

Classical Philadelphia chromosome-negative myeloproliferative neoplasms (Ph-neg MPN) are considered to originate from somatic mutation(s) of a hematopoietic stem cell leading to clonal hematopoiesis, as shown by X-chromosome allelic usage in females. Somatic mutations in JAK2, cMPL and calreticulin (CALR) genes are the three main disease-defining mutations. Mutations in the JAK2 gene are found in almost all polycythemia vera (PV) patients and in 45–50% of essential thrombocythemia (ET) and primary myelofibrosis (PMF) patients, while mutations in cMPL are found in about 2–5% of ET and PMF. CALR mutations (CALRMUT) have been identified in the majority of ET and PMF patients who lack JAK2 and cMPL mutations, but are generally not found in PV patients. Compared with JAK2- and cMPL-mutated patients, CALRMUT patients have higher platelet counts, lower hemoglobin and leukocyte counts, a lower risk of thrombosis(1) and a more favorable prognosis, but no difference in the rate of transformation to post-ET PMF.(2)
Determination of clonality based on X-chromosome inactivation has helped to differentiate reactive processes from MPNs. However, prior to the discovery of MPN disease-defining mutations, it was reported that some patients with a clinical phenotype of ET had polyclonal hematopoiesis.(3)
There are two widely used clonality assays based on X-chromosome inactivation (XCI), the HUMARA assay and the transcriptional-based clonality assay (TCA). The HUMARA clonality assay is based on assessing the DNA methylation status of adjacent CpG sites close to trinucleotide repeats in exon 1 of the human androgen receptor gene (AR).(4) Because some investigators observed unexpected results using the HUMARA assay, the second method was developed by our group based on analysis of transcripts of X-chromosome polymorphic genes subjected to X-chromosome inactivation, i.e. transcriptional-based clonality assays (TCA).(5, 6) Using both assays, discrepant data have been obtained in some females; however TCA revealed only a single transcript in individual burst forming units-erythroid (BFU-E), while heterogeneous methylation at the AR locus was obtained by the HUMARA assay.(6)
It has been reported recently that JAK2V617F ET is more likely to have polyclonal hematopoiesis than CALRMUT ET; this study was conducted using the HUMARA assay.(7) We evaluated 96 ET, 33 PMF and 65 PV females for JAK2, cMPL, and CALR mutation status, and using the TCA clonality assay, we examined the existence of polyclonal hematopoiesis in our cohort of MPN females seen over the last three decades. We also report two previously undescribed exon 9 CALR mutations and our analyses of CALRMUT burden in granulocytes (GNC), CD3+ T-cells, and some CD34+ progenitors.
The 194 female patients with ET, PMF or PV were diagnosed according to the revised WHO criteria and had been treated with hydroxyurea or JAK2 inhibitors, but not pegylated interferon-α. Informed consent was obtained from all patients and 10mL of peripheral blood was used for the study. Platelets and granulocytes were isolated according to previously published protocols.(5)
Genomic DNA was isolated from GNC using the Puregene DNA purification kit (Gentra, MN, USA). JAK2V617F and MPLW515L/K were detected by quantitative allele-specific PCR (qRT-PCR)(8) and CALRMUT by semi-quantitative fragment analysis.(9) CALRMUT was categorized as CALRMUT type 1 (52-bp deletion; c.1092_1143del), CALRMUT type 2 (5-bp insertion; c.1154_1155insTTGTC) and other previously described variants. Samples with an unreported CALR profile were sequenced.
The frequency of JAK2V617F was 57% (N=55) in ET and 42% (N=14) in PMF; all PV females were JAK2V617F mutated. CALRMUT was present in 18% (N=17) of ET and 39% (N=13) of PMF. MPLW515L was found in only one PMF patient. Triple-negatives accounted for 25% (N=24) of ET and 15% (N=5) of PMF (Figure 1a). This distribution of mutations is consistent with previous studies.(9–11)
Figure 1.

Distribution of mutations in all females according the diagnosis (a); Clonality status in JAK2V617F, CALRMUT and triple negative MPN female patients: ET (b), PMF (c) and PV (d). Each bar represents the relative number of individuals. Frequencies were compared by Fisher’s exact test.
