

Abstract
Epidemiological and clinical studies have suggested that the pathogenesis of inflammatory bowel disease (IBD) is strongly influenced by genetic predisposition. Beyond the limitations of linkage analysis, multiple genome-wide association studies, their meta-analyses, and targeted genotyping array techniques have broadened our understanding of the genetic architecture of IBD. Currently, over 200 single nucleotide polymorphisms are known to be associated with susceptibility to IBD and through functional analysis of genes and loci, a substantial proportion of pathophysiologic mechanisms have been revealed. However, because only a modest fraction of predicted heritability can be explained by known genes/loci, additional strategies are needed including the identification of rare variants with large effect sizes to help explain the missing heritability. Considerable progress is also being made on applying outcomes of genetic research in diagnostics, classification, prognostics, and the development of new therapeutics of IBD.
Keywords: Inflammatory bowel disease, Crohn’s disease, Ulcerative colitis, Disease susceptibility, Heritability, Genetics, Sequencing, Genome-wide association study, Pharmacogenetics
1. Introduction
The inflammatory bowel diseases (IBD), including Crohn’s disease (CD) and ulcerative colitis (UC), are chronic inflammatory diseases of the gastrointestinal tract of unknown pathogenesis. As described in the landmark article by Crohn et al., familial aggregation of IBD implicates genetic background in the development of IBD.1 Although not yet fully understood, dysregulated mucosal immune response to microbes in a genetically susceptible individual is thought to be pathogenic mechanism of IBD.2 It is hoped that genetic studies will possibly answer questions including which individuals are destined to suffer from IBD and which IBD patients will more likely suffer a disabling course of disease. Unlike classical Mendelian disorders, IBD is genetically complex disorder, where traditional genetic analytics are not able to shape the real features of disease. However, with rapid technologic developments, such as gene chip and computational/statistical techniques over the past a few decades, very significant progress has been made in our understanding of the genetic architecture of IBD. This review will cover both historical and the current status of IBD genetic research, as well as identified genes/loci associated with IBD, and potential clinical application of our knowledge on IBD genetics.
2. History of genetic research for IBD
Epidemiologic observations showing clear familial clustering of IBD and higher risk of CD in Jews, especially in Ashkenazi Jews prompted researchers to be interested in heritability and genetic risk of IBD.3,4 In addition, when combing 6 twin studies from Europe, the concordance rates of 30.3% in 112 monozygotic twins vs. 3.6% in 196 dizygotic twins for CD and 15.4% in 143 monozygotic twins vs. 3.9% in 206 dizygotic twins for UC supported further the impact of genetics in IBD risk.5 According to previous genetic epidemiologic studies, a lifetime risk of developing IBD for first-degree relatives of a CD patient was estimated to be 4.8%–5.2% for non-Jews and 7.8% for Jews.3,6,7 The corresponding figures for first-degree relatives of a patient with UC are 1.6% for non-Jews and 5.2% for Jews.3 Accordingly, the familial aggregation of IBD, the observed concordance in twin studies, and increased risk of developing IBD in relatives of proband triggered molecular genetic studies on IBD.
3. Linkage studies
Due to the technical limitation and high cost of sequencing, early IBD genetic studies were inevitably restricted to studying a number of genetic variants in a small number of individuals.8 Therefore, the earliest studies were designed around understanding the patterns of disease inheritance within a specific family having diseases of interest. By tracing the DNA segments that segregated depending on disease status within families, sections of the genome that were likely to confer risk to the specific disease could be identified.8 This approach called linkage analysis was useful for detecting variants with high penetrance that segregate well with disease status.8 In 1996, the first linkage study in IBD identified a portion on chromosome 16 (IBD1 locus) with CD,9 a finding supported in subsequent studies.10–15 Further studies also identified and replicated areas of significant linkage on additional chromosomes which were designated as IBD2-IBD9.16 A natural extension of identifying these regions was to perform fine-mapping and through this approach CARD15/NOD2 was identified as the underlying gene at IBD1 in 2001.17 At the same time, two independent studies, using a candidate gene approach, also identified the role of CARD15/NOD2 in CD susceptibility.18 Three single nucleotide polymorphisms (SNPs), R702W, G908R, and L1007fsinsC, were revealed to represent 81% of disease-causing mutations within CARD15/NOD2.19 Further replication studies confirmed that these three SNPS are independently associated with CD.20–22 Identification of CARD15/NOD2 remained one of the few robustly replicated genetic risk loci discovered through following up linkage analysis signals, not just in IBD, but across common diseases.8 The widespread failure of linkage analyses, in general, strongly suggested that common diseases do not have a single, highly penetrant genetic causes, but that they are likely driven by the accumulation of multiple risk factors of only modest effect (the ‘common disease, common variant’ hypothesis).23 Discovering genetic associations via linkage analysis under this scenario is very difficult, as the genetic risk may be spread throughout the genome rather than concentrated in a single locus.23 Therefore, alternative association analyses approaches which test if the population-level allele frequencies of cases and controls are statistically different, were thought to be much more powerful because it would be possible to select the right variant to test among millions of variants existing in human population.8,23
4. Genome-wide association studies
The development of publicly available databases such as the SNP Consortium and the International HapMap Consortium, that contain increasingly comprehensive information about SNPs across the genome, together with an increased understanding of linkage disequilibrium structures in humans as well as the development of new microarray technologies leading to genome-wide SNP chip, opened the way to genome-wide association studies (GWAS).8,24–27 GWAS compare the allele frequency of a particular variant between unrelated cases and controls. These new approaches were correctly hailed as the next step to unravel the genetic architecture of complex diseases like IBD. GWAS-based approaches avoid underlying assumptions for biological or positional candidate loci, genes, and variants.28 Therefore, GWAS have been labeled as a “hypothesis-free or unbiased” approach, overcoming the limitations imposed by our incomplete understanding of the pathophysiology of complex diseases.28 In the first GWAS for IBD, Yamazaki and colleagues explored 72,738 SNPs and identified several associated SNPs with CD in Japanese populations at the TNFSF15 gene, a finding replicated in European cohorts.29 In the first European ancestry GWAS study, CD-risk variants were identified in the interleukin 23 receptor (IL23R) gene.30 Further independent replication studies confirmed IL23R CD associations and also extended the association to UC.30 A study adopting a slightly different approach, through studying non-synonymous variants across the genome, identified an association to a protein-coding variant in ATG16L1 with CD, providing the first evidence for the importance of autophagy in CD.31 GWAS approaches also revealed a pair of associations on chromosomes 5p13 and 10q21 located in gene deserts, thereby suggesting the important role of regulatory and non-coding elements in CD.32,33 Further GWAS have shown association of other genes related with innate (TLR4, STAT3, NKX2-3, CARD9) and adaptive (TNFSF15, PTPN2, IL-12B, IRF5) immune response pathways and highlight the role of autophagy and intracellular bacterial handing (CARD15/NOD2, ATG16L1, IRGM) in CD.8,34 These initial CD studies also suggested a partial overlap between CD and other immune-related disorders. Around 30% of associated variants in these initial studies on CD were shared with UC, while close to 50% of loci were revealed to be shared with at least one other immune-mediated disease such as type 1 diabetes, celiac disease or rheumatoid arthritis.35,36 Additional GWAS in UC cohorts lead to the discovery of multiple novel UC-specific loci.37–40 Three loci associated with biologically relevant candidate genes, HNF4A, CDH1, and LAMB1, suggested a possible role of epithelial barrier defects in UC pathogenesis.8,39 GWAS on UC also confirmed the association between UC and human leukocyte antigen (HLA) locus.41 In contrast to UC, where several variants in HLA-B make the largest contribution to genetic risk (ORs 1.4–1.5), genes in the HLA region confer only a modest effect on CD risk (ORs 1.1–1.2).36,42
5. Current status of genetic research for IBD
5.1 GWAS meta-analysis
Although early GWAS identified multiple loci associated with CD and UC, thereby generating new biological hypothesis for IBD, the relatively weak associations only explained a fraction of the heritability expected from twin studies.8 This ‘missing heritability’ was partially attributed to types of variation not adequately captured by GWAS (e.g. non-European ethnicity, as well as rare and structural variations).8,43–45 In addition, it was recognized that a substantial number of additional and yet-unidentified common variants with even smaller effect size (e.g. ORs < 1.2 or even 1.1) than those identified by early GWAS, requiring much larger sample sizes, existed.8,36,44 The need for large sample sizes re-energized The International IBD Genetics Consortium (IIBDGC) (http://www.ibdgenetics.org/) with the aim of bringing together investigators and GWAS datasets from IBD genetics groups around the world in order to search for variants with small effect size not detected by underpowered GWAS.8,36 The first meta-analysis combined data from over 13,000 individuals from three previously published GWAS and identified 21 new CD loci including another autophagy gene, LRRK2.46 Two years later, a second CD meta-analysis of six GWAS with a total sample size of over 50,000 individuals identified 30 new loci, bringing the total number of CD susceptibility loci to 71, explaining 23.3% of the estimated heritability for CD in European ancestry populations.47 The first GWAS meta-analysis of UC patients combined 3 studies in a discovery set and performed replication in the independent population.48 As a result, thirteen novel loci were identified and multiple previously reported UC- and CD-associated loci were confirmed, increasing the number of UC loci to approximately 30.48 Additional meta-analysis of six UC GWAS datasets, comprising 48,950 individuals, identified 29 additional risk loci with genome-wide significance, increasing the number of UC-associated loci to 47, with an estimated 16% of heritability explained.49 The recent meta-analysis of 15 GWAS for CD and/or UC as well as additional typing on the Immunochip (totalling over 75,000 subjects) identified 71 novel IBD loci, increasing the number of IBD-associated loci to 163 (110 associated with both CD and UC, 30 CD-specific, and 23 UC-specific).42 Out of 53 disease-specific loci, 43 showed the same direction of effect in both CD and UC, suggesting that nearly all of the biological mechanisms involved in one disease play some role in the other.4,42 Collectively, multiple pathways were implicated as IBD-related mechanisms, including those involved in innate immunity, JAK/STAT signaling pathway, cytokine production (interferon-gamma, IL-12, tumor-necrosis-factor-alpha, and IL-10 signaling) and lymphocyte activation.8,42 Approximately 70% of IBD risk loci (113 out of 163) were revealed to be shared with other complex diseases or traits, including 66 loci shared with other immune-mediated disorders (especially ankylosing spondylitis and psoriasis).42 Moreover, six out of 8 genes linked to Mendelian susceptibility to mycobacterial disease overlapped with IBD and seven out of 8 loci known to be associated with leprosy by GWAS were also shared with IBD.42 These overlaps suggest that selection pressures arising from mycobacterial infection may have shaped the genetic architecture of IBD.36,42
5.2 IBD genetic studies in non-European populations
Historically, GWAS in IBD had centered around subjects of European ancestry with the exception of the first ever GWAS which was a Japanese CD study.29 Although TNFSF15 was identified as a CD gene in Japanese, previous replication studies have shown lack of common CARD15/NOD2 variants in Japanese CD patients.29,50,51 This lack of association between CARD15/NOD2 variants and CD in Japanese was replicated in Koreans.52,53 Replication studies on the association between IL23R and IBD have shown conflicting results. A Japanese study showed that none of the 10 IL23R SNPs from the original study by Duerr et al. was associated with CD.54 However, in a Korean study, two variants (rs1004819 and rs1495465) were associated with CD.55 Of the 35 known IL23R SNPs studied in Han Chinese cohort, only one non-synonymous SNP, rs11465788 (C>T), was associated with CD.56 This association had not been identified in previous studies but was replicated in a subsequent Korean study.57 Following the first GWAS in Asians,29 subsequent GWAS on Japanese and Korean CD patients demonstrated that some risk loci for CD are shared between East Asians and European ancestry populations, while some appear to be peculiar to the East Asian populations including a loci on chromosome 4p14, 10q25, in the ATG16L2-FCHSD2 region on11q13, and in the SLC25A15-ELF1-WBP4 region on 13q14.58,59 GWAS on Asian UC patients have shown a tendency to more extensive genetic overlap with European ancestry populations than that seen in CD.60,61 Recent GWAS from northern India also identified shared contribution of a proportion of UC-susceptibility genes between northern Indians and Europeans as well as identifying 3 HLA-independent loci.62 In a similar fashion to studies performed on European populations, researchers have utilized other platforms in East Asians including a study utilizing the Immunochip in Koreans, which identified six additional CD loci thereby increasing the total explained genetic variance for CD from 5.31% to 7.27%.63 A similar approach in UC has increased the risk loci to 13 including 3 previously reported by GWAS in Koreans.64 Immunochip analysis of African Americans showed overlaps with Caucasian and Asian studies, through replication of 5 (IL23R, FCGR2A, PTGER4, CARD15/NOD2, and IKZF3) out of 163 SNPs from the Caucasian study and showing the strongest associations between UC and HLA rs9271366, replicating an association previously observed in both Japanese and Korean UC GWAS.42,61,65,66 Most recently, the first trans-ethnic association study of both CD and UC by the IIBDGC, with GWAS or Immunochip data on 86,640 Europeans and Immunochip data from 9,846 individuals of East Asian, Indian or Iranian descent was published.67 As a result, 38 newly associated loci (27 with both CD and UC, 7 specific to CD, and 4 specific to UC) were identified, increasing the number of known IBD risk loci to 231 independent SNPs within 200 loci.67 Among those, the majority were shared across diverse ancestry groups, with only a handful demonstrating population-specific effects driven by heterogeneity in risk allele frequency (for example, CARD15/NOD2) or effect size (for example, TNFSF15-TNFSF8).67 Still, the number of genes/loci associated with IBD in Asian population is currently fewer than that of Western patients due, at least in part, to the smaller studies performed to date in Asians and other populations. It was suggested that genetic factors could play a critical role in shaping the microbiota in IBD patients as well as healthy subjects.68,69 Our speculation for a lower incidence of IBD in Asians is that differences in genetic architecture between Western and Eastern IBD populations could result in differences in microbial milieu, thereby causing different incidences of IBD depending on ethnicity. However, regarding host genetics, microbiome, their crosstalk, and the contribution of other environmental factors, more extensive research is needed, especially in Asians. The CD, UC, or IBD-associated loci revealed by GWAS, GWAS meta-analysis, and Immunochip analysis are summarized in Table 1. The number of IBD-associated loci identified among various ethnic groups over the past 15 years is presented in Figure 1.
Table 1.
