

ABSTRACT
Many metagenomic sequencing studies have observed the presence of closely related bacterial species or genotypes in the same microbiome. Previous attempts to explain these patterns of microdiversity have focused on the abiotic environment, but few have considered how biotic interactions could drive patterns of microbiome diversity. We dissected the patterns, processes, and mechanisms shaping the ecological distributions of three closely related Staphylococcus species in cheese rind biofilms. Paradoxically, the most abundant species (S. equorum) is the slowest colonizer and weakest competitor based on growth and competition assays in the laboratory. Through in vitro community reconstructions, we determined that biotic interactions with neighboring fungi help resolve this paradox. Species-specific stimulation of the poor competitor by fungi of the genus Scopulariopsis allows S. equorum to dominate communities in vitro as it does in situ. Results of comparative genomic and transcriptomic experiments indicate that iron utilization pathways, including a homolog of the S. aureus staphyloferrin B siderophore operon pathway, are potential molecular mechanisms underlying Staphylococcus-Scopulariopsis interactions. Our integrated approach demonstrates that fungi can structure the ecological distributions of closely related bacterial species, and the data highlight the importance of bacterium-fungus interactions in attempts to design and manipulate microbiomes.
IMPORTANCE
Decades of culture-based studies and more recent metagenomic studies have demonstrated that bacterial species in agriculture, medicine, industry, and nature are unevenly distributed across time and space. The ecological processes and molecular mechanisms that shape these distributions are not well understood because it is challenging to connect in situ patterns of diversity with mechanistic in vitro studies in the laboratory. Using tractable cheese rind biofilms and a focus on coagulase-negative Staphylococcus (CNS) species, we demonstrate that fungi can mediate the ecological distributions of closely related bacterial species. One of the Staphylococcus species studied, S. saprophyticus, is a common cause of urinary tract infections. By identifying processes that control the abundance of undesirable CNS species, cheese producers will have more precise control on the safety and quality of their products. More generally, Staphylococcus species frequently co-occur with fungi in mammalian microbiomes, and similar bacterium-fungus interactions may structure bacterial diversity in these systems.
INTRODUCTION
From the early days of 16S rRNA clone libraries to the current groundswell of high-throughput metagenomics, one common pattern to emerge in studies of microbial community structure is the presence of numerous closely related species or strains of prokaryotes in the same habitat (1–3). These phylogenetic clusters have been observed in a wide range of environments, from marine microbiomes (4, 5) to our own human microbial landscapes (6). Many widespread bacterial species, including Vibrio, Staphylococcus, and Streptococcus species, commonly co-occur with closely related species or strains (6–8). Explaining these phylogenetic clusters has remained an important puzzle in microbial ecology because theory predicts that competitive exclusion should lead to dominance of only one major lineage when multiple phylogenetically similar lineages co-occur (9, 10). Identifying the ecological processes and molecular mechanisms that shape the distributions of closely related bacterial species can provide important tools to better manage and manipulate microbiomes in agriculture, medicine, and natural ecosystems.
Most previous attempts to explain the ecological distributions of bacterial species have focused on the role of abiotic niche partitioning and intraspecific or intrageneric biotic interactions (8, 11, 12), with much of our understanding coming from marine Vibrio species (5, 13–15) and oral biofilms (16, 17). For example, in coastal waters of the Atlantic Ocean, the coexistence of numerous genotypes of several species of Vibrio can be explained by temporal niche partitioning across seasons, preferences for particular microhabitats within seawater, and antagonistic interactions between ecologically defined populations (5, 18).
While niche specialization and intraspecific or intrageneric interactions may play important roles in driving coexistence in some microbiomes, biotic interactions between distantly related neighbors that span domains of life could also drive the composition within phylogenetic clusters. For example, in many microbiomes, bacteria coexist with diverse eukaryotic microbes, including fungi (19). Just as different plant species can mediate interactions between closely related insect herbivores that specialize on different resources (20), the distributions of coexisting bacterial species could be strongly regulated by the biotic niches created by co-occurring fungi. However, the causes and consequences of biotic interactions within microbiomes are poorly understood relative to impacts of the abiotic environment (3, 21). Examples of pairwise interactions between microbial species have been documented, including bacterium-bacterium interactions (22) and bacterium-fungus interactions (19). But it is unclear if these pairwise interactions dissected in the laboratory can help explain patterns of microbial diversity in multispecies microbial communities found in nature.
Cheese rind biofilms (Fig. 1A) provide a tractable system for exploring the role of biotic interactions in shaping distributions of closely related bacterial species. These simple communities are composed of various species of bacteria, yeasts, and molds that colonize the cheese surface during the aging process (23–25). Previous work has demonstrated that the accessibility, reproducibility, and tractability of these communities are ideal for dissecting the ecology of microbial communities (24). Here, we further demonstrate the utility of these communities by linking in situ observations of species abundance with in vitro experiments to determine what ecological processes shape the distribution of species within these microbial communities.
FIG 1 .

Ecological distributions of Staphylococcus species in cheese rind microbiomes. (A) A natural rind cheese showing the surface biofilm and curd. (B) Colonies of three of the most abundant CNS species found in cheese rinds were grown on plate count agar and photographed in natural light. Most strains of S. saprophyticus and S. xylosus are yellow or orange, while most strains of S. equorum are white. (C) Relative abundances of four CNS species in 25 cheese rind communities determined via metagenomic mapping of reads to reference genomes. Data are presented as relative abundances of reads that mapped to a Staphylococcus genome. Numbers at the bottom of the figure panel represent cheese sample identification numbers. (D) Relative abundances of four CNS species across a temporal sampling of 3 wheels of cheese aged over 56 days. Data for fungal and bacterial communities are from amplicon data presented in reference 26. Data for Staphylococcus communities were determined and are presented as described for panel C. (E) A model of Staphylococcus interactions with the biotic and abiotic environment during cheese rind development.
Coagulase-negative Staphylococcus (CNS) species are widespread but poorly understood members of cheese rind microbial communities. In a large-scale survey of 137 cheese rind communities, we found that Staphylococcus species were abundant (15% abundance on average across all cheese styles) and frequent (greater than 1% of the bacterial community in 67% of the cheeses surveyed) (24). Previously published studies performed in Europe and preliminary culture-based work performed in our laboratory have demonstrated that, across naturally aged cheeses and meats, four species of CNS are common: S. equorum, S. xylosus, S. saprophyticus, and S. succinus (26) (Fig. 1B). These species can also be commonly found co-occurring in animal microbiomes, including cow teats (27). Interestingly, these commonly co-occurring CNS species are very closely related members of the “saprophyticus cluster group” within the genus Staphylococcus (28–30), suggesting that they may have similar traits and share overlapping niches in the cheese rind microbiome. Ecological processes that shape the distribution of these commonly co-occurring species have not been identified.
By combining metagenomic data from in situ communities, in vitro reconstructions of experimental communities, and comparative genomics and transcriptomics, we address the following questions. (i) What are the ecological distributions of Staphylococcus species across cheese rind communities and during the development of cheese microbiomes? (ii) What ecological processes, including abiotic and biotic selection, determine the distributions of Staphylococcus species? (iii) Which potential molecular mechanisms underlie these ecological processes? The integrated ecological framework that we develop for dissecting the ecological distributions of these nonpathogenic Staphylococcus species could be applied to pathogenic Staphylococcus species in less-tractable microbiomes.
RESULTS
Staphylococcus equorum is the most abundant and widespread Staphylococcus species in cheese rind biofilms.
To measure the relative abundances of the four CNS species previously isolated from cheeses (S. equorum, S. xylosus, S. saprophyticus, and S. succinus), we used whole-genome shotgun metagenomic sequencing of rind microbiomes from 25 cheeses. Of these 25 metagenomes, 7 were previously published (24) and 18 were new metagenomes sequenced for this study (see Table S1 in the supplemental material). From each metagenome, we mapped 5.2 million 100-bp reads to an ~40-kb region of the reference genomes that provides careful discrimination between the four Staphylococcus species (see Materials and Methods). Percent relative abundance of each species was calculated as the number of reads mapped to a specific Staphylococcus species divided by the total number of reads mapped to all four Staphylococcus species genomes.
Across the 25 cheeses analyzed, S. equorum was the most abundant species (Fig. 1C), making up an average of 68.2% of the Staphylococcus reads detected across these rind communities. While 4 of 25 rind microbiomes were dominated by either S. saprophyticus or S. xylosus, S. equorum was dominant in 84% of the rind samples. S. succinus was very rare across the rinds sampled; it was detected in only 4 of the 25 rind communities. This metagenomic view of CNS in cheese supports previous culture-based surveys (26) suggesting that S. equorum is the most frequently encountered and locally abundant CNS species across cheeses. Given its widespread distribution, we hypothesized that certain traits in S. equorum promote its dominance in cheese and other fermented food microbiomes.
In addition to distribution across cheeses, we also investigated temporal patterns of abundance of the different Staphylococcus species during the development of a rind community. We chose a cheese (sample 491 in Fig. 1C) that had multiple species of Staphylococcus present so that we could identify any differences in colonization dynamics across the three species. Successional dynamics of all bacteria and fungi for this cheese have been previously described using amplicon metagenomic sequencing (24). Three replicate wheels of cheese were sampled at 1, 14, 28, 42, and 56 days after production, and the whole-genome shotgun metagenomic sequencing approach described above was used to determine relative abundances of Staphylococcus species over time. Unlike many of the previously described metagenomes, S. equorum did not dominate the Staphylococcus community at final stages of community development but showed a pattern of increasing relative abundance over time. At the early time points (day 1), reads of S. equorum were not detected whereas reads of S. saprophyticus and S. xylosus were detected. Over time, the relative abundance of S. equorum increased as the cheese aged, with the highest relative abundances detected at the final sampling time (day 56) (Fig. 1D). Note that these are relative abundance data, so it is impossible to distinguish between increasing abundance of S. equorum over time and decreasing abundance of the other species. But these data do suggest that S. equorum may be a late-colonizing species in cheese rind microbiomes.
