

ABSTRACT
Macroautophagy/autophagy is a conserved catabolic process that recycles cytoplasmic material during low energy conditions. BECN1/Beclin1 (Beclin 1, autophagy related) is an essential protein for function of the class 3 phosphatidylinositol 3-kinase (PtdIns3K) complexes that play a key role in autophagy nucleation and elongation. Here, we show that AMP-activated protein kinase (AMPK) regulates autophagy by phosphorylating BECN1 at Thr388. Phosphorylation of BECN1 is required for autophagy upon glucose withdrawal. BECN1T388A, a phosphorylation defective mutant, suppresses autophagy through decreasing the interaction between PIK3C3 (phosphatidylinositol 3-kinase catalytic subunit type 3) and ATG14 (autophagy-related 14). The BECN1T388A mutant has a higher affinity for BCL2 than its wild-type counterpart; the mutant is more prone to dimer formation. Conversely, a BECN1 phosphorylation mimic mutant, T388D, has stronger binding to PIK3C3 and ATG14, and promotes higher autophagy activity than the wild-type control. These findings uncover a novel mechanism of autophagy regulation.
KEYWORDS: AMPK, autophagy, BECN1, phosphorylation, regulation
Introduction
Macroautophagy, commonly referred to as autophagy, is an evolutionarily conserved catabolic process that protects cells against a variety of stresses, such as nutrient starvation, organelle damage, protein aggregates and infection.1-4 Autophagy involves the sequestration of cytoplasmic “cargo” into a phagophore, which expands and matures into a double-membrane autophagosome; fusion of the latter with the lysosome exposes the engulfed cytoplamic material to lysosomal hydrolases for degradation and subsequent recycling.5-7 More than 40 autophagy-related proteins have been identified in yeast and various mammalian species.8 These proteins are involved in autophagy initiation, phagophore nucleation and elongation, autophagosome maturation, and fusion with the lysosome. In all steps, autophagy is regulated by different molecules.
BECN1 is one of the first identified mammalian autophagy-related proteins.9 It contains 3 structural domains: a BH3 domain at the N terminus (amino acids 114–123, which mainly bind BCL2), a central coiled-coil domain (CCD, amino acids 144–269, a domain binds ATG14 and UVRAG [UV radiation resistance associated]) and an evolutionarily conserved domain (ECD, amino acids 244–337, which binds PIK3C3). Crystal structure of the C-terminal domain of BECN1 reveals 3 β-sheet-α-helix repeats, which are required for targeting the PIK3C3-BECN1-ATG14 complex to the phagophore. This part of the protein is named the β-α repeated, autophagy-specific (BARA) domain. The N-terminal region of BECN1 is responsible for vacuolar protein sorting.10 BECN1 is a component of the PtdIns3K complexes that participate in phagophore nucleation and elongation.11 BECN1 recruits ATG14 or UVRAG to the PtdIns3K complexes to promote autophagy.12,13 Under normal steady-state growth conditions, BCL2 directly binds to BECN1 through its BH3 domain and suppresses autophagy.14,15
AMPK is a key energy sensor that maintains cellular energy homeostasis upon nutrient starvation.16 AMPK activity is dependent on the AMP:ATP or ADP:ATP ratio. AMPK is a heterotrimetric complex comprised of a catalytic α-subunit and 2 regulatory β- and γ-subunits.17 AMPK catalytic subunits contain a conventional Ser/Thr kinase domain at the N terminus. AMPK activity is increased when its Thr172 residue in the activation loop is phosphorylated by upstream kinases.18 AMPK phosphorylates its substrates on a consensus sequence, RXXS/T.19
Autophagy can be stimulated under multiple stresses, such as glucose deprivation, serum starvation, or ER stress. Post-translational modifications of proteins, including phosphorylation, play critical roles in cellular stress responses. For example, the mechanistic target of rapamycin (serine/threonine kinase) complex 1 (MTORC1) or AMPK phosphorylate ULK1 (unc-51 like autophagy activating kinase 1) to suppress or stimulate autophagy, respectively.20-23 AMPK also phosphorylates BECN1 at S90 and S93, and PIK3C3 to regulate autophagy or cell trafficking.21,24-26 Although BECN1 phosphorylation has been reported in autophagy regulation, the effects of BARA domain phosphorylation are still not understood.10 This study demonstrates that AMPK phosphorylates BECN1 at Thr388 in the BARA domain to induce autophagy.
Results
AMPK is required for autophagy induction
Although AMPK has been identified as a regulator of autophagy, the precise mechanism of how AMPK controls the process still remains controversial.24-30 To address this issue, we examined AMPK-modulated autophagy by determining the LC3-II level, which is a marker of autophagy. The amount of LC3-II is higher in HEK293T cells treated with AICAR, a known AMPK activator (Fig. 1A). Autophagy was also induced by glucose deprivation (Fig. 1B). Conversely, autophagy was suppressed when cells were pretreated with compound C (an AMPK inhibitor) even with glucose starvation (Fig. 1C). Similarly, autophagy was attenuated when endogenous PRKAA1 (an AMPK catalytic subunit) was depleted regardless of glucose deprivation (Fig. 1D, E, F). LC3-II was augmented in HEK293T cells transfected with a plasmid encoding GFP-PRKAA1-CA (a constitutively active mutant form of PRKAA1). In contrast, LC3-II obviously decreased in GFP-PRKAA1-KD (kinase dead PRKAA1)-transfected cells (Fig. 1G). A higher autophagy level was found in Prkaa wild-type (WT) than Prkaa knockout (KO) mouse embryonic fibroblast (MEF) cells (Fig. 1H). These results indicate that autophagy induction is closely related to AMPK activity.
Figure 1.

AMPK is essential for the induction of autophagy. HEK293T cells were treated with 0.25 mM AICAR for 1 h (A) or subjected to glucose deprivation for 3 h (B). Autophagy was determined by LC3 immunoblotting. p-AMPK was determined indicating AMPK activation. (C) After pretreatment with 10 μM compound C for 0.5 h, glucose starvation was performed in HEK293T cells for 3 h. LC3 and AMPK activity were then assayed using immunoblotting. (D) HEK293T cells were transfected with pGE1-shPRKAA and grown under nutrient-rich conditions for 36 h before being lysed for western blotting. LC3 and PRKAA were detected by immunoblotting. (E) Representative images of cells were fixed and examined by immunofluorescence after transfection with pGE1-shPRKAA in HeLa cells for 48 h, and glucose starvation (3 h). The red dots are LC3 puncta. Scale bars: 10 μm. (F) Quantification of GFP-LC3 puncta shown in (E). Bars are mean ± SEM of triplicate samples (≥ 10 cells analyzed per sample). The comparison of different groups was carried out using 2-tailed unpaired Student t test by Graphpad Prism5 (Graphpad Software, San Diego, CA, USA). Differences were considered statistically significant (***, ###) at P < 0.05. (G) GFP-PRKAA1 WT, CA and KD were transfected into HEK293T cells to examine the role of AMPK in autophagy. (H) Prkaa WT and KO MEF cells were subjected to glucose starvation for 3 h. Cell lysates were probed with the indicated antibodies.
AMPK phosphorylates BECN1
BECN1 can be phosphorylated by a number of different kinases.31-33 Because AMPK is a protein serine/threonine kinase, we hypothesized that AMPK may phosphorylate BECN1 to initiate autophagy. Thus, we first tested whether AMPK could interact with BECN1. Binding of AMPK with BECN1 could be readily found in cells co-expressing GFP-PRKAA1 and GST-BECN1 (Fig. 2A). GST-BECN1 was cotransfected with GFP-PRKAA1-WT, constitutively active (CA), kinase dead (KD) or control vector. Phosphorylation of BECN1 was tested using phosphorylated (p)-AMPK substrate antibody after glutathione bead affinity isolation. The phosphorylation of GST-BECN1 was obviously detected after GFP-PRKAA1-WT and CA overexpression compared to GFP-PRKAA1-KD and control (Fig. 2B).