Summary of significant CD, UC, and IBD-associated loci by GWAS, GWAS meta-analysis, and Immunochip analysis
| SNP | Key genes (+ no. additional genes in locus)* | Associated disease | European P-value | European OR | 95% CI | Source(s) |
|---|---|---|---|---|---|---|
| rs2066847 | NOD2, ADCY7, (5) | CD | 5.86E-209 | 3.103 | 1.497–1.618 | Jostins et al.† |
| rs12994997 | ATG16L1, INPP5D, (7) | CD | 4.14E-70 | 1.233 | 1.193–1.274 | Jostins et al. |
| rs9264942 | HLA-C, PSORS1C1, NFKBIL1, MICB, (18) | CD | 4.96E-28 | 1.145 | 1.107–1.184 | Jostins et al. |
| rs9286879 | FASLG, TNFSF18, (0) | CD | 5.53E-22 | 1.125 | 1.083–1.167 | Jostins et al. |
| rs2024092 | GPX4, HMHA1, (20) | CD | 8.26E-22 | 1.156 | 1.112–1.201 | Jostins et al. |
| rs3764147 | LACC1, (3) | CD | 2.19E-21 | 1.155 | 1.112–1.199 | Jostins et al. |
| rs2945412 | LGALS9, NOS2, (3) | CD | 8.68E-17 | 1.137 | 1.1–1.175 | Jostins et al. |
| rs6716753 | SP140, (5) | CD | 1.17E-16 | 1.134 | 1.089–1.18 | Jostins et al. |
| rs6651252 | 0 | CD | 1.45E-16 | 1.185 | 1.128–1.246 | Jostins et al. |
| rs2284553 | IFNGR2, IFNAR1, IFNAR2, IL10RB, GART, TMEM50B, (6) | CD | 2.14E-16 | 1.123 | 1.086–1.162 | Jostins et al. |
| rs1728918 | UCN, (23) | CD | 4.86E-16 | 1.123 | 1.086–1.16 | Jostins et al. |
| rs516246 | DBP, SPHK2, IZUMO1, FUT2, (22) | CD | 1.00E-15 | 1.107 | 1.071–1.143 | Jostins et al. |
| rs6679677 | PTPN22, DCLRE1B, (7) | CD | 2.03E-15 | 1.196 | 1.129–1.268 | Jostins et al. |
| rs13204742 | (2) | CD | 8.38E-15 | 1.173 | 1.118–1.23 | Jostins et al. |
| rs212388 | TAGAP, (5) | CD | 3.04E-14 | 1.105 | 1.069–1.141 | Jostins et al. |
| rs13126505 | (1) | CD | 1.84E-12 | 1.172 | 1.1–1.248 | Jostins et al. |
| rs10065637 | IL6ST, IL31RA, (1) | CD | 3.68E-12 | 1.123 | 1.079–1.17 | Jostins et al. |
| rs12663356 | (3) | CD | 4.01E-12 | 1.095 | 1.06–1.131 | Jostins et al. |
| rs3897478 | ADAM30, (5) | CD | 1.97E-11 | 1.161 | 1.101–1.224 | Jostins et al. |
| rs11681525 | - | CD | 4.08E-11 | 0.86 | 0.83–0.90 | Liu et al.‡ |
| rs3853824 | - | CD | 1.17E-10 | 0.92 | 0.9–0.94 | Liu et al. |
| rs7555082 | PTPRC | CD | 1.47E-10 | 1.13 | 1.09–1.18 | Liu et al. |
| rs727563 | TEF, NHP2L1, PMM1, L3MBTL2, CHADL | CD | 1.88E-10 | 1.10 | 1.07–1.13 | Liu et al. |
| rs4802307 | (9) | CD | 2.00E-10 | 1.099 | 1.06–1.139 | Jostins et al. |
| rs9491697 | (3) | CD | 3.79E-10 | 1.077 | 1.042–1.112 | Jostins et al. |
| rs7702331 | (4) | CD | 5.63E-10 | 1.088 | 1.05–1.126 | Jostins et al. |
| rs10865331 | (3) | CD | 9.77E-10 | 1.098 | 1.062–1.134 | Jostins et al. |
| rs35320439 | PDCD1, ATG4B | CD | 9.89E-10 | 1.09 | 1.06–1.12 | Liu et al. |
| rs7773324 | IRF4, DUSP22 | CD | 1.06E-09 | 0.92 | 0.90–0.95 | Liu et al. |
| rs7954567 | CD27, TNFRSF1A, LTBR | CD | 1.30E-09 | 1.09 | 1.06–1.11 | Liu et al. |
| rs9525625 | AKAP1, TFSF11 | CD | 1.41E-09 | 1.08 | 1.05–1.10 | Liu et al. |
| rs17391694 | (5) | CD | 2.96E-09 | 1.134 | 1.077–1.194 | Jostins et al. |
| rs864745 | CREB5, JAZF1,(1) | CD | 3.65E-09 | 1.087 | 1.052–1.123 | Jostins et al. |
| rs16967103 | RASGRP1, SPRED1, (2) | CD | 3.88E-09 | 1.088 | 1.045–1.132 | Jostins et al. |
| rs10798069 | PTGS2, PLA2G4A | CD | 4.25E-09 | 0.93 | 0.91–0.95 | Liu et al. |
| rs17695092 | CPEB4, (2) | CD | 4.68E-09 | 1.095 | 1.055–1.136 | Jostins et al. |
| rs7758080 | MAP3K7IP2 | CD | 7.27E-09 | 1.08 | 1.05–1.11 | Liu et al. |
| rs7236492 | NFATC1, TST | CD | 9.09E-09 | 0.91 | 0.87–0.94 | Liu et al. |
| rs7015630 | RIPK2, (4) | CD | 1.42E-08 | 1.075 | 1.035–1.116 | Jostins et al. |
| rs6837335 | TXK, TEC, SLC10A4, (3) | CD | 1.75E-08 | 1.086 | 1.049–1.123 | Jostins et al. |
| rs13204048 | - | CD | 2.89E-08 | 0.93 | 0.91–0.96 | Liu et al. |
| rs10486483 | (2) | CD | 3.48E-08 | 1.089 | 1.048–1.13 | Jostins et al. |
| rs1748195 | USP1 | CD | 7.13E-08 | 1.07 | 1.04–1.10 | Liu et al. |
| rs9319943 | - | CD | 9.05E-07 | 1.08 | 1.05–1.11 | Liu et al. |
| rs724016 | - | CD | 3.36E-06 | 1.06 | 1.03–1.08 | Liu et al. |
| rs6927022 | HLA-DQB1, HLA-DRB1, HLA-DQA1, HLA-DRA, (12) | UC | 4.71E-133 | 1.444 | 1.387–1.503 | Jostins et al. |
| rs6426833 | (9) | UC | 2.39E-68 | 1.265 | 1.221–1.31 | Jostins et al. |
| rs6017342 | ADA, HNF4A, (9) | UC | 1.43E-43 | 1.228 | 1.185–1.273 | Jostins et al. |
| rs4380874 | DLD, (9) | UC | 2.07E-26 | 1.137 | 1.097–1.177 | Jostins et al. |
| rs2816958 | (3) | UC | 1.98E-17 | 1.23 | 1.161–1.302 | Jostins et al. |
| rs561722 | FAM55A, FAM55D, (5) | UC | 5.15E-17 | 1.12 | 1.079–1.163 | Jostins et al. |
| rs798502 | CARD11, GNA12, TTYH3, (4) | UC | 6.09E-17 | 1.127 | 1.084–1.171 | Jostins et al. |
| rs4728142 | IRF5, TNPO3, TSPAN33, (11) | UC | 4.37E-14 | 1.104 | 1.066–1.143 | Jostins et al. |
| rs17229285 | 0 | UC | 1.73E-13 | 1.117 | 1.079–1.157 | Jostins et al. |
| rs10797432 | TNFRSF14, MMEL1, PLCH2, (8) | UC | 2.62E-12 | 1.078 | 1.041–1.116 | Jostins et al. |
| rs3774959 | NFKB1, MANBA, (2) | UC | 3.66E-12 | 1.118 | 1.077–1.159 | Jostins et al. |
| rs2189234 | - | UC | 1.95E-10 | 1.08 | 1.06–1.11 | Liu et al. |
| rs11150589 | ITGAL, (20) | UC | 6.04E-10 | 1.09 | 1.052–1.128 | Jostins et al. |
| rs113010081 | FLJ78302, LTF, CCR1, CCR2, CCR3, CCR5 | UC | 9.02E-10 | 1.14 | 1.09–1.18 | Liu et al. |
| rs1126510 | CALM3, (14) | UC | 1.55E-09 | 1.075 | 1.037–1.113 | Jostins et al. |
| rs7210086 | (3) | UC | 1.89E-09 | 1.111 | 1.062–1.163 | Jostins et al. |
| rs254560 | (6) | UC | 2.55E-09 | 1.056 | 1.019–1.093 | Jostins et al. |
| rs1077773 | AHR | UC | 5.96E-09 | 0.93 | 0.91–0.95 | Liu et al. |
| rs9847710 | PRKCD, ITIH4, (8) | UC | 1.05E-08 | 1.064 | 1.027–1.102 | Jostins et al. |
| rs483905 | JRKL, MAML2, (2) | UC | 1.21E-08 | 1.056 | 1.017–1.096 | Jostins et al. |
| rs11739663 | SLC9A3, (8) | UC | 1.81E-08 | 1.071 | 1.027–1.117 | Jostins et al. |
| rs4722672 | (14) | UC | 2.06E-08 | 1.091 | 1.043–1.14 | Jostins et al. |
| rs6088765 | PROCR, UQCC, CEP250, (8) | UC | 2.21E-08 | 1.079 | 1.041–1.117 | Jostins et al. |
| rs28374715 | ITPKA, NDUFAF1, NUSAP1, (8) | UC | 2.43E-08 | 1.082 | 1.04–1.126 | Jostins et al. |
| rs1016883 | RFTN2, PLCL1, (7) | UC | 2.87E-08 | 1.1 | 1.051–1.15 | Jostins et al. |
| rs1728785 | ZFP90, (6) | UC | 3.71E-08 | 1.075 | 1.031–1.121 | Jostins et al. |
| rs17736589 | - | UC | 4.34E-08 | 1.09 | 1.05–1.12 | Liu et al. |
| rs11583043 | SLC30A, EDG1 | UC | 6.05E-08 | 1.08 | 1.05–1.11 | Liu et al. |
| rs7011507 | - | UC | 6.40E-08 | 0.90 | 0.87–0.94 | Liu et al. |
| rs3116494 | ICOS, CD28, CTLA4 | UC | 1.30E-07 | 1.08 | 1.05–1.11 | Liu et al. |
| rs616597 | NFKBIZ | UC | 9.34E-06 | 0.93 | 0.91–0.96 | Liu et al. |
| rs11209026 | IL23R, IL12RB2, (4) | IBD | 8.12E-161 | 2.013 | 1.885–2.15 | Jostins et al. |
| rs11742570 | PTGER4, (1) | IBD | 1.81E-82 | 1.198 | 1.164–1.234 | Jostins et al. |
| rs10781499 | CARD9, PMPCA, SDCCAG3, INPP5E, (19) | IBD | 4.38E-56 | 1.188 | 1.154–1.222 | Jostins et al. |
| rs4409764 | NKX2–3, (6) | IBD | 1.03E-54 | 1.182 | 1.149–1.217 | Jostins et al. |
| rs2188962 | IRF1, IL13, CSF2, SLC22A4, IL4, IL3, IL5, PDLIM4, SLC22A5, ACSL6, (8) | IBD | 1.35E-52 | 1.158 | 1.125–1.191 | Jostins et al. |
| rs2836878 | (3) | IBD | 4.62E-48 | 1.18 | 1.142–1.219 | Jostins et al. |
| rs3197999 | MST1, PFKFB4, MST1R, UCN2, GPX1, IP6K2, BSN, IP6K1, USP4, (56) | IBD | 1.01E-47 | 1.18 | 1.144–1.216 | Jostins et al. |
| rs10761659 | (3) | IBD | 6.37E-46 | 1.166 | 1.134–1.2 | Jostins et al. |
| rs10758669 | JAK2, (4) | IBD | 7.88E-45 | 1.174 | 1.139–1.209 | Jostins et al. |
| rs6871626 | IL12B, (3) | IBD | 1.43E-42 | 1.181 | 1.146–1.216 | Jostins et al. |
| rs3024505 | IL10, IL20, IL19, IL24, PIGR, MAPKAPK2, FAIM3, RASSF5, (3) | IBD | 6.66E-42 | 1.208 | 1.163–1.254 | Jostins et al. |
| rs1801274 | FCGR2A, FCGR2B, FCGR3A, HSPA6, FCGR3B, FCRLA, (9) | IBD | 2.12E-38 | 1.124 | 1.092–1.157 | Jostins et al. |
| rs12946510 | IKZF3, ZPBP2, GSDMB, ORMDL3, GSDMA, (12) | IBD | 4.10E-38 | 1.157 | 1.124–1.19 | Jostins et al. |
| rs11741861 | TNIP1, IRGM, ZNF300P1, (8) | IBD | 2.94E-37 | 1.249 | 1.186–1.314 | Jostins et al. |
| rs2155219 | (5) | IBD | 4.24E-36 | 1.151 | 1.119–1.185 | Jostins et al. |
| rs2413583 | ATF4, TAB1, APOBEC3G, (16) | IBD | 4.40E-33 | 1.209 | 1.163–1.257 | Jostins et al. |
| rs7554511 | KIF21B, (6) | IBD | 1.24E-32 | 1.164 | 1.128–1.202 | Jostins et al. |
| rs4246905 | TNFSF8, TNFSF15, TNC, (2) | IBD | 2.80E-32 | 1.142 | 1.106–1.178 | Jostins et al. |
| rs7134599 | IFNG, IL26, IL22, (1) | IBD | 8.51E-32 | 1.096 | 1.064–1.128 | Jostins et al. |
| rs7608910 | REL, C2orf74, KIAA1841, AHSA2, (6) | IBD | 8.65E-32 | 1.138 | 1.105–1.171 | Jostins et al. |
| rs2823286 | 0 | IBD | 9.28E-30 | 1.157 | 1.121–1.194 | Jostins et al. |
| rs11564258 | LRRK2, MUC19 | IBD | 6.38E-29 | 1.334 | 1.217–1.461 | Jostins et al. |
| rs3091316 | CCL13, CCL2, CCL11, (4) | IBD | 1.22E-26 | 1.122 | 1.087–1.158 | Jostins et al. |
| rs7282490 | ICOSLG, (9) | IBD | 2.35E-26 | 1.105 | 1.072–1.138 | Jostins et al. |
| rs1893217 | (6) | IBD | 3.05E-26 | 1.171 | 1.127–1.216 | Jostins et al. |
| rs11010067 | CREM, (3) | IBD | 2.49E-25 | 1.115 | 1.082–1.148 | Jostins et al. |
| rs6062504 | TNFRSF6B, LIME1, SLC2A4RG, ZGPAT, (23) | IBD | 1.09E-23 | 1.104 | 1.071–1.139 | Jostins et al. |
| rs12942547 | STAT3, STAT5B, STAT5A, (13) | IBD | 5.51E-22 | 1.103 | 1.072–1.136 | Jostins et al. |
| rs26528 | RABEP2, IL27, EIF3C, SULT1A1, SULT1A2, NUPR1, (9) | IBD | 9.65E-22 | 1.099 | 1.067–1.13 | Jostins et al. |
| rs6920220 | TNFAIP3, (1) | IBD | 1.40E-21 | 1.102 | 1.064–1.141 | Jostins et al. |
| rs3749171 | GPR35, (12) | IBD | 3.07E-21 | 1.135 | 1.093–1.177 | Jostins et al. |