The most abundant species (Staphylococcus equorum) is a slow colonizer and a weak competitor.
To explain the distributions of Staphylococcus species, we began to test the role of major abiotic and biotic components that have been previously identified in cheese rinds, including interactions between Staphylococcus species (congeneric competition), interactions between Staphylococcus species and the abiotic environment (abiotic selection), and interactions between Staphylococcus species and other microbial species present in cheese rinds (biotic selection) (Fig. 1E).
One mechanism commonly used to explain coexistence in multispecies communities is the competition-colonization tradeoff (31–33). Theoretical models and experimental data have both shown that poor competitors are generally better at quickly colonizing an open niche before competitors arrive, while rapid colonizers are generally poor at competition with neighboring species. This competition-colonization tradeoff can lead to coexistence of the two types of species within a community (31–33). Based on the low rate of colonization by S. equorum observed in the temporal metagenomic survey, we predicted that S. equorum would be a poor colonizer of cheese. We also predicted that it would be a strong competitor to compensate for its poor colonization ability. To assess competition and colonization, we used in vitro cheese rind assays, where cheese curd agar (CCA) was used to mimic the cheese environment (24). We focused only on the three most frequently detected species (S. equorum, S. xylosus, S. saprophyticus) because S. succinus is rarely detected (<1% abundance across all cheeses sampled and across the time series).
As expected based on the temporal metagenomic data, S. equorum was a slow colonizer of cheese compared to S. xylosus and S. saprophyticus (Fig. 2A). Over a 4-day period, 2 of 6 strains of S. equorum did not grow, while the remaining 4 strains grew much more slowly than S. saprophyticus and S. xylosus, with significant differences in growth between S. equorum and both S. saprophyticus and S. xylosus at 24 h of growth. These data corroborate the in situ late-colonization pattern observed in the temporal metagenomic study (Fig. 1D).
FIG 2 .

Ecological processes impacting the distributions of Staphylococcus (Staph) species in cheese rinds. (A) Colonization of different strains of S. equorum, S. saprophyticus, and S. xylosus on cheese curd agar. Points represent means (n = 3) ± 1 standard error. Strain (F11,24 = 115.19, P < 0.0001) and time of harvest (F3,72 = 151.09, P < 0.001) and their interaction (F33,72 = 16.187, P < 0.001) were significant based on a repeated-measures analysis of variance (ANOVA), with all S. equorum isolates having significantly lower growth than the S. saprophyticus and S. xylosus isolates based on Tukey’s honestly significant difference (HSD) test. Red asterisks next to strain names indicate the strains of each species used for the remaining experiments. (B) Pairwise competition of the three species of Staphylococcus (S. equorum [S.eq] strain BC9, S. saprophyticus [S.sap] strain BC4, and S. xylosus [S.xyl] strain BC10), with colors as defined for panel A. Bars represent means (n = 3) ± 1 standard error. Asterisks indicate significant differences in growth rates (P < 0.01) in the presence of an interspecific competitor compared to growth in the absence of an interspecific competitor (Dunnett’s test). (C) Growth of same species/strains as described for panel B over a range of pH and salt concentrations in BHI medium as measured by OD600 after 48 h. Points represent means (n = 3) ± 1 standard error. S. xylosus has a greater niche breadth (OD600 sum = 21.47 ± 0.50) than S. saprophyticus (OD600 sum = 19.187 ± 0.28) or S. equorum (OD600 sum = 19.08 ± 0.33) based on ANOVA with Tukey’s HSD test (F2,5 = 13.336, P < 0.01). (D) Relative abundances of the three species of Staphylococcus in experimental in vitro communities over 21 days. The three species were inoculated at initially identical densities and grown alone or with 5 single-species neighbors or an entire community containing all 5 neighboring species. Both biotic treatment (F6,20 = 48.268, P < 0.0001) and time of harvest (F2,40 = 29.310, P < 0.001) and their interaction (F12,40 = 12.528, P < 0.001) had significant effects on the abundance of S. equorum. Asterisks indicate significant stimulation of S. equorum compared to Staph-alone treatment based on Tukey’s HSD tests of absolute numbers of CFU. (E) Absolute abundances of the three species of Staphylococcus in the same experimental in vitro communities as described for panel D but only for molds Penicillium and Scopulariopsis. Solid lines represent the species growth in treatments without neighbors, while dashed lines represent the species growth in treatments with the fungal partner. Points represent means (n = 4) ± 1 standard error.
While the growth of S. equorum alone suggested that it is a slow colonizer of the cheese environment and may be at a disadvantage compared to the other two species, synergistic effects when grown with neighbors may allow it to outcompete the other two Staphylococcus species. Surprisingly, when grown in pairwise competition assays with the other two species of Staphylococcus, S. equorum was also a poor competitor. S. xylosus growth was not inhibited when grown with neighbors, but growth of both S. saprophyticus and S. equorum was significantly inhibited in the presence of neighboring species (Fig. 2B). S. xylosus had the greatest negative impact on neighboring species, suggesting that it is the strongest competitor in these communities. These in vitro growth and competition experiments suggest that S. equorum, the most widespread CNS species in cheese rinds, is the slowest colonizer and the weakest competitor of the three CNS species studied.
In some systems, specialization to different abiotic niches can allow species to coexist, and widespread species tend to have the largest abiotic niche breadth (34). We predicted that S. equorum might have a broader environmental niche breadth than the other two CNS species, and in environments that limit the growth of S. xylosus and S. saprophyticus, S. equorum can thrive. To test this prediction, we measured the growth of the three CNS species across gradients of salt and pH that span those found during the aging of cheeses (Fig. 2C; see also Fig. S1 in the supplemental material). Salt and pH are two of the most important drivers of bacterial growth in cheese microbiomes, and both of these environmental variables change during the aging of cheese as S. equorum becomes more abundant (24, 35). In contrast to our prediction, S. xylosus had the broadest niche breadth as determined by total growth across the range of pH and salt concentrations, with S. equorum and S. saprophyticus having more restricted niches (Fig. 2C; see also Fig. S1). Although the salt tolerance of S. equorum is greater than that of S. saprophyticus and comparable to that of S. xylosus, S. saprophyticus can grow at lower pH than both of the other species, which may explain why it dominates in the early stages of cheese rind development. These data suggest that, while these different Staphylococcus species do have different environmental tolerances, S. equorum does not have the broadest niche breadth and does not specialize in a particular abiotic niche. Therefore, abiotic niche breadth does not explain the widespread distribution of S. equorum in cheese rinds.
Biotic interactions explain discordant in situ and in vitro patterns of abundance.
The contrasting in situ observations and in vitro experiments present an interesting ecological conundrum: how can a slow colonizer and weak competitor become the most abundant species in most cheese rind communities? We hypothesized that neighboring bacterial or fungal species could mediate competition between the Staphylococcus species through specific promotion of S. equorum or specific inhibition of S. xylosus and S. saprophyticus.
To test how biotic interactions impact the abundance of the three species, we repeated the same three-way competition experiment described above but added common bacterial (Brachybacterium and Brevibacterium) and fungal (Scopulariopsis, Penicillium, and Debaryomyces) species from cheese rinds to the 3-species Staphylococcus community. Addition of bacterial neighbors did not shift the abundance of S. equorum in the community (Fig. 2D). However, addition of fungi dramatically shifted the composition of the Staphylococcus community. Most strikingly, the mold Scopulariopsis shifted the community composition such that S. equorum became the dominant Staphylococcus species at the final time point (day 21), as was observed in situ (Fig. 2C). These shifts in relative abundance could be due to species-specific growth promotion of S. equorum or inhibition of the strong competitors S. xylosus and S. saprophyticus. Absolute data show the former; Scopulariopsis strongly stimulates growth of S. equorum but not growth of the other Staphylococcus species (Fig. 2E) (S. equorum abundance [F1,20] = 49.51, P < 0.001; S. saprophyticus F1,20 = 0.76, P = 0.39; S. xylosus F1,20 = 4.77, P = 0.04).
On the basis of our in vitro studies showing that Scopulariopsis strongly promotes S. equorum growth, we predicted that the presence of S. equorum and that of Scopulariopsis would be strongly positively correlated in naturally forming rind microbiomes. In situ data collected across the development of a cheese rind community (Fig. 1D) do show strong positive associations between S. equorum and the fungi Scopulariopsis (Spearman’s rho = 0.77, P = 0.006) and Penicillium (Spearman’s rho = 0.78, P = 0.005) (see Fig. S2 in the supplemental material). The presence of species of the Actinobacteria genera Brevibacterium (Spearman’s rho = 0.82, P < 0.001) and Brachybacterium (Spearman’s rho = 0.85, P < 0.001) is also strongly positively correlated with S. equorum abundance, even though these bacteria did not promote the growth of S. equorum experimentally (Fig. 2D), suggesting that in situ correlations of abundance do not always correctly predict experimental outcomes. Given that these data come from repeated sampling of the same three wheels of cheese, albeit from different locations across the rind surface (see Materials and Methods), these strong correlations should be interpreted cautiously due to lack of complete independence. But the data do provide a preliminary indication of the correspondence between the patterns of abundance in experimental interactions observed in vitro and in situ.
Comparative genomics and transcriptomics point to potential molecular mechanisms underlying distributions of Staphylococcus species.
A specific interaction between the mold Scopulariopsis and S. equorum shifts the composition of the Staphylococcus community from dominance by a strong competitor, S. xylosus, to dominance by a weak competitor, S. equorum. To determine the potential molecular mechanism(s) underlying the bacterium-fungus interaction, we used comparative genomics of each of the Staphylococcus species alone as well as transcriptomics (RNA-sequencing or RNA-seq) of cocultures. Comparative genomics can pinpoint gene sets that are unique to species/strains that might explain ecological traits specific to a species (36, 37), while RNA-seq has the power to reveal the subset of genes and pathways that may underlie the outcomes of species interactions (38, 39).
