.
Moreover, our study used comprehensive systems and approaches and demonstrated an unexpected function of BBC3, suggesting a functional property of BBC3 to exhibit apoptotic or autophagic activities under different conditions. A previous study indicated that current cancer therapies induce a tight interplay between autophagy and apoptosis.47 Autophagy plays a dual role, either promoting cell survival or cell death in response to cancer treatments depending on the cellular context and on the nature of the treatments.48 Consistent with these findings in cancer cells, methamphetamine increased BBC3 expression with concomitant increases in apoptosis and autophagy. The explanation for the consequent decrease in cell viability was because apoptosis overwhelmed autophagy, resulting in decreased cell viability in microglia treated with methamphetamine, as both apoptosis and autophagy cooperatively regulated cell survival. However, why did BBC3 deficiency fail to reverse the decrease in cell viability induced by methamphetamine? A possible explanation for this observation could be that although BBC3 deficiency decreased methamphetamine-induced apoptosis, it also decreased autophagy, as autophagy plays a prosurvival role in microglia (Fig. 5). Using both pharmacological and genetic approaches, we demonstrated that autophagy inhibition augmented the methamphetamine-induced decrease in cell viability, whereas the autophagy inducer rapamycin attenuated the methamphetamine-induced cell death. Our findings are consistent with that of a previous study that showed that methamphetamine induces autophagy as a prosurvival response in endothelial cells.49 Our findings suggested that BBC3 exhibits an unexpected complex function in different contexts, acting as a survival and proliferative factor by modulating apoptosis and autophagy cooperatively. Because of its potent apoptotic activity, BBC3 has been commonly considered a promising therapeutic molecule for cancer treatment;15 however, the knockout or knockdown of BBC3 expression worsened the methamphetamine-induced decrease in microglial survival. Therefore, mechanisms of the observed pro-survival role of BBC3 could vary in different cellular environments, helping to advance our understanding of the intricate relationship between apoptosis and autophagy.
Lending credence to the role of Mir143 in cell proliferation,24-26 our findings suggested that the transduction of primary microglia with Mir143 decreased microglial survival. Intriguingly, Mir143 further potentiated the methamphetamine-induced decrease in microglial viability. Reciprocally, inhibition of Mir143 activity via anti-Mir143 attenuated the decrease in cell viability in the presence of methamphetamine. Notably, Bbc3 is not the only target gene of Mir143. Previous studies have indicated that Mir143 exerts its tumor-suppressive function through targeting oncogenes such as SDC1, KRAS and CEBPA.24,25,50 Moreover, astrocyte-enriched Mir29a regulates BBC3 expression and reduces neuronal vulnerability to ischemia in the forebrain.51 Additionally, Mir125b exerts neuroprotection against ethanol-induced apoptosis by targeting Bbc3.52 To confirm whether BBC3 was involved in anti-Mir143-induced cell survival, we cotransduced microglia with lentiviral vectors expressing anti-Mir143 and Bbc3 siRNA. Intriguingly, the increased microglial survival induced by anti-Mir143 was abrogated by cotransduction with Bbc3 siRNA lentivirus. This finding was further confirmed by the cotransduction of cells with Mir143 and BBC3 OE (lacking the 3′-UTR) as evidenced by the fact that Mir143 failed to decrease cell survival in the presence of BBC3 OE lentivirus. These findings further underscore the role of the Mir143-BBC3 axis in mediating the methamphetamine-induced decrease in cell viability. Consistent with the role of the Mir143-BBC3 cascade in the survival of microglia, Mir143-BBC3 coregulated apoptosis and autophagy. These findings not only support the notion that the interplay of apoptosis and autophagy is a common mechanism of microglial survival with different effects47,48 but also add a novel molecular link between apoptosis and autophagy in the context of drug abuse.
These findings were validated in vivo, where a genetic approach using anti-Mir143 lentivirus was employed. Microinjection of anti-Mir143 lentivirus into the hippocampus of mice followed by methamphetamine injection resulted in the restoration of microglial survival, thus validating the role of Mir143 in the survival of microglia. Taken together, our findings implicate Mir143 as a regulator of microglia survival via its suppression of the target gene Bbc3. In the context of neurotoxicity induced by methamphetamine, when Mir143 expression is upregulated, treatment with anti-Mir143 may have a beneficial effect by dampening the decreased survival of microglia. Indeed, we observed that microinjecting anti-Mir143 into the hippocampus attenuated the methamphetamine-induced decrease in microglial survival. Although this finding was further confirmed in heterozygous Mir143+/- mice, one of the caveats of our study is the fact that we failed to obtain homozygous mir143−/- mice from 35 littermates by breeding heterozygous Mir143−/+ mice, whereas a previous study successfully generated homozygous mir143−/- mice.53 This discrepancy could be attributed to the different source of embryonic stem cells or the different strain of mice. The reason for this discrepancy regarding the homozygous mir143−/- mice is currently under investigation.
In response to cellular damage due to methamphetamine, the host is also capable of generating BBC3, which exerts somewhat paradoxically diverse roles as an inducer of both apoptosis and autophagy. Thus, depending on the specific context, the same host factor can manifest diverse activation responses that lead to diverse outcomes. The ultimate outcome of methamphetamine in the CNS is thus a result of the ensuing shift in balance between apoptosis and autophagy manifested over time following methamphetamine treatment. Here, we reported the diverse function of BBC3 as an inducer of both apoptosis and autophagy in the setting of methamphetamine abuse. Because this paradoxical regulation is a common theme of this type of factor, caution has to be exercised in the development of therapeutic targets involving these mediators.
In summary, Mir143 plays a crucial role in microglial survival via the suppression of BBC3 expression and the subsequent cooperation of apoptosis and autophagy. Thus, the Mir143-BBC3 axis mediates a regulatory pathway that is critical for regulating microglial survival.