| rs1819333 | CCR6, RPS6KA2, RNASET2, (3) | IBD | 6.76E-21 | 1.081 | 1.051–1.113 | Jostins et al. |
| rs395157 | OSMR, FYB, LIFR | IBD | 2.22E-20 | 1.10 | 1.07–1.12 | Liu et al. |
| rs917997 | IL1R2, IL18RAP, IL18R1, IL1R1, IL1RL1, IL1RL2, (3) | IBD | 3.12E-20 | 1.103 | 1.067–1.14 | Jostins et al. |
| rs6568421 | (2) | IBD | 8.24E-20 | 1.108 | 1.074–1.142 | Jostins et al. |
| rs921720 | TRIB1, (1) | IBD | 8.30E-20 | 1.081 | 1.049–1.113 | Jostins et al. |
| rs17085007 | (2) | IBD | 2.79E-19 | 1.106 | 1.065–1.147 | Jostins et al. |
| rs11879191 | TYK2, PPAN-P2RY11, ICAM1, (25) | IBD | 2.04E-18 | 1.136 | 1.096–1.177 | Jostins et al. |
| rs1250546 | (5) | IBD | 3.15E-18 | 1.096 | 1.065–1.128 | Jostins et al. |
| rs12568930 | (3) | IBD | 1.26E-17 | 1.095 | 1.054–1.138 | Jostins et al. |
| rs2266959 | MAPK1, YDJC, UBE2L3, RIMBP3, CCDC116, (8) | IBD | 1.39E-16 | 1.105 | 1.066–1.145 | Jostins et al. |
| rs529866 | SOCS1, LITAF, RMI2, (10) | IBD | 1.73E-16 | 1.124 | 1.085–1.166 | Jostins et al. |
| rs4845604 | RORC, (14) | IBD | 3.52E-16 | 1.144 | 1.098–1.192 | Jostins et al. |
| rs17293632 | SMAD3, (2) | IBD | 5.97E-16 | 1.067 | 1.032–1.102 | Jostins et al. |
| rs6545800 | ADCY3, (6) | IBD | 6.14E-16 | 1.109 | 1.077–1.141 | Jostins et al. |
| rs6586030 | TSPAN14, C10orf58, (4) | IBD | 9.24E-16 | 1.115 | 1.076–1.156 | Jostins et al. |
| rs35675666 | TNFRSF9, (6) | IBD | 1.12E-15 | 1.112 | 1.07–1.156 | Jostins et al. |
| rs4246215 | C11orf9, FADS1, FADS2,(12) | IBD | 1.93E-15 | 1.079 | 1.046–1.112 | Jostins et al. |
| rs925255 | FOSL2, BRE, (1) | IBD | 2.67E-15 | 1.092 | 1.061–1.124 | Jostins et al. |
| rs17694108 | CEBPG, (8) | IBD | 5.85E-15 | 1.1 | 1.065–1.135 | Jostins et al. |
| rs1456896 | ZPBP, IKZF1, (4) | IBD | 7.28E-15 | 1.088 | 1.055–1.123 | Jostins et al. |
| rs4256159 | 0 | IBD | 9.00E-15 | 1.107 | 1.063–1.152 | Jostins et al. |
| rs941823 | (3) | IBD | 2.07E-14 | 1.071 | 1.036–1.107 | Jostins et al. |
| rs8005161 | GPR65, GALC, (1) | IBD | 2.35E-14 | 1.153 | 1.097–1.211 | Jostins et al. |
| rs9557195 | GPR183, GPR18, (6) | IBD | 2.37E-14 | 1.112 | 1.075–1.151 | Jostins et al. |
| rs2412970 | LIF, OSM, MTMR3, (8) | IBD | 2.70E-14 | 1.08 | 1.049–1.111 | Jostins et al. |
| rs6863411 | SPRY4, NDFIP1, (5) | IBD | 3.59E-14 | 1.089 | 1.057–1.121 | Jostins et al. |
| rs9358372 | (2) | IBD | 8.66E-14 | 1.089 | 1.057–1.121 | Jostins et al. |
| rs1569723 | CD40, MMP9, PLTP, (11) | IBD | 9.95E-14 | 1.091 | 1.056–1.126 | Jostins et al. |
| rs3851228 | TRAF3IP2, FYN, REV3L, (2) | IBD | 1.08E-13 | 1.153 | 1.089–1.219 | Jostins et al. |
| rs1734907 | EPO, (21) | IBD | 1.67E-13 | 1.114 | 1.071–1.158 | Jostins et al. |
| rs7657746 | IL2, IL21, (2) | IBD | 2.76E-13 | 1.116 | 1.08–1.154 | Jostins et al. |
| rs1363907 | ERAP2, ERAP1, LNPEP, (2) | IBD | 5.62E-13 | 1.068 | 1.037–1.099 | Jostins et al. |
| rs12103 | TNFRSF18, TNFRSF4, (30) | IBD | 7.66E-13 | 1.099 | 1.059–1.139 | Jostins et al. |
| rs2488389 | C1orf53, (2) | IBD | 8.45E-13 | 1.115 | 1.077–1.153 | Jostins et al. |
| rs1292053 | TUBD1, RPS6KB1, (9) | IBD | 8.85E-13 | 1.076 | 1.045–1.106 | Jostins et al. |
| rs11230563 | CD6, CD5, PTGDR2, (12) | IBD | 9.03E-13 | 1.085 | 1.053–1.118 | Jostins et al. |
| rs259964 | ZNF831, CTSZ, (5) | IBD | 1.01E-12 | 1.085 | 1.054–1.116 | Jostins et al. |
| rs2382817 | SLC11A1, CXCR2, CXCR1, PNKD, ARPC2, TMBIM1, CTDSP1, (8) | IBD | 3.70E-12 | 1.073 | 1.042–1.104 | Jostins et al. |
| rs10495903 | (5) | IBD | 8.03E-12 | 1.086 | 1.041–1.131 | Jostins et al. |
| rs9297145 | SMURF1, (6) | IBD | 8.21E-12 | 1.082 | 1.047–1.117 | Jostins et al. |
| rs2538470 | CNTNAP2 | IBD | 3.00E-11 | 1.07 | 1.05–1.09 | Liu et al. |
| rs17119 | 0 | IBD | 3.08E-11 | 1.071 | 1.032–1.11 | Jostins et al. |
| rs1517352 | STAT1, STAT4, (2) | IBD | 3.28E-11 | 1.077 | 1.046–1.109 | Jostins et al. |
| rs559928 | CCDC88B, RPS6KA4, TRPT1, FLRT1, (20) | IBD | 4.19E-11 | 1.101 | 1.061–1.142 | Jostins et al. |
| rs670523 | UBQLN4, RIT1, MSTO1, (28) | IBD | 5.79E-11 | 1.06 | 1.028–1.092 | Jostins et al. |
| rs1042058 | MAP3K8, (3) | IBD | 5.93E-11 | 1.075 | 1.044–1.106 | Jostins et al. |
| rs11672983 | NLRP7, NLRP2, KIR2DL1, LILRB4, (15) | IBD | 6.50E-11 | 1.087 | 1.055–1.119 | Jostins et al. |
| rs4703855 | - | IBD | 7.16E-11 | 0.93 | 0.91–0.95 | Liu et al. |
| rs7495132 | CRTC3, (3) | IBD | 9.48E-11 | 1.134 | 1.082–1.189 | Jostins et al. |
| rs1847472 | (1) | IBD | 1.57E-10 | 1.06 | 1.029–1.092 | Jostins et al. |
| rs111781203 | CCL20 | IBD | 2.16E-10 | 0.94 | 0.92–0.96 | Liu et al. |
| rs907611 | TNNI2, LSP1, (17) | IBD | 2.70E-10 | 1.068 | 1.035–1.101 | Jostins et al. |
| rs194749 | ZFP36L1, (4) | IBD | 2.70E-10 | 1.075 | 1.039–1.111 | Jostins et al. |
| rs2231884 | RELA, FOSL1, CTSW, SNX32, (22) | IBD | 2.91E-10 | 1.083 | 1.044–1.122 | Jostins et al. |
| rs12722515 | IL2RA, IL15RA, (6) | IBD | 3.76E-10 | 1.102 | 1.06–1.147 | Jostins et al. |
| rs4836519 | (1) | IBD | 4.24E-10 | 1.072 | 1.039–1.106 | Jostins et al. |
| rs6142618 | HCK, (10) | IBD | 6.05E-10 | 1.072 | 1.041–1.103 | Jostins et al. |
| rs2227564 | (13) | IBD | 6.75E-10 | 1.082 | 1.048–1.118 | Jostins et al. |
| rs10896794 | CNTF, LPXN, (8) | IBD | 6.80E-10 | 1.08 | 1.045–1.116 | Jostins et al. |
| rs7404095 | PRKCB, (5) | IBD | 9.68E-10 | 1.06 | 1.03–1.091 | Jostins et al. |
| rs4911259 | DNMT3B, (8) | IBD | 1.20E-09 | 1.075 | 1.044–1.106 | Jostins et al. |
| rs7240004 | SMAD7, (2) | IBD | 1.31E-09 | 1.057 | 1.026–1.088 | Jostins et al. |
| rs10521318 | IRF8, (4) | IBD | 1.41E-09 | 1.155 | 1.094–1.219 | Jostins et al. |
| rs1991866 | (2) | IBD | 1.65E-09 | 1.054 | 1.024–1.084 | Jostins et al. |
| rs4743820 | NFIL3, (2) | IBD | 3.60E-09 | 1.056 | 1.023–1.089 | Jostins et al. |
| rs727088 | CD226, (2) | IBD | 4.65E-09 | 1.077 | 1.046–1.108 | Jostins et al. |
| rs4656958 | CD48, SLAMF1, ITLN1, CD244, F11R, USF1, SLAMF7, ARHGAP30, (8) | IBD | 6.80E-09 | 1.061 | 1.029–1.094 | Jostins et al. |
| rs630923 | CXCR5, (17) | IBD | 7.07E-09 | 1.074 | 1.039–1.11 | Jostins et al. |
| rs11168249 | VDR, (8) | IBD | 7.78E-09 | 1.054 | 1.024–1.084 | Jostins et al. |
| rs2790216 | CISD1, IPMK, (2) | IBD | 8.07E-09 | 1.066 | 1.029–1.104 | Jostins et al. |
| rs34856868 | BTBD8 | IBD | 9.80E-09 | 0.82 | 0.77–0.88 | Liu et al. |
| rs2930047 | DAP, (2) | IBD | 1.03E-08 | 1.065 | 1.034–1.096 | Jostins et al. |
| rs11612508 | LOH12CR1, (8) | IBD | 1.06E-08 | 1.058 | 1.025–1.091 | Jostins et al. |
| rs653178 | SH2B3, ALDH2, ATXN2 | IBD | 1.11E-08 | 1.06 | 1.04–1.08 | Liu et al. |
| rs4692386 | - | IBD | 1.21E-08 | 0.94 | 0.93–0.96 | Liu et al. |
| rs38904 | (6) | IBD | 1.31E-08 | 1.054 | 1.025–1.085 | Jostins et al. |
| rs12654812 | DOK3, (17) | IBD | 1.68E-08 | 1.068 | 1.036–1.1 | Jostins et al. |
| rs2111485 | IFIH1, (5) | IBD | 1.93E-08 | 1.066 | 1.035–1.097 | Jostins et al. |
| rs12199775 | PHACTR2, (5) | IBD | 1.99E-08 | 1.129 | 1.066–1.195 | Jostins et al. |
| rs2651244 | (3) | IBD | 2.29E-08 | 1.015 | 0.986–1.044 | Jostins et al. |
| rs6740462 | SPRED2, (1) | IBD | 2.35E-08 | 1.081 | 1.046–1.116 | Jostins et al. |
| rs6025 | SELP, SELE, SELL | IBD | 2.51E-08 | 0.84 | 0.79–0.89 | Liu et al. |
| rs2472649 | CXCL5, CXCL1, CXCL3, IL8, CXCL6, PF4, CXCL2, PF4V1, (3) | IBD | 2.57E-08 | 1.095 | 1.046–1.146 | Jostins et al. |
| rs4664304 | MARCH7, LY75, PLA2R1 | IBD | 2.61E-08 | 1.06 | 1.04–1.08 | Liu et al. |
| rs4899554 | FOS, MLH3, (6) | IBD | 2.71E-08 | 1.083 | 1.042–1.125 | Jostins et al. |
| rs7911264 | (4) | IBD | 2.98E-08 | 1.066 | 1.035–1.097 | Jostins et al. |
| rs913678 | CEBPB, (5) | IBD | 4.59E-08 | 1.056 | 1.024–1.088 | Jostins et al. |
| rs17057051 | PTK2B, TRIM35, EPHX2 | IBD | 5.50E-08 | 0.94 | 0.92–0.96 | Liu et al. |
| rs11064881 | PRKAB1 | IBD | 5.95E-08 | 1.10 | 1.07–1.14 | Liu et al. |
| rs3740415 | NFKB2, TRIM8, TMEM180 | IBD | 1.03E-07 | 0.95 | 0.93–0.97 | Liu et al. |
| rs2073505 | HGFAC | IBD | 1.46E-07 | 1.10 | 1.06–1.14 | Liu et al. |
| rs564349 | C5orf4, DUSP1 | IBD | 1.54E-07 | 1.06 | 1.04–1.08 | Liu et al. |
| rs6856616 | - | IBD | 9.72E-07 | 1.10 | 1.06–1.14 | Liu et al. |
CD, Crohn’s disease; CI; confidence interval; GWAS, Genome-wide association study; IBD, inflammatory bowel disease;
OR, odds ratio; SNP, single nucleotide polymorphism; UC, ulcerative colitis.
Numbers in parentheses refer to the number of additional genes in the locus.
Refers to the study by Jostins et al.42
Refers to the study by Liu et al.67
Figure 1.
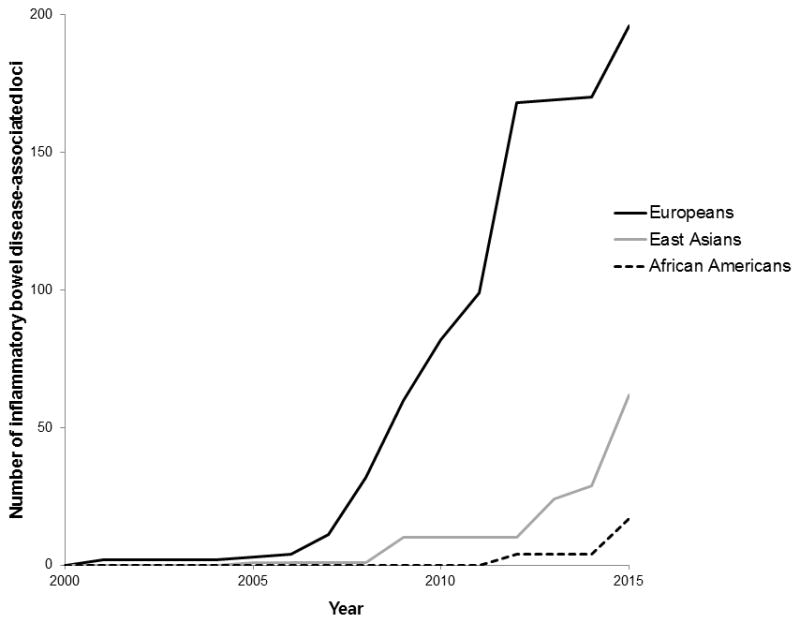
Progress of discovery of inflammatory bowel disease-associated loci in three ethnic groups over the past 15 years.
5.3 Beyond GWAS
With GWAS, Immunochip, their meta-analysis and the recently published trans-ethnic study, the number of identified IBD-associated loci has increased approximately 100-fold during the past 15 years.67 However, this approach is still based on the ‘common disease, common variant’ hypothesis,23 and is mostly capturing variants identified from European ancestry populations.8,67 The latest estimates suggest that the Immunochip and GWAS identified variants listed above only explain 19% and 26% of the heritability, respectively for CD and corresponding figures of 15% and 19%, respectively, for UC.70 This missing heritability could be attributed to the regions overlooked by GWAS, such as the sex chromosomes, as suggested by recent studies which identified ARHGEF6 and XIAP as IBD-related genes.8,71–75 It is also important to recognize that heritability estimates are prone to error and are an inexact science.