Figure 2.

BECN1 is a physiological target of AMPK. (A) HEK293T cells were cotransfected with plasmids encoding GFP-PRKAA1WT and GST-BECN1. GST-BECN1 was immunoprecipitated using glutathione agarose beads and detected with immunoblots using the indicated antibodies. (B) GST-BECN1 was affinity isolated in various PRKAA1 mutant-expressing HEK293T cells. Phosphorylated BECN1 was detected using p-AMPK substrate antibody and anti-GST to test GST-BECN1 expression. Glucose starvation (3 h) (C) or AICAR treatment (0.25 mM, 1 h) (D) were performed in pCDH1-Flag- BECN1-transfected HEK293T cells. p-AMPK substrate antibody was used to detect the p-BECN1. Whole cell lysates were immunoblotted with anti-Flag, p-AMPK, PRKAA and ACTIN antibodies. (E) After transfection with Flag-BECN1, HEK293T cells were with treated with compound C (Com. C), followed by glucose deprivation for 3 h. Flag-BECN1 was immunoprecipitated and immunoblotted with p-AMPK substrate antibody. Expression of Flag proteins was used as a loading control. IP, immunoprecipitation; WCL, whole cell lysate.
To further elucidate the role of AMPK in BECN1 phosphorylation, we examined BECN1 phosphorylation using p-AMPK substrate antibody after glucose deprivation for 3 h. Our results showed that BECN1 was phosphorylated by AMPK when the kinase was activated by glucose deprivation (Fig. 2C). Consistent with glucose starvation, increased phosphorylation of BECN1 was observed in cells treated with AICAR (Fig. 2D). In contrast, phosphorylation of BECN1 was suppressed when cells were pretreated with compound C even when the cells were challenged with glucose starvation (Fig. 2E). These data indicated that BECN1 could be phosphorylated by AMPK.
AMPK phosphorylates BECN1 at Thr388
Kim et al. previously reported 2 unconventional AMPK phosphorylation sites (S90 and S93 in human; S91 and S94 in mouse) on BECN1.34 However, whether BECN1 is phosphorylated by AMPK in a conventional manner is unknown. Based on the alignment of consensus substrate phosphorylation motifs,34 Thr29, Ser90 and Thr388 in BECN1 were predicted as potential phosphorylation sites of AMPK (Fig. 3A). We then coexpressed GFP-PRKAA1-CA and GST-BECN1 WT, T29A, S90A and T388A in HEK293T cells to determine whether AMPK phosphorylates BECN1 or these mutant forms. The anti-p-AMPK substrate antibody showed that BECN1 T388, but not T29, could be phosphorylated by AMPK (Fig. 3B). Although S90 is a phosphorylation site of AMPK reported by Kim et al.,26 we could not find the difference between BECN1 WT and S90A while using p-AMPK substrate antibody to detect the phosphorylation. Human BECN1 T388 is a highly conserved site in different species, including Drosophila melanogaster, Danio rerio, Xenopus tropicalis, Mus musculus, and Macaca mulatta (Fig. 3C). To investigate BECN1 T388 phosphorylation by AMPK, a plasmid encoding BECN1 WT or phosphorylation-defective mutant (T388A) was transfected into HEK293T cells and immunoprecipitated after glucose deprivation. Notably, AMPK phosphorylation was nearly completely abolished upon glucose deprivation when BECN1T388A was overexpressed (Fig. 3D).
Figure 3.
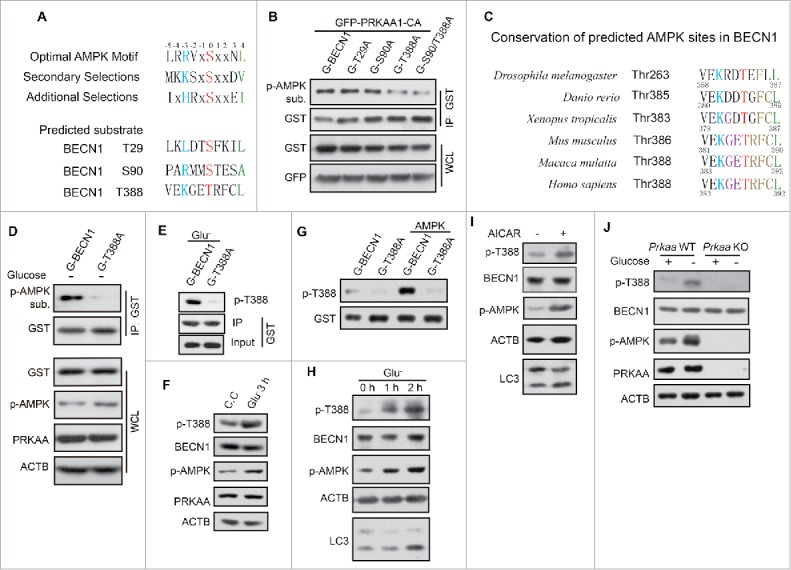
BECN1 Thr388 is the phosphorylated site of AMPK. (A) The putative phosphorylation sites of BECN1 by AMPK were predicted from the consensus motif of AMPK substrates. (B) GST (G)-BECN1 wild type and mutants (T29A, S90A, T388A, S90A/T388A) were cotransfected with GFP-PRKAA1-CA in HEK293T cells and affinity isolated with glutathione beads. Phosphorylated proteins were detected by p-AMPK substrate antibody. The lower panels demonstrate the expression of the proteins by western blot analysis (WB) with anti-GST and anti-GFP antibodies. (C) Conservation of the potential AMPK-phosphorylated site Thr388. (D) Detection of phosphorylated BECN1 by immunoblot analysis of immunoprecipitated GST-BECN1 using AMPK-substrate antibody. The lower panels show BECN1 expression and AMPK activation for comparison. (E) AMPK is required for glucose starvation-induced phosphorylation of GST-BECN1 T388. Phosphospecific antibody recognized p-GST-BECN1 T388 well but not the mutant T388A. (F) Activation of AMPK is sufficient to induce phosphorylation of endogenous BECN1. 293T cells were incubated with compound C (C.C, 20 μM, 30 min) and then shifted into complete medium for 3 h before harvesting. In parallel, cells were deprived of glucose for 3 h. (G) In vitro AMPK kinase assay. GST-BECN1WT and GST-BECN1T388A were affinity isolated and then incubated with AMPK for 30 min in vitro. p-BECN1 T388 antibody was used to test phosphorylation of BECN1. (H) Time course of glucose starvation in HEK293T cells. Lysates were immunoblotted with the antibody indicated. (I) HEK293T cells incubated with AICAR (0.25 mM, 1 h), then tested with p-BECN1 T388 antibody. (J) Determination of the phosphorylation of endogenous BECN1 protein after subjecting Prkaa WT and KO MEF cells to glucose starvation for 3 h.
To further confirm BECN1 phosphorylation, we generated a phosphospecific antibody for BECN1 T388. We verified that the antibody preferentially recognized the exogenous GST-BECN1WT but not GST-BECN1T388A (Fig. 3E). This antibody also could recognize the endogenous p-BECN1 when AMPK was activated by glucose deprivation (Fig. 3F). In vitro kinase assay verified that AMPK phosphorylated BECN1 T388 directly (Fig. 3G). The amount of p-BECN1 T388 accumulated gradually following the time course of glucose starvation and AICAR treatment (Fig. 3H, I). p-BECN1 was augmented in Prkaa WT MEFs during glucose starvation, but not in Prkaa KO MEFs even with glucose depletion (Fig. 3J). Thus, our data demonstrate that BECN1 T388 could be one phosphorylation site that is a target of AMPK.