Methods
Reagents
Lentiviral vectors carrying Mir143 OE (HB-LP140MI143), anti-Mir143 (HB-LP240MA143), mouse Bbc3 siRNA (HB-LP140240) and Atg7 siRNA (HB-LP400si717) and adenoviral vector carrying mRFP-GFP-MAP1LC3 (HB-AP2100001) were purchased from HANBIO. BBC3 OE plasmid (pHA-BBC3; 16588 deposited by Bert Vogelstein) was obtained from Addgene. According to the sequence of Bbc3, the BBC3 OE lentivirus was packaged by HANBIO. Methamphetamine was ordered from the National Institute for the Control of Pharmaceutical and Biological Products. Control siRNA (sc-37007), mouse Trp53 siRNA (sc-29436), mouse Bbc3 siRNA (sc-37154), mouse Becn1 siRNA (sc-29798) and pifithrin-α hydrobromide (sc-45050) were obtained from Santa Cruz Biotechnology. Rapamycin (R0395) and 3-methyladenine (M9281) were purchased from Sigma-Aldrich.
Animals
Mir143 mutant mice (Mutant Mouse Resource &Research Centers supported by NIH; https://www.mmrrc.org/catalog/cellLineSDS.php?mmrrc_id=43338) were generated using a standard mirKO targeting vector containing a PGK-EM7 promoter puroDtk selection cassette flanked by recombinase sites as described in a previous study.54 These mice were obtained from a model animal resource information platform (http://www.nbri-nju.com/strain-view-T000498?search=mi&description=true&page=3). Genotyping of mice or embryos was routinely performed with tail specimens or embryonic tissue by PCR using a mixture of 2 primers. The primer sequences used were as follows: Mir143-3p common forward, 5′-TCTAGAAAGTATAGGAACTTCCATGGTC-3′ (p1), and Mir143-3′ gsp reverse-1, 5′-CCCAGAATGGGTAGATGTAGCGTCTT-3′ (p2); Mir143-5′ gsp forward, 5′-TGGGTTCCATCTTTAGCACTGAAA-3′ (p3), and Mir143-5′ common reverse, 5′-ATAGCATACATTATACGAAGTTATCACTGG-3′ (p4).The PCR with p1-p2 primers yields a 272-base pair (bp) product and the PCR with p3-p4 primers yields 220-bp products for heterozygous mice and negative bands for WT mice. The PCR with p3-p2 primers yields 3163-bp and 565-bp products for heterozygous and WT mice, respectively. Heterozygous animals were used to obtain homozygous mir143−/- mice. Unfortunately, we failed to generate homozygous mir143−/- mice from 35 littermates thus far; therefore, heterozygous mice were used in our current study. However, a previous study successfully generated mir143 mutant mice using a different source of embryonic stem cells.53
Bbc3 KO mice were kindly shared by Dr. Gerard Zambettiat St. Jude Children's Research Hospital, Inc., and by Dr. Tao Cheng from State Key Laboratory of Experimental Hematology, Institute of Hematology and Blood Disease Hospital, Chinese Academy of Medical Sciences and Peking Union Medical College. C57BL/6J mice (male, 6-8 wk) were purchased from the Comparative Medicine Center, Yangzhou University (Yangzhou, China). All animals were housed under conditions of constant temperature and humidity on a 12h light: 12h dark cycle, with lights on at 07:00 a.m. Food and water were available ad libitum. Animals were deeply anesthetized by an overdose of isoflurane followed by pneumothorax before perfusion. All animal procedures were performed according to the protocols approved by the Institutional Animal Care and Use Committee of the Medical School, Southeast University.
Cell cultures
Primary mouse microglia cells were obtained from postnatal (P1 to P2) C57BL/6J mice, which were purchased from the Comparative Medicine Center, Yangzhou University. After the membranes and large blood vessels were dissociated, the dissected brain cortices in phosphate-buffered saline (PBS; 137 mM NaCl, 2.7 mM KCl, 10 mM Na2HPO4, 2 mM KH2PO4) supplemented with brain tissues were digested by trypsin-EDTA (Gibco, 25200056). Cells were seeded in 25-cm2 cell culture flasks pre-coated with poly-D-lysine (Sigma-Aldrich, P0296) with Dulbecco's modified Eagle's medium (DMEM) supplemented with fetal bovine serum (10% v/v) and penicillin-streptomycin (1% v/v). After 3 d, the medium was changed for the first time. Seven days later, the medium was replaced every 3 d, and CSF2/GM-CSF (colony stimulating factor 2 [granulocyte-macrophage]; 0.25 ng/ml; PeproTech, 315-03) was added to the flasks to promote microglial proliferation. The microglia were detached from the flasks by shaking and collected from the cell medium by centrifugation at 1500×g for 5 min.
BV-2 cells are a murine immortalized microglial cell line. These cells were used to characterize the effects of methamphetamine on brain microglia. BV-2 cells were obtained from the China Center for Type Culture Collection, routinely maintained in DMEM (10% fetal bovine serum, 1% penicillin-streptomycin) and incubated in 5% CO2 at 37°C.
Luciferase activity assays
The 990-bp human BBC3 gene 3′-UTR containing the putative Mir143 target site was PCR-amplified from human genomic DNA using forward (5′-GCGGCTCGAGAGCCCAATTAGGTGCCTG-3′) and reverse (5′-AATGCGGCCG CCCACTGTTCCAATCTGATTTTAT-3′) primers, and the DNA fragment was cloned into the XhoI and NotI sites on the 3′ end of the luc2 gene in the pmiR-RB-REPORTTM vector (RiboBio, UR100000). For the pmiR-RB-Bbc3 3′-UTR-Mir143-target-mutant vector, the Mir143 target site (CATCTCA) within the BBC3 3′-UTR was changed to GTAGAGT by PCR mutagenesis with primers Bbc3-Mir143T-F (5′-CCCACCACGTAGAGTGGAAAGGCTGTTGTGCTG-3′) and Bbc3-Mir143T-R (5′-GCCTTTCCACTCTACGTGGTGGGCGGCAGAGGC-3′). Briefly, 293T cells were transfected with Mir143 OE pLV-[hsa-Mir143] vector and the target plasmid pmiR-RB-Bbc3-3′-UTR or pmiR-RB-Bbc3-3′-UTR-Mir143-target-mutant in a molar ratio of 50:1. The Mir control pLV-[Mir control] plasmid was used as a negative control. Luciferase activity was determined 24 h post-transfection, and the reporter assay was performed following the manufacturer's protocol (Promega, E2920). Renilla luciferase activity was normalized to firefly luciferase activity and expressed as a percentage of the control (n > 3, performed in 5 wells each time).