Rare or low frequency variants
Generally, rare variants have a low correlation with the marker SNPs used in the traditional genotyping platforms that are, on the whole, designed to capture common variation with a minor allele frequency over 0.05.8,76 Therefore, to discover a rare or low frequency disease-associated allele, direct testing of the variants is necessary. In addition, with the infrequency of such alleles in the population, even the largest catalogues of known human variation will not contain all variants of interest as novel variants are discovered every time a human genome is sequenced.8 Therefore, sequencing of an entire region, not just the known variable sites would be required to discover rare or low frequency variations. With the introduction of next generation sequencing (NGS) technology, progress in this area will soon be realized. Early NGS studies for IBD were focused on early-onset IBD, under the hypothesis that these are more severe cases and may be closer to single gene or Mendelian disorders than adult-onset IBD.8 Exome sequencing in a male child who presented at 15 months with very severe CD-like presentation identified a XIAP mutation leading to immunodeficiency with bowel manifestations that improved significantly after allogenic hematopoietic progenitor cell transplant.72 Subsequently, according to a recent German study, private variants in XIAP were commonly observed in about 4% of male patients with pediatric onset CD.75 Similar examples of rare mutations found in very early onset IBD (VEO IBD) subjects were identified in genes encoding the IL-10R subunit proteins,77 and a deletion mutation in ADAM17 that was homozygous in both of two children born to consanguineous parents.78 Further application of NGS technology for searching rare and low frequency variation in candidate IBD loci across cases and controls will likely reveal additional variants. Deep resequencing of GWAS loci in 350 cases of CD and 350 controls, followed by genotyping of 70 rare and low-frequency protein-altering variants in independent case-control series identified four additional independent risk loci in CARD15/NOD2, two additional protective variants in IL23R, and a highly significant association with a protective splice variant in CARD9.79 A similar targeted sequencing approach on 55 candidate genes in 200 cases of UC and 150 controls with follow-up genotyping of 42 rare non-synonymous variants in independent case-control cohorts confirmed significant association of rare variants in both IL23R and CARD9, previously identified from sequencing of CD loci and identified a novel association in RNF186.80 Ideally, the next step will be the deep sequencing of whole genome across sufficient number of cases and controls, not limited to candidate genes. However, minor allele frequency of a targeted rare variant is very low, thereby needing extremely large number of cases and controls to obtain a meaningfully significant difference unless variants with a very strong effect are identified. In addition to difficulty in enrolling large number of study subjects, the cost of deep sequencing of the whole genome remains a barrier as does the intensity of the subsequent analyses. Before whole genome sequencing becomes common place as costs continue to drop, a compromise approach of whole exome (coding regions in the genome) sequencing will likely become common place, as exomes represent only 1% of the complete genome.81 One example of successful exome sequencing is the discovery of a novel A-to-C missense variant (c.694A>C) in exon 6 of the FOXP3 gene on chromosome X in VEO IBD.82 Interestingly, GWAS-based studies have shown that a substantial proportion of IBD-associated loci are located in non-coding regions, suggesting rare variants regulating gene expression may also be important in IBD pathogenesis.8 The roles of non-coding regions on their pathogenic effects through modulation of gene expression are being identified by expression quantitative trait loci (eQTL)-GWAS mapping analysis.27
A further example of regions potentially missed by GWAS-based approaches was also suggested by a recent functional methylome map of UC colonic mucosal tissues which identified sixty-one genomic regions with differential methylations patterns that were also associated with nearby differentially expressed transcripts, implicating epigenetic regulation of gene expression in contributing to UC pathogenesis.27,83 Various types of micro-RNAs have also been implicated in IBD pathogenesis through the regulation of several IBD-associated genes, such as CARD15/NOD2, ATG16L1, and IL-23R.84–86 Another suggested approach is sequencing many individuals at low depth and combining data across individuals to generate accurate calls in shared stretches of chromosome.87 This approach may also provide useful disease-specific reference panels that can drive imputation into additional samples to increase power further.87
5.4 Identifying the causal variants and genes
Although genome-wide scanning, meta-analyses, and Immunochip analyses have identified over 200 SNPs associated with CD and/or UC, the question on ‘functionality’ and ‘causality’ of these findings remains elusive. Increasing resources are being directed to identifying causal variants and thereby implicating genes within known loci.8 Traditional approaches to ‘prove’ causality included experimental validation of suggested genes using in vitro cellular or animal models. This approach is difficult and a time-consuming process, but can be complemented by fine-mapping strategies capitalizing on the large number of samples already genotyped by GWAS and Immunochip platforms. It is believed that the majority of GWAS-identified risk variants are not likely themselves to be causally affecting the trait, but rather, are correlated to the true causal variant through linkage disequilibrium.88 Therefore, real causal variants could be identified in fine-mapping studies through targeted sequencing followed by prioritization of variants for functional validation.88 For prioritization purpose, a novel framework integrating association strength with functional genomic annotation data has been proposed recently.88 In addition, an algorithm which is integrating fine-mapped genetic and epigenetic data to identify candidate causal variants for 21 autoimmune diseases has also been suggested.89
6. Genes/loci associated with IBD risk
6.1 CARD15/NOD2
The identification of CARD15/NOD2 in the IBD1 locus as a CD-associated gene was a major breakthrough for IBD genetic research. NOD2 consists of two amino-terminal caspase recruitment domains (CARDs), a centrally located nucleotide binding domain and multiple leucine rich repeats (LRRs) at its carboxyterminal end.17 The LRR serves as a receptor for muramyl dipeptide, a small molecule derived from the cell wall peptidoglycan of gram-positive and gram-negative bacteria.90 Three different variants located in or near the LRR region are independently associated with CD in European ancestry populations: the frameshift mutation (L1007fsinsC), which causes a truncated protein transcript and two nonsynonymous polymorphisms (Arg702Trp and Gly908Arg).17,20–22 Carriage of one copy of the risk allele increases the risk of developing CD 2- to 4-fold and carriage of two risk alleles increases the risk of disease 20- to 40-fold in adults.16 However, the allele frequency of the 3 SNPs, and thus the population-attributable risk for CD from these mutations varies widely depending on ethnicity, showing no effect in East Asians.19,20,51–53,91–96 Studies on the function of CARD15/NOD2 have shown that variants are associated with an impairment in intracellular killing of Salmonella, increased susceptibility to infection with Listeria monocytogenes, and decreased expression of certain cryptdins.97,98 In addition, CARD15/NOD2-mutant mouse showed elevated NF-κB activation in response to muramyl dipeptide, more severe colitis induced by dextran sodium sulphate, and increased macrophage apoptosis and levels of IL-1β.99 Recently, nod1 and nod2 proteins were also implicated as having a role in autophagy by recruiting atg16l1 protein to the plasma membrane at the point of bacterial entry, and by inducing autophagosome formation.100–102 In CD patients with CARD15/NOD2 variants, ileal expression of α-defensin was more pronouncedly diminished than in those with wild-type.103 CARD15/NOD2 variants are not associated with UC and some data suggest that they may decrease the risk of colonic inflammation.19,104 In summary, CARD15/NOD2 mutations which are functionally linked to a deficient antimicrobial defense, are significantly associated with risk of CD in European ancestry, but not in East Asians.
6.2 Autophagy genes
Autophagy is a natural cellular mechanism removing unnecessary or dysfunctional cellular components for maintaining cellular and tissue homeostasis.105 The representative autophagy genes, ATG16L1 and IRGM have been shown to play roles as restriction factors for pathogens through autophagy.105 The discovery of the association of the Thr300Ala (rs2241880) in ATG16L1 with CD was a significant advance in our understanding of the pathogenesis of IBD. In 2007, three GWAS from Germany, UK, and North America identified a significant association of ATG16L1 with CD.31,33,106 Subsequent studies from multiple European cohorts confirmed this association.107–111 A Japanese study of 1311 CD cases and 6585 controls did not find any association between ATG16L1 variants and CD.96 However, a subsequent Korean study of 1809 cases and 2436 controls, did demonstrate Thr300Ala association with CD.112 Interestingly, a Korean GWAS suggested an association between ATG16L2 and CD.59 Atg16l2 protein is a homologue of atg16l1, but its role in autophagy has not been confirmed yet.59,112,113 The association of another autophagy gene, IRGM, with CD was also reported in European population studies.106,107,114 However, replication studies on Asians showed conflicting results.96,115–118 Immunochip and GWAS-based studies in UC have not shown any significant association in Europeans and Asians.37,38,116,117 A knock-in mouse model expressing the ATG16L1 Thr300Ala variant showed morphological defects in Paneth and goblet cells.119 Selective autophagy was also reduced in multiple cell types from Thr300Ala knock-in mice compared with wild type mice.119 ATG16L1 Thr300Ala protein showed more susceptibility to caspase 3- and caspase 7-mediated cleavage than wild type atg16l1 protein, resulting in decreased protein stability and effects on antibacterial autophagy and inflammatory cytokine production.119 In a subsequent study, increased proportions of abnormal Paneth cells were associated with the presence of Thr300Ala risk allele.120 The cumulative number of ATG16L1 Thr300A and CD-associated CARD15/NOD2 variants had an additive effect on the proportion of abnormal Paneth cells as well.120 Abnormal Paneth cells were also associated with a faster time to disease recurrence after surgical resection in CD cases.120 Based on evidences from these studies, therapeutic approaches influencing autophagy could be a rational and promising agent for a subset of CD in the near future.121
6.3 IL23R
The first published European ancestry GWAS identified association between several common variants at IL23R and IBD,30 an association confirmed in subsequent GWAS and replication studies.106,122 Among multiple associated alleles in the IL23R gene region, the most significant association was observed at rs11209096 (Arg181Gln) where having the minor glutamine allele gives a 2- to 3-fold protection against developing IBD.27,30 Subsequent functional studies revealed that the glutamine is a loss-of-function allele, and confers attenuated IL-23-mediated Th17 effector function, decrease of circulating CD4+ Th17 cells and CD8+ Tc17 cells, and reduced levels of proinflammatory cytokine secretion.123–125 These findings suggested that blocking the IL-23 signaling pathway may be effective in treating IBD and studies blocking both the IL-12/23 pathway with monoclonal antibody to p40 subunit of IL-12/23 and targeting IL-23 pathway through blocking p19 (subunit of IL-23) have shown promise in active CD patients, especially for patients who have failed anti-TNF therapy. 124,127
6.4 TNFSF15
The first ever GWAS on IBD which was performed in a Japanese population identified TNFSF15 as an IBD locus. Tnfsf15_28 (14,340T→C in intron 3 of TNFSF15) was the most significantly associated variant with CD among several associated SNPs at this locus.29 In the UK cohort from the same study, five polymorphic markers (tnfsf15_26, 31, 35, 36 and 41) were associated with IBD.29 Subsequently, the association of TNFSF15 with CD was confirmed in replication studies, GWAS and deep resequencing from both Japan and Korea.58,59,128–130 In European ancestry populations, the association of TNFSF15 with CD was confirmed, although the observed effect size was smaller than that seen in East Asians.46,131,132 Tnfsf15 protein, also known as TL1A is a proinflammatory molecule which stimulates proliferation and effector functions of CD8(+) cytotoxic T cells as well as Th1,Th2, and Th17 cells in the presence of TCR stimulation.133 Currently, TL1A is believed to be involved in the pathogenesis of IBD by stimulation of effector T cells and upregulation of proinflammatory cytokine production, defective generation of peripheral Tregs, and dampening suppressive activity of preexisting Tregs.133 A recent study revealed that monocyte-derived macrophages from rs6478108 A allele CD risk-carriers in the TNFSF15 region showed increased tnfsf15 expression, and increased pattern-recognition receptor-induced signaling and cytokines compared with GG carriers.134 TNFSF15 variants have also been associated with stricturing phenotype in CD.135 Collectively, these findings suggest that targeting tnfsf15 could be a promising therapeutic area for IBD patients. In a mouse model, antibody to TL1A prevented DSS-induced chronic colitis and T cell-mediated chronic colitis, as well as attenuating established DSS-induced chronic colitis by down-regulating of both Th1 and Th17 activation.136 In the near future, it is likely that TL1A blocking agents will be trialed in IBD patients.
6.5 HLA
The HLA region on chromosome 6p21 contains many genes related to immune function. The association between the HLA region and IBD has been studied for many years, and multiple associated loci have been reported including confirming early serological-based associations with HLA. The recent GWAS/Immunochip meta-analysis demonstrated that SNP rs6927022 near the class I gene HLA-DQA1 is the strongest UC-associated locus in the genome, and rs9264942 located in the HLA-B gene locus of the HLA class I region is the strongest CD-associated HLA locus.42 The recent high-density genotyping for 7406 SNPs within the HLA region in a total of 66954 individuals (18,405 with CD, 14,308 with UC, and 34,241 controls) identified multiple risk alleles associated with one or both diseases, with HLA-DRB1*01:03 being the most strongly associated with both CD and UC.137 They also showed that the contribution of class I and class II HLA variants to disease risk is relatively equivalent in CD, but HLA class II variation has a more important role in UC.137 In addition, most associated HLA alleles had a predominant role in either CD or UC, with very few conferring shared IBD risk.137
7. Clinical application of genetics in IBD
Considering significant heterogeneity in phenotype, natural history, and therapeutic response, IBD could be an ideal model for personalized or precision medicine, which incorporates individual variability in building prevention and treatment strategies.138 The significant advances in understanding the genetic architecture of IBD may be helpful in clinical practice in areas such as diagnostics, prognostics, and therapeutics.
8. Diagnosis of IBD
Despite abundant knowledge acquired from multiple studies on genetic traits associated with IBD, low pre-test probability (i.e. low prevalence of IBD in the general population) and moderate genotype-relative risks of IBD-associated loci limit the utility of genetic testing for new diagnosis of IBD.27,139 One example of this limitation is poor agreement on direction of risk of CD between two direct-to-consumer (DTC) genetic testing products with 3 out of 5 individuals receiving discordant CD risk-estimates from the 2 DTCs.140 An alternative strategy of combining serological, genetic, and inflammatory markers to differentiate non-IBD, CD, and UC has been attempted.141 Using a total of 17 markers (8 serological markers, 4 genetic markers, and 5 inflammatory markers) for a diagnostic random forest algorithm, the IBD vs. non-IBD discrimination area under the curve (AUC) was 0.87, which was significantly higher than the AUC for serology-only panel, 0.80 (P = 0.0001).141 Similarly, the AUC for CD vs. UC increased from 0.78 of serology-only panel to 0.93 with the combined panel (P = 0.0001).141 However, the contribution of genetic markers on this model needs to be evaluated further and the validity of this approach remains to be confirmed in additional cohorts.27 Although there is a long way to go for applying genetic tests of common variants as a screening or diagnostic tool of IBD, tests for single genes causing IBD could contribute to both diagnosis and intervention based on pathogenesis as well as genetic counseling.27 Known examples of single genes resulting in VEO IBD are the previously discussed XIAP, IL-10R, ADAM17, and FOXP3.72,77,78.82 Because 50 or more single genes causing IBD are implicated in VEO IBD, a useful IBD gene panel test including multiple candidate genes may guide clinicians to aid diagnosis and management of VEO IBD.