Phosphorylation of Thr388 in BECN1 is required for induction of autophagy
To investigate the biological function of BECN1 phosphorylation by AMPK, we overexpressed BECN1 WT and mutants in HEK293 cells. Since BECN1 S93 is a reported AMPK phosphorylation site, we used BECN1S93A as a positive control in our assay. Compared with BECN1WT and BECN1S93A, BECN1T388A obviously suppressed autophagy (Fig. 4A), whereas the BECN1T388D mutant enhanced autophagy, which was indicated by SQSTM1 reduction and LC3-II augmentation (Fig. 4B). To determine whether phosphorylation of T388 plays a role in autophagy flux, we treated the cells with or without the vacuolar-type H+-ATPase inhibitor bafilomycin A1 (Baf. A1). More LC3-II was observed in BECN1WT- and BECN1T388D-expressing cells than control and BECN1T388A-expressing cells in normal conditions. A similar result was obtained in all cells treated with Baf. A1 (Fig. 4C).
Figure 4.

Phosphorylation of BECN1 Thr388 plays a key role in autophagy induced by AMPK. (A) BECN1 T388A mutant represses autophagy. Autophagy was detected by LC3 immunoblot of GST (G)-BECN1, S93A (positive control, reported previously) and T388A-transfected HEK293T cell lysates. BECN1 expression was detected with GST antibody. (B) The BECN1T388D mutant promotes autophagy. HEK293T cells were transfected with a plasmid encoding GST-BECN1WT, GST-BECN1T388A or GST-BECN1T388D. Lysates were immunoblotted with the antibodies indicated. (C) Plasmids encoding BECN1WT, BECN1T388A or BECN1T388D were transfected into HEK293T cells. The cells were treated or not with the inhibitor Baf. A1. LC3-II was detected to explore the autophagy flux. Expression of BECN1 variants was detected by blotting with anti-GST antibody; the expression of ACTIN was monitored as a loading control. L.E., long exposure. (D) Representative images of endogenous LC3 puncta in HeLa cells transfected with plasmids encoding Flag-BECN1WT, Flag-BECN1T388A, or Flag-BECN1T388D. Scale bars: 10 μm. (E) Quantification of endogenous LC3 puncta is shown in (D). (F) Co-transfection of pCDH1-Flag (F)-BECN1, T388A or T388D with a plasmid encoding GFP-LC3 into HeLa cells. Representative images of GFP-LC3 puncta are shown as indicated. Scale bars: 10 μm. (G) Quantification of GFP-LC3 puncta shown in (F). (H) Endogenous BECN1 was knocked down by shBECN1. (I) Autophagy level was determined in stable cell lines after endogenous BECN1 knockdown.
To further validate these findings, we examined endogenous LC3 puncta by immunofluorescence when BECN1 mutants were transfected into HeLa cells. LC3 puncta accumulation was observed when BECN1WT was overexpressed. More puncta were found in BECN1T388D-transfected cells, whereas the numbers of LC3 puncta were reduced in BECN1T388A-overexpressing cells (Fig. 4D, E). Similar results were observed in stable cell lines expressing BECN1 and different mutants (Fig. 4F, G). We knocked down endogenous BECN1 by targeting its 3′ untranslated region (UTR), which could not affect exogenous BECN1 only containing CDS (coding DNA Sequence) (Fig 4H). BECN1WT and BECN1T388D were overexpressed in knockdown cells and incubated with/without glucose. The data showed that LC3-II levels were enhanced in BECN1WT cells, but not in control cells when cells subjected to glucose starvation. LC3-II levels were higher in T388D cells regardless of glucose deprivation (Fig. 4I). These results demonstrated that phosphorylation of BECN1 T388 is crucial for autophagy.
AMPK is necessary for BECN1 Thr388 phosphorylation-induced autophagy
Glucose starvation activates AMPK and consequently promotes autophagy. Therefore, we tested whether AMPK is essential for the autophagy induction regulated by BECN1 T388 phosphorylation. Endogenous PRKAA1 was knocked down and then LC3 was examined by western blot. Notably, autophagy was suppressed in cells overexpressing GST and GST-BECN1WT but not GST-BECN1T388D when PRKAA was knocked down (Fig. 5A). Moreover, we deprived cells of glucose for 3 h when Prkaa WT and Prkaa KO MEF cells were cultured. We found that p-BECN1 was enhanced after Prkaa WT MEFs suffered glucose starvation, but the phosphorylation was abolished in Prkaa KO MEF cells. LC3-II was increased when Prkaa WT MEF cells were subjected to glucose starvation (Fig. 5B). Flag, Flag-BECN1WT and Flag-BECN1T388D were overexpressed in Prkaa WT or KO MEF cells to further validate that AMPK-phosphorylated BECN1 T388 regulates autophagy. The results demonstrated that Flag-BECN1T388D promoted autophagy in both Prkaa WT and KO MEF cells, but Flag-BECN1WT failed to induce autophagy in Prkaa KO MEF cells (Fig. 5C). The immunofluorescence experiments led to a similar conclusion (Fig. 5D, E). These data established a physiological role for AMPK in the phosphorylation of BECN1 T388-induced autophagy in response to glucose starvation.
Figure 5.
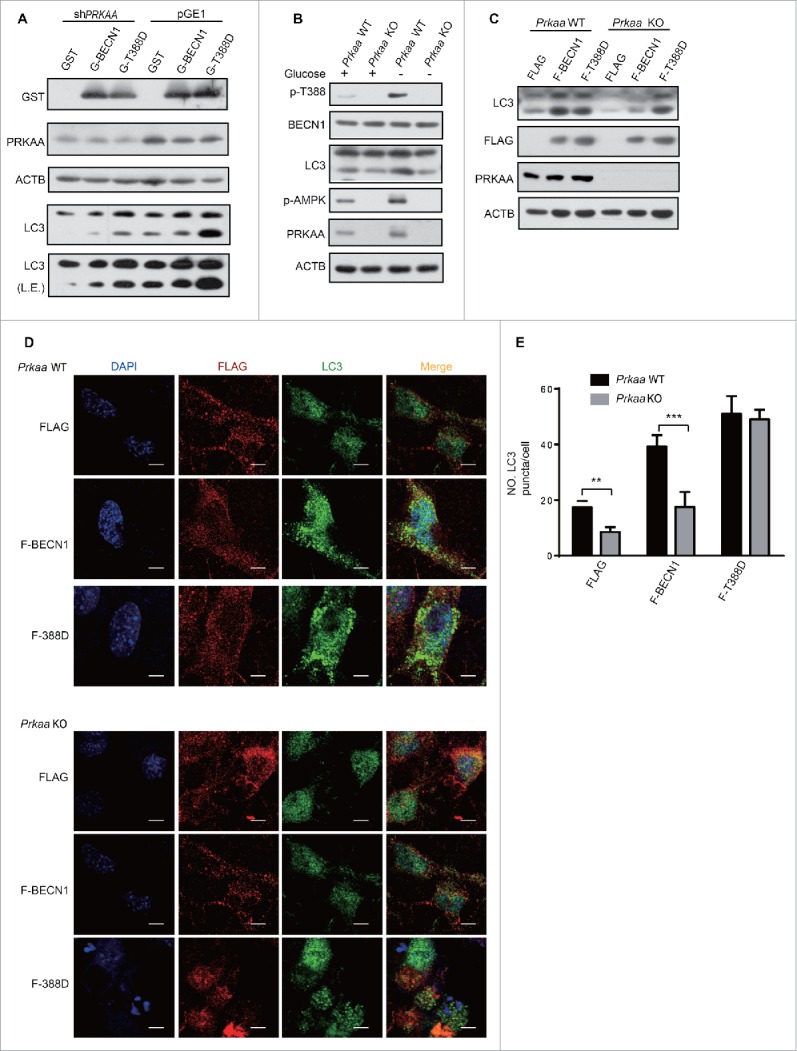
AMPK is necessary for BECN1 Thr388 phosphorylation-induced autophagy. (A) PRKAA knockdown decreased autophagy in cells transfected with GST and GST (G)-BECN1. After coexpression of pGE1 or pGE1-shPRKAA and GST, GST-BECN1WT or GST-BECN1T388D in HEK293T cells for 48 h, cell lysates were probed with the indicated antibodies. L.E., long exposure. (B) Decreased BECN1 T388 and attenuated autophagy were found in Prkaa KO MEF cells. Prkaa WT and KO MEF cells were subjected to glucose starvation for 3 h. p-BECN1 and LC3-II were detected. (C) Autophagy enhanced by phosphorylation of BECN1 is specifically detected in Prkaa WT MEF cells. MEFs were infected with lentivirus and lysed after 48 h. F, Flag. (D) Immunofluorescence was performed after MEFs were infected with the indicated virus. Representative images of endogenous LC3 puncta are shown as indicated. Scale bars: 10 μm. (E) Quantification of GFP-LC3 puncta. Bars are mean ± SEM of triplicate samples (≥ 10 cells analyzed per sample).