To determine gene sets that are unique to S. equorum, we compared protein-coding genes of 15 new genomes sequenced for this study that span the three Staphylococcus species, as well as 7 existing genomes (see Table S2 in the supplemental material). We found 76 protein-coding genes that were both present in all S. equorum strains and absent in S. saprophyticus and S. xylosus genomes (Fig. 3A; see also Table S3). One striking pattern to emerge is the enrichment of genes in S. equorum associated with iron acquisition and metabolism (Fig. 3A). Iron is one of the most limiting nutrients in cheese rinds (40), and Staphylococcus species have evolved various mechanisms for coping with low iron availability, including the production of siderophores (41). Much of the iron enrichment signal in S. equorum is due to the presence of a siderophore production and uptake operon that is homologous to the staphyloferrin B operon in Staphylococcus aureus. Staphyloferrin B is an alpha-hydroxycarboxylate siderophore that has been shown to be important in the growth and virulence of S. aureus (41). This operon is uncommon in the genus Staphylococcus; the entire operon is found only in S. aureus, S. pseudintermedius, and now S. equorum.
FIG 3 .

Putative molecular mechanisms underlying interactions between Staphylococcus equorum and fungi. (A) Comparative genomics of Staphylococcus species reveals species-specific genes, which are unevenly distributed across functional categories. Data are from 12 S. equorum (gray), 5 S. saprophyticus (yellow), and 5 S. xylosus (orange) strains. The Venn diagram shows overlap of clusters of protein-coding genes across the three species. The bar graph shows the distribution of species-specific genes (found in all strains of one species but not other species) across SEED subsystems. (B) Genome size estimates based on genome assemblies and percentages of prophage with intact genomes, as determined through PHAST annotations. (C) Changes in genome expression of S. equorum grown with the fungus Penicillium (left panel) or Scopulariopsis (right panel). Panels show scatter plots of global gene expression for S. equorum alone (x axis) versus with a fungus (y axis). Each dot represents a transcript from across the genome. Orange dots represent those transcripts that were differentially expressed when grown with the fungus (>5-fold change in expression; Rockhopper q value < 0.05). (D) Distribution of differentially expressed genes represented in panel C across SEED subsystems. (E) Representative read mapping at the staphyloferrin B operon from treatments with S. equorum alone (top), S. equorum plus Penicillium (middle), and S. equorum plus Scopulariopsis (bottom). Blue-shaded regions represent average coverage from read mappings. (F) Mean (± standard error) free iron concentration in cheese curd agar (CCA), CCA plus Scopulariopsis, and CCA plus Penicillium. All three conditions had significantly different iron concentrations based on ANOVA with Tukey’s HSD test (F2,10 = 10.30, P < 0.01). (G) Lyophilized and concentrated supernatants of CCA incubated without a fungus, with Penicillium, and with Scopulariopsis were spotted onto cellulose disks on chrome azurol S (CAS) medium. Shown is a representative plate after 1 day of incubation. (H) The supernatants used in the experiment whose results are shown in panel G were spotted onto cheese curd agar plates seeded with the three Staphylococcus species. The radius of zones of stimulation was measured around the cellulose disk after 3 days. (I) Mean methionine concentrations (± standard error) determined in cheese curd alone and cheese curd plus Scopulariopsis using capillary electrophoresis-time of flight mass spectrometry (CE-TOF MS). See Table S5 in the supplemental material for more details on metabolomics.
We also identified genomic features in S. equorum that might help explain why it is competitively inferior to S. saprophyticus and S. xylosus. The composition of genes that are absent in S. equorum, but present in S. saprophyticus and S. xylosus, does not suggest any obvious nutrition stress response or other deficiencies (see Table S3 in the supplemental material). But we did observe substantial variation in the distributions of prophage-encoding genes across the three species. The abundance of prophages within a genome has been shown to decrease the growth of some bacterial species, leading to the notion of a prophage burden (42–44). While S. equorum does have a significantly greater total genome size (F2,19 = 26.79, P < 0.001) and a higher total number of coding sequences (F2,19 = 15.70, P < 0.001), it also has a higher potential prophage burden, with a significantly higher percentage of total coding sequences being intact phage in S. equorum compared to S. xylosus and S. saprophyticus (Fig. 3B) (F2,19 = 4.04, P = 0.034). The high abundance of these prophages in S. equorum cannot not be explained by the lack of restriction-modification (R-M) systems that might help prevent phage infection; of the three species, S. equorum actually has the highest number of genes predicted to be components of R-M systems (see Table S2). None of these three CNS species are predicted to have functional clustered regularly interspaced short palindromic repeat (CRISPR) systems. It is unknown if and how these prophage can be activated to lyse cells, but the burden with respect to copying phage DNA during chromosome replication and the potential for phage-mediated lysis of cells may be greater for S. equorum than for S. saprophyticus and S. xylosus.
To better understand specific genes and pathways within S. equorum that respond to the strong stimulation by the mold Scopulariopsis, we used RNA-seq to identify genes that were up- and downregulated in the genome of S. equorum in the presence of fungi compared to the levels seen with growth of S. equorum alone. We compared the effect of Scopulariopsis (strongly promoted growth) on the S. equorum transcriptome to the effect of Penicillium (weakly promoted growth) to examine differences in the transcriptional responses to these two fungi. Complementing our comparative genomics findings, the RNA-seq data also point to a key role of iron acquisition in the biology of S. equorum on cheese, in particular when grown with Scopulariopsis. While the two fungal species had similar impacts on the transcriptional profiles of S. equorum in terms of the distributions of genes that were up- and downregulated (Fig. 3C; see also Table S4 in the supplemental material), Scopulariopsis had a unique impact on genes involved with iron acquisition and metabolism. A total of 23 genes associated with iron acquisition were downregulated when S. equorum was grown with Scopulariopsis, while no genes associated with iron acquisition were downregulated when S. equorum was grown with Penicillium (Fig. 3D; see also Table S4). No genes associated with iron acquisition were upregulated in either fungal treatment. The staphyloferrin B operon identified with comparative genomics was a major part of the strong iron response in our RNA-seq data set. Mean expression across the staphyloferrin B operon was significantly lower for both the biosynthesis components (sbnABCDEFGHI; t = 5.22, P < 0.001) and the uptake components (sirABC; t = 3.82, P < 0.01) of the operon when S. equorum was grown with Scopulariopsis (Fig. 3E).
To determine if and how fungi might alter iron availability in cheese, we measured free iron availability in our cheese curd medium. Both fungal species caused a decrease in free iron availability, but Scopulariopsis decreased iron availability to a lesser extent (Fig. 3F), potentially explaining the differences in iron-associated gene expression patterns in the RNA-seq datasets. This assay does not measure chelated forms of iron, such as iron in siderophore complexes. Siderophores produced by fungi may sequester free iron and may be used in place of endogenously produced staphyloferrin B. Supernatants of Scopulariopsis and Penicillium contained siderophores (Fig. 3G), as detected with the chrome azurol S (CAS) assay (45), and these supernatants stimulated the growth of S. equorum, but not S. saprophyticus or S. xylosus, on cheese curd agar (Fig. 3H), highlighting the specificity of these bacterium-fungus interactions. The fact that Penicillium supernatants also strongly stimulate S. equorum is surprising given that we did not see strong stimulation of S. equorum when Penicillium was grown with the three-species Staphylococcus community (Fig. 2E). This may be because actively growing Penicillium fungi have other impacts on neighboring species not captured by these supernatant assays.
Two other strong transcriptional responses were observed in the presence of both fungal species: expression of genes involved with methionine biosynthesis pathways strongly decreased, and expression of genes involved with thiamine biosynthesis increased (see Fig. S3 in the supplemental material). Filamentous fungi produce large amounts of extracellular proteases, and these proteases may release free amino acids that could stimulate the growth of neighboring bacterial species (35, 46). Using a global metabolomics screen, we observed a substantial increase in the levels of many free amino acids when Scopulariopsis was grown on cheese curd agar, including a greater than 10-fold increase in levels of methionine (see Table S5). The reason for an increase in expression of thiamine biosynthesis genes in S. equorum is less clear, but previous studies of bacterium-fungus interactions have observed that bacterially produced thiamine can support the growth of fungi that are thiamine auxotrophs (47).
DISCUSSION
Biotic interactions between microbial species have been frequently proposed as potential drivers of microbiome diversity (21, 48–50). Putative biotic interactions have been inferred from observational metagenomic studies, where positive or negative associations between microbial taxa are used as a proxy for positive or negative interactions (49–51). This approach can be confounded by the structure of relative abundance data of most metagenomic studies and by shared niche preferences of noninteracting organisms (52). Microbial interactions within microbiomes have rarely been experimentally validated due to the challenges of creating ecologically relevant conditions in a laboratory environment and the limited ability to culture all members in complex multispecies microbiomes. When potential pairwise interactions have been carefully measured and dissected mechanistically (19, 53–55), they have rarely been linked to potential patterns of biodiversity and ecological processes occurring in naturally forming microbiomes (56). By integrating observations from metagenomics with in vitro community reconstructions and comparative omics approaches, we have demonstrated that very specific biotic interactions between bacteria and fungi can shape the composition of widely distributed and closely related bacterial species.