9. IBD subphenotypes and prognosis
In addition to the genetic variation associated with overall IBD risk, there has been considerable interest in the association between genetic variation and IBD subphenotypes, prognosis, and natural history. There have been multiple studies on the association between IBD susceptibility genes and prognosis, mainly among European ancestry subjects. The CARD15/NOD2 SNPs have been associated with ileal CD, stricturing disease, penetrating disease, familial disease, and earlier onset of disease.19–21,92,142–146 CARD15/NOD2 was also significantly associated with both bowel resection and complicated disease course (defined as one or more of the following: stenosing or internal penetrating behavior; perianal disease; or bowel resection) from a recent European multinational, multicenter study.145 Subphenotypic associations of CARD15/NOD2 for CD was also confirmed in a study involving 49 sites in 16 countries in Europe, North America, and Australia, which suggested that CARD15/NOD2 primary association was predominantly with disease location (ileal) and not with a stricturing phenotype.147 Among autophagy genes, the ATG16L1 T300A variant has been associated with ileal CD,55,110,148,149 and IRGM was associated with internal and perianal fistula in Italian CD patients.150 IL23R variation was also associated with ileal involvement (rs7517848),151 and with stricturing or penetrating phenotype (rs1004819, rs1495965, and rs11465804).55,145 For HLA, the DR3 DQ2 haplotype was predictive of extensive UC rather than distal disease.152 For CD, HLA-DRB1*01:03 was associated with pure colonic CD rather than ileal involvement and also with later age of diagnosis.144,153 In contrast, HLA-DRB1*07:01 and DRB1*04 were associated with ileal CD.144 The most recent multicenter study also confirmed the association between HLA and IBD subphenotypes, especially disease location.147 Three SNPs in the TNFSF15 (rs6478109, rs7848647, and rs4979462) were associated with perianal lesions in Japanese patients with CD.128 In Korean CD patients, rs6478108 CC genotype was associated with stricture and non-perianal penetrating complications, and rs4574921 CC genotype with perianal fistula.135 The rs4263839 in the TNFSF15 was related with bowel resection in CD patients with European ancestry.145 Comparing medically refractory UC (MRUC) and controls, the contribution of the HLA region to severe disease was confirmed and 2 additional loci reached a suggestive level of responsibility including TNFSF15.154
Understanding the limitation of a prognostic test based on a single variant with limited effect size, researchers have combined genetic variants to produce composite or gene-risk scores. Previously, a genetic risk score from the total number of risk alleles (0, 1, or 2) across 46 risk SNPs associated with MRUC ‘explained’ 48% of the variance for colectomy risk.154 In addition, when UC patients were grouped into four categories based on quarterly-divided risk score, the proportion of MRUC for the four groups was: less than 1%; 17%; 74%; and 100%, respectively (P < 2.2 × 10−16).154 In the most recent genotype-phenotype association study from the IIBDGC, information from 193 SNPs and 23 HLA types which are known to be associated with IBD was accumulated and used to generate genetic risk scores (CD score and UC score).147 As a result, CD vs. UC risk score showed very strong correlations with CD location and behavior (P = 1.65 × 10−78, or P = 9.23 × 10−18 after excluding the individual loci that achieved genome-wide, CARD15/NOD2, HLA, and 3p21).147 Moreover, predictive models based on genetic risk score strongly distinguished colonic from ileal CD and based on genetic risk score, patients with IBD could be much better characterized into 3 groups (ileal CD, colonic CD, and UC) rather than CD and UC as currently defined.147 This modeling could be clinically useful for differential diagnosis of IBD, especially for colonic IBD. Genetically, it appears that colonic CD is ‘closer’ to UC than it is to pure ileal CD.147 Further work using ‘finer’ phenotyping characteristics and classification may be able to delineate this further.147 Another approach is developing a composite model including genetic variables and other variables such as clinical and serologic markers to predict the course of IBD. For example, a combination of phenotypic, serologic, and genetic variables ‘predicted’ time to first surgery in CD more accurately than clinical only, genetics only, and clinical + serology models.155 In that study, variation at the IL12B locus showed a consistent association with both the need for surgery and the time to surgery in all models, thereby suggesting IL12B may be a potential target for therapeutic intervention.155 A group of researchers combined demographics, clinical characteristics such as disease location and perianal involvement, serologic markers, and CARD15/NOD2 genotypes to build a validated, individualized, and web-based tool to visualize individual risks for developing CD complication.156 As a result, a multivariate model including disease location, serologic markers (ASCA, CBir1, ANCA), the CARD15/NOD2 frameshift mutation was associated with the risk of complication, and a web-based tool based on the multivariate model was able to individualized disease outcomes in a patient-friendly format.156
10. Pharmacogenetics associated with IBD therapy
Identification of genetic variations related with effects or adverse events related with IBD therapy are recognized as an area that is most likely to benefit from advances in genetic technologies. The thiopurines are commonly-used drugs for treating IBD and it is well established in Caucasians that genetic polymorphisms in the thiopurine S-methyltransferase (TPMT) gene leading to reduced TPMT activity and high 6-TGN concentration are associated with the development of myelotoxicity.157 However, despite lower frequency of TPMT mutations in Asians including Koreans, Japanese, and Chinese than in European ancestry, the frequency of thiopurine-associated leukopenia was considerably higher in Asians.158 Moreover, even in Europeans, only around 25% of myelosuppressive episodes during thiopurine therapy are associated with a TPMT deficient genotype.159,160 These findings suggest that additional factors involved in thiopurine-associated myelosuppression exist. A recent Korean Immunochip-based association study in 978 Korean patients with CD treated with thiopurines identified association between a nonsynonymous SNP in NUDT15, rs116855232 (encoding p.Arg139Cys) and early leukopenia (allele frequency of 55.3% in early leukopenia cases vs. 3.4% in thiopurine-treated cases without leukopenia, OR 35.6; Pcombined = 4.88 × 10−94).158 The presence of this NUDT15 allele also showed a high sensitivity and specificity for early leukopenia (89.4% and 93.2%, respectively), with an area under the curve value of 0.92 (95% CI 0.88–0.97).158 This SNP is rare in European ancestry individuals but is also associated with thiopurine-induced leukopenia in Europeans and the association with bone marrow toxicity has also been reproduced in Japanese IBD patients.158,161,162 Pancreatitis is one of the dose-independent adverse reactions related with thiopurines limiting their use in IBD.157 In a recent worldwide study to identify genetic markers predicting pancreatitis within 3 months of starting thiopurines in patients with IBD, a GWAS on 172 cases and 2035 controls as well as additional validation was performed.163 As a result, strong evidence of association within the class II HLA region, with the most significant association identified at rs2647087 (OR 2.59, P = 2 × 10−16), with further replication in an independent set, was observed.163 Clinically, individuals homozygous for the risk allele at rs2647087 had an approximate 17% risk for pancreatitis, and a risk of 9% in rs2647087 heterozygotes was observed.163 Therefore, genetic information on the risk of myelosuppression (including both NUDT15 and TPMT variants) and pancreatitis development could be utilized as a screening panel that could aid clinicians to reduce the risk of serious adverse events related with thiopurines for IBD patients through choosing alternative approaches such as methotrexate for those at high-risk for these events.158,163
Given the failure of anti-TNF agents for a substantial number of IBD patients, multiple attempts to reveal genetic factors in predicting responses to anti-TNF agents have been attempted. Early studies showed inconsistent results and no association was observed between CARD15/NOD2 and non-response.139 In a study on 90 patients with UC, who were treated with infliximab as an induction therapy, patients homozygous for IBD risk-increasing IL23R variants (rs1004819, rs2201841, rs10889677, rs11209032, rs1495965) were more likely to have a response than those homozygous for risk-decreasing IL23R variants (rs7517847, rs10489629, rs11465804, rs1343151) (74.1 vs. 34.6 % ; P = 0.001).164 Based on the finding that infliximab acts partly through inducing apoptosis of activated T cells, whether SNPs involved in apoptosis-related pathway is associated with infliximab response was investigated by the Leuven group. They constructed a novel ‘apoptotic pharmacogenetic index’ (ranging from 0 to 3) by assigning points for 3 SNPs in the apoptosis-related genes (Fas ligand −843 C>T, Fas −670 G >A, and caspase-9 93 C>T) based on their association with response to infliximab in CD.165,166 Response and remission rates after infliximab administration significantly increased with apoptotic pharmacogenetic index score in both luminal CD and fistulizing CD with some influence by age, medication, or CRP levels in some subgroups.166 Another approach of searching for genetic variation related with anti-TNF therapeutic response is analyzing gene expression profile in tissues such as bowel mucosa.139 A Belgian study performed microarray analysis of total RNA from pre-treatment rectal mucosal biopsy samples from refractory UC patients.167 As a result, the top five differentially expressed genes (osteoprotegerin, stanniocalcin-1, prostaglandin-endoperoxide synthase 2, IL-13 receptor alpha 2 and IL-11) could separate infliximab responders from non-responders with 89.1% overall accuracy, 95.0% sensitivity, and 84.6% specificity.167 Using similar methods for CD, the top 5 differentially expressed genes (TNFAIP6, S100A8, IL11, G0S2, S100A9) could completely separate responders and non-responders to infliximab in the colonic CD group, but not in an ileal CD group.168 Interestingly, predictive genes from UC and colonic CD datasets showed near complete overlap, and the top five differentially expressed gene panel in UC predicted response to infliximab in colonic CD with 94.7% of accuracy.167,168 These findings provide insight into the molecular mechanisms involved in anti-TNF responsiveness, which appear similar between UC and colonic CD, but not with ileal CD.167,168 A small study of 94 pediatric IBD patients attempted to build a model to predict primary nonresponse to anti-TNFs.169 A composite model with three novel loci (rs975664 in the TACR1, rs4855535 in the FAM19A4, rs6100556 in the PHACTR3), one known IBD susceptibility loci (rs2836878 in the BRWDI), pANCA positivity, and UC diagnosis could predict the primary nonresponse to anti-TNFs with an R2 of 0.82 and an AUC of 0.98.169 In conclusion, genetic variations involved in the immune pathways and drug metabolism seem to have a greater effect on TNF responsiveness than IBD susceptibility genes. Moreover, the combination of genotype, serotype, and phenotype may be useful in predicting response to anti-TNFs, resulting in proper use of anti-TNF drugs for right patients.139,169 However, it is important to stress that these underpowered study findings’ need validation in additional cohorts. More recently it has become apparent that there is considerable pharmacokinetic variation between individuals exposed to anti-TNFs and it is likely that true non-responders can only be determined after adequate drug exposure has been confirmed.
11. Conclusions
During the past decades, the research community has achieved remarkable advances in the understanding of the genetics of IBD and various other related immune-mediated disorders through approaches including linkage analysis, GWAS, and GWAS meta-analysis, etc. Through experience with working on individual cohort GWAS sets, it became clear that large numbers were essential for advancing the field towards an understanding of the molecular architecture of IBD. This recognition led to the collaboration of researchers from multiple centers in Europe, North America, and Australia. This multinational collaborative approach should be a model for other groups such as those studying Asian populations, in whom there is a rising incidence of IBD. Despite multiple susceptibility genes/loci discovered through chip-based technologies such as GWAS and Immunochip, only a modest portion of the expected heritability has been explained, which may be inherent in the basic concept of ‘common disease, common variant’ hypothesis. Therefore, rare variants with greater effect sizes on IBD development need investigating and various strategies are currently being pursued to achieve this. The ultimate goal of genetic studies is to make advances in the management of IBD, including diagnosis, subclassification, predicting course of disease, and the development of new therapeutics. These fields are evolving rapidly in parallel with other ‘omic’ advances which, it is anticipated, will lead to an increasingly personalized management of IBD patients and a realization of precision medicine in the near future.
12. Expert commentary
In European ancestry populations, more than half of predicted heritability of IBD cannot be explained by genes/loci discovered to date, a figure that is considerably higher in Asian IBD populations. Furthermore, the majority of IBD-associated loci involve non-coding variation, which is in contrast to the traditional concept of disease-causing nonsynonymous, coding region variations. Well-designed, adequately powered fine mapping and deep sequencing strategies coupled with gene expression studies in appropriate tissues, will help define the functional role of these multiple non-coding variants. One interesting phenomenon is that the majority of people carrying IBD-associated risk variants remain healthy, others may develop another immune-mediated disease such as spondyloarthropathy, while a third, unfortunate, group may develop more than one condition. These observations suggest that other factors including gene-environment, gene-gut microbome, and even perhaps gene-gene interactions need to be studied to determine why some subjects get disease and others remain healthy (even within a family sharing similar environmental exposures). Furthermore, additional studies on epigenetic regulation of genes are necessary to extend our knowledge on the pathogenesis and natural history of IBD.
13. 5-year view
Our current knowledge on the architecture of IBD genetics has been achieved by embracing advances such as microarray technology, novel analytic programs, and statistical methodologies handling huge volumes of data. The same will be true for future development of IBD genetics, and next generation sequencing technology and further rapidly developing advanced technologies are expected to reveal more variants associated with IBD susceptibility and its prognosis. The collegiate and collaborative nature of the IIBDGC has been pivotal in driving the advances in this arena and it is evident that large sample sizes with well-defined subjects’ characteristics are required for more accurately characterizing the genetic architecture of IBD populations. It is imperative that the potential benefits of these genetic advances are available to all populations and the establishment of an Asian IBD genetic consortium as well as additional efforts in African-American populations together with trans-ethnic studies will be necessary to achieve these aims. Furthermore, designing new genotyping chips containing loci specifically designed to capture common and rare variants in these populations will also be beneficial. These populations with a rapidly expanding prevalence are crucial to study both environmental influences and therefore also gene by environmental associations. Findings from these studies will be of benefit to all IBD populations.
14. Key issues
Genetic contribution on IBD development was strongly suggested by epidemiologic observation such as familial disease clustering, high concordance rates in twins, and the different prevalences of IBD observed in different ethnicities.
GWAS opened a new era into IBD genetic research, helping overcome the limitation of linkage analysis for discovering causal variants responsible for complex immune-mediated chronic disorders.
GWAS, GWAS meta-analysis and Immunochip analyses expanded the number of IBD-associated loci to 163 (110 with IBD, 30 CD-specific, and 23 UC-specific) in European ancestry populations.
The recent genetic association studies on Asian and African ancestry are showing both overlap and some ethnic specificity in the molecular architecture of IBD.
The trans-ethnic Immunochip analysis increased the number of IBD risk loci to 231 independent SNPs within 200 loci, with the majority shared across diverse ancestry groups.
Even with IBD-associated loci from multiple genetic association studies and meta-analyses, only a modest fraction of predicted heritability can be explained in both European ancestry and Asian populations. To overcome this limitation, searching for rare or low frequency disease-associated alleles with deep resequencing, whole genome sequencing, and whole exome sequencing approaches using next generation sequencing technology will extend our knowledge on the genetic architecture of IBD.
From a screening and diagnostic point of view, the clinical application of IBD genetics is still limited.
Multiple loci have been known to be associated with specific IBD subphenotypes and prognosis. A genetic risk scoring system could be more helpful for classification, subphenotyping, and prognostication of IBD in the future especially if therapeutic approaches targeting some of the implicated genetic pathways are developed.
Genetic knowledge on genes associated with thiopurine-related adverse events such as myelosuppression and pancreatitis is expanding, leading to direct clinical benefit in IBD.
History has taught us that collaborative approaches are needed to assemble adequately powered cohorts for genetic research and these principles should be continued to be followed as investigators study non-European ancestry populations and also extend the studies to other modalities such as sequencing or the study of epigenetics.
Footnotes
Declaration of interests
The authors are supported by the European Union Seventh framework program 305479, the Leona M. and Harry B. Helmsley Charitable Trust and the National Institutes of Health (National Institute of Diabetes and Digestive and Kidney Diseases) DK062413. D McGovern is on the advisory board for Merck, UCB and has research support from Amgen. B Duk Ye is on the advisory board for Shire Korea, Abbvie Korea and acts as a consultant for Celltrion. The authors have no other relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript apart from those disclosed.