Phosphorylation of BECN1 Thr388 enhances the interaction between BECN1 and PIK3C3
It has been reported that BECN1 interacts with PIK3C3-PIK3R4/VPS15 to form protein complexes that play a key role in autophagy regulation.35 To determine whether the association of BECN1 with PIK3C3 was affected by AMPK activity, we cotransfected plasmids encoding PIK3C3 along with GST-BECN1 and performed co-immunoprecipitation experiments after glucose starvation. As anticipated, the results showed that interaction between BECN1 and PIK3C3 was augmented after AMPK was activated by glucose deprivation (Fig. 6A). Similarly, GST-BECN1 bound more PIK3C3 in HEK293T cells treated with AICAR (Fig. 6B). Conversely, depletion of endogenous PRKAA decreased the interaction between BECN1 and PIK3C3 (Fig. 6C).
Figure 6.
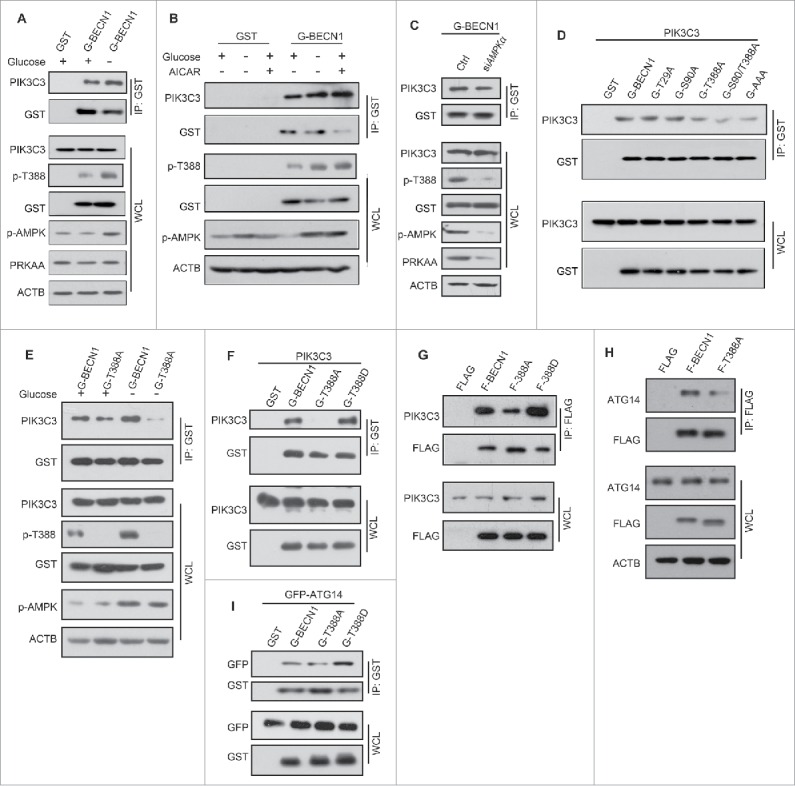
Phosphorylation of BECN1 Thr388 is required for PIK3C3-BECN1-ATG14 complex formation. Overexpression of GST (G)-BECN1 in HEK293T cells, followed 24 h later by glucose deprivation for 3 h (A) or with AICAR treatment for 1 h (B), or endogenous PRKAA depletion (C). Endogenous PIK3C3 was detected in GST-BECN1 immunoprecipitates. Whole cell lysates and immunoprecipitates were immunoblotted with the indicated antibodies (D). Recombinant BECN1T388A coimmunoprecipitates less recombinant PIK3C3. pcDNA3.1-PIK3C3 was cotransfected with plasmids encoding various BECN1 mutants (T29A, S90A, T388A, S90A/T388A, T29A/S90A/T399A) and WT in HEK293T cells. Immunoblots of glutathione bead immunoprecipitates from HEK293T cells expressing PIK3C3 and GST-BECN1 proteins, as indicated. (E-G) BECN1 Thr388 phosphorylation affects the interaction between BECN1 and PIK3C3. (E) GST-BECN1WT- or GST-BECN1T388A mutant-transfected HEK293T cells were incubated with or without glucose for 3 h. Immunoblots of glutathione bead immunoprecipitates from HEK293T cells expressing GST-BECN1 proteins, as indicated. (F) Plasmids encoding GST-BECN1WT, GST-BECN1T388A or GST-BECN1T388D were transfected into HEK293T cells with pCDNA3-PIK3C3. T388A served as a negative control. Immunoblots of glutathione bead immunoprecipitates from HEK293T cells expressing PIK3C3 and GST-BECN1 proteins, as indicated. (G) BECN1 variant stable cell lysates were immunoprecipted by Flag (F) beads, the immunoprecipitates were resolved by SDS-PAGE and detected by western blot with the indicated antibodies. (H and I) BECN1 Thr388 phosphorylation decreases PIK3C3-BECN1-ATG14 complex formation. (H) A plasmid encoding GFP-ATG14 was cotransfected with plasmids encoding GST-BECN1WT or either of 2 mutants (T388A, T388D) into HEK293T cells. Immunoblots of glutathione bead immunoprecipitates from HEK293T cells expressing GFP-ATG14 and GST-BECN1 proteins, as indicated. (I) Flag-BECN1WT or 2 mutants (T388A, T388D) were overexpressed in HEK293T cells. Immunoblots of anti-Flag immunoprecipitates from HEK293T cells expressing Flag-BECN1 proteins, as indicated.
To further investigate how AMPK regulates the formation of the BECN1-PIK3C3 complex, we coexpressed PIK3C3 and GST-BECN1 mutants in HEK293T cells. Subsequently, GST-BECN1 was immunoprecipitated after transfection for 24 h. Interestingly, all GST-BECN1 mutants containing T388A bound less PIK3C3 than BECN1WT, but the other mutants pulled down the same amount of PIK3C3 as the wild-type counterpart (Fig. 6D).
Previously, we demonstrated that glucose starvation leads to an enhanced interaction between BECN1 and PIK3C3. To study how phosphorylation of BECN1 T388 influences PIK3C3 complex formation, we cotransfected plasmids encoding PIK3C3 with GST-BECN1WT or the GST-BECN1T388A mutant, and then subjected the cells to glucose starvation. The results showed that less BECN1T388A-PIK3C3 complex formed even during glucose starvation (Fig. 6E). In contrast, BECN1T388D bound more PIK3C3 than its wild-type counterpart (Fig. 6F). Similar results were obtained in stable cell lines overexpressing Flag-BECN1WT and Flag-BECN1T388A (Fig. 6G).