Western blot analysis
Proteins were extracted in RIPA lysis buffer (Beyotime, P0013B). The nuclear protein fraction was extracted using a KeyGen Nuclear and Cytoplasmic Protein Extraction Kit (KeyGenBiotech, KGP150). For cytosolic and mitochondrial protein extraction, a Cell Mitochondria Isolation Kit (Beyotime, C3601) was used according to the manufacturer's instructions. Proteins were separated by sodium dodecyl sulfate-polyacrylamide gel electrophoresis (12%) and electrophoretically transferred onto polyvinylidene fluoride membranes. The membranes were blocked with 5% nonfat dry milk in Tris-buffered saline (50 mM Tris, 150 mM NaCl, pH 7.6) with Tween-20 (0.05%; Beyotime, ST825), probed with antibodies overnight at 4°C, and then incubated with horseradish peroxidase-conjugated goat anti-mouse/rabbit IgG secondary antibody (1:2000). The antibodies used were as follows: anti-MAP1LC3 (L7543) from Sigma-Aldrich; anti-BAX (2772), anti-BCL2L1 (2764), anti-cleaved CASP9 (9509), anti-CASP9 (9504), anti-cleaved CASP3 (9664), anti-CASP3 (9662), anti-H3F3/Histone H3 (9715), anti-ATG7 (2631), anti-phosphorylated (p)-AKT1 (9271), anti-AKT1 (9272), anti-p-MTOR (2971), anti-MTOR (2983), anti-p-RPS6KB1(9206), anti-p-EIF4EBP1 (9455), anti-EIF4EBP1 (9644), anti-p-RPS6 (2211), anti-p-MAPK1 (9101), and anti-MAPK1 (9107) from Cell Signaling Technology; anti-BBC3 (sc-28226), anti-TRP53 (FL-393), anti-GAPDH (sc-32233), and anti-BECN1 (sc-4834) from Santa Cruz Biotechnology; anti-SQSTM1 (18420), anti-BCL2 (12789), anti-CYCS (10993), andanti-COX4I1/COXIV (11242) from Proteintech; anti-NOS2 (ab3523) from Abcam and anti-ACTB/β-actin (BS6007M) from Bioworld. Detection was performed using a MicroChemi4.2® (DNR, Israel) digital image scanner. Band intensity was quantified by ImageJ software (NIH).
Real-time PCR
Total RNA isolated from cells was subjected to reverse transcription using a PrimeScript RT Master Mix Kit (TaKaRa, RR036). Real-time PCR analysis was performed for Bbc3 (forward primer: 5′-ACGACCTCAACGCGCAGTA-3′; reverse primer: 5′-CTAGTTGGGCTCCATTTCTGG-3′) and Actb (forward primer: 5′-CTAGCACCATGAAGATCAAGAT-3′; reverse primer: 5′-CCAGGATAGAGCCAC CAA-3′). Relative quantification was performed using TaKaRa SYBR® Premix Taq™ (TbIRNase H Plus; TaKaRa, RR420). Specific primers and probes for mature Mir143 and Rnu6 snRNA were obtained from RiboBio. All reactions were run in triplicate. The amount of Mir143 was obtained by normalizing to Rnu6 snRNA relative to the control as previously reported.55
Fluorescence in situ hybridization combined with immunostaining
Treated primary mouse microglia were fixed in 4% paraformaldehyde for 15 min and incubated in PBS overnight at 4°C. Cells were permeabilized with 0.3% Triton X-100 (Biosharp, BS363A) in PBS for 15 min, pre-hybridized in hybridization buffer (50% formamide, 10 mM Tris-HCl, pH 8.0, 200 μg/ml yeast tRNA [Invitrogen, 15401-029], 1X Denhardt's solution [Sigma-Aldrich, D2532], 600 mM NaCl, 0.25% SDS [Invitrogen, #15553-027], 1 mM EDTA, 10% dextran sulfate [Sigma-Aldrich, 8906-100]) for 1 h at 37°C. Then, the slides were heated to 65°C for 5 min in hybridization buffer with 2 nM commercially available digoxigenin-labeled Mir143 probe (Exiqon, 38515-15) followed by hybridization at 37°C overnight. The next day, the slides were washed 3 times in 2X SSC (3 M NaCl, 0.3 M sodium citrate, pH 7.4) and twice in 0.2X SSC at 42°C. The slides were then blocked with 1% BSA (Biosharp, BS043D) and 3% normal goat serum (ZSGB-BIO, ZLI-9056) in PBS for 1 h at room temperature and incubated with horseradish peroxidase-conjugated anti-digoxigenin antibody (Roche Diagnostics GmbH, 11207733910) overnight at 4°C. After the slides were washed 3 times with TBS, they were incubated with a TSA Cy5 kit (PerkinElmer, NEL745001KT) for 10 min at room temperature. The samples were washed once with 0.1% Tween-20 in PBS, twice in PBS and once in DEPC water (Sigma-Aldrich, V900882-10ML); blocked with 1% BSA and 3% normal goat serum in 1X PBS for 1 h at room temperature; and incubated with anti-AIF1 antibody (Wako, 019-19741) overnight at 4°C. The slides were washed twice with PBS and incubated with Alexa Fluor 488-conjugated goat anti-rabbit IgG (Invitrogen, A11008) antibody for 1 h at room temperature. Then, the slides were washed twice with PBS and mounted with Prolong gold anti-fade reagent containing DAPI (SouthernBiotech, 0100-20). The specificity of the Mir143 signal in FISH experiments was confirmed by comparison with a scrambled control. Unlike the Mir143 probe, the scramble probe showed no signal in microglia.