References
Reference annotations
* Of interest
** Of considerable interest
- 1.Crohn BB, Ginzburg L, Oppenheimer GD. Regional ileitis: A pathologic and clinical entity. JAMA. 1932;99:1323–9. [PubMed] [Google Scholar]
- 2.Cho JH. The genetics and immunopathogenesis of inflammatory bowel disease. Nat Rev Immunol. 2008;8:458–66. doi: 10.1038/nri2340. [DOI] [PubMed] [Google Scholar]
- 3.Yang H, McElree C, Roth MP, Shanahan F, Targan SR, Rotter JI. Familial empirical risks for inflammatory bowel disease: differences between Jews and non-Jews. Gut. 1993;34:517–24. doi: 10.1136/gut.34.4.517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ek WE, D’Amato M, Halfvarson J. The history of genetics in inflammatory bowel disease. Ann Gastroenterol. 2014;27:294–303. [PMC free article] [PubMed] [Google Scholar]
- 5.Brant SR. Update on the heritability of inflammatory bowel disease: the importance of twin studies. Inflamm Bowel Dis. 2011;17:1–5. doi: 10.1002/ibd.21385. [DOI] [PubMed] [Google Scholar]
- 6.Roth MP, Petersen GM, McElree C, Vadheim CM, Panish JF, Rotter JI. Familial empiric risk estimates of inflammatory bowel disease in Ashkenazi Jews. Gastroenterology. 1989;96:1016–20. doi: 10.1016/0016-5085(89)91618-1. [DOI] [PubMed] [Google Scholar]
- 7.Peeters M, Nevens H, Baert F, et al. Familial aggregation in Crohn’s disease: increased age-adjusted risk and concordance in clinical characteristics. Gastroenterology. 1996;111:597–603. doi: 10.1053/gast.1996.v111.pm8780562. [DOI] [PubMed] [Google Scholar]
- 8.de Lange KM, Barrett JC. Understanding inflammatory bowel disease via immunogenetics. J Autoimmun. 2015;64:91–100. doi: 10.1016/j.jaut.2015.07.013. [DOI] [PubMed] [Google Scholar]
- 9.Hugot JP, Laurent-Puig P, Gower-Rousseau C, et al. Mapping of a susceptibility locus for Crohn’s disease on chromosome 16. Nature. 1996;379:821–823. doi: 10.1038/379821a0. [DOI] [PubMed] [Google Scholar]
- 10.Ohmen JD, Yang HY, Yamamoto KK, et al. Susceptibility Locus for Inflammatory Bowel Disease on Chromosome 16 has a Role in Crohn’s disease, but Not in Ulcerative Colitis. Human Molecular Genetics. 1996;5:1679–83. doi: 10.1093/hmg/5.10.1679. [DOI] [PubMed] [Google Scholar]
- 11.Parkes M, Satsangi J, Lathrop GM, Bell JI, Jewell DP. Susceptibility loci in inflammatory bowel disease. Lancet. 1996;348:1588. doi: 10.1016/S0140-6736(05)66204-6. [DOI] [PubMed] [Google Scholar]
- 12.Curran ME, Lau KF, Hampe J, et al. Genetic analysis of inflammatory bowel disease in a large European cohort supports linkage to chromosomes 12 and 16. Gastroenterology. 1998;115:1066–71. doi: 10.1016/s0016-5085(98)70075-7. [DOI] [PubMed] [Google Scholar]
- 13.Brant SR, Fu Y, Fields CT, et al. American families with Crohn’s disease have strong evidence for linkage to chromosome 16 but not chromosome 12. Gastroenterology. 1998;115:1056–61. doi: 10.1016/s0016-5085(98)70073-3. [DOI] [PubMed] [Google Scholar]
- 14.Cavanaugh JA, Callen DF, Wilson SR, et al. Analysis of Australian Crohn’s disease pedigrees refines the localization for susceptibility to inflammatory bowel disease on chromosome 16. Ann Hum Genet. 1998;62:291–8. doi: 10.1046/j.1469-1809.1998.6240291.x. [DOI] [PubMed] [Google Scholar]
- 15.Cavanaugh J. International collaboration provides convincing linkage replication in complex disease through analysis of a large pooled data set: Crohn disease and chromosome 16. Am J Hum Genet. 2001;68:1165–71. doi: 10.1086/320119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Cooney R, Jewell D. The genetic basis of inflammatory bowel disease. Dig Dis. 2009;27:428–42. doi: 10.1159/000234909. [DOI] [PubMed] [Google Scholar]
- 17*.Hugot JP, Chamaillard M, Zouali H, et al. Association of NOD2 leucine-rich repeat variants with susceptibility to Crohn’s disease. Nature. 2001;411:599–603. doi: 10.1038/35079107. This pivotal study discovered three new variants, a frameshift variant (L1007fsinsC) and two missense variants (R702W and G908R) of CARD15/NOD2 associated with Crohn’s disease. [DOI] [PubMed] [Google Scholar]
- 18.Ogura Y, Bonen DK, Inohara N, et al. A frameshift mutation in NOD2 associated with susceptibility to Crohn’s disease. Nature. 2001;411:603–6. doi: 10.1038/35079114. [DOI] [PubMed] [Google Scholar]
- 19.Lesage S, Zouali H, Cezard JP, et al. CARD15/NOD2 mutational analysis and genotype-phenotype correlation in 612 patients with inflammatory bowel disease. Am J Hum Genet. 2002;70:845–57. doi: 10.1086/339432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Cuthbert AP, Fisher SA, Mirza MM, et al. The contribution of NOD2 gene mutations to the risk and site of disease in inflammatory bowel disease. Gastroenterology. 2002;122:867–74. doi: 10.1053/gast.2002.32415. [DOI] [PubMed] [Google Scholar]
- 21.Hampe J, Grebe J, Nikolaus S, et al. Association of NOD2 (CARD 15) genotype with clinical course of Crohn’s disease: a cohort study. Lancet. 2002;359:1661–5. doi: 10.1016/S0140-6736(02)08590-2. [DOI] [PubMed] [Google Scholar]
- 22.Vermeire S, Wild G, Kocher K, et al. CARD15 genetic variation in a Quebec population: prevalence, genotype-phenotype relationship, and haplotype structure. Am J Hum Genet. 2002;71:74–83. doi: 10.1086/341124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Risch N, Merikangas K. The future of genetic studies of complex human diseases. Science. 1996;273:1516–7. doi: 10.1126/science.273.5281.1516. [DOI] [PubMed] [Google Scholar]
- 24.Sachidanandam R, Weissman D, Schmidt SC, et al. A map of human genome sequence variation containing 1. 42 million single nucleotide polymorphisms. Nature. 2001;409:928–33. doi: 10.1038/35057149. [DOI] [PubMed] [Google Scholar]
- 25.The International HapMap Consortium. A haplotype map of the human genome. Nature. 2005;437:1299–1320. doi: 10.1038/nature04226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.McVean GAT, Myers SR, Hunt S, Deloukas P, Bentley DR, Donnelly P. The fine-scale structure of recombination rate variation in the human genome. Science. 2004;304:581–4. doi: 10.1126/science.1092500. [DOI] [PubMed] [Google Scholar]
- 27.McGovern DP, Kugathasan S, Cho JH. Genetics of Inflammatory Bowel Diseases. Gastroenterology. 2015;149:1163–76. doi: 10.1053/j.gastro.2015.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kitsios GD, Zintzaras E. Genome-wide association studies: hypothesis-”free” or “engaged”? Transl Res. 2009;154:161–4. doi: 10.1016/j.trsl.2009.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29*.Yamazaki K, McGovern D, Ragoussis J, et al. Single nucleotide polymorphisms in TNFSF15 confer susceptibility to Crohn’s disease. Hum Mol Genet. 2005;14:3499–506. doi: 10.1093/hmg/ddi379. This first genome-wide association study on inflammatory bowel disease revealed the association between TNFSF15 with Crohn’s disease in the Japanese and European populations. [DOI] [PubMed] [Google Scholar]
- 30*.Duerr RH, Taylor KD, Brant SR, et al. A genome-wide association study identifies IL23R as an inflammatory bowel disease gene. Science. 2006;314:1461–3. doi: 10.1126/science.1135245. Through genome-wide association study, the association of IL23R with inflammatory bowel disease was identified, suggesting IL-23 signaling pathway as a therapeutic target in inflammatory bowel disease. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31*.Hampe J, Franke A, Rosenstiel P, et al. A genome-wide association scan of nonsynonymous SNPs identifies a susceptibility variant for Crohn disease in ATG16L1. Nat Genet. 2007;39:207–11. doi: 10.1038/ng1954. This is the first study revealing ATG16L1 and autophagy pathway as being associated with Crohn’s disease pathogenesis. [DOI] [PubMed] [Google Scholar]
- 32.Libioulle C, Louis E, Hansoul S, et al. Novel Crohn disease locus identified by genome-wide association maps to a gene desert on 5p13. 1 and modulates expression of PTGER4. PLoS Genet. 2007;3:e58. doi: 10.1371/journal.pgen.0030058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Rioux JD, Xavier RJ, Taylor KD, et al. Genome-wide association study identifies new susceptibility loci for Crohn disease and implicates autophagy in disease pathogenesis. Nat Genet. 2007;39:596–604. doi: 10.1038/ng2032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Van Limbergen J, Wilson DC, Satsangi J. The genetics of Crohn’s disease. Annu Rev Genomics Hum Genet. 2009;10:89–116. doi: 10.1146/annurev-genom-082908-150013. [DOI] [PubMed] [Google Scholar]
- 35.Zhernakova A, van Diemen CC, Wijmenga C. Detecting shared pathogenesis from the shared genetics of immune-related diseases. Nat Rev Genet. 2009;10:43–55. doi: 10.1038/nrg2489. [DOI] [PubMed] [Google Scholar]
- 36.Liu JZ, Anderson CA. Genetic studies of Crohn’s disease: Past, present and future. Best Pract Res Clin Gastroenterol. 2014;28:373–86. doi: 10.1016/j.bpg.2014.04.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Fisher SA, Tremelling M, Anderson CA, et al. Genetic determinants of ulcerative colitis include the ECM1 locus and five loci implicated in Crohn’s disease. Nat Genet. 2008;40:710–712. doi: 10.1038/ng.145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Franke A, Balschun T, Karlsen TH, et al. Sequence variants in IL10, ARPC2 and multiple other loci contribute to ulcerative colitis susceptibility. Nat Genet. 2008;40:1319–1323. doi: 10.1038/ng.221. [DOI] [PubMed] [Google Scholar]
- 39.Barrett JC, Lee JC, Lees CW, et al. Genome-wide association study of ulcerative colitis identifies three new susceptibility loci, including the HNF4A region. Nat Genet. 2009;41:1330–1334. doi: 10.1038/ng.483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Silverberg MS, Cho JH, Rioux JD, et al. Ulcerative colitis-risk loci on chromosomes 1p36 and 12q15 found by genome-wide association study. Nat Genet. 2009;41:216–20. doi: 10.1038/ng.275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Satsangl J, Farrant JM, Jewell DP, et al. Contribution of genes of the major histocompatibility complex to susceptibility and disease phenotype in inflammatory bowel disease. Lancet. 1996;347:1212–7. doi: 10.1016/s0140-6736(96)90734-5. [DOI] [PubMed] [Google Scholar]
- 42**.Jostins L, Ripke S, Weersma RK, et al. Host-microbe interactions have shaped the genetic architecture of inflammatory bowel disease. Nature. 2012;491:119–24. doi: 10.1038/nature11582. Though analyses of GWAS meta-analysis and Immunochip in over 75,000 inflammatory bowel disease and control subjects, a total of 163 loci associated with inflammatory bowel disease were identified. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Maher B. Personal genomes: The case of the missing heritability. Nature. 2008;456:18–21. doi: 10.1038/456018a. [DOI] [PubMed] [Google Scholar]
- 44.Manolio TA, Collins FS, Cox NJ, et al. Finding the missing heritability of complex diseases. Nature. 2009;461:747–53. doi: 10.1038/nature08494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Cortes A, Brown MA. Promise and pitfalls of the Immunochip. Arthritis Res Ther. 2011;13:101. doi: 10.1186/ar3204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Barrett JC, Hansoul S, Nicolae DL, et al. Genome-wide association defines more than 30 distinct susceptibility loci for Crohn’s disease. Nat Genet. 2008;40:955–62. doi: 10.1038/NG.175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Franke A, McGovern DPB, Barrett JC, et al. Genome-wide meta-analysis increases to 71 the number of confirmed Crohn’s disease susceptibility loci. Nat Genet. 2010;42:1118–25. doi: 10.1038/ng.717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.McGovern DP, Gardet A, Torkvist L, et al. Genome-wide association identifies multiple ulcerative colitis susceptibility loci. Nat Genet. 2010;42:332–7. doi: 10.1038/ng.549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Anderson CA, Boucher G, Lees CW, et al. Meta-analysis identifies 29 additional ulcerative colitis risk loci, increasing the number of confirmed associations to 47. Nat Genet. 2011;43:246–52. doi: 10.1038/ng.764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Yamazaki K, Takazoe M, Tanaka T, Kazumori T, Nakamura Y. Absence of mutation in the NOD2/CARD15 gene among 483 Japanese patients with Crohn’s disease. J Hum Genet. 2002;47:469–72. doi: 10.1007/s100380200067. [DOI] [PubMed] [Google Scholar]
- 51.Inoue N, Tamura K, Kinouchi Y, et al. Lack of common NOD2 variants in Japanese patients with Crohn’s disease. Gastroenterology. 2002;123:86–91. doi: 10.1053/gast.2002.34155. [DOI] [PubMed] [Google Scholar]
- 52.Croucher PJ, Mascheretti S, Hampe J, et al. Haplotype structure and association to Crohn’s disease of CARD15 mutations in two ethnically divergent populations. Eur J Hum Genet. 2003;11:6–16. doi: 10.1038/sj.ejhg.5200897. [DOI] [PubMed] [Google Scholar]
- 53.Lee GH, Kim CG, Kim JS, Jung HC, Song IS. Frequency analysis of NOD2 gene mutations in Korean patients with Crohn’s disease. Korean J Gastroenterol. 2005;45:162–8. [PubMed] [Google Scholar]
- 54.Yamazaki K, Onouchi Y, Takazoe M, Kubo M, Nakamura Y, Hata A. Association analysis of genetic variants in IL23R, ATG16L1 and 5p13. 1 loci with Crohn’s disease in Japanese patients. J Hum Genet. 2007;52:575–83. doi: 10.1007/s10038-007-0156-z. [DOI] [PubMed] [Google Scholar]
- 55.Yang SK, Park M, Lim J, et al. Contribution of IL23R but not ATG16L1 to Crohn’s disease susceptibility in Koreans. Inflamm Bowel Dis. 2009;15:1385–90. doi: 10.1002/ibd.20921. [DOI] [PubMed] [Google Scholar]
- 56.Bin C, Zhirong Z, Xiaoqin W, et al. Contribution of rs11465788 in IL23R gene to Crohn’s disease susceptibility and phenotype in Chinese population. J Genet. 2009;88:191–6. doi: 10.1007/s12041-009-0027-9. [DOI] [PubMed] [Google Scholar]
- 57.Kim SW, Kim ES, Moon CM, et al. Genetic polymorphisms of IL-23R and IL-17A and novel insights into their associations with inflammatory bowel disease. Gut. 2011;60:1527–36. doi: 10.1136/gut.2011.238477. [DOI] [PubMed] [Google Scholar]