ATG14, a crucial part of the PIK3C3-BECN1 complex, is important for autophagy activation.36 To study whether phosphorylation of BECN1 affects its interaction with ATG14, a plasmid expressing GFP-ATG14 was cotransfected with Flag-BECN1WT, Flag-BECN1T388A or Flag-BECN1T388D. Immunoprecipitation results showed that Flag-BECN1T388D had a stronger binding affinity to GFP-ATG14 than wild-type Flag-BECN1. Conversely, in stable cell lines, less GFP-ATG14 was immunoprecipitated by Flag-BECN1T388A (Fig. 6H). Similarly, less endogenous ATG14 was detected in the immunoprecipitated Flag-BECN1T388A-expressing cells than those with Flag-BECN1WT (Fig. 6I). Therefore, AMPK phopshorylation facilitates BECN1 interaction with PIK3C3 and ATG14 to promote autophagy.
Association of BECN1 and BCL2 is decreased when BECN1 is phosphorylated by AMPK
As a BH3 domain-containing protein,37 BECN1 determines the initiation of autophagy or apoptosis by selectively interacting with BCL2. It has been reported that DAPK (death associated protein kinase) phosphorylates BECN1 to induce the dissociation of the BECN1-BCL2 complex.38 To investigate whether AMPK-induced BECN1 phosphorylation could lead to a similar outcome, we assessed the GST-BECN1 and GFP-BCL2 interaction in transfected HEK293T cells. Fewer BECN1-BCL2 complexes were detected under glucose-starvation conditions (Fig. 7A).
Figure 7.

BCL2-BECN1 complex formation is attenuated by phosphorylation of BECN1 Thr388. (A) GST (G)-BECN1 and GFP-BCL2 were cotransfected into HEK293T cells and subjected or not to glucose starvation for 3 h (right panel). Immunoblots of glutathione immunoprecipitates from HEK293T cells expressing GFP-BCL2 and GST-BECN1 proteins, as indicated. (B) Plasmids expressing GST-BECN1, MYC-BCL2 and pGE1-shPRKAA were transfected into HEK293T cells and cultured for 48 h. Immunoblots of glutathione immunoprecipitates from HEK293T cells expressing MYC-BCL2 and GST-BECN1 proteins, as indicated. (C) A plasmid expressing MYC-BCL2 was cotransfected with various GST-BECN1 constructs into HEK293T cells. Immunoblots of glutathione immunoprecipitates from HEK293T cells expressing MYC-BCL2 and GST-BECN1 proteins, as indicated. (D) Compound C treatment increases the interaction of BCL2 and BECN1WT but not the BECN1T388D mutant. A plasmid expressing GFP-BCL2 was cotransfected with plasmids expressing GST-BECN1WT or GST-BECN1T388D or GST control into HEK293T cells. After 24-h transfection, the cells were treated with or without compound C (Com. C). Immunoblots of glutathione immunoprecipitates from HEK293T cells expressing GFP-BCL2 and GST-BECN1 proteins, as indicated. (E and F) The Thr388 phosphorylation of BECN1 decreased the formation of BECN1 homodimers. (E) Plasmids expressing the GST- or Flag-tagged BECN1 variants were transfected and affinity isolated with GST beads, followed by westerns blot detection with Flag antibody. L.E., long exposure. (F) The co-immunoprecipitates from anti-Flag beads in lysates of Flag (F)-BECN1WT-, Flag-BECN1T388A- and Flag-BECN1T388D-expressing stable cells were detected with anti-BECN1 antibody. (G) Model: autophagy is induced by AMPK phosphorylation of BECN1 during glucose deprivation.
We speculated that AMPK could modulate the association of BECN1 and BCL2. Therefore, we first tested the amount of BECN1-BCL2 complexes in PRKAA-depleted cells. The results illustrated that PRKAA knockdown enhanced the interaction between BECN1 and BCL2 (Fig. 7B). To further explore whether phosphorylation of BECN1 T388 affected its interaction with BCL2, we cotransfected GST-BECN1 variants with MYC-BCL2 in HEK293T cells. Our results showed that BECN1T388A and 3A mutant (S90/93/T388A) bound more BCL2 than the wild-type control (Fig. 7C). After tansfection, GFP-BCL2 and GST-BECN1WT or GST-BECN1T388D cells were treated with the AMPK inhibitor compound C. Immunoblotting showed that more BECN1-BCL2 complexes formed when AMPK activity was inhibited; however, GST-BECN1T388D consistently bound less BCL2 (Fig. 7D). Compound C treatment decreased BECN1 T388 phosphorylation by inhibiting AMPK activity in WT cells, but the T388D mutant binding to BCL2 was not changed.
The crystal structure of the BECN1 CCD revealed that BECN1 can form a homodimer.39 The homodimer would increase affinity to BCL2 and attenuate ATG14 recruitment. To dissect the detailed mechanism of this modification, we studied whether this phosphorylation of BECN1 could influence its homodimerization. We transfected different tagged BECN1 and mutants in HEK293T cells, and the results showed that T388A, a phosphorylation defective mutant, could form more homodimers. In contrast, fewer homodimers were detected in T388D-overexpressing cells (Fig. 7E). We obtained similar results that BECN1T388A pulled down more endogenous BECN1 in stable cell lines (Fig. 7F). These data indicated that phosphorylation of BECN1 T388 by activated AMPK decreases formation of the BECN1-BCL2 complex.
Discussion
In this study, we demonstrate that AMPK phosphorylates BECN1 to induce autophagy (Fig. 7G). AMPK is activated and then phosphorylates BECN1 at Thr388 during glucose deprivation. BECN1 phosphorylation is required for autophagy induction and LC3 puncta accumulation. Our findings highlight a more precise mechanism on how AMPK regulates autophagy when cells encounter serious stress.
Proteins accomplish a variety of functions by different post-translational modifications, including acetylation, ubiquitination and phosphorylation. These modifications are involved in the entire process of autophagy. For example, when cells are treated with the pan-HDAC inhibitor panobinostat, acetylated HSPA/HSP70 proteins induce SUMOylation of PIK3C3 and increases PIK3C3s binding to BECN1.39 Wang et al. and Wei et al. found that both EGFR (epidermal growth factor receptor) and AKT can individually regulate the activity of autophagy by phosphorylating BECN1.31,40 Kim et al. also showed that AMPK phosphorylates BECN1 S91/S94 (S90/S93 in human) to induce autophagy.26 These 2 sites do not correspond to classical AMPK phosphorylation motifs. In this study, we found a classic AMPK phosphorylation site, Thr388, in BECN1 that regulates autophagy. Kim et al. showed S93 phosphorylation does not affect the binding of ATG14 to the PtdIns3K complex, but our results indicate that phosphorylation at Thr388 increases ATG14 binding. These data suggest that AMPK may regulate autophagy via BECN1 through different molecular mechanisms.
Our findings reveal that phosphorylation of BECN1 Thr388 also increases its interaction with PIK3C3. The BECN1T388D mutant has a higher binding activity toward PIK3C3 whereas the T388A mutant has reduced binding between PIK3C3 and BECN1. BECN1T388A also has less binding with ATG14. Although the function of the BARA domain is not clear, structural analysis shows that the BARA domain of BECN1 is essential for autophagy and for targeting of the PtdIns3K to the site of phagophore formation.10 Presumably, it may play a key role in promoting the formation of the PIK3C3-BECN1-ATG14 complex. Because Thr388 is located in repeat 3 of the BECN1 BARA domain it is possible that BECN1 Thr388 is important to the formation of the PIK3C3-BECN1 complex and nucleation of phagophores. Our data thus provide a feasible mechanism to explain how the BARA domain of BECN1 regulates autophagy during cellular stress.