After comparing in situ distribution patterns of commonly isolated Staphylococcus species obtained through metagenomic sequencing with in vitro competition, colonization, and niche breadth data, we were presented with a striking paradox. Across many independently assembled cheese microbiomes sampled in the United States and Europe, the most widely distributed and locally abundant Staphylococcus species was a slow colonizer and a poor competitor and did not have the largest niche breadth. By measuring the impacts of neighboring species from the cheese community on the distributions of these Staphylococcus species, we identified fungal facilitation as a mechanism to explain the distribution of the weakest species, S. equorum. One specific fungus, the widely distributed Scopulariopsis, had the strongest effect on the composition of Staphylococcus biofilms. Our findings demonstrate a clear example of an interdomain interaction shaping the distribution of bacterial species within a microbiome and suggest that bacterium-fungus interactions can determine the distributions of widely distributed microbes.
Our comparative genomics and transcriptomics data suggest several putative mechanisms that shape the ecological distribution of S. equorum in cheese rinds. Comparative genomics did not reveal nutritional deficiencies that may explain lower growth rates of S. equorum in cheese, but the accumulation of prophages and large genome sizes of S. equorum may pose constraints on potential maximum growth rates. The fungi Scopulariopsis and Penicillium had different impacts on the S. equorum transcriptome, likely due to differences in metabolites produced by these two fungi or their impacts on the cheese environment (e.g., pH, nutrient availability). Both fungal species caused a decrease in expression of genes associated with amino acid acquisition and metabolism, while only Scopulariopsis caused a decrease in expression of iron acquisition genes. Much of the iron in cheese is bound by lactoferrin, an iron chelator present in milk (35), and microbes that colonize cheese produce siderophores to cope with iron limitation (40). Free amino acids are also in low supply in fresh cheese, as they are bound up in casein (35). Scopulariopsis may alter the availability of both iron and free amino acids by a variety of mechanisms (Fig. 4). Direct production of fungal siderophores by Scopulariopsis may relieve S. equorum of the costly production of the siderophore staphyloferrin B and potentially provide a unique iron source through cross-feeding. Scopulariopsis may also increase iron availability through release of chelated iron from lactoferrin through proteolysis, as has been suggested to occur with proteases of Pseudomonas aeruginosa (57–59). Proteolysis could also increase availability of free amino acids through casein degradation, potentially promoting the growth of slow-growing S. equorum. This current work highlights only a few potential molecular mechanisms, and the study did not fully test all potential mechanisms. More-detailed chemical analyses, including characterization of the fungal siderophores, are required to fully understand the molecular mechanisms driving the Staphylococcus-Scopulariopsis interaction.
FIG 4 .
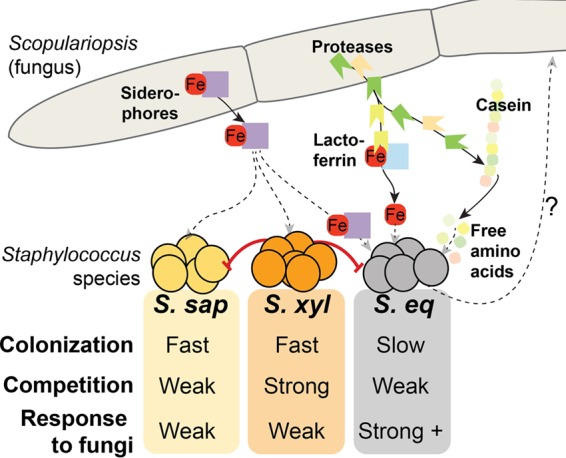
An overview of traits of cheese rind Staphylococcus species and potential mechanisms driving their interactions with fungi. Solid lines indicate interactions/mechanisms established in this work or in previous studies. Dashed lines indicate potential or unknown interactions/mechanisms.
Biotic interactions strongly altered the ecological distributions of the Staphylococcus species in these cheese biofilms, but other factors not manipulated in this study may also shape the abundance of S. equorum in microbiomes. High dispersal rates may counteract the slow colonization and poor competition traits of S. equorum and allow it to dominate cheese rind microbiomes. In the cheese production environment, high abundances of S. equorum in the raw milk used for cheesemaking or addition of S. equorum as a starter culture might allow high dispersal rates during the initial phase of cheese development. Previous surveys of microbial communities in raw milk have not indicated that S. equorum is particularly dominant relative to S. saprophyticus and S. xylosus (60–64), and S. equorum is not globally distributed as a starter culture in the cheese industry, suggesting that dispersal may not be important in explaining the widespread distribution of this species in cheese rinds. Future studies should examine the relative impacts of dispersal and biotic interactions in shaping the distribution of S. equorum.
While our work is driven by the search for basic principles of microbial community assembly, these results also have direct implications for the management of cheese microbiomes. One species of CNS studied, S. saprophyticus, causes urinary tract infections (UTIs) in healthy adults (65–69). While there is some confusion over whether the strains found in cheese and other foods can actually cause UTIs (65) and while there are no published reports of UTIs caused by eating cheeses, keeping S. saprophyticus out of cheese products should be a goal of cheese producers due to the potential for transfer of virulence factors or other mobile elements to the microbiome of cheese consumers. S. equorum is not considered a pathogen and has been suggested to be a potential starter culture given its previously described antilisterial activities (70, 71). Our work suggests that cheesemakers need to consider the management of co-occurring fungi in order to favor the growth of S. equorum over S. saprophyticus.
Fungi co-occur with bacteria in many microbiomes, including soils and the human body, but the ecology of bacteria and the ecology of fungi have traditionally been studied separately in the fields of bacteriology and mycology (19). Here we show that bacterium-fungus interactions can be important for explaining patterns of diversity within widespread microbial communities. Just as eukaryotic hosts (animals) can select for specific bacterial communities in host-associated microbiomes (72–74), fungi can select for specific bacterial communities in free-living microbiomes. While our work was performed using a synthetic community growing on an artificial substrate created by humans, fungal selection of bacterial diversity is likely to play a role in other microbiomes. For example, closely related Staphylococcus species co-occur in many human body sites, including the respiratory tract and skin microbiomes (6, 75–77). Various fungi can also co-occur in these same body habitats and may drive the distributions of pathogens such as Staphylococcus aureus. Future work exploring how fungi can shape the ecological distributions of bacterial species at fine phylogenetic scales can further elucidate the molecular mechanisms driving ecological patterns and processes in microbiomes.
MATERIALS AND METHODS
Isolation of strains.
Strains were isolated from rinds of cheeses or raw milk used for cheesemaking by serially diluting rind scrapings or milk samples on plate count agar with milk and salt (PCAMS) (24) with cycloheximide at a final concentration of 100 µg/ml. DNA was extracted from cells streaked on PCAMS using a PowerSoil DNA isolation kit (Mo Bio, Carlsbad, CA), and strains were initially identified to the species level using Sanger sequencing of the16S rRNA gene as previously described (24).
Whole shotgun metagenomic library construction and mapping.
Whole shotgun metagenomic libraries from cheese rinds were constructed as described in reference 26 except that a New England Biolabs (Ipswich, MA) NEBNext Ultra DNA Library prep kit for Illumina was used according the manufacturer’s instructions. DNA was extracted from rinds using a PowerSoil DNA isolation kit, standardized to 1 µg in 50 µl of molecular-grade water, sheared to approximately 400 bp using a Covaris S220 focused ultrasonicator (Covaris, Woburn, MA), and used for library construction.
To determine the abundance of Staphylococcus species within each metagenome, 2.6 million 100-bp reads (for the survey across cheeses) or 1.7 million reads (for the survey over time from one cheese) were mapped to a conserved 39,648-kb region of the reference genome for each of the four CNS species (S. equorum strain BC9, S. saprophyticus strain BC4, S. xylosus strain BC10, and S. succinus strain BC15). This 39,648-kb region contains 44 protein-coding sequences and was chosen because preliminary cross-mapping studies for comparisons between the different Staphylococcus species demonstrated that this region could distinguish between these closely related species whereas the use of most other regions of the genome with highly conserved regions leads to false positives. Shotgun metagenomic data were mapped to reference genomes using the read mapper in Geneious 8.1.6. Reads were mapped with the following settings: minimum overlap identity = 97%; index word length = 14; map multiple best matches to none. Error rates based on mapping reads from individual genomes to all possible reference genomes were very low, ranging from 0.007% to 0.02%, demonstrating that this mapping approach provides precise discrimination of these four closely related species.
In vitro growth, competition, and community assembly experiments.
Inocula for all in vitro experiments came from frozen glycerol stocks of brain heart infusion (BHI) broth liquid cultures that had been previously plated to determine CFU per microliter of inoculum. The main three strains used for experiments comparing Staphylococcus species were BC9 for S. equorum, BC4 for S. saprophyticus, and BC10 for S. xylosus. As our data in Fig. 2A demonstrate, each of these strains accurately represents the colonization and growth rates of each species. Depending on the in vitro experiment, different combinations of microbes were pooled in 1× phosphate-buffered saline (PBS) and inoculated onto the surface of 96-well microplates, with each well containing 150 µl of cheese curd agar (CCA) as previously described (24). For growth, competition, and community assembly experiments, inoculation for each species was approximately 200 CFU per well. For pairwise competition experiments, the total number of CFU of each species, even when grown alone, was 200 CFU. Growth-alone treatments had 200 CFU, while two-species treatments had 400 CFU in total. Postinoculation, microplates were sealed with a sterile, breathable film (Aeraseal; Excel Scientific, Victorville, CA). In vitro communities were incubated at 24°C for 3 days (to simulate cheese production facility conditions) and then at 14°C (to simulate cheese aging conditions) for the remainder of the incubation period (up to 21 days, depending on the experiment). To determine the CFU of each community member, communities were harvested by removing the entire CCA plug from a 96-well plate, homogenizing, and serial dilution plating as previously described (24).
Niche screens.
The niche breadth of the three Staphylococcus species was determined by growing strains of each species in 200 µl of BHI broth under 24 different sets of pH and salt conditions that were distributed in grids across 96-well plates (see Fig. S1 in the supplemental material). Ten microliters of standardized inoculum at 20,000 CFU/µl of the appropriate strain was added to each well. Three replicate grids were assayed for each strain. After 48 h of growth with shaking at 200 rpm on a platform-shaking incubator at 24°C, the full volume of BHI broth in each well was pipetted up and down 5 times to remove any clumps, and optical density at 600 nm (OD600) was determined on a SpectraMax M5 plate reader.