- 58.Yamazaki K, Umeno J, Takahashi A, et al. A genome-wide association study identifies 2 susceptibility loci for Crohn’s disease in a Japanese population. Gastroenterology. 2013;144:781–8. doi: 10.1053/j.gastro.2012.12.021. [DOI] [PubMed] [Google Scholar]
- 59.Yang SK, Hong M, Zhao W, et al. Genome-wide association study of Crohn’s disease in Koreans revealed three new susceptibility loci and common attributes of genetic susceptibility across ethnic populations. Gut. 2014;63:80–7. doi: 10.1136/gutjnl-2013-305193. [DOI] [PubMed] [Google Scholar]
- 60.Asano K, Matsushita T, Umeno J, et al. A genome-wide association study identifies three new susceptibility loci for ulcerative colitis in the Japanese population. Nat Genet. 2009;41:1325–9. doi: 10.1038/ng.482. [DOI] [PubMed] [Google Scholar]
- 61.Yang SK, Hong M, Zhao W, et al. Genome-wide association study of ulcerative colitis in Koreans suggests extensive overlapping of genetic susceptibility with Caucasians. Inflamm Bowel Dis. 2013;19:954–66. doi: 10.1097/MIB.0b013e3182802ab6. [DOI] [PubMed] [Google Scholar]
- 62.Juyal G, Negi S, Sood A, et al. Genome-wide association scan in north Indians reveals three novel HLA-independent risk loci for ulcerative colitis. Gut. 2015;64:571–9. doi: 10.1136/gutjnl-2013-306625. [DOI] [PubMed] [Google Scholar]
- 63.Yang SK, Hong M, Choi H, et al. Immunochip analysis identification of 6 additional susceptibility loci for Crohn’s disease in Koreans. Inflamm Bowel Dis. 2015;21:1–7. doi: 10.1097/MIB.0000000000000268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Ye BD, Choi H, Hong M, et al. Identification of ten additional susceptibility loci for ulcerative colitis through Immunochip analysis in Koreans. Inflamm Bowel Dis. 2016;22:13–9. doi: 10.1097/MIB.0000000000000584. [DOI] [PubMed] [Google Scholar]
- 65.Huang C, Haritunians T, Okou DT, et al. Characterization of genetic loci that affect susceptibility to inflammatory bowel diseases in African Americans. Gastroenterology. 2015;149:1575–86. doi: 10.1053/j.gastro.2015.07.065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Okada Y, Yamazaki K, Umeno J, et al. HLA-Cw*1202-B*5201-DRB1*1502 haplotype increases risk for ulcerative colitis but reduces risk for Crohn’s disease. Gastroenterology. 2011;141:864–71. doi: 10.1053/j.gastro.2011.05.048. [DOI] [PubMed] [Google Scholar]
- 67**.Liu JZ, van Sommeren S, Huang H, et al. Association analyses identify 38 susceptibility loci for inflammatory bowel disease and highlight shared genetic risk across populations. Nat Genet. 2015;47:979–86. doi: 10.1038/ng.3359. This IIBDGC study analyzed genetic data of 9,846 individuals of East Asian, Indian, or Iranian descent in addition to 86,640 European individuals and increased the number of inflammatory bowel disease-realted loci to 200, and demonstrated differences in effect sizes for some loci across populations. This highlights the importance of transancestry association studies. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.De Cruz P, Prideaux L, Wagner J, et al. Characterization of the gastrointestinal microbiota in health and inflammatory bowel disease. Inflamm Bowel Dis. 2012;18:372–90. doi: 10.1002/ibd.21751. [DOI] [PubMed] [Google Scholar]
- 69.Tong M, McHardy I, Ruegger P, et al. Reprograming of gut microbiome energy metabolism by the FUT2 Crohn’s disease risk polymorphism. ISME J. 2014;8:2193–206. doi: 10.1038/ismej.2014.64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Chen GB, Lee SH, Brion MJ, et al. Estimation and partitioning of (co)heritability of inflammatory bowel disease from GWAS and immunochip data. Hum Mol Genet. 2014;23:4710–20. doi: 10.1093/hmg/ddu174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Rigaud S, Fondaneche MC, Lambert N, et al. XIAP deficiency in humans causes an X-linked lymphoproliferative syndrome. Nature. 2006;444:110–4. doi: 10.1038/nature05257. [DOI] [PubMed] [Google Scholar]
- 72.Worthey EA, Mayer AN, Syverson GD, et al. Making a definitive diagnosis: successful clinical application of whole exome sequencing in a child with intractable inflammatory bowel disease. Genet Med. 2011;13:255–62. doi: 10.1097/GIM.0b013e3182088158. [DOI] [PubMed] [Google Scholar]
- 73.Pachlopnik Schmid J, Canioni D, Moshous D, et al. Clinical similarities and differences of patients with X-linked lymphoproliferative syndrome type 1 (XLP-1/SAP deficiency) versus type 2 (XLP-2/XIAP deficiency) Blood. 2011;117:1522–9. doi: 10.1182/blood-2010-07-298372. [DOI] [PubMed] [Google Scholar]
- 74.Chang D, Gao F, Slavney A, et al. Accounting for eXentricities: analysis of the X chromosome in GWAS reveals X-linked genes implicated in autoimmune diseases. PLoS One. 2014;9:e113684. doi: 10.1371/journal.pone.0113684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Zeissig Y, Petersen B-S, Milutinovic S, et al. XIAP variants in male Crohn’s disease. Gut. 2015;64:66–76. doi: 10.1136/gutjnl-2013-306520. [DOI] [PubMed] [Google Scholar]
- 76.Li B, Liu DJ, Leal SM. Identifying rare variants associated with complex traits via sequencing. Curr Protoc Hum Genet. 2013;Chapter 1(Unit 1.26) doi: 10.1002/0471142905.hg0126s78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Glocker EO, Kotlarz D, Boztug K, et al. Inflammatory bowel disease and mutations affecting the interleukin-10 receptor. N Engl J Med. 2009;361:2033–45. doi: 10.1056/NEJMoa0907206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Blaydon DC, Biancheri P, Di WL, et al. Inflammatory skin and bowel disease linked to ADAM17 deletion. N Engl J Med. 2011;365:1502–8. doi: 10.1056/NEJMoa1100721. [DOI] [PubMed] [Google Scholar]
- 79.Rivas MA, Beaudoin M, Gardet A, et al. Deep resequencing of GWAS loci identifies independent rare variants associated with inflammatory bowel disease. Nat Genet. 2011;43:1066–73. doi: 10.1038/ng.952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Beaudoin M, Goyette P, Boucher G, et al. Deep resequencing of GWAS loci identifies rare variants in CARD9, IL23R and RNF186 that are associated with ulcerative colitis. PLoS Genet. 2013;9:e1003723. doi: 10.1371/journal.pgen.1003723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Ng SB, Turner EH, Robertson PD, et al. Targeted capture and massively parallel sequencing of 12 human exomes. Nature. 2009;461:272–6. doi: 10.1038/nature08250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Okou DT, Mondal K, Faubion WA, et al. Exome sequencing identifies a novel FOXP3 mutation in a 2-generation family with inflammatory bowel disease. J Pediatr Gastroenterol Nutr. 2014;58:561–8. doi: 10.1097/MPG.0000000000000302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Hasler R, Feng Z, Backdahl L, et al. A functional methylome map of ulcerative colitis. Genome Res. 2012;22:2130–7. doi: 10.1101/gr.138347.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Chen Y, Wang C, Liu Y, et al. miR-122 targets NOD2 to decrease intestinal epithelial cell injury in Crohn’s disease. Biochem Biophys Res Commun. 2013;438:133–9. doi: 10.1016/j.bbrc.2013.07.040. [DOI] [PubMed] [Google Scholar]
- 85.Brain O, Owens Benjamin MJ, Pichulik T, et al. The intracellular sensor NOD2 induces microRNA-29 expression in human dendritic cells to limit IL-23 release. Immunity. 2013;39:521–36. doi: 10.1016/j.immuni.2013.08.035. [DOI] [PubMed] [Google Scholar]
- 86.Lu C, Chen J, Xu HG, et al. MIR106B and MIR93 prevent removal of bacteria from epithelial cells by disrupting ATG16L1-mediated autophagy. Gastroenterology. 2014;146:188–99. doi: 10.1053/j.gastro.2013.09.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Li Y, Sidore C, Kang HM, Boehnke M, Abecasis GR. Low-coverage sequencing: implications for design of complex trait association studies. Genome Res. 2011;21:940–51. doi: 10.1101/gr.117259.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Kichaev G, Yang WY, Lindstrom S, et al. Integrating functional data to prioritize causal variants in statistical fine-mapping studies. PLoS Genet. 2014;10:e1004722. doi: 10.1371/journal.pgen.1004722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Farh KK, Marson A, Zhu J, et al. Genetic and epigenetic fine mapping of causal autoimmune disease variants. Nature. 2015;518:337–43. doi: 10.1038/nature13835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Inohara N, Ogura Y, Fontalba A, et al. Host recognition of bacterial muramyl dipeptide mediated through NOD2. Implications for Crohn’s disease. J Biol Chem. 2003;278:5509–12. doi: 10.1074/jbc.C200673200. [DOI] [PubMed] [Google Scholar]
- 91.Arnott ID, Nimmo ER, Drummond HE, et al. NOD2/CARD15, TLR4 and CD14 mutations in Scottish and Irish Crohn’s disease patients: evidence for genetic heterogeneity within Europe? Genes Immun. 2004;5:417–25. doi: 10.1038/sj.gene.6364111. [DOI] [PubMed] [Google Scholar]
- 92.Economou M, Trikalinos TA, Loizou KT, Tsianos EV, Ioannidis JP. Differential effects of NOD2 variants on Crohn’s disease risk and phenotype in diverse populations: a metaanalysis. Am J Gastroenterol. 2004;99:2393–404. doi: 10.1111/j.1572-0241.2004.40304.x. [DOI] [PubMed] [Google Scholar]
- 93.Ng SC, Tsoi KK, Kamm MA, et al. Genetics of inflammatory bowel disease in Asia: Systematic review and meta-analysis. Inflamm Bowel Dis. 2012;18:1164–76. doi: 10.1002/ibd.21845. [DOI] [PubMed] [Google Scholar]
- 94.Bonen DK, Ogura Y, Nicolae DL, et al. Crohn’s disease-associated NOD2 variants share a signaling defect in response to lipopolysaccharide and peptidoglycan. Gastroenterology. 2003;124:140–6. doi: 10.1053/gast.2003.50019. [DOI] [PubMed] [Google Scholar]
- 95.Mathew CG, Lewis CM. Genetics of inflammatory bowel disease: progress and prospects. Hum Mol Genet. 2004;13(Spec No 1):R161–8. doi: 10.1093/hmg/ddh079. [DOI] [PubMed] [Google Scholar]
- 96.Hirano A, Yamazaki K, Umeno J, et al. Association study of 71 European Crohn’s disease susceptibility loci in a Japanese population. Inflamm Bowel Dis. 2013;19:526–33. doi: 10.1097/MIB.0b013e31828075e7. [DOI] [PubMed] [Google Scholar]
- 97.Hisamatsu T, Suzuki M, Reinecker HC, Nadeau WJ, McCormick BA, Podolsky DK. CARD15/NOD2 functions as an antibacterial factor in human intestinal epithelial cells. Gastroenterology. 2003;124:993–1000. doi: 10.1053/gast.2003.50153. [DOI] [PubMed] [Google Scholar]
- 98.Kobayashi KS, Chamaillard M, Ogura Y, et al. Nod2-dependent regulation of innate and adaptive immunity in the intestinal tract. Science. 2005;307:731–4. doi: 10.1126/science.1104911. [DOI] [PubMed] [Google Scholar]
- 99.Maeda S, Hsu LC, Liu H, et al. Nod2 mutation in Crohn’s disease potentiates NF-kappaB activity and IL-1β processing. Science. 2005;307:734–8. doi: 10.1126/science.1103685. [DOI] [PubMed] [Google Scholar]
- 100.Travassos LH, Carneiro LAM, Ramjeet M, et al. Nod1 and Nod2 direct autophagy by recruiting ATG16L1 to the plasma membrane at the site of bacterial entry. Nat Immunol. 2010;11:55–62. doi: 10.1038/ni.1823. [DOI] [PubMed] [Google Scholar]
- 101.Homer CR, Richmond AL, Rebert NA, Achkar JP, McDonald C. ATG16L1 and NOD2 interact in an autophagy-dependent antibacterial pathway implicated in Crohn’s disease pathogenesis. Gastroenterology. 2010;139:1630–41. doi: 10.1053/j.gastro.2010.07.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Cooney R, Baker J, Brain O, et al. NOD2 stimulation induces autophagy in dendritic cells influencing bacterial handling and antigen presentation. Nat Med. 2010;16:90–7. doi: 10.1038/nm.2069. [DOI] [PubMed] [Google Scholar]
- 103.Wehkamp J, Harder J, Weichenthal M, et al. NOD2 (CARD15) mutations in Crohn’s disease are associated with diminished mucosal alpha-defensin expression. Gut. 2004;53:1658–64. doi: 10.1136/gut.2003.032805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Hampe J, Cuthbert A, Croucher PJ, et al. Association between insertion mutation in NOD2 gene and Crohn’s disease in German and British populations. Lancet. 2001;357:1925–8. doi: 10.1016/S0140-6736(00)05063-7. [DOI] [PubMed] [Google Scholar]
- 105.Baxt LA, Xavier RJ. Role of autophagy in the maintenance of intestinal homeostasis. Gastroenterology. 2015;149:553–62. doi: 10.1053/j.gastro.2015.06.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Burton PR, Clayton DG, Cardon LR, et al. Genome-wide association study of 14,000 cases of seven common diseases and 3,000 shared controls. Nature. 2007;447:661–78. doi: 10.1038/nature05911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Parkes M, Barrett JC, Prescott NJ, et al. Sequence variants in the autophagy gene IRGM and multiple other replicating loci contribute to Crohn’s disease susceptibility. Nat Genet. 2007;39:830–2. doi: 10.1038/ng2061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Prescott NJ, Fisher SA, Franke A, et al. A nonsynonymous SNP in ATG16L1 predisposes to ileal Crohn’s disease and is independent of CARD15 and IBD5. Gastroenterology. 2007;132:1665–71. doi: 10.1053/j.gastro.2007.03.034. [DOI] [PubMed] [Google Scholar]
- 109.Glas J, Konrad A, Schmechel S, et al. The ATG16L1 gene variants rs2241879 and rs2241880 (T300A) are strongly associated with susceptibility to Crohn’s disease in the German population. Am J Gastroenterol. 2008;103:682–91. doi: 10.1111/j.1572-0241.2007.01694.x. [DOI] [PubMed] [Google Scholar]
- 110.Van Limbergen J, Russell RK, Nimmo ER, et al. Autophagy gene ATG16L1 influences susceptibility and disease location but not childhood-onset in Crohn’s disease in Northern Europe. Inflamm Bowel Dis. 2008;14:338–46. doi: 10.1002/ibd.20340. [DOI] [PubMed] [Google Scholar]
- 111.Weersma RK, Zhernakova A, Nolte IM, et al. ATG16L1 and IL23R are associated with inflammatory bowel diseases but not with celiac disease in the Netherlands. Am J Gastroenterol. 2008;103:621–7. doi: 10.1111/j.1572-0241.2007.01660.x. [DOI] [PubMed] [Google Scholar]
- 112.Yang SK, Ye BD, Song K. ATG16L1 contributes to Crohn’s disease susceptibility in Koreans: overmuch concern for ethnic difference? Gut. 2015;64:687–8. doi: 10.1136/gutjnl-2014-308242. [DOI] [PubMed] [Google Scholar]