As a BH3-containing protein, BECN1 can directly interact with BCL2. Indeed, the BCL2-BECN1 complex inhibits autophagy under starvation conditions.41 Our study further demonstrates that AMPK phoshophylates BECN1 on Thr388 to decrease its interaction with BCL2. These data indicate that AMPK regulates autophagy not only by promoting formation of the PIK3C3-BECN1 complex, but also by inhibiting the interaction of BECN1 and BCL2 to release more available BECN1 to form complexes including PIK3C3.
BECN1 Thr388 phosphorylation promotes autophagy by inhibiting BECN1-BCL2 complex formation and increasing PIK3C3-BECN1-ATG14 complex formation at the same time after glucose deprivation. Then, a question arises: how does Thr388 in the BARA domain influence both the BH3 domain (BCL2 binding domain) and CCD (ATG14 binding domain)? A possible explanation is that repeat 3 of the BARA domain may be near to the BH3 and CCD in the tertiary structure of BECN1. The phosphorylation of Thr388 thus causes conformational changes and induces interaction changes between different BECN1 complexes. Yet, a detailed structural analysis of the full-length BECN1 protein is required to confirm this hypothesis.
In conclusion, AMPK promotes phagophore nucleation by phosphorylating BECN1 Thr388 to regulate PIK3C3-BECN1 complex formation. These findings provide a new molecular mechanism of AMPK-regulated autophagy.
Materials and methods
Antibodies and reagents
Anti-PIK3C3 (4263), phospho-PRKAA/AMPKα T172 (2531), PRKAA1/AMPKα1 (2532), PRKAA2/AMPKα2 (2757), phospho-(Ser/Thr) AMPK substrate (5759), MYC (2278), ATG14 (5504) and GFP (2956) antibodies were from Cell Signaling Technology. LC3 antibody (NB100-2220) was from NOVUS Biologicals. PRKAA1/AMPKα1 (sc-1929) and ACTB/β-ACTIN antibodies (sc-47778) were from Santa Cruz Biotechnology. SQSTM1 antibody (ab56416) was from Abcam. GST (E022040-01) was from EARTHOX, compound C (P5499) and Flag (F3165) were purchased from SIGMA. Anti-phospho-BECN1 T388 antibody was generated by immunizing rabbits with the corresponding phosphopeptides coupled with keyhole limpet hemocyanin. The phosphospecific antibody was affinity purified (ABclonal Technology).
DNA constructions
The constructs encoding GFP-PRKAA1-WT, GFP-PRKAA1-CA and GFP-PRKAA1-KD were obtained from Prof. Xiaolong Liu (Institute of Biochemistry and Cell Biology, Shanghai, China). Wild type, mutants (T29A, S90A, T388A, S90A T388A) of BECN1 were obtained by PCR amplification and subcloned into the GST(modified from pRK5) or pCDH1-Flag vectors (which is modified based on pCDH1; System Biosciences,CD810A-1). The constructs were sequenced to confirm that no secondary mutation was introduced. GFP-BCL2, GFP-ATG14 and pCDNA3.1-PIK3C3 were also from PCR amplification and subcloned into EGFP-C1 and pcDNA3.1+ vectors (Clontech,6084–1; Thermofisher, V79020). shRNAs against PRKAA1 (sense: GCACAGACAATTGCAGTAAAT) and PRKAA2 (sense: CCCACTGAAACGAGCAACTAT) were cloned into the pGE1 backbone (Stratagene, 240094). shRNAs against BECN1 5′UTR-1 (sense: GAGCGATGGTAGTTCTGGA), 5′UTR-2 (sense: AGACAGAGCGATGGTAGTT) and 3′UTR (sense: CATGCCATCTATAGTTGCC) were cloned into the pGIPZ backbone (Dharmacon, RHS4346) by direct mutagenesis to introduce the necessary sites.
Cell culture and transfection
HEK293T, HeLa, Prkaa WT and KO MEF cells were cultured in high-glucose (25 mM) DMEM (Hyclone, SH30243.01B) supplemented with 10% fetal bovine serum (Hyclone, SV30084.03) and 50 μg/ml penicillin/streptromycin (Gibco, 15140–122). HepG2 stable cell lines were cultured in high-glucose DMEM supplemented with 10% fetal bovine serum, puromycin (SIGMA, 58-60-6; final concentration 1 μg/μl ) and 50 μg/ml penicillin/streptromycin. Glucose starvation performed in this study was carried out by switching the culture medium from complete medium to the same medium lacking glucose (Gibco, 11966–025). For transient expression of proteins, cells were transfected with recombinant DNA plasmids using polyethylenimine (SIGMA, 408727) performed as previously reported.42 Cells were harvested 24 h after transfection for co-immunoprecipitation assay or western blot analysis unless otherwise stated.
Immunoprecipitation and immunoblotting
Cells were lysed with Lysis Buffer (20 mM Tris-HCl, pH 7.2, 1 mM EDTA, 100 mM NaCl, 0.1% NP-40 [Calbiochem, 492016], 1% Triton X-100 [Sigma, X100]) containing a protease inhibitor cocktail (Roche, 04693132001) and phosphatase inhibitor cocktails (Roche, 04906837001). The clarified lysates were subjected to immunoprecipitation using glutathione agarose (Thermo Scientific, 16102) or anti-Flag M2 affinity gel (Sigma, A2220) and incubated at 4°C for 5 h. The resulting immunocomplexes were washed 5 times with lysis buffer and boiled in SDS-sample buffer. Samples were subsequently separated by SDS-PAGE and transferred onto PVDF membranes (Bio-Rad). Immunoblot analysis was performed with the indicated antibodies and visualized with Super-Signal West Pico Chemiluminescent substrate (Pierce Chemical, 34080).
Immunofluorescence staining
HeLa cells transfected with GST-BECN1 or mutants (T388A, T388D) and GFP-LC3 plasmid using polyethylenimine and cultured on 4-well chamber slides. After 24-h transfection, the cells were fixed with 4% paraformaldehyde in PBS for 15 min at room temperature (RT), and permeabilized with 0.1% Triton X-100 in PBS for 10 min at RT. The cells were washed 3 times with PBS, then blocked with 5% BSA (Sigma, A2058) in PBS for 1 h at RT. Cells were incubated with anti-LC3 (1:500) and anti-GST (1:1000) antibodies for 1 h at RT. The cells were washed with PBS and then incubated with Alexa Fluor 488-conjugated anti-rabbit IgG antibody or Alexa Fluor 568-conjugated anti-mouse IgG (Invitrogen, A-11004; 1:1000) for 1 h. ProLong Gold antifade reagent with DAPI (Invitrogen, D1306) was used to resist photobleaching. Images were acquired on an Olympus microscope (Olympus; IX71) coupled to the DPS-PSW software. Statistical significance (P-value) was determined by 2-tailed unpaired Student t test by Graphpad Prism5.
In vitro kinase assay
GST and GST-BECN1 were overexpressed in HEK293T cells, which were lysed and subjected to centrifugation (10 min, 21130 × g). The clarified lysates were then subjected to immunoprecipitation using glutathione agarose and incubated at 4°C for 5 h. After washing the beads (cold PBS) 3 times, dephosphorylates the pulldown by CIP (Alkaline Phosphatase, Calf Intestinal, New England Biolabs, M0290) at 37°C for 1 h. Using cold PBS wash 3 times and then wash beads with AMPK kinase assay buffer (50 mM HEPES, pH 7.5, 1 mM DTT, 0.02% Brij35 [Fisher Scientific, BP345], AMP [300 μM; Sigma, A2252):ATP [150 μM;Sigma, A1852)). The beads were resuspended with 25 μl kinase assay buffer, followed by the addition of 5 μg AMPK (Creative Biomart, AMPK(A1/B2/G1)-244H) and incubation at 37°C for 30 min. The beads were then washed with cold PBS 3 times, followed by the addition of SDS loading buffer and boiled at 95°C for 8 min. Phosphorylation of BECN1 was detected using the specific antibody p-BECN1 T388.