To assess clonality, we first genotyped single nucleotide exonic polymorphisms from five X-chromosome genes subjected to X-chromosome inactivation [BTK; FHL1; IDS; G6PD; MPP1] using TaqMan allele-discrimination assays. Females heterozygous for any X-chromosome loci were considered informative for quantitative transcriptional clonality assay (qTCA). Total RNA was isolated from GNC using Tri-Reagent (Molecular Research Center, OH) and allelic usage ratio of informative X-chromosome polymorphic genes determined. Aliquots of 200 ng of DNA-free RNA were reverse-transcribed using SuperScript VILO for qRT-PCR (Invitrogen, CA, USA). Quantitative allele-specific PCR (qRT-PCR) was performed as described previously(12). A ratio of ≥80:20 was used to define clonality, accepting that ~5% of normal females will have skewing of X-inactivation sufficient to be classified as clonal hematopoiesis.(6)
qTCA evaluations of ET. Of 96 ET females, 80 were informative for TCA. Forty-seven had JAK2V617F (30% mean allele burden) and 45 (96%) of these were clonal, whereas 12 (75%) of 16 CALRMUT ET females had clonal hematopoiesis (p=0.032) (Figure 1b). Of the 17 triple-negatives, 14 (82%) were clonal. In contrast to the study using the HUMARA assay, we found a higher frequency of clonal hematopoiesis in JAK2V617F (96% versus 26%, p<0.001) and triple-negative ET (82% versus 9%, p<0.001), but a similar frequency of clonal CALRMUT ET.(7)
qTCA evaluations of PMF. Thirty of 33 PMF females were informative for TCA; 12 of 13 (94%) with JAK2V617F (39% mean allele burden) and 11 of 12 (92%) with CALRMUT were clonal (Figure 1c). In four triple negative PMF, three were clonal and one polyclonal. The single MPLW515L PMF female was clonal.
qTCA evaluations of PV. All 57 informative JAK2V617F (44% mean allele burden) PV females were clonal by qTCA (Figure 1d).
Distribution of CALRMUT variants. In the 16 CALRMUT ET females, seven (44%) had type 1 CALRMUT, eight (50%) had type 2 CALRMUT and one had another CALRMUT mutation. Among 13 PMF females, six (46%) had type 1 CALRMUT, five had type 2 CALRMUT (50%) and two had other CALRMUT mutations. Both of these “other” CALRMUT mutations are previously undescribed 46bp and 37bp deletions [chr19:13,054,570-13,054,615 and chr19:13,054,570-13,054,606, respectively]. These sequences were deposited using the human reference genome (http://genome.ucsc.edu/cgi-bin/hgGateway) (hg19). Clonality status was not associated with the type of CALRMUT.
Relative allelic burden of CALRMUT. We also examined the relative allelic burden of CALRMUT in GNC, T-cells and CD34+ progenitors using fragment analysis, a method which has a 3–5% allelic burden detection limit.(13) CD34+ cells and T-lymphocytes were sorted by FACS using antibodies against CD34 or CD3 (FACS Vantage SE, BD Bioscience, CA). Among 29 CALRMUT ET and PMF female patients, the mean CALRMUT allelic burden was ~50% in GNC and was undetectable in CD3+ T-cells. This is consistent with previously published data, which suggested that CALRMUT is a somatic mutation favoring differentiation to myeloid progenitors.(14) Among six ET females patients from whom CD34+ cells were available, the CALRMUT allelic burden was lower in CD34+ cells than in GNC (p=0.005, Figure 2), similar to previously reported in JAK2V617F PV patients.(15)
Figure 2.
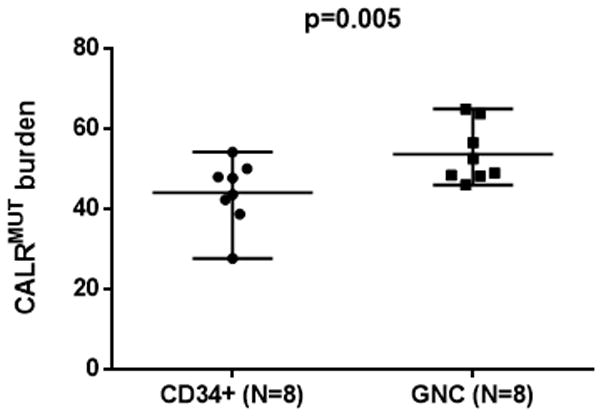
CALRMUT burden in CD34+ cells and granulocytes (GNC). The lines represent mean and range. The paired t-test applied to compare the means of CALRMUT burden in the two matched groups.
In conclusion, we report analyses of 194 Ph- MPN females and confirm that polyclonal hematopoiesis exists in a minority of ET females. However, we found a marked difference between a >95% clonal prevalence in our JAK2V617F ET cohort versus 26% reported by another group, and a similar discrepancy in triple negative ET.(7) We submit this difference is very likely due to different XCI clonality assays, as methylation of the AR locus in the HUMARA assay does not always reflect the X-chromosome inactivation state.(6) The prevalence of clonal hematopoiesis in CALRMUT was lower than JAK2V617F ET in our cohort (75% versus 96%,) suggesting that CALRMUT clones have a weaker suppressive effect on residual normal hematopoietic stem cells than JAK2 mutated clones, which may contribute to the more benign course of CALRMUT ET.
Acknowledgments
This study was supported by the Myeloproliferative Disorders (MPD) Consortium (Project #1; J.T.P. Principal Investigator) research funding from the National Institutes of Health (Grant number: NIH-P01CA108671);
LTL is supported by a doctoral fellowship from the CAPES Foundation, Ministry of Education of Brazil, Brasília – DF 70040-020, Brazil.