- 113.Ishibashi K, Fujita N, Kanno E, et al. Atg16L2, a novel isoform of mammalian Atg16L that is not essential for canonical autophagy despite forming an Atg12-5-16L2 complex. Autophagy. 2011;7:1500–13. doi: 10.4161/auto.7.12.18025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Barrett JC, Hansoul S, Nicolae DL, et al. Genome-wide association defines more than 30 distinct susceptibility loci for Crohn’s disease. Nat Genet. 2008;40:955–62. doi: 10.1038/NG.175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Prescott NJ, Dominy KM, Kubo M, et al. Independent and population-specific association of risk variants at the IRGM locus with Crohn’s disease. Hum Mol Genet. 2010;19:1828–39. doi: 10.1093/hmg/ddq041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Moon CM, Shin DJ, Kim SW, et al. Associations between genetic variants in the IRGM gene and inflammatory bowel diseases in the Korean population. Inflamm Bowel Dis. 2013;19:106–14. doi: 10.1002/ibd.22972. [DOI] [PubMed] [Google Scholar]
- 117.Fuyuno Y, Yamazaki K, Takahashi A, et al. Genetic characteristics of inflammatory bowel disease in a Japanese population. J Gastroenterol. doi: 10.1007/s00535-015-1135-3. (in press) [DOI] [PubMed] [Google Scholar]
- 118.Baskaran K, Pugazhendhi S, Ramakrishna BS. Association of IRGM gene mutations with inflammatory bowel disease in the Indian population. PLoS One. 2014;9:e106863. doi: 10.1371/journal.pone.0106863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Lassen KG, Kuballa P, Conway KL, et al. Atg16L1 T300A variant decreases selective autophagy resulting in altered cytokine signaling and decreased antibacterial defense. Proc Natl Acad Sci U S A. 2014;111:7741–6. doi: 10.1073/pnas.1407001111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.VanDussen KL, Liu TC, Li D, et al. Genetic variants synthesize to produce paneth cell phenotypes that define subtypes of Crohn’s disease. Gastroenterology. 2014;146:200–9. doi: 10.1053/j.gastro.2013.09.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Levine B, Packer M, Codogno P. Development of autophagy inducers in clinical medicine. J Clin Invest. 2015;125:14–24. doi: 10.1172/JCI73938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Raelson JV, Little RD, Ruether A, et al. Genome-wide association study for Crohn’s disease in the Quebec Founder Population identifies multiple validated disease loci. Proc Natl Acad Sci U S A. 2007;104:14747–52. doi: 10.1073/pnas.0706645104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Di Meglio P, Di Cesare A, Laggner U, et al. The IL23R R381Q gene variant protects against immune-mediated diseases by impairing IL-23-induced Th17 effector response in humans. PLoS One. 2011;6:e17160. doi: 10.1371/journal.pone.0017160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Sarin R, Wu X, Abraham C. Inflammatory disease protective R381Q IL23 receptor polymorphism results in decreased primary CD4+ and CD8+ human T-cell functional responses. Proc Natl Acad Sci U S A. 2011;108:9560–5. doi: 10.1073/pnas.1017854108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Pidasheva S, Trifari S, Phillips A, et al. Functional studies on the IBD susceptibility gene IL23R implicate reduced receptor function in the protective genetic variant R381Q. PLoS One. 2011;6:e25038. doi: 10.1371/journal.pone.0025038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Sandborn WJ, Feagan BG, Fedorak RN, et al. A Randomized Trial of Ustekinumab, a Human Interleukin-12/23 Monoclonal Antibody, in Patients With Moderate-to-Severe Crohn’s Disease. Gastroenterology. 2008;135:1130–41. doi: 10.1053/j.gastro.2008.07.014. [DOI] [PubMed] [Google Scholar]
- 127.Sands B, Chen J, Penney M, et al. A randomized, double-blind placebo-controlled phase 2a induction study of MEDI2070 (anti-p19 antibody) in patients with active Crohn’s disease who have failed anti-TNF antibody therapy. J Crohns Colitis. 2015;9:S15–6. [Google Scholar]
- 128.Kakuta Y, Kinouchi Y, Negoro K, Takahashi S, Shimosegawa T. Association study of TNFSF15 polymorphisms in Japanese patients with inflammatory bowel disease. Gut. 2006;55:1527–8. doi: 10.1136/gut.2006.100297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Yang SK, Lim J, Chang HS, et al. Association of TNFSF15 with Crohn’s disease in Koreans. Am J Gastroenterol. 2008;103:1437–42. doi: 10.1111/j.1572-0241.2007.01752.x. [DOI] [PubMed] [Google Scholar]
- 130.Hong SN, Park C, Park SJ, et al. Deep resequencing of 131 Crohn’s disease associated genes in pooled DNA confirmed three reported variants and identified eight novel variants. Gut. 2016;65:788–96. doi: 10.1136/gutjnl-2014-308617. [DOI] [PubMed] [Google Scholar]
- 131.Thiebaut R, Kotti S, Jung C, et al. TNFSF15 polymorphisms are associated with susceptibility to inflammatory bowel disease in a new European cohort. Am J Gastroenterol. 2009;104:384–91. doi: 10.1038/ajg.2008.36. [DOI] [PubMed] [Google Scholar]
- 132.Latiano A, Palmieri O, Latiano T, et al. Investigation of multiple susceptibility loci for inflammatory bowel disease in an Italian cohort of patients. PLoS One. 2011;6:e22688. doi: 10.1371/journal.pone.0022688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Slebioda TJ, Kmiec Z. Tumour necrosis factor superfamily members in the pathogenesis of inflammatory bowel disease. Mediators Inflamm. 2014;2014:325129. doi: 10.1155/2014/325129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Hedl M, Abraham C. A TNFSF15 disease-risk polymorphism increases pattern-recognition receptor-induced signaling through caspase-8-induced IL-1. Proc Natl Acad Sci U S A. 2014;111:13451–6. doi: 10.1073/pnas.1404178111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Yang DH, Yang SK, Song K, et al. TNFSF15 is an independent predictor for the development of Crohn’s disease-related complications in Koreans. J Crohns Colitis. 2014;8:1315–26. doi: 10.1016/j.crohns.2014.04.002. [DOI] [PubMed] [Google Scholar]
- 136.Takedatsu H, Michelsen KS, Wei B, et al. TL1A (TNFSF15) regulates the development of chronic colitis by modulating both T-helper 1 and T-helper 17 activation. Gastroenterology. 2008;135:552–67. doi: 10.1053/j.gastro.2008.04.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137*.Goyette P, Boucher G, Mallon D, et al. High-density mapping of the MHC identifies a shared role for HLA-DRB1*01:03 in inflammatory bowel diseases and heterozygous advantage in ulcerative colitis. Nat Genet. 2015;47:172–9. doi: 10.1038/ng.3176. This recent large-scaled high-density mapping of the MHC region implicates the role of multiple HLA alleles in both Crohn’s disease and ulcerative colitis, with a primary role for HLA-DRB1*01:03. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Collins FS, Varmus H. A new initiative on precision medicine. N Engl J Med. 2015;372:793–5. doi: 10.1056/NEJMp1500523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Gerich ME, McGovern DP. Towards personalized care in IBD. Nat Rev Gastroenterol Hepatol. 2014;11:287–99. doi: 10.1038/nrgastro.2013.242. [DOI] [PubMed] [Google Scholar]
- 140.Ng PC, Murray SS, Levy S, Venter JC. An agenda for personalized medicine. Nature. 2009;461:724–6. doi: 10.1038/461724a. [DOI] [PubMed] [Google Scholar]
- 141.Plevy S, Silverberg MS, Lockton S, et al. Combined serological, genetic, and inflammatory markers differentiate non-IBD, Crohn’s disease, and ulcerative colitis patients. Inflamm Bowel Dis. 2013;19:1139–48. doi: 10.1097/MIB.0b013e318280b19e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Ahmad T, Armuzzi A, Bunce M, et al. The molecular classification of the clinical manifestations of Crohn’s disease. Gastroenterology. 2002;122:854–66. doi: 10.1053/gast.2002.32413. [DOI] [PubMed] [Google Scholar]
- 143.Brant SR, Picco MF, Achkar JP, et al. Defining complex contributions of NOD2/CARD15 gene mutations, age at onset, and tobacco use on Crohn’s disease phenotypes. Inflamm Bowel Dis. 2003;9:281–289. doi: 10.1097/00054725-200309000-00001. [DOI] [PubMed] [Google Scholar]
- 144.Newman B, Silverberg MS, Gu X, et al. CARD15 and HLA DRB1 alleles influence susceptibility and disease localization in Crohn’s disease. Am J Gastroenterol. 2004;99:306–15. doi: 10.1111/j.1572-0241.2004.04038.x. [DOI] [PubMed] [Google Scholar]
- 145.Cleynen I, Gonzalez JR, Figueroa C, et al. Genetic factors conferring an increased susceptibility to develop Crohn’s disease also influence disease phenotype: results from the IBDchip European Project. Gut. 2013;62:1556–65. doi: 10.1136/gutjnl-2011-300777. [DOI] [PubMed] [Google Scholar]
- 146.Adler J, Rangwalla SC, Dwamena BA, Higgins PD. The prognostic power of the NOD2 genotype for complicated Crohn’s disease: a meta-analysis. Am J Gastroenterol. 2011;106:699–712. doi: 10.1038/ajg.2011.19. [DOI] [PubMed] [Google Scholar]
- 147**.Cleynen I, Boucher G, Jostins L, et al. Inherited determinants of Crohn’s disease and ulcerative colitis phenotypes: a genetic association study. Lancet. 2016;387:156–67. doi: 10.1016/S0140-6736(15)00465-1. Using gene risk scoring system, this large-scaled multinational study shows that inflammatory bowel disease could be much better explained by three groups (ileal Crohn’s disease, colonic Crohn’s disease, and ulcerative colitis) rather than by Crohn’s disease and ulcerative colitis. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Amre DK, Mack DR, Morgan K, et al. Autophagy gene ATG16L1 but not IRGM is associated with Crohn’s disease in Canadian children. Inflamm Bowel Dis. 2009;15:501–7. doi: 10.1002/ibd.20785. [DOI] [PubMed] [Google Scholar]
- 149.Fowler EV, Doecke J, Simms LA, et al. ATG16L1 T300A shows strong associations with disease subgroups in a large Australian IBD population: further support for significant disease heterogeneity. Am J Gastroenterol. 2008;103:2519–26. doi: 10.1111/j.1572-0241.2008.02023.x. [DOI] [PubMed] [Google Scholar]
- 150.Latiano A, Palmieri O, Cucchiara S, et al. Polymorphism of the IRGM gene might predispose to fistulizing behavior in Crohn’s disease. Am J Gastroenterol. 2009;104:110–6. doi: 10.1038/ajg.2008.3. [DOI] [PubMed] [Google Scholar]
- 151.Okazaki T, Wang MH, Rawsthorne P, et al. Contributions of IBD5, IL23R, ATG16L1, and NOD2 to Crohn’s disease risk in a population-based case-control study: evidence of gene-gene interactions. Inflamm Bowel Dis. 2008;14:1528–41. doi: 10.1002/ibd.20512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Satsangi J, Welsh KI, Bunce M, et al. Contribution of genes of the major histocompatibility complex to susceptibility and disease phenotype in inflammatory bowel disease. Lancet. 1996;347:1212–7. doi: 10.1016/s0140-6736(96)90734-5. [DOI] [PubMed] [Google Scholar]
- 153.Silverberg MS, Mirea L, Bull SB, et al. A population- and family-based study of Canadian families reveals association of HLA DRB1*0103 with colonic involvement in inflammatory bowel disease. Inflamm Bowel Dis. 2003;9:1–9. doi: 10.1097/00054725-200301000-00001. [DOI] [PubMed] [Google Scholar]
- 154.Haritunians T, Taylor KD, Targan SR, et al. Genetic predictors of medically refractory ulcerative colitis. Inflamm Bowel Dis. 2010;16:1830–40. doi: 10.1002/ibd.21293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Dubinsky MC, Kugathasan S, Kwon S, et al. Multidimensional prognostic risk assessment identifies association between IL12B variation and surgery in Crohn’s disease. Inflamm Bowel Dis. 2013;19:1662–70. doi: 10.1097/MIB.0b013e318281f275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Siegel CA, Horton H, Siegel LS, et al. A validated web-based tool to display individualised Crohn’s disease predicted outcomes based on clinical, serologic and genetic variables. Aliment Pharmacol Ther. 2016;43:262–71. doi: 10.1111/apt.13460. [DOI] [PubMed] [Google Scholar]
- 157.Chouchana L, Narjoz C, Beaune P, Loriot MA, Roblin X. Review article: the benefits of pharmacogenetics for improving thiopurine therapy in inflammatory bowel disease. Aliment Pharmacol Ther. 2012;35:15–36. doi: 10.1111/j.1365-2036.2011.04905.x. [DOI] [PubMed] [Google Scholar]
- 158*.Yang SK, Hong M, Baek J, et al. A common missense variant in NUDT15 confers susceptibility to thiopurine-induced leukopenia. Nat Genet. 2014;46:1017–20. doi: 10.1038/ng.3060. A novel NUDT15 variant (p.Arg139Cys) was discovered to be a significantly associated with thiopurine-induced leukopenia in patients with inflammatory bowel disease, especially in Koreans. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Colombel JF, Ferrari N, Debuysere H, et al. Genotypic analysis of thiopurine S-methyltransferase in patients with Crohn’s disease and severe myelosuppression during azathioprine therapy. Gastroenterology. 2000;118:1025–30. doi: 10.1016/s0016-5085(00)70354-4. [DOI] [PubMed] [Google Scholar]
- 160.Dewit O, Moreels T, Baert F, et al. Limitations of extensive TPMT genotyping in the management of azathioprine-induced myelosuppression in IBD patients. Clin Biochem. 2011;44:1062–6. doi: 10.1016/j.clinbiochem.2011.06.079. [DOI] [PubMed] [Google Scholar]
- 161.Kakuta Y, Naito T, Onodera M, et al. NUDT15 R139C causes thiopurine-induced early severe hair loss and leukopenia in Japanese patients with IBD. Pharmacogenomics J. doi: 10.1038/tpj.2015.43. (in press) [DOI] [PubMed] [Google Scholar]
- 162.Asada A, Nishida A, Shioya M, et al. NUDT15 R139C-related thiopurine leukocytopenia is mediated by 6-thioguanine nucleotide-independent mechanism in Japanese patients with inflammatory bowel disease. J Gastroenterol. 2016;51:22–9. doi: 10.1007/s00535-015-1142-4. [DOI] [PubMed] [Google Scholar]
- 163.Heap GA, Weedon MN, Bewshea CM, et al. HLA-DQA1-HLA-DRB1 variants confer susceptibility to pancreatitis induced by thiopurine immunosuppressants. Nat Genet. 2014;46:1131–4. doi: 10.1038/ng.3093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Jurgens M, Laubender RP, Hartl F, et al. Disease activity, ANCA, and IL23R genotype status determine early response to infliximab in patients with ulcerative colitis. Am J Gastroenterol. 2010;105:1811–9. doi: 10.1038/ajg.2010.95. [DOI] [PubMed] [Google Scholar]
- 165.Hlavaty T, Pierik M, Henckaerts L, et al. Polymorphisms in