Genome sequencing and comparative genomics.
DNA was extracted using Mo Bio PowerSoil DNA extraction kits and streaks generated from a single bacterial colony grown for 2 to 3 days on PCAMS. Approximately 1 µg of purified genomic DNA (gDNA) was sheared using a Covaris S220 instrument to approximately 450 bp and was used as the input for a New England Biolabs (Ipswitch, MA) NEBNext Ultra DNA Library prep kit for Illumina. Libraries were spread across multiple sequencing lanes with other projects and were sequenced using 100-bp paired-end reads on an Illumina HiSeq 2500 instrument. Approximately 10 million reads were sequenced for each genome. Failed reads were removed from libraries, and reads were trimmed to remove low-quality bases and were assembled to create draft genomes using the de novo assembler in CLC Genomics workbench 8.0. Assembled genomes were annotated using RAST (78).
To compare the presence and absence of genes across strains and species, core and accessory genes were identified by assigning protein-coding sequences to functionally orthologous groups using the MultiParanoid method of the PanGenome Analysis Pipeline (PGAP) (79). Species-to-species orthologs were identified by pairwise strain comparison using BLAST with PGAP defaults: a minimum local coverage of 25% of the longer group and a global match of no less than 50% of the longer group, a minimum score value of 50, and a maximum E value of 1E−8. Multistrain orthologs were then found using MultiParanoid (80). Genes uniquely associated with each species were determined by filtering gene families to identify those present in all strains of a species and not present in any others, and functional annotations were determined using the RAST server and SEED subsystem annotations (78). Assessment of restriction-modification systems was conducted using SEED annotations, and assessment of potential CRISPR systems was conducted using CRISPRfinder (http://crispr.i2bc.paris-saclay.fr/).
RNA-seq experimental setup, RNA isolation, library construction, and sequencing.
Experimental biofilms of S. equorum grown alone and with the two neighboring fungi were constructed by inoculating 20 ml of cheese curd agar in 100-mm-diameter petri dishes with 37,000 CFU of S. equorum. In the treatment using S. equorum alone, 100 µl of PBS was added to the S. equorum inoculum. In the fungal treatments, 100 µl of fungal inoculum was added to the S. equorum inoculum. Scopulariopsis was inoculated at ~3,700 CFU, while Penicillium was inoculated at ~370 CFU. The inoculation densities of the S. equorum and the fungi represented levels of bacterial and fungal densities similar to those that have been observed on cheese rinds (81, 82). We used different inoculation densities for the two fungi to take into account their very different growth rates; Penicillium reaches stationary growth in about 3 days whereas Scopulariopsis takes about 6 days to reach stationary growth. Previous preliminary experiments had demonstrated that using 1/10 the amount of the Penicillium inoculum produced similar densities of fungal growth across the S. equorum biofilms.
After 4 days of growth at 24°C, three replicate plates of each treatment (S. equorum alone, S. equorum plus Penicillium, and S. equorum plus Scopulariopsis) were harvested and used to create replicate RNA-seq libraries. Rind biofilms were harvested by scraping the cheese curd surface with a sterile razor blade to remove most of the microbial biomass. Harvested biofilms were stored in RNAProtect reagent (Qiagen) to stabilize mRNA and were frozen at −80°C. RNA was extracted using a standard phenol-chloroform protocol adopted from transcriptomics work in gut microbiomes (83). This protocol uses a standard bead-beating step in a lysis buffer to release cell contents from pelleted biofilms in RNAProtect. To ensure that the RNA was of high quality and not degraded, 500 ng of each RNA preparation was run and visualized on a 1.5% agarose gel. DNA was removed from the nucleic acid pool using a TURBO DNA-free kit (Life Technologies), and 5S/tRNA and rRNA were depleted using MEGAClear (Life Technologies) and RiboZero (Illumina) kits, respectively. To remove both yeast and bacterial rRNA, yeast and bacterial rRNA probes from the RiboZero kits were mixed 1:2 and used for rRNA depletion. To confirm that the samples were free of DNA contaminants, a PCR of the 16S rRNA was conducted with standard 16S primers (27f and 1492r).
RNA-seq libraries were constructed from purified mRNA using a NEBNext Ultra RNA Library prep kit for Illumina (New England Biolabs) and the manufacturer’s instructions and were sequenced using paired-end 100-bp reads on an Illumina HiSeq system in rapid run mode by the Harvard Bauer Core Facility. About 16 million reads were sequenced for each library. Only forward reads of the paired ends were used for analysis.
Analysis of RNA-seq libraries, including read mapping, normalization, and quantification of transcript abundances, was done using Rockhopper version 1.3.0 (84) with default settings. The S. equorum strain BC9 genome was concatenated and used as the reference genome for read mapping. Expression values were normalized using the upper quartile of gene expression. We considered differentially expressed genes to be those that exhibited a greater than 5-fold change in expression and that were deemed significantly different based on a q value of <0.05. Rockhopper’s q values are P values adjusted for the false-discovery rate using the Benjamini-Hochberg procedure.
Fungal supernatant siderophore assay and growth stimulation assays.
To determine if fungi that promoted the growth of S. equorum produced siderophores and if supernatants of these fungi contained compounds that could stimulate bacterial growth, we grew the strains of Penicillium and Scopulariopsis described above in static liquid cheese curd (2% cheese curd, as opposed to 10% in solid medium) for 2 weeks at 24°C to obtain fungal supernatants. The cleared supernatant from this liquid was filter sterilized, frozen at −80°C, lyophilized, and then resuspended in sterile water. As a control, we also lyophilized 2% liquid cheese curd medium that had not been conditioned by any organism. For each of the three supernatant powders (control, Penicillium, Scopulariopsis), we added 2 mg of powder to 2 ml of sterile water for the experiments described below.
The chrome azurol S (CAS) assay was used to determine if fungal supernatants contained siderophores. We placed three sterile cellulose disks on the surface of a petri dish containing 20 ml of solid CAS assay medium (45). To the sterile cellulose disks, we added 20 µl of the hydrated cheese curd control powder that was not conditioned, the Scopulariopsis supernatant, or the Penicillium supernatant. After incubation in the dark at 24°C for 48 h was performed, we noted the presence of orange halos around the cellulose disks, indicating iron removal from the CAS assay blue dye complex.
We used the same approach to detect stimulation of Staphylococcus species by supernatants except that PCAMS or 10% cheese curd agar plates were used instead of the CAS assay medium. Standard (100-mm-diameter) petri dishes were seeded with approximately 75,000 CFU of one of the three Staphylococcus species to create a lawn of bacteria that could respond to the supernatant treatments. Three sterile cellulose disks were placed on the lawns, and 20 µl of control, Scopulariopsis supernatant, or Penicillium supernatant was added to one of the three disks. Three replicate plates for each species were used. Zones of stimulation were recorded after 3 days of growth, and the radius of stimulation zones was measured for the CCA plates after inverting photos of the plates to improve the ability to see colonies on the opaque cheese curd surface.
Metabolomics and iron assays.
Cheese curd distributed in standard petri dishes was inoculated with either 100 µl of PBS or 100 µl of PBS containing ~3,700 CFU of Scopulariopsis (as in the RNA-seq experiments described above). After 10 days of growth, three replicate plates were harvested from each treatment and frozen until analysis by Human Metabolome Technologies (HMT) Inc. (Yamagata, Japan). Approximately 30 mg of each sample was mixed with 50% (vol/vol) acetonitrile in water, the mixture was homogenized, and the supernatant was filtered through a 5-kDa-cutoff filter (Ultrafree-MC-PLHCC; Human Metabolome Technologies Inc.) to remove macromolecules. The samples were then subjected to capillary electrophoresis-time of flight mass spectrometry (CE-TOF MS) for analysis of sugars, amino acids, nucleotides, and other ionic metabolites or to liquid chromatography-time of flight mass spectrometry (LT-TOF MS) for analysis of fatty acids, steroids, and other lipids using previously described analytical conditions and internal standards (85–87). Peaks were assigned to putative metabolites based on HMT’s standard library on the basis of m/z data and migration time (in CE-TOF MS) or retention time (in LT-TOF MS). Welch’s t test was used with false-discovery-rate correction of P values using the Benjamini-Hochberg procedure to identify metabolites that were significantly increased or decreased in abundance in the presence of Scopulariopsis on the basis of the relative peak area (metabolite peak area/ internal standard peak area × sample amount) for each metabolite.
To determine the concentration of available iron in the cheese curd medium, replicate plugs of cheese curd from the 96-well assays described above were removed from wells without microbes present (Control), where Scopulariopsis was inoculated (+Scopulariopsis), and where Penicillium was inoculated (+Penicillium) at the densities used in the community experiments described above. Iron concentrations in homogenates of these cheese curd samples were determined using an iron colorimetric assay kit (BioVision Inc.) according to the manufacturer’s instructions, except that 0.1 vol of 20% SDS was added to each sample to minimize interference from protein precipitation.
Accession number(s).
Metagenomic libraries have been deposited in MG-RAST with accession numbers listed in Table S1 in the supplemental material. Assembled genome sequences have been deposited as whole-genome-sequencing (WGS) submissions to NCBI with accession numbers listed in Table S2. Raw RNA-seq reads and differential expression data have been submitted to the NCBI GEO database with GEO accession number GSE75505 (samples GSM1958050 through GSM1958058).