Microinjection of anti-Mir143
Eight-wk-old C57BL/6J mice were microinjected bilaterally with either anti-Mir control-RFP lentivirus or the anti-Mir143-RFP lentivirus (2 μl of 109 viral genomes µl−1, HANBIO, HB-LP240MA143) into the hippocampus using the following microinjection coordinates: 2.06 mm behind the bregma, ±1.5 mm lateral from the sagittal midline, at the depth of 1.7 mm to skull surface. To evaluate the effect of anti-Mir143 on the methamphetamine-induced decrease in microglial survival, 2 wk after microinjection of lentivirus, 8 mice were divided into 2 groups (N = 5; male). One group was treated with saline, and the other group was treated with methamphetamine (intraperitoneal, 30 mg/kg). The methamphetamine was injected every 2 h for a total of 4times/day. Two d later, these animals were perfused and sectioned followed by AIF1 staining to assess microglial activation.
Immunostaining and image analysis
According our previous study,56 the sections encompassing the entire hippocampus were cut into 35 μm sections on a cryostat. The sections were incubated with H2O2 for 10 min, permeabilized with 0.3% Triton X-100 in PBS for 15 min and blocked with 10% normal goat serum in 0.3% Triton X-100 for 1 h at room temperature. The sections were then incubated with rabbit anti-AIF1 antibody (Wako, 019-19741) overnight at 4°C. The next day, the sections were washed, incubated with biotinylated goat anti-rabbit IgG (Vector Laboratories, BA-1000) in PBS for 1 h at room temperature, and then incubated with VECTASTAIN® (VECTASTAIN®ABC Kit, Vector Laboratories, PK-6200) for 1 h. The horseradish peroxidase reaction product was visualized using an enhanced DAB peroxidase substrate kit (Vector Laboratories, SK-4100). The immunostaining signals were quantitatively analyzed using the optical Fractionator method with Microbrightfield Stereo-Investigator software (Stereo Investigator software, Microbrightfield, VT, USA). The total number of AIF1 microglia in the entire area of the hippocampus was counted from 4 or 5 samples per group.
MTT assay
Cell viability was measured by MTT assay. Cells were seeded in 96-well plates for 2 d and treated with different concentrations of methamphetamine for 24 h. MTT dye (20 µl, 5 mg/ml; Biosharp, BS030C) was added to each well, and the cells were then incubated in a CO2 incubator at 37°C for 1.5 h. The medium was aspirated from each well, and DMSO (200 µl) was added to dissolve the formazan crystals. The absorbance of each well was obtained using a Synergy H1 Multi-Mode Reader (BioTek, Winooski, VT, USA) with a reference wavelength of 570 nm.
CCK8 assay
Cell viability was also measured using a CCK8 kit from Yeasen (40203ES80). Cells were plated at a density of 2×104 cells/well in a 96-well plate. After the cells were exposed to methamphetamine for 24 h, CCK-8 (10 µl) was added to each well of the 96-well plate, and the plate was incubated for 1.5 h at 37°C. Viable cells were counted by absorbance measurements at 450 nm using a Synergy H1 Multi-Mode Reader (BioTek, Winooski, VT, USA).
Trypan blue exclusion assay
Cells were cultured in 24-well plates and treated with 1.5 Mm methamphetamine. After 24 h, the cells were collected and then stained with trypan blue dye for 5 min at room temperature. Cells were counted by 4 independent hemocytometer counts. Cell death was determined as the percentage of dead cells over the total number of cells according to a previous study.57
Measurement of the mitochondrial membrane potential
The integrity of the inner mitochondrial membrane was measured by observing the potential gradient across this membrane using a mitochondrial membrane potential assay kit with JC-1 (Beyotime, C2006). Cells were grown on cover slips or in 96-well plates and incubated with JC-1 dye at 37°C for 20 min. The cells were then washed twice with PBS. The cells on coverslips or in 96-well plates were analyzed immediately using a confocal microscope (Olympus FV1000, Center Valley, PA, USA) or a Synergy H1 Multi-Mode Reader (BioTek, Winooski, VT, USA). Red emission indicates membrane potential-dependent JC-1 aggregates in mitochondria. Green fluorescence reflects the monomeric form of JC-1 appearing in the cytoplasm after mitochondrial membrane depolarization.
Transmission electron microscopy assay
Cells were cultured in 24-well plates and treated with 1.5 mM methamphetamine. After the cells were incubated with methamphetamine for 24 h, the treated cells were harvested in a 1.5 ml microcentrifuge tube for each sample. Cells were washed with cold PBS once and fixed in a mixture of 2% glutaraldehyde and 2% formaldehyde in 0.1 M sodium cacodylate buffer (pH 7.4) at 4°C overnight. All specimens were washed 4 times in 0.1 M sodium cacodylate buffer (pH 7.4) containing 264 mM sucrose (Sinopharm Chemical Reagent Co., Ltd, 10021418) before being transferred to 1% osmium tetroxide in 0.1 M sodium cacodylate (pH 7.4) for 2 h. The specimens were then washed 3 times in ddH2O and stained in 2% aqueous uranyl acetate for 2 h. The specimens were dehydrated in an ethanol series, infiltrated in propylene oxide 2 times, and embedded in Epon812 (SPI Science, 02659-AB). Ultra-thin sections (90-nm thick) were cut using a Leica UC7 with a diamond knife. Grids were post-stained with 2% saturated uranyl acetate in 50% ethanol and 1% lead citrate (pH 12), examined usingH-7650 electron microscope (Hitachi, Japan), and recorded with a Ganton830 digital camera.