Footnotes
Conflict of Interest: The authors declare no conflict of interest.
References
- 1.Tefferi A, Wassie EA, Lasho TL, Finke C, Belachew AA, Ketterling RP, et al. Calreticulin mutations and long-term survival in essential thrombocythemia. Leukemia. 2014;28(12):2300–3. doi: 10.1038/leu.2014.148. [DOI] [PubMed] [Google Scholar]
- 2.Tefferi A, Barbui T. Polycythemia vera and essential thrombocythemia: 2015 update on diagnosis, risk-stratification and management. Am J Hematol. 2015;90(2):162–73. doi: 10.1002/ajh.23895. [DOI] [PubMed] [Google Scholar]
- 3.Harrison CN, Gale RE, Machin SJ, Linch DC. A Large Proportion of Patients With a Diagnosis of Essential Thrombocythemia Do Not Have a Clonal Disorder and May Be at Lower Risk of Thrombotic Complications. Blood [Internet] 1999;93(2):417–24. [PubMed] [Google Scholar]
- 4.Busque L, Zhu J, DeHart D, Griffith B, Willman C, Carroll R, et al. An expression based clonality assay at the human androgen receptor locus (HUMARA) on chromosome X. Nucleic Acids Res. 1994;22(4):697–8. doi: 10.1093/nar/22.4.697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Prchal JT, Guan YL. A novel clonality assay based on transcriptional analysis of the active X chromosome. Stem Cells. 1993;11(Suppl 1):62–5. doi: 10.1002/stem.5530110613. [DOI] [PubMed] [Google Scholar]
- 6.Swierczek SI, Piterkova L, Jelinek J, Agarwal N, Hammoud S, Wilson A, et al. Methylation of AR locus does not always reflect X chromosome inactivation state. Blood. 2012;119(13):e100–9. doi: 10.1182/blood-2011-11-390351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Allen C, Lambert JR, Linch DC, Gale RE. X chromosome inactivation analysis reveals a difference in the biology of ET patients with JAK2 and CALR mutations. Blood. 2014;124(13):2091–3. doi: 10.1182/blood-2014-06-580183. [DOI] [PubMed] [Google Scholar]
- 8.Nussenzveig RH, Swierczek SI, Jelinek J, Gaikwad A, Liu E, Verstovsek S, et al. Polycythemia vera is not initiated by JAK2V617F mutation. Exp Hematol. 2007;35(1):32–8. doi: 10.1016/j.exphem.2006.11.012. [DOI] [PubMed] [Google Scholar]
- 9.Klampfl T, Gisslinger H, Harutyunyan AS, Nivarthi H, Rumi E, Milosevic JD, et al. Somatic mutations of calreticulin in myeloproliferative neoplasms. N Engl J Med. 2013;369(25):2379–90. doi: 10.1056/NEJMoa1311347. [DOI] [PubMed] [Google Scholar]
- 10.Nangalia J, Massie CE, Baxter EJ, Nice FL, Gundem G, Wedge DC, et al. Somatic CALR mutations in myeloproliferative neoplasms with nonmutated JAK2. N Engl J Med. 2013;369(25):2391–405. doi: 10.1056/NEJMoa1312542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Rumi E, Pietra D, Ferretti V, Klampfl T, Harutyunyan AS, Milosevic JD, et al. JAK2 or CALR mutation status defines subtypes of essential thrombocythemia with substantially different clinical course and outcomes. Blood. 2014;123(10):1544–51. doi: 10.1182/blood-2013-11-539098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Swierczek SI, Agarwal N, Nussenzveig RH, Rothstein G, Wilson A, Artz A, et al. Hematopoiesis is not clonal in healthy elderly women. Blood. 2008;112(8):3186–93. doi: 10.1182/blood-2008-03-143925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Jones AV, Ward D, Lyon M, Leung W, Callaway A, Chase A, et al. Evaluation of methods to detect CALR mutations in myeloproliferative neoplasms. Leuk Res. 2015;39(1):82–7. doi: 10.1016/j.leukres.2014.11.019. [DOI] [PubMed] [Google Scholar]
- 14.Rumi E, Harutyunyan AS, Pietra D, Milosevic JD, Casetti IC, Bellini M, et al. CALR exon 9 mutations are somatically acquired events in familial cases of essential thrombocythemia or primary myelofibrosis. Blood. 2014;123(15):2416–9. doi: 10.1182/blood-2014-01-550434. [DOI] [PubMed] [Google Scholar]
- 15.Stein BL, Williams DM, Rogers O, Isaacs MA, Spivak JL, Moliterno AR. Disease burden at the progenitor level is a feature of primary myelofibrosis: a multivariable analysis of 164 JAK2 V617F-positive myeloproliferative neoplasm patients. Exp Hematol. 2011;39(1):95–101. doi: 10.1016/j.exphem.2010.09.008. [DOI] [PMC free article] [PubMed] [Google Scholar]