SUPPLEMENTAL MATERIAL
Abiotic niche assays for more strains of each Staphylococcus species. Heat maps represent the growth of each strain across a gradient of salt and pH values as measured by OD600 after 48 h. Data for BC9, BC4, and BC10 are presented in more detail in Fig. 2. Data represent means of results of 3 replicates. Figure S1 relates to Fig. 2. Download
Spearman’s rank correlation coefficients of relative abundances of Staphylococcus species (from shotgun metagenomic data) and relative abundances of other members of the cheese rind community (from amplicon sequencing data). Correlations highlighted in bold are statistically significant (P < 0.01). See Results for discussion of caveats with respect to interpreting these data. Figure S2 relates to Fig. 1 and 2. Download
(A) The thiamine biosynthesis pathway. The part of the pathway highlighted with thick lines was differentially expressed (green = increased expression, red = decreased expression) in S. equorum grown with fungi. Small yellow boxes indicate if differential expression was observed with just Penicillium (P), with just Scopulariopsis (S), or with both Penicillium and Scopulariopsis (PS). (B) The methionine biosynthesis pathway. Notations are the same as those described for panel A. Figure S3 relates to Fig. 3. Download
Shotgun metagenome metadata and features.
Genome metadata and statistics.
Gene clusters identified by pan-genome analysis pipeline (PGAP) present in all strains from each species.
Significantly differentially expressed genes in plus-Penicillium and plus-Scopulariopsis treatments based on results of RNA-seq experiments.
Metabolites detected in cheese curd agar (CCA) with and without Scopulariopsis by the use of capillary electrophoresis-time of flight mass spectrometry (CE-TOF MS) and liquid chromatography-time of flight mass spectrometry (LT-TOF MS).
ACKNOWLEDGMENTS
Elizabeth Landis, Esther Miller, Robert Schaeffer, Ina Bodinaku, Jason Shaffer, and Eric Scott provided helpful feedback on earlier versions of the manuscript. Sanjukta Ghosh and Michael Doire provided laboratory management support.
Footnotes
Citation Kastman EK, Kamelamela N, Norville JW, Cosetta CM, Dutton RJ, Wolfe BE. 2016. Biotic interactions shape the ecological distributions of Staphylococcus species. mBio 7(5):e01157-16. doi:10.1128/mBio.01157-16.
REFERENCES
- 1.Horner-Devine MC, Bohannan BJ. 2006. Phylogenetic clustering and overdispersion in bacterial communities. Ecology 87:S100–S108. doi: 10.1890/0012-9658(2006)87[100:PCAOIB]2.0.CO;2. [DOI] [PubMed] [Google Scholar]
- 2.Bryant JA, Lamanna C, Morlon H, Kerkhoff AJ, Enquist BJ, Green JL. 2008. Microbes on mountainsides: contrasting elevational patterns of bacterial and plant diversity. Proc Natl Acad Sci 105:11505–11511. doi: 10.1073/pnas.0801920105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Konopka A, Lindemann S, Fredrickson J. 2015. Dynamics in microbial communities: unraveling mechanisms to identify principles. ISME J 9:1488–1495. doi: 10.1038/ismej.2014.251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Acinas SG, Klepac-Ceraj V, Hunt DE, Pharino C, Ceraj I, Distel DL, Polz MF. 2004. Fine-scale phylogenetic architecture of a complex bacterial community. Nature 430:551–554. doi: 10.1038/nature02649. [DOI] [PubMed] [Google Scholar]
- 5.Hunt DE, David LA, Gevers D, Preheim SP, Alm EJ, Polz MF. 2008. Resource partitioning and sympatric differentiation among closely related bacterioplankton. Science 320:1081–1085. doi: 10.1126/science.1157890. [DOI] [PubMed] [Google Scholar]
- 6.Oh J, Byrd AL, Deming C, Conlan S, NISC Comparative Sequencing Program, Kong HH, Segre JA. 2014. Biogeography and individuality shape function in the human skin metagenome. Nature 514:59–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Nishiguchi MK. 2000. Temperature affects species distribution in symbiotic populations of Vibrio spp. Appl Environ Microbiol 66:3550–3555. doi: 10.1128/AEM.66.8.3550-3555.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kreth J, Merritt J, Shi W, Qi F. 2005. Competition and coexistence between Streptococcus mutans and Streptococcus sanguinis in the dental biofilm. J Bacteriol 187:7193–7203. doi: 10.1128/JB.187.21.7193-7203.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Violle C, Nemergut DR, Pu Z, Jiang L. 2011. Phylogenetic limiting similarity and competitive exclusion. Ecol Lett 14:782–787. doi: 10.1111/j.1461-0248.2011.01644.x. [DOI] [PubMed] [Google Scholar]
- 10.Fritschie KJ, Cardinale BJ, Alexandrou MA, Oakley TH. 2014. Evolutionary history and the strength of species interactions: testing the phylogenetic limiting similarity hypothesis. Ecology 95:1407–1417. doi: 10.1890/13-0986.1. [DOI] [PubMed] [Google Scholar]
- 11.Pérez-Gutiérrez R-A, López-Ramírez V, Islas Á, Alcaraz LD, Hernández-González I, Olivera BC, Santillán M, Eguiarte LE, Souza V, Travisano M, Olmedo-Alvarez G. 2013. Antagonism influences assembly of a Bacillus guild in a local community and is depicted as a food-chain network. ISME J 7:487–497. doi: 10.1038/ismej.2012.119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kinkel LL, Schlatter DC, Xiao K, Baines AD. 2014. Sympatric inhibition and niche differentiation suggest alternative coevolutionary trajectories among Streptomycetes. ISME J 8:249–256. doi: 10.1038/ismej.2013.175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Thompson JR, Pacocha S, Pharino C, Klepac-Ceraj V, Hunt DE, Benoit J, Sarma-Rupavtarm R, Distel DL, Polz MF. 2005. Genotypic diversity within a natural coastal bacterioplankton population. Science 307:1311–1313. doi: 10.1126/science.1106028. [DOI] [PubMed] [Google Scholar]
- 14.Thompson JR, Randa MA, Marcelino LA, Tomita-Mitchell A, Lim E, Polz MF. 2004. Diversity and dynamics of a north Atlantic coastal Vibrio community. Appl Environ Microbiol 70:4103–4110. doi: 10.1128/AEM.70.7.4103-4110.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Polz MF, Hunt DE, Preheim SP, Weinreich DM. 2006. Patterns and mechanisms of genetic and phenotypic differentiation in marine microbes. Philos Trans R Soc Lond B Biol Sci 361:2009–2021. doi: 10.1098/rstb.2006.1928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kreth J, Zhang Y, Herzberg MC. 2008. Streptococcal antagonism in oral biofilms: Streptococcus sanguinis and Streptococcus gordonii interference with Streptococcus mutans. J Bacteriol 190:4632–4640. doi: 10.1128/JB.00276-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kuramitsu HK, He X, Lux R, Anderson MH, Shi W. 2007. Interspecies interactions within oral microbial communities. Microbiol Mol Biol Rev 71:653–670. doi: 10.1128/MMBR.00024-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Cordero OX, Wildschutte H, Kirkup B, Proehl S, Ngo L, Hussain F, Le Roux F, Mincer T, Polz MF. 2012. Ecological populations of bacteria act as socially cohesive units of antibiotic production and resistance. Science 337:1228–1231. doi: 10.1126/science.1219385. [DOI] [PubMed] [Google Scholar]
- 19.Frey-Klett P, Burlinson P, Deveau A, Barret M, Tarkka M, Sarniguet A. 2011. Bacterial-fungal interactions: hyphens between agricultural, clinical, environmental, and food microbiologists. Microbiol Mol Biol Rev 75:583–609. doi: 10.1128/MMBR.00020-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Denno RF, McClure MS, Ott JR. 1995. Interspecific interactions in phytophagous insects: competition reexamined and resurrected. Annu Rev Entomol 40:297–331. doi: 10.1146/annurev.en.40.010195.001501. [DOI] [Google Scholar]
- 21.Nemergut DR, Schmidt SK, Fukami T, O’Neill SP, Bilinski TM, Stanish LF, Knelman JE, Darcy JL, Lynch RC, Wickey P, Ferrenberg S. 2013. Patterns and processes of microbial community assembly. Microbiol Mol Biol Rev 77:342–356. doi: 10.1128/MMBR.00051-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hibbing ME, Fuqua C, Parsek MR, Peterson SB. 2010. Bacterial competition: surviving and thriving in the microbial jungle. Nat Rev Microbiol 8:15–25. doi: 10.1038/nrmicro2259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Button JE, Dutton RJ. 2012. Cheese microbes. Curr Biol 22:R587–R589. doi: 10.1016/j.cub.2012.06.014. [DOI] [PubMed] [Google Scholar]
- 24.Wolfe BE, Button JE, Santarelli M, Dutton RJ. 2014. Cheese rind communities provide tractable systems for in situ and in vitro studies of microbial diversity. Cell 158:422–433. doi: 10.1016/j.cell.2014.05.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wolfe BE, Dutton RJ. 2015. Fermented foods as experimentally tractable microbial ecosystems. Cell 161:49–55. doi: 10.1016/j.cell.2015.02.034. [DOI] [PubMed] [Google Scholar]
- 26.Coton E, Desmonts M-H, Leroy S, Coton M, Jamet E, Christieans S, Donnio P-Y, Lebert I, Talon R. 2010. Biodiversity of coagulase-negative staphylococci in French cheeses, dry fermented sausages, processing environments and clinical samples. Int J Food Microbiol 137:221–229. doi: 10.1016/j.ijfoodmicro.2009.11.023. [DOI] [PubMed] [Google Scholar]