Immunoprecipitation
To investigate the interaction between BECN1 with BCL2, cells were treated with 1.5 mM methamphetamine and lysed with RIPA buffer. In total, 100 μl protein was incubated with 1 μl BECN1 antibody overnight at 4°C and precipitated with 20 μl Protein A/G PLUS-Agarose (Santa Cruz Biotechnology, sc-2003). After the cell pellets were rotated at 4°C for 90 min, the cell pellets were rinsed twice with 1 ml RIPA lysis buffer and boiled for 5 min with 5XSDS loading buffer (6 μl) and RIPA lysis buffer (24 μl). Then, the protein complexes were detected using BCL2 antibody; the input protein (without antibody addition) served as a control to confirm that equal amounts of total protein were used.
Statistical analyses
Statistical analysis was performed using Student t test or one-way analysis of variance (ANOVA) followed by the Holm-Sidak test (SigmaPlot 11.0). The appropriate tests are indicated in the figure legends. The results were considered significant if p < 0.05 by ANOVA.
Supplementary Material
Abbreviations
- 3-MA
3-methyladenine
- ACTB
actin, beta
- AICD
activation-induced cell death
- AIF1
allograft inflammatory factor 1
- AKT1
thymoma viral proto-oncogene 1
- ATG7
autophagy- related 7
- BafA1
bafilomycinA1
- BAX
BCL2-associated X protein
- BBC3
BCL2 binding component 3
- BCL2
B-cell leukemia/lymphoma 2
- BCL2L1
BCL2-like 1
- BECN1
Beclin 1, autophagy related
- BH3
BCL2-homology 3
- CASP3
caspase 3
- CASP9
caspase 9
- CCK8
Cell Counting Kit-8
- CNS
central nervous system
- COX4I1
cytochrome c oxidase subunit IV isoform 1
- CQ
chloroquine
- CSF2/GM-CSF
colony stimulating factor 2 (granulocyte-macrophage)
- CYCS
cytochrome c, somatic
- DAPI
4′,6-diamidino-2-phenylindole
- DMEM
Dulbecco's modified Eagle's medium
- EIF4EBP1
eukaryotic translation initiation factor 4E binding protein 1
- FISH
fluorescence in situ hybridization
- GAPDH
glyceraldehyde-3-phosphate dehydrogenase
- H3F3
H3 histone, family 3
- KO
knockout
- MAP1LC3
microtubule-associated protein 1 light chain 3
- MAPK1
mitogen-activated protein kinase 1
- Meth
methamphetamine
- Mir143
microRNA143
- miRs
microRNAs
- MTOR
mechanistic target of rapamycin (serine/threonine kinase)
- NO
nitric oxide
- NOS2
nitric oxide synthase 2, inducible
- OE
overexpression
- PCR
polymerase chain reaction
- RPS6
ribosomal protein S6
- RPS6KB1
ribosomal protein S6 kinase, polypeptide 1
- SQSTM1
sequestosome 1
- TRP53
transformation related protein 53
- TUNEL
terminal deoxynucleotidyl transferase-mediated dUTP nick end-labeling
- UTR
untranslated region
- WT
wild type
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
Funding
This work was supported by grants from the National Natural Science Foundation of China (No. 81322048 and No. 81473190). We were also awarded grants from the Jiangsu Specially Appointed Professor and the Major State Basic Research Development Program of China (973 Program) (2013CB733800, 2013CB733803) and the Postdoctoral Research Start Foundation of Southeast University (1124000342).
References
- [1].Schepers RJ, Oyler JM, Joseph RE Jr., Cone EJ, Moolchan ET, Huestis MA. Methamphetamine and amphetamine pharmacokinetics in oral fluid and plasma after controlled oral methamphetamine administration to human volunteers. Clin Chem 2003; 49:121-32; PMID:12507968; http://dx.doi.org/ 10.1373/49.1.121 [DOI] [PubMed] [Google Scholar]
- [2].Kaushal N, Matsumoto RR. Role of sigma receptors in methamphetamine-induced neurotoxicity. Curr Neuropharmacol 2011; 9:54-7; PMID:21886562; http://dx.doi.org/ 10.2174/157015911795016930 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Davalos D, Grutzendler J, Yang G, Kim JV, Zuo Y, Jung S, Littman DR, Dustin ML, Gan WB. ATP mediates rapid microglial response to local brain injury in vivo. Nat Neurosci 2005; 8:752-8; PMID:15895084; http://dx.doi.org/ 10.1038/nn1472 [DOI] [PubMed] [Google Scholar]
- [4].Nimmerjahn A, Kirchhoff F, Helmchen F. Resting microglial cells are highly dynamic surveillants of brain parenchyma in vivo. Science 2005; 308:1314-8; PMID:15831717; http://dx.doi.org/ 10.1126/science.1110647 [DOI] [PubMed] [Google Scholar]
- [5].Escubedo E, Guitart L, Sureda FX, Jimenez A, Pubill D, Pallas M, Camins A, Camarasa J. Microgliosis and down-regulation of adenosine transporter induced by methamphetamine in rats. Brain Res 1998; 814:120-6; PMID:9838075; http://dx.doi.org/ 10.1016/S0006-8993(98)01065-8 [DOI] [PubMed] [Google Scholar]
- [6].Asanuma M, Tsuji T, Miyazaki I, Miyoshi K, Ogawa N. Methamphetamine-induced neurotoxicity in mouse brain is attenuated by ketoprofen, a non-steroidal anti-inflammatory drug. Neurosci Lett 2003; 352:13-6; PMID:14615038; http://dx.doi.org/ 10.1016/j.neulet.2003.08.015 [DOI] [PubMed] [Google Scholar]
- [7].LaVoie MJ, Card JP, Hastings TG. Microglial activation precedes dopamine terminal pathology in methamphetamine-induced neurotoxicity. Exp Neurol 2004; 187:47-57; PMID:15081587; http://dx.doi.org/ 10.1016/j.expneurol.2004.01.010 [DOI] [PubMed] [Google Scholar]