Abbreviations
- AICAR
5-amino-1-β-D-ribofuranosyl-imidazole-4-carboxamide
- AMPK
AMP-activated protein kinase
- BCL2
B-cell CLL/lymphoma 2
- BECN1
Beclin 1, autophagy related
- CA
constitutively active
- KD
kinase dead
- KO
knockout
- MAP1LC3/LC3
microtubule associated protein 1 light chain 3
- MEF
mouse embryonic fibroblast
- PtdIns3K
phosphatidylinositol 3-kinase
- UTR
untranslated region
- WT
wild type
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
Acknowledgements
We thank C. Chan (University of Oklahoma Health Sciences Center) for critical comments and English editing of the manuscript.
Funding
This work was supported by grants from the Ministry of Science and Technology of China (973 Program; 2014CB910500 and 2011CB910900), the National Natural Science Foundation of China (81172231) and the Chinese Academy of Sciences (Hundred Talents Program No.2010OHTP07) to Z Liu. The authors declare no competing financial interests.
Reference
- [1].Scott RC, Schuldiner O, Neufeld TP. Role and regulation of starvation-induced autophagy in the Drosophila fat body. Dev Cell 2004; 7:167-78; PMID:15296714; http://dx.doi.org/ 10.1016/j.devcel.2004.07.009 [DOI] [PubMed] [Google Scholar]
- [2].Kanki T, Klionsky DJ. Mitophagy in yeast occurs through a selective mechanism. J Biol Chem 2008; 283:32386-93; PMID:18818209; http://dx.doi.org/ 10.1074/jbc.M802403200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Ogata M, Hino S, Saito A, Morikawa K, Kondo S, Kanemoto S, Murakami T, Taniguchi M, Tanii I, Yoshinaga K, et al.. Autophagy is activated for cell survival after endoplasmic reticulum stress. Mol Cell Biol 2006; 26:9220-31; PMID:17030611; http://dx.doi.org/ 10.1128/MCB.01453-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Birmingham CL, Smith AC, Bakowski MA, Yoshimori T, Brumell JH. Autophagy controls Salmonella infection in response to damage to the Salmonella-containing vacuole. J Biol Chem 2006; 281:11374-83; PMID:16495224; http://dx.doi.org/ 10.1074/jbc.M509157200 [DOI] [PubMed] [Google Scholar]
- [5].Berry DL, Baehrecke EH. Growth arrest and autophagy are required for salivary gland cell degradation in Drosophila. Cell 2007; 131:1137-48; PMID:18083103; http://dx.doi.org/ 10.1016/j.cell.2007.10.048 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Gozuacik D, Kimchi A. Autophagy and cell death. Curr Top Dev Biol 2007; 78:217-45; PMID:17338918; http://dx.doi.org/ 10.1016/S0070-2153(06)78006-1 [DOI] [PubMed] [Google Scholar]
- [7].Gozuacik D, Bialik S, Raveh T, Mitou G, Shohat G, Sabanay H, Mizushima N, Yoshimori T, Kimchi A. DAP-kinase is a mediator of endoplasmic reticulum stress-induced caspase activation and autophagic cell death. Cell Death Differ 2008; 15:1875-86; PMID:18806755; http://dx.doi.org/ 10.1038/cdd.2008.121 [DOI] [PubMed] [Google Scholar]
- [8].Codogno P. Shining light on autophagy. Nat Rev Mol Cell Biol 2014; 15:153; PMID:24496390; http://dx.doi.org/ 10.1038/nrm3751 [DOI] [PubMed] [Google Scholar]
- [9].Liang XH, Jackson S, Seaman M, Brown K, Kempkes B, Hibshoosh H, Levine B. Induction of autophagy and inhibition of tumorigenesis by Beclin1. Nature 1999; 402:672-6; PMID:10604474; http://dx.doi.org/ 10.1038/45257 [DOI] [PubMed] [Google Scholar]
- [10].Noda NN, Kobayashi T, Adachi W, Fujioka Y, Ohsumi Y, Inagaki F. Structure of the novel C-terminal domain of vacuolar protein sorting 30/autophagy-related protein 6 and its specific role in autophagy. J Biol Chem 2012; 287:16256-66; PMID:22437838; http://dx.doi.org/ 10.1074/jbc.M112.348250 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Cao Y, Klionsky DJ. Physiological functions of Atg6/Beclin1: a unique autophagy-related protein. Cell Res 2007; 17:839-49; PMID:17893711; http://dx.doi.org/ 10.1038/cr.2007.78 [DOI] [PubMed] [Google Scholar]
- [12].Matsunaga K, Saitoh T, Tabata K, Omori H, Satoh T, Kurotori N, Maejima I, Shirahama-Noda K, Ichimura T, Isobe T, et al.. Two Beclin1-binding proteins, ATG14L and Rubicon, reciprocally regulate autophagy at different stages. Nat Cell Biol 2009; 11:385-96; PMID:19270696; http://dx.doi.org/ 10.1038/ncb1846 [DOI] [PubMed] [Google Scholar]
- [13].Liang C, Feng P, Ku B, Dotan I, Canaani D, Oh BH, Jung JU. Autophagic and tumour suppressor activity of a novel Beclin1-binding protein UVRAG. Nat Cell Biol 2006; 8:688-99; PMID:16799551; http://dx.doi.org/ 10.1038/ncb1426 [DOI] [PubMed] [Google Scholar]
- [14].Pattingre S, Tassa A, Qu X, Garuti R, Liang XH, Mizushima N, Packer M, Schneider MD, Levine B. BCL2 antiapoptotic proteins inhibit Beclin1-dependent autophagy. Cell 2005; 122:927-39; PMID:16179260; http://dx.doi.org/ 10.1016/j.cell.2005.07.002 [DOI] [PubMed] [Google Scholar]
- [15].Erlich S, Mizrachy L, Segev O, Lindenboim L, Zmira O, Adi-Harel S, Hirsch JA, Stein R, Pinkas-Kramarski R. Differential interactions between Beclin1 and BCL2 family members. Autophagy 2007; 3:561-8; PMID:17643073; http://dx.doi.org/ 10.4161/auto.4713 [DOI] [PubMed] [Google Scholar]
- [16].Hardie DG. AMP-activated protein kinase as a drug target. Ann Rev Pharmacol Toxicol 2007; 47:185-210; PMID:16879084; http://dx.doi.org/ 10.1146/annurev.pharmtox.47.120505.105304 [DOI] [PubMed] [Google Scholar]
- [17].Hardie DG, Ross FA, Hawley SA. AMPK: a nutrient and energy sensor that maintains energy homeostasis. Nat Rev Mol Cell Biol 2012; 13:251-62; PMID:22436748; http://dx.doi.org/ 10.1038/nrm3311 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Woods A, Vertommen D, Neumann D, Turk R, Bayliss J, Schlattner U, Wallimann T, Carling D, Rider MH. Identification of phosphorylation sites in AMP-activated protein kinase (AMPK) for upstream AMPK kinases and study of their roles by site-directed mutagenesis. J Biol Chem 2003; 278:28434-42; PMID:12764152; http://dx.doi.org/ 10.1074/jbc.M303946200 [DOI] [PubMed] [Google Scholar]
- [19].Vautard-Mey G, Fevre M. Mutation of a putative AMPK phosphorylation site abolishes the repressor activity but not the nuclear targeting of the fungal glucose regulator CRE1. Curr Genet 2000; 37:328-32; PMID:10853770; http://dx.doi.org/ 10.1007/s002940050535 [DOI] [PubMed] [Google Scholar]
- [20].Chan EY. mTORC1 phosphorylates the ULK1-mAtg13-FIP200 autophagy regulatory complex. Sci Signal 2009; 2:pe51; PMID:19690328; http://dx.doi.org/ 10.1126/scisignal.284pe51 [DOI] [PubMed] [Google Scholar]