- 27.Verdier-Metz I, Gagne G, Bornes S, Monsallier F, Veisseire P, Delbès-Paus C, Montel M-C. 2012. Cow teat skin, a potential source of diverse microbial populations for cheese production. Appl Environ Microbiol 78:326–333. doi: 10.1128/AEM.06229-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Shah MM, Iihara H, Noda M, Song SX, Nhung PH, Ohkusu K, Kawamura Y, Ezaki T. 2007. dnaJ gene sequence-based assay for species identification and phylogenetic grouping in the genus Staphylococcus. Int J Syst Evol Microbiol 57:25–30. doi: 10.1099/ijs.0.64205-0. [DOI] [PubMed] [Google Scholar]
- 29.Ghebremedhin B, Layer F, König W, König B. 2008. Genetic classification and distinguishing of Staphylococcus species based on different partial gap, 16S rRNA, hsp60, rpoB, sodA, and tuf gene sequences. J Clin Microbiol 46:1019–1025. doi: 10.1128/JCM.02058-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lamers RP, Muthukrishnan G, Castoe TA, Tafur S, Cole AM, Parkinson CL. 2012. Phylogenetic relationships among Staphylococcus species and refinement of cluster groups based on multilocus data. BMC Evol Biol 12:171. doi: 10.1186/1471-2148-12-171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Levins R, Culver D. 1971. Regional coexistence of species and competition between rare species. Proc Natl Acad Sci U S A 68:1246–1248. doi: 10.1073/pnas.68.6.1246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Tilman D. 1994. Competition and biodiversity in spatially structured habitats. Ecology 75:2–16. doi: 10.2307/1939377. [DOI] [Google Scholar]
- 33.Chesson P. 2000. General theory of competitive coexistence in spatially varying environments. Theor Popul Biol 58:211–237. doi: 10.1006/tpbi.2000.1486. [DOI] [PubMed] [Google Scholar]
- 34.Slatyer RA, Hirst M, Sexton JP. 2013. Niche breadth predicts geographical range size: a general ecological pattern. Ecol Lett 16:1104–1114. doi: 10.1111/ele.12140. [DOI] [PubMed] [Google Scholar]
- 35.Monnet C, Landaud S, Bonnarme P, Swennen D. 2015. Growth and adaptation of microorganisms on the cheese surface. FEMS Microbiol Lett 362:1–9. doi: 10.1093/femsle/fnu025. [DOI] [PubMed] [Google Scholar]
- 36.Cai H, Rodríguez BT, Zhang W, Broadbent JR, Steele JL. 2007. Genotypic and phenotypic characterization of Lactobacillus casei strains isolated from different ecological niches suggests frequent recombination and niche specificity. Microbiology 153:2655–2665. doi: 10.1099/mic.0.2007/006452-0. [DOI] [PubMed] [Google Scholar]
- 37.Dutilh BE, Thompson CC, Vicente AC, Marin MA, Lee C, Silva GG, Schmieder R, Andrade BG, Chimetto L, Cuevas D, Garza DR, Okeke IN, Aboderin AO, Spangler J, Ross T, Dinsdale EA, Thompson FL, Harkins TT, Edwards RA. 2014. Comparative genomics of 274 Vibrio cholerae genomes reveals mobile functions structuring three niche dimensions. BMC Genomics 15:654. doi: 10.1186/1471-2164-15-654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Rosenthal AZ, Matson EG, Eldar A, Leadbetter JR. 2011. RNA-seq reveals cooperative metabolic interactions between two termite-gut spirochete species in co-culture. ISME J 5:1133–1142. doi: 10.1038/ismej.2011.3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Neupane S, Finlay RD, Alström S, Elfstrand M, Högberg N. 2015. Transcriptional responses of the bacterial antagonist Serratia plymuthica to the fungal phytopathogen Rhizoctonia solani. Environ Microbiol Rep 7:123–127. doi: 10.1111/1758-2229.12203. [DOI] [PubMed] [Google Scholar]
- 40.Monnet C, Back A, Irlinger F. 2012. Growth of aerobic ripening bacteria at the cheese surface is limited by the availability of iron. Appl Environ Microbiol 78:3185–3192. doi: 10.1128/AEM.00085-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Hammer ND, Skaar EP. 2011. Molecular mechanisms of Staphylococcus aureus iron acquisition. Annu Rev Microbiol 65:129–147. doi: 10.1146/annurev-micro-090110-102851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Chen Y, Golding I, Sawai S, Guo L, Cox EC. 2005. Population fitness and the regulation of Escherichia coli genes by bacterial viruses. PLoS Biol 3:e229. doi: 10.1371/journal.pbio.0030229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Quesada JM, Soriano MI, Espinosa-Urgel M. 2012. Stability of a Pseudomonas putida KT2440 bacteriophage-carried genomic island and its impact on rhizosphere fitness. Appl Environ Microbiol 78:6963–6974. doi: 10.1128/AEM.00901-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Martínez-García E, Jatsenko T, Kivisaar M, de Lorenzo V. 2015. Freeing Pseudomonas putida KT2440 of its proviral load strengthens endurance to environmental stresses. Environ Microbiol 17:76–90. doi: 10.1111/1462-2920.12492. [DOI] [PubMed] [Google Scholar]
- 45.Schwyn B, Neilands JB. 1987. Universal chemical assay for the detection and determination of siderophores. Anal Biochem 160:47–56. doi: 10.1016/0003-2697(87)90612-9. [DOI] [PubMed] [Google Scholar]
- 46.Gripon JC. 1999. Mould-ripened cheeses, p 111–136. In Cheese: chemistry, physics and microbiology. Springer, New York, NY. [Google Scholar]
- 47.Deveau A, Brulé C, Palin B, Champmartin D, Rubini P, Garbaye J, Sarniguet A, Frey-Klett P. 2010. Role of fungal trehalose and bacterial thiamine in the improved survival and growth of the ectomycorrhizal fungus Laccaria bicolor S238N and the helper bacterium Pseudomonas fluorescens BBc6R8. Environ Microbiol Rep 2:560–568. doi: 10.1111/j.1758-2229.2010.00145.x. [DOI] [PubMed] [Google Scholar]
- 48.Burmølle M, Ren D, Bjarnsholt T, Sørensen SJ. 2014. Interactions in multispecies biofilms: do they actually matter? Trends Microbiol 22:84–91. doi: 10.1016/j.tim.2013.12.004. [DOI] [PubMed] [Google Scholar]
- 49.Faust K, Raes J. 2012. Microbial interactions: from networks to models. Nat Rev Microbiol 10:538–550. doi: 10.1038/nrmicro2832. [DOI] [PubMed] [Google Scholar]
- 50.Barberán A, Bates ST, Casamayor EO, Fierer N. 2012. Using network analysis to explore co-occurrence patterns in soil microbial communities. ISME J 6:343–351. doi: 10.1038/ismej.2011.119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Faust K, Lima-Mendez G, Lerat J-S, Sathirapongsasuti JF, Knight R, Huttenhower C, Lenaerts T, Raes J. 2015. Cross-biome comparison of microbial association networks. Front Microbiol 6:1200. doi: 10.3389/fmicb.2015.01200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Widder S, Allen RJ, Pfeiffer T, Curtis TP, Wiuf C, Sloan WT, Cordero OX, Brown SP, Momeni B, Shou W, Kettle H, Flint HJ, Haas AF, Laroche B, Kreft J-U, Rainey PB, Freilich S, Schuster S, Milferstedt K, van der Meer JR, Groszkopf T, Huisman J, Free A, Picioreanu C, Quince C, Klapper I, Labarthe S, Smets BF, Wang H, Isaac Newton Institute Fellows, Soyer OS. 29 March 2016. Challenges in microbial ecology: building predictive understanding of community function and dynamics. ISME J doi: 10.1038/ismej.2016.45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Lindsay AK, Hogan DA. 2014. Candida albicans: molecular interactions with Pseudomonas aeruginosa and Staphylococcus aureus. Fungal Biol Rev 28:85–96. doi: 10.1016/j.fbr.2014.10.002. [DOI] [Google Scholar]
- 54.Peleg AY, Hogan DA, Mylonakis E. 2010. Medically important bacterial-fungal interactions. Nat Rev Microbiol 8:340–349. doi: 10.1038/nrmicro2313. [DOI] [PubMed] [Google Scholar]
- 55.Wargo MJ, Hogan DA. 2006. Fungal—bacterial interactions: a mixed bag of mingling microbes. Curr Opin Microbiol 9:359–364. doi: 10.1016/j.mib.2006.06.001. [DOI] [PubMed] [Google Scholar]
- 56.Stopnisek N, Zühlke D, Carlier A, Barberán A, Fierer N, Becher D, Riedel K, Eberl L, Weisskopf L. 2016. Molecular mechanisms underlying the close association between soil Burkholderia and fungi. ISME J 10:253–264. doi: 10.1038/ismej.2015.73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Xiao R, Kisaalita WS. 1997. Iron acquisition from transferrin and lactoferrin by Pseudomonas aeruginosa pyoverdin. Microbiology 143:2509–2515. doi: 10.1099/00221287-143-7-2509. [DOI] [PubMed] [Google Scholar]
- 58.Döring G, Pfestorf M, Botzenhart K, Abdallah MA. 1988. Impact of proteases on iron uptake of Pseudomonas aeruginosa pyverdin from transferrin and lactoferrin. Infect Immun 56:291–293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Britigan BE, Hayek MB, Doebbeling BN, Fick RB. 1993. Transferring and lactoferrin undergo proteolytic cleavage in the Pseudomonas aeruginosa-infected lungs of patients with cystic fibrosis. Infect Immun 61:5049–5055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.De Visscher A, Supré K, Haesebrouck F, Zadoks RN, Piessens V, Van Coillie E, Piepers S, De Vliegher S. 2014. Further evidence for the existence of environmental and host-associated species of coagulase-negative staphylococci in dairy cattle. Vet Microbiol 172:466–474. doi: 10.1016/j.vetmic.2014.06.011. [DOI] [PubMed] [Google Scholar]