- [8].Thomas DM, Dowgiert J, Geddes TJ, Francescutti-Verbeem D, Liu X, Kuhn DM. Microglial activation is a pharmacologically specific marker for the neurotoxic amphetamines. Neurosci Lett 2004; 367:349-54; PMID:15337264; http://dx.doi.org/ 10.1016/j.neulet.2004.06.065 [DOI] [PubMed] [Google Scholar]
- [9].Thomas DM, Kuhn DM. Attenuated microglial activation mediates tolerance to the neurotoxic effects of methamphetamine. J Neurochem 2005; 92:790-7; PMID:15686480; http://dx.doi.org/ 10.1111/j.1471-4159.2004.02906.x [DOI] [PubMed] [Google Scholar]
- [10].Sekine Y, Ouchi Y, Sugihara G, Takei N, Yoshikawa E, Nakamura K, Iwata Y, Tsuchiya KJ, Suda S, Suzuki K, et al.. Methamphetamine causes microglial activation in the brains of human abusers. J Neurosci 2008; 28:5756-61; PMID:18509037; http://dx.doi.org/ 10.1523/JNEUROSCI.1179-08.2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Yun HJ, Yoon JH, Lee JK, Noh KT, Yoon KW, Oh SP, Oh HJ, Chae JS, Hwang SG, Kim EH, et al.. Daxx mediates activation-induced cell death in microglia by triggering MST1 signalling. Embo J 2011; 30:2465-76; PMID:21572393; http://dx.doi.org/ 10.1038/emboj.2011.152 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Chipuk JE, Green DR. PUMA cooperates with direct activator proteins to promote mitochondrial outer membrane permeabilization and apoptosis. Cell Cycle 2009; 8:2692-6; PMID:19652530; http://dx.doi.org/ 10.4161/cc.8.17.9412 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Thakur VS, Ruhul Amin AR, Paul RK, Gupta K, Hastak K, Agarwal MK, Jackson MW, Wald DN, Mukhtar H, Agarwal ML. p53-Dependent p21-mediated growth arrest pre-empts and protects HCT116 cells from PUMA-mediated apoptosis induced by EGCG. Cancer Lett 2010; 296:225-32; PMID:20444544; http://dx.doi.org/ 10.1016/j.canlet.2010.04.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Yu J, Zhang L. PUMA, a potent killer with or without p53. Oncogene 2008; 27 Suppl 1: S71-83; PMID:19641508; http://dx.doi.org/ 10.1038/onc.2009.45 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Yu J, Zhang L, Hwang PM, Kinzler KW, Vogelstein B. PUMA induces the rapid apoptosis of colorectal cancer cells. Mol Cell 2001; 7:673-82; PMID:11463391; http://dx.doi.org/ 10.1016/S1097-2765(01)00213-1 [DOI] [PubMed] [Google Scholar]
- [16].Nakano K, Vousden KH. PUMA, a novel proapoptotic gene, is induced by p53. Mol Cell 2001; 7:683-94; PMID:11463392; http://dx.doi.org/ 10.1016/S1097-2765(01)00214-3 [DOI] [PubMed] [Google Scholar]
- [17].O'Neill LA, Sheedy FJ, McCoy CE. MicroRNAs: the fine-tuners of Toll-like receptor signalling. Nat Rev Immunol 2011; 11:163-75; PMID:21331081; http://dx.doi.org/ 10.1038/nri2957 [DOI] [PubMed] [Google Scholar]
- [18].Johnnidis JB, Harris MH, Wheeler RT, Stehling-Sun S, Lam MH, Kirak O, Brummelkamp TR, Fleming MD, Camargo FD. Regulation of progenitor cell proliferation and granulocyte function by microRNA-223. Nature 2008; 451:1125-9; PMID:18278031; http://dx.doi.org/ 10.1038/nature06607 [DOI] [PubMed] [Google Scholar]
- [19].Ponomarev ED, Veremeyko T, Barteneva N, Krichevsky AM, Weiner HL. MicroRNA-124 promotes microglia quiescence and suppresses EAE by deactivating macrophages via the C/EBP-alpha-PU.1 pathway. Nat Med 2011; 17:64-70; PMID:21131957; http://dx.doi.org/ 10.1038/nm.2266 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].O'Connell RM, Rao DS, Chaudhuri AA, Baltimore D. Physiological and pathological roles for microRNAs in the immune system. Nat Rev Immunol 2010; 10:111-22; PMID:20098459; http://dx.doi.org/ 10.1038/nri2708 [DOI] [PubMed] [Google Scholar]
- [21].Bazzoni F, Rossato M, Fabbri M, Gaudiosi D, Mirolo M, Mori L, Tamassia N, Mantovani A, Cassatella MA, Locati M. Induction and regulatory function of miR-9 in human monocytes and neutrophils exposed to proinflammatory signals. Proc Natl Acad Sci U S A 2009; 106:5282-7; PMID:19289835; http://dx.doi.org/ 10.1073/pnas.0810909106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Cardoso AL, Guedes JR, Pereira de Almeida L, Pedroso de Lima MC. miR-155 modulates microglia-mediated immune response by down-regulating SOCS-1 and promoting cytokine and nitric oxide production. Immunology 2012; 135:73-88; PMID:22043967; http://dx.doi.org/ 10.1111/j.1365-2567.2011.03514.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Ponomarev ED, Veremeyko T, Weiner HL. MicroRNAs are universal regulators of differentiation, activation, and polarization of microglia and macrophages in normal and diseased CNS. Glia 2013; 61:91-103; PMID:22653784; http://dx.doi.org/ 10.1002/glia.22363 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Li H, Zhang Z, Zhou X, Wang Z, Wang G, Han Z. Effects of microRNA-143 in the differentiation and proliferation of bovine intramuscular preadipocytes. Mol Biol Rep 2011; 38:4273-80; PMID:21113671; http://dx.doi.org/ 10.1007/s11033-010-0550-z [DOI] [PubMed] [Google Scholar]