- [21].Egan DF, Shackelford DB, Mihaylova MM, Gelino S, Kohnz RA, Mair W, Vasquez DS, Joshi A, Gwinn DM, Taylor R, et al.. Phosphorylation of ULK1 (hATG1) by AMP-activated protein kinase connects energy sensing to mitophagy. Science 2011; 331:456-61; PMID:21205641; http://dx.doi.org/ 10.1126/science.1196371 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Mao K, Klionsky DJ. AMPK activates autophagy by phosphorylating ULK1. Circ Res 2011; 108:787-8; PMID:21454792; http://dx.doi.org/ 10.1161/RES.0b013e3182194c29 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Kim J, Guan KL. Regulation of the autophagy initiating kinase ULK1 by nutrients: roles of mTORC1 and AMPK. Cell Cycle 2011; 10:1337-8; PMID:21403467; http://dx.doi.org/ 10.4161/cc.10.9.15291 [DOI] [PubMed] [Google Scholar]
- [24].Kim J, Kundu M, Viollet B, Guan KL. AMPK and mTOR regulate autophagy through direct phosphorylation of Ulk1. Nat Cell Biol 2011; 13:132-41; PMID:21258367; http://dx.doi.org/ 10.1038/ncb2152 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Mack HI, Zheng B, Asara J, Thomas SM. AMPK-dependent phosphorylation of ULK1 regulates ATG9 localization. Autophagy 2012; 8:1197-214; PMID:22932492; http://dx.doi.org/ 10.4161/auto.20586 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Kim J, Kim YC, Fang C, Russell RC, Kim JH, Fan W, Liu R, Zhong Q, Guan KL. Differential regulation of distinct VPS34 complexes by AMPK in nutrient stress and autophagy. Cell 2013; 152:290-303; PMID:23332761; http://dx.doi.org/ 10.1016/j.cell.2012.12.016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Meley D, Bauvy C, Houben-Weerts JH, Dubbelhuis PF, Helmond MT, Codogno P, Meijer AJ. AMP-activated protein kinase and the regulation of autophagic proteolysis. J Biol Chem 2006; 281:34870-9; PMID:16990266; http://dx.doi.org/ 10.1074/jbc.M605488200 [DOI] [PubMed] [Google Scholar]
- [28].Grotemeier A, Alers S, Pfisterer SG, Paasch F, Daubrawa M, Dieterle A, Viollet B, Wesselborg S, Proikas-Cezanne T, Stork B. AMPK-independent induction of autophagy by cytosolic Ca2+ increase. Cell Signal 2010; 22:914-25; PMID:20114074; http://dx.doi.org/ 10.1016/j.cellsig.2010.01.015 [DOI] [PubMed] [Google Scholar]
- [29].Williams T, Forsberg LJ, Viollet B, Brenman JE. Basal autophagy induction without AMP-activated protein kinase under low glucose conditions. Autophagy 2009; 5:1155-65; PMID:19844161; http://dx.doi.org/ 10.4161/auto.5.8.10090 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Shang L, Wang X. AMPK and mTOR coordinate the regulation of Ulk1 and mammalian autophagy initiation. Autophagy 2011; 7:924-6; PMID:21521945; http://dx.doi.org/ 10.4161/auto.7.8.15860 [DOI] [PubMed] [Google Scholar]
- [31].Wang RC, Wei Y, An Z, Zou Z, Xiao G, Bhagat G, White M, Reichelt J, Levine B. Akt-mediated regulation of autophagy and tumorigenesis through Beclin1 phosphorylation. Science 2012; 338:956-9; PMID:23112296; http://dx.doi.org/ 10.1126/science.1225967 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Bao XX, Xie BS, Li Q, Li XP, Wei LH, Wang JL. Nifedipine induced autophagy through Beclin1 and mTOR pathway in endometrial carcinoma cells. Chin Med J 2012; 125:3120-6; PMID:22932192 [PubMed] [Google Scholar]
- [33].Maejima Y, Kyoi S, Zhai P, Liu T, Li H, Ivessa A, Sciarretta S, Del Re DP, Zablocki DK, Hsu CP, et al.. Mst1 inhibits autophagy by promoting the interaction between Beclin1 and BCL2. Nat Med 2013; 19:1478-88; PMID:24141421; http://dx.doi.org/ 10.1038/nm.3322 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Gwinn DM, Shackelford DB, Egan DF, Mihaylova MM, Mery A, Vasquez DS, Turk BE, Shaw RJ. AMPK phosphorylation of raptor mediates a metabolic checkpoint. Mol Cell 2008; 30:214-26; PMID:18439900; http://dx.doi.org/ 10.1016/j.molcel.2008.03.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Kihara A, Kabeya Y, Ohsumi Y, Yoshimori T. Beclin-phosphatidylinositol 3-kinase complex functions at the trans-Golgi network. EMBO Rep 2001; 2:330-5; PMID:11306555; http://dx.doi.org/ 10.1093/embo-reports/kve061 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Zhong Y, Wang QJ, Li X, Yan Y, Backer JM, Chait BT, Heintz N, Yue Z. Distinct regulation of autophagic activity by ATG14L and Rubicon associated with Beclin1-phosphatidylinositol-3-kinase complex. Nat Cell Biol 2009; 11:468-76; PMID:19270693; http://dx.doi.org/ 10.1038/ncb1854 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Oberstein A, Jeffrey PD, Shi Y. Crystal structure of the Bcl-XL-Beclin1 peptide complex: Beclin1 is a novel BH3-only protein. J Biol Chem 2007; 282:13123-32; PMID:17337444; http://dx.doi.org/ 10.1074/jbc.M700492200 [DOI] [PubMed] [Google Scholar]
- [38].Zalckvar E, Berissi H, Mizrachy L, Idelchuk Y, Koren I, Eisenstein M, Sabanay H, Pinkas-Kramarski R, Kimchi A. DAP-kinase-mediated phosphorylation on the BH3 domain of Beclin1 promotes dissociation of Beclin1 from Bcl-XL and induction of autophagy. EMBO Rep 2009; 10:285-92; PMID:19180116; http://dx.doi.org/ 10.1038/embor.2008.246 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Yang Y, Fiskus W, Yong B, Atadja P, Takahashi Y, Pandita TK, Wang HG, Bhalla KN. Acetylated hsp70 and KAP1-mediated VPS34 SUMOylation is required for autophagosome creation in autophagy. Proc Natl Acad Sci U S A 2013; 110:6841-6; PMID:23569248; http://dx.doi.org/ 10.1073/pnas.1217692110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Wei Y, Zou Z, Becker N, Anderson M, Sumpter R, Xiao G, Kinch L, Koduru P, Christudass CS, Veltri RW, et al.. EGFR-mediated Beclin1 phosphorylation in autophagy suppression, tumor progression, and tumor chemoresistance. Cell 2013; 154:1269-84; PMID:24034250; http://dx.doi.org/ 10.1016/j.cell.2013.08.015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Wei Y, Pattingre S, Sinha S, Bassik M, Levine B. JNK1-mediated phosphorylation of BCL2 regulates starvation-induced autophagy. Mol Cell 2008; 30:678-88; PMID:18570871; http://dx.doi.org/ 10.1016/j.molcel.2008.06.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Horbinski C, Stachowiak MK, Higgins D, Finnegan SG. Polyethyleneimine-mediated transfection of cultured postmitotic neurons from rat sympathetic ganglia and adult human retina. BMC Neurosci 2001; 2:2; PMID:11231879; http://dx.doi.org/ 10.1186/1471-2202-2-2 [DOI] [PMC free article] [PubMed] [Google Scholar]