- 61.Tremblay YD, Lamarche D, Chever P, Haine D, Messier S, Jacques M. 2013. Characterization of the ability of coagulase-negative staphylococci isolated from the milk of Canadian farms to form biofilms. J Dairy Sci 96:234–246. doi: 10.3168/jds.2012-5795. [DOI] [PubMed] [Google Scholar]
- 62.Braem G, De Vliegher S, Verbist B, Piessens V, Van Coillie E, De Vuyst L, Leroy F. 2013. Unraveling the microbiota of teat apices of clinically healthy lactating dairy cows, with special emphasis on coagulase-negative staphylococci. J Dairy Sci 96:1499–1510. doi: 10.3168/jds.2012-5493. [DOI] [PubMed] [Google Scholar]
- 63.Xu J, Tan X, Zhang X, Xia X, Sun H. 2015. The diversities of staphylococcal species, virulence and antibiotic resistance genes in the subclinical mastitis milk from a single Chinese cow herd. Microb Pathog 88:29–38. doi: 10.1016/j.micpath.2015.08.004. [DOI] [PubMed] [Google Scholar]
- 64.Vanderhaeghen W, Piepers S, Leroy F, Van Coillie E, Haesebrouck F, De Vliegher S. 2015. Identification, typing, ecology and epidemiology of coagulase negative staphylococci associated with ruminants. Vet J 203:44–51. doi: 10.1016/j.tvjl.2014.11.001. [DOI] [PubMed] [Google Scholar]
- 65.Raz R, Colodner R, Kunin CM. 2005. Who are you—Staphylococcus saprophyticus? Clin Infect Dis 40:896–898. doi: 10.1086/428353. [DOI] [PubMed] [Google Scholar]
- 66.Latham RH, Running K, Stamm WE. 1983. Urinary tract infections in young adult women caused by Staphylococcus saprophyticus. JAMA 250:3063–3066. [PubMed] [Google Scholar]
- 67.Rupp ME, Soper DE, Archer GL. 1992. Colonization of the female genital tract with Staphylococcus saprophyticus. J Clin Microbiol 30:2975–2979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Hovelius B, Mårdh PA. 1984. Staphylococcus saprophyticus as a common cause of urinary tract infections. Rev Infect Dis 6:328–337. doi: 10.1093/clinids/6.3.328. [DOI] [PubMed] [Google Scholar]
- 69.Flores-Mireles AL, Walker JN, Caparon M, Hultgren SJ. 2015. Urinary tract infections: epidemiology, mechanisms of infection and treatment options. Nat Rev Microbiol 13:269–284. doi: 10.1038/nrmicro3432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Carnio MC, Höltzel A, Rudolf M, Henle T, Jung G, Scherer S. 2000. The macrocyclic peptide antibiotic micrococcin P1 is secreted by the foodborne bacterium Staphylococcus equorum WS 2733 and inhibits Listeria monocytogenes on soft cheese. Appl Environ Microbiol 66:2378–2384. doi: 10.1128/AEM.66.6.2378-2384.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Place RB, Hiestand D, Gallmann HR, Teuber M. 2003. Staphylococcus equorum subsp. linens, subsp. nov., a starter culture component for surface ripened semi-hard cheeses. Syst Appl Microbiol 26:30–37. doi: 10.1078/072320203322337281. [DOI] [PubMed] [Google Scholar]
- 72.Nyholm SV, McFall-Ngai M. 2004. The winnowing: establishing the squid-vibrio symbiosis. Nat Rev Microbiol 2:632–642. doi: 10.1038/nrmicro957. [DOI] [PubMed] [Google Scholar]
- 73.Bäckhed F, Ley RE, Sonnenburg JL, Peterson DA, Gordon JI. 2005. Host-bacterial mutualism in the human intestine. Science 307:1915–1920. doi: 10.1126/science.1104816. [DOI] [PubMed] [Google Scholar]
- 74.Cullen TW, Schofield WB, Barry NA, Putnam EE, Rundell EA, Trent MS, Degnan PH, Booth CJ, Yu H, Goodman AL. 2015. Gut microbiota. Antimicrobial peptide resistance mediates resilience of prominent gut commensals during inflammation. Science 347:170–175. doi: 10.1126/science.1260580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Dutre T, Al Dousary S, Zhang N, Bachert C. 2013. Allergic fungal rhinosinusitis—more than a fungal disease? J Allergy Clin Immunol 132:487–489. doi: 10.1016/j.jaci.2013.02.040. [DOI] [PubMed] [Google Scholar]
- 76.Nguyen LD, Viscogliosi E, Delhaes L. 2015. The lung mycobiome: an emerging field of the human respiratory microbiome. Front Microbiol 6:89. doi: 10.3389/fmicb.2015.00089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Mahdavinia M, Keshavarzian A, Tobin MC, Landay AL, Schleimer RP. 2016. A comprehensive review of the nasal microbiome in chronic rhinosinusitis (CRS). Clin Exp Allergy 46:21–41. doi: 10.1111/cea.12666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Aziz RK, Bartels D, Best AA, DeJongh M, Disz T, Edwards RA, Formsma K, Gerdes S, Glass EM, Kubal M, Meyer F, Olsen GJ, Olson R, Osterman AL, Overbeek RA, McNeil LK, Paarmann D, Paczian T, Parrello B, Pusch GD, Reich C, Stevens R, Vassieva O, Vonstein V, Wilke A, Zagnitko O. 2008. The RAST server: rapid annotations using subsystems technology. BMC Genomics 9:75. doi: 10.1186/1471-2164-9-75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Zhao Y, Wu J, Yang J, Sun S, Xiao J, Yu J. 2012. PGAP: pan-genomes analysis pipeline. Bioinformatics 28:416–418. doi: 10.1093/bioinformatics/btr655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Östlund G, Schmitt T, Forslund K, Köstler T, Messina DN, Roopra S, Frings O, Sonnhammer EL. 2010. InParanoid 7: new algorithms and tools for eukaryotic orthology analysis. Nucleic Acids Res 38:D196–D203. doi: 10.1093/nar/gkp931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Addis E, Fleet GH, Cox JM, Kolak D, Leung T. 2001. The growth, properties and interactions of yeasts and bacteria associated with the maturation of Camembert and blue-veined cheeses. Int J Food Microbiol 69:25–36. doi: 10.1016/S0168-1605(01)00569-4. [DOI] [PubMed] [Google Scholar]
- 82.Flórez AB, Mayo B. 2006. PCR–DGGE as a tool for characterizing dominant microbial populations in the Spanish blue-veined Cabrales cheese. Int Dairy J 16:1205–1210. doi: 10.1016/j.idairyj.2005.11.008. [DOI] [Google Scholar]
- 83.David LA, Maurice CF, Carmody RN, Gootenberg DB, Button JE, Wolfe BE, Ling AV, Devlin AS, Varma Y, Fischbach MA, Biddinger SB, Dutton RJ, Turnbaugh PJ. 2014. Diet rapidly and reproducibly alters the human gut microbiome. Nature 505:559–563. doi: 10.1038/nature12820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.McClure R, Balasubramanian D, Sun Y, Bobrovskyy M, Sumby P, Genco CA, Vanderpool CK, Tjaden B. 2013. Computational analysis of bacterial RNA-Seq data. Nucleic Acids Res 41:e140. doi: 10.1093/nar/gkt444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Soga T, Igarashi K, Ito C, Mizobuchi K, Zimmermann HP, Tomita M. 2009. Metabolomic profiling of anionic metabolites by capillary electrophoresis mass spectrometry. Anal Chem 81:6165–6174. doi: 10.1021/ac900675k. [DOI] [PubMed] [Google Scholar]
- 86.Sugimoto M, Kaneko M, Onuma H, Sakaguchi Y, Mori M, Abe S, Soga T, Tomita M. 2012. Changes in the charged metabolite and sugar profiles of pasteurized and unpasteurized Japanese sake with storage. J Agric Food Chem 60:2586–2593. doi: 10.1021/jf2048993. [DOI] [PubMed] [Google Scholar]
- 87.Soga T, Ueno Y, Naraoka H, Ohashi Y, Tomita M, Nishioka T. 2002. Simultaneous determination of anionic intermediates for Bacillus subtilis metabolic pathways by capillary electrophoresis electrospray ionization mass spectrometry. Anal Chem 74:2233–2239. doi: 10.1021/ac020064n. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Abiotic niche assays for more strains of each Staphylococcus species. Heat maps represent the growth of each strain across a gradient of salt and pH values as measured by OD600 after 48 h. Data for BC9, BC4, and BC10 are presented in more detail in Fig. 2. Data represent means of results of 3 replicates. Figure S1 relates to Fig. 2. Download
Spearman’s rank correlation coefficients of relative abundances of Staphylococcus species (from shotgun metagenomic data) and relative abundances of other members of the cheese rind community (from amplicon sequencing data). Correlations highlighted in bold are statistically significant (P < 0.01). See Results for discussion of caveats with respect to interpreting these data. Figure S2 relates to Fig. 1 and 2. Download
(A) The thiamine biosynthesis pathway. The part of the pathway highlighted with thick lines was differentially expressed (green = increased expression, red = decreased expression) in S. equorum grown with fungi. Small yellow boxes indicate if differential expression was observed with just Penicillium (P), with just Scopulariopsis (S), or with both Penicillium and Scopulariopsis (PS). (B) The methionine biosynthesis pathway. Notations are the same as those described for panel A. Figure S3 relates to Fig. 3. Download
Shotgun metagenome metadata and features.
Genome metadata and statistics.
Gene clusters identified by pan-genome analysis pipeline (PGAP) present in all strains from each species.
Significantly differentially expressed genes in plus-Penicillium and plus-Scopulariopsis treatments based on results of RNA-seq experiments.
Metabolites detected in cheese curd agar (CCA) with and without Scopulariopsis by the use of capillary electrophoresis-time of flight mass spectrometry (CE-TOF MS) and liquid chromatography-time of flight mass spectrometry (LT-TOF MS).