- [25].Li R, Zhang L, Jia L, Duan Y, Li Y, Wang J, Bao L, Sha N. MicroRNA-143 targets Syndecan-1 to repress cell growth in melanoma. PLoS One 2014; 9:e94855; PMID:24722758; http://dx.doi.org/ 10.1371/journal.pone.0094855 [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- [26].Borralho PM, Simoes AE, Gomes SE, Lima RT, Carvalho T, Ferreira DM, Vasconcelos MH, Castro RE, Rodrigues CM. miR-143 overexpression impairs growth of human colon carcinoma xenografts in mice with induction of apoptosis and inhibition of proliferation. PLoS One 2011; 6:e23787; PMID:21901135; http://dx.doi.org/ 10.1371/journal.pone.0023787 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Jordan SD, Kruger M, Willmes DM, Redemann N, Wunderlich FT, Bronneke HS, Merkwirth C, Kashkar H, Olkkonen VM, Bottger T, et al.. Obesity-induced overexpression of miRNA-143 inhibits insulin-stimulated AKT activation and impairs glucose metabolism. Nat Cell Biol 2011; 13:434-46; PMID:21441927; http://dx.doi.org/ 10.1038/ncb2211 [DOI] [PubMed] [Google Scholar]
- [28].Sharma A, Hu XT, Napier TC, Al-Harthi L. Methamphetamine and HIV-1 Tat down regulate beta-catenin signaling: implications for methampetamine abuse and HIV-1 co-morbidity. J Neuroimmune Pharmacol 2011; 6:597-607; PMID:21744004; http://dx.doi.org/ 10.1007/s11481-011-9295-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Talloczy Z, Martinez J, Joset D, Ray Y, Gacser A, Toussi S, Mizushima N, Nosanchuk JD, Goldstein H, Loike J, et al.. Methamphetamine inhibits antigen processing, presentation, and phagocytosis. PLoS Pathog 2008; 4:e28; PMID:18282092; http://dx.doi.org/ 10.1371/journal.ppat.0040028 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Lee J, Hur J, Lee P, Kim JY, Cho N, Kim SY, Kim H, Lee MS, Suk K. Dual role of inflammatory stimuli in activation-induced cell death of mouse microglial cells. Initiation of two separate apoptotic pathways via induction of interferon regulatory factor-1 and caspase-11. J Biol Chem 2001; 276:32956-65; PMID:11402054; http://dx.doi.org/ 10.1074/jbc.M104700200 [DOI] [PubMed] [Google Scholar]
- [31].Lee P, Lee J, Kim S, Lee MS, Yagita H, Kim SY, Kim H, Suk K. NO as an autocrine mediator in the apoptosis of activated microglial cells: correlation between activation and apoptosis of microglial cells. Brain Res 2001; 892:380-5; PMID:11172787; http://dx.doi.org/ 10.1016/S0006-8993(00)03257-1 [DOI] [PubMed] [Google Scholar]
- [32].Rabinowitz JD, White E. Autophagy and metabolism. Science 2010; 330:1344-8; PMID:21127245; http://dx.doi.org/ 10.1126/science.1193497 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell 2011; 144:646-74; PMID:21376230; http://dx.doi.org/ 10.1016/j.cell.2011.02.013 [DOI] [PubMed] [Google Scholar]
- [34].Saiki S, Sasazawa Y, Imamichi Y, Kawajiri S, Fujimaki T, Tanida I, Kobayashi H, Sato F, Sato S, Ishikawa K, et al.. Caffeine induces apoptosis by enhancement of autophagy via PI3K/Akt/mTOR/p70S6K inhibition. Autophagy 2011; 7:176-87; PMID:21081844; http://dx.doi.org/ 10.4161/auto.7.2.14074 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Aoki H, Takada Y, Kondo S, Sawaya R, Aggarwal BB, Kondo Y. Evidence that curcumin suppresses the growth of malignant gliomas in vitro and in vivo through induction of autophagy: role of Akt and extracellular signal-regulated kinase signaling pathways. Mol Pharmacol 2007; 72:29-39; PMID:17395690; http://dx.doi.org/ 10.1124/mol.106.033167 [DOI] [PubMed] [Google Scholar]
- [36].Ellington AA, Berhow MA, Singletary KW. Inhibition of Akt signaling and enhanced ERK1/2 activity are involved in induction of macroautophagy by triterpenoid B-group soyasaponins in colon cancer cells. Carcinogenesis 2006; 27:298-306; PMID:16113053; http://dx.doi.org/ 10.1093/carcin/bgi214 [DOI] [PubMed] [Google Scholar]
- [37].Klionsky DJ, Cuervo AM, Seglen PO. Methods for monitoring autophagy from yeast to human. Autophagy 2007; 3:181-206; PMID:17224625; http://dx.doi.org/ 10.4161/auto.3678 [DOI] [PubMed] [Google Scholar]
- [38].Kroemer G, Galluzzi L, Vandenabeele P, Abrams J, Alnemri ES, Baehrecke EH, Blagosklonny MV, El-Deiry WS, Golstein P, Green DR, et al.. Classification of cell death: recommendations of the Nomenclature Committee on Cell Death 2009. Cell Death Differ 2009; 16:3-11; PMID:18846107; http://dx.doi.org/ 10.1038/cdd.2008.150 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Klionsky DJ, Abdalla FC, Abeliovich H, Abraham RT, Acevedo-Arozena A, Adeli K, Agholme L, Agnello M, Agostinis P, Aguirre-Ghiso JA, et al.. Guidelines for the use and interpretation of assays for monitoring autophagy. Autophagy 2012; 8:445-544; PMID:22966490; http://dx.doi.org/ 10.4161/auto.19496 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Zalckvar E, Berissi H, Mizrachy L, Idelchuk Y, Koren I, Eisenstein M, Sabanay H, Pinkas-Kramarski R, Kimchi A. DAP-kinase-mediated phosphorylation on the BH3 domain of beclin 1 promotes dissociation of beclin 1 from Bcl-XL and induction of autophagy. EMBO Rep 2009; 10:285-92; PMID:19180116; http://dx.doi.org/ 10.1038/embor.2008.246 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Wirawan E, Lippens S, Vanden Berghe T, Romagnoli A, Fimia GM, Piacentini M, Vandenabeele P. Beclin1: a role in membrane dynamics and beyond. Autophagy 2012; 8:6-17; PMID:22170155; http://dx.doi.org/ 10.4161/auto.8.1.16645 [DOI] [PubMed] [Google Scholar]
- [42].Li DD, Wang LL, Deng R, Tang J, Shen Y, Guo JF, Wang Y, Xia LP, Feng GK, Liu QQ, et al.. The pivotal role of c-Jun NH2-terminal kinase-mediated Beclin 1 expression during anticancer agents-induced autophagy in cancer cells. Oncogene 2009; 28:886-98; PMID:19060920; http://dx.doi.org/ 10.1038/onc.2008.441 [DOI] [PubMed] [Google Scholar]
- [43].Zhang F, Li Y, Tang Z, Kumar A, Lee C, Zhang L, Zhu C, Klotzsche-von Ameln A, Wang B, Gao Z, et al.. Proliferative and survival effects of PUMA promote angiogenesis. Cell Rep 2012; 2:1272-85; PMID:23122957; http://dx.doi.org/ 10.1016/j.celrep.2012.09.023 [DOI] [PubMed] [Google Scholar]
- [44].Gilmore TD. Introduction to NF-kappaB: players, pathways, perspectives. Oncogene 2006; 25:6680-4; PMID:17072321; http://dx.doi.org/ 10.1038/sj.onc.1209954 [DOI] [PubMed] [Google Scholar]
- [45].Jeffers JR, Parganas E, Lee Y, Yang C, Wang J, Brennan J, MacLean KH, Han J, Chittenden T, Ihle JN, et al.. Puma is an essential mediator of p53-dependent and -independent apoptotic pathways. Cancer Cell 2003; 4:321-8; PMID:14585359; http://dx.doi.org/ 10.1016/S1535-6108(03)00244-7 [DOI] [PubMed] [Google Scholar]
- [46].Villunger A, Michalak EM, Coultas L, Mullauer F, Bock G, Ausserlechner MJ, Adams JM, Strasser A. p53- and drug-induced apoptotic responses mediated by BH3-only proteins puma and noxa. Science 2003; 302:1036-8; PMID:14500851; http://dx.doi.org/ 10.1126/science.1090072 [DOI] [PubMed] [Google Scholar]
- [47].Maiuri MC, Zalckvar E, Kimchi A, Kroemer G. Self-eating and self-killing: crosstalk between autophagy and apoptosis. Nat Rev Mol Cell Biol 2007; 8:741-52; PMID:17717517; http://dx.doi.org/ 10.1038/nrm2239 [DOI] [PubMed] [Google Scholar]
- [48].White E. Deconvoluting the context-dependent role for autophagy in cancer. Nat Rev Cancer 2012; 12:401-10; PMID:22534666; http://dx.doi.org/ 10.1038/nrc3262 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Ma J, Wan J, Meng J, Banerjee S, Ramakrishnan S, Roy S. Methamphetamine induces autophagy as a pro-survival response against apoptotic endothelial cell death through the Kappa opioid receptor. Cell Death Dis 2014; 5:e1099; PMID:24603327; http://dx.doi.org/ 10.1038/cddis.2014.64 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Chen X, Guo X, Zhang H, Xiang Y, Chen J, Yin Y, Cai X, Wang K, Wang G, Ba Y, et al.. Role of miR-143 targeting KRAS in colorectal tumorigenesis. Oncogene 2009; 28:1385-92; PMID:19137007; http://dx.doi.org/ 10.1038/onc.2008.474 [DOI] [PubMed] [Google Scholar]
- [51].Ouyang YB, Xu L, Lu Y, Sun X, Yue S, Xiong XX, Giffard RG. Astrocyte-enriched miR-29a targets PUMA and reduces neuronal vulnerability to forebrain ischemia. Glia 2013; 61:1784-94; PMID:24038396; http://dx.doi.org/ 10.1002/glia.22556 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Chen X, Liu J, Feng WK, Wu X, Chen SY. MiR-125b protects against ethanol-induced apoptosis in neural crest cells and mouse embryos by targeting Bak 1 and PUMA. Exp Neurol 2015; 271:104-11; PMID:26024858; http://dx.doi.org/ 10.1016/j.expneurol.2015.04.026 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Xin M, Small EM, Sutherland LB, Qi X, McAnally J, Plato CF, Richardson JA, Bassel-Duby R, Olson EN. MicroRNAs miR-143 and miR-145 modulate cytoskeletal dynamics and responsiveness of smooth muscle cells to injury. Genes Dev 2009; 23:2166-78; PMID:19720868; http://dx.doi.org/ 10.1101/gad.1842409 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Prosser HM, Koike-Yusa H, Cooper JD, Law FC, Bradley A. A resource of vectors and ES cells for targeted deletion of microRNAs in mice. Nat Biotechnol 2011; 29:840-5; PMID:21822254; http://dx.doi.org/ 10.1038/nbt.1929 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Hu G, Zhou R, Liu J, Gong AY, Chen XM. MicroRNA-98 and let-7 regulate expression of suppressor of cytokine signaling 4 in biliary epithelial cells in response to Cryptosporidium parvum infection. J Infect Dis 2010; 202:125-35; PMID:20486857; http://dx.doi.org/ 10.1086/653212 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Yao H, Ma R, Yang L, Hu G, Chen X, Duan M, Kook Y, Niu F, Liao K, Fu M, et al.. MiR-9 promotes microglial activation by targeting MCPIP1. Nat Commun 2014; 5:4386; PMID:25019481 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Guo WJ, Zhang YM, Zhang L, Huang B, Tao FF, Chen W, Guo ZJ, Xu Q, Sun Y. Novel monofunctional platinum (II) complex Mono-Pt induces apoptosis-independent autophagic cell death in human ovarian carcinoma cells, distinct from cisplatin. Autophagy 2013; 9:996-1008; PMID:23580233; http://dx.doi.org/ 10.4161/auto.24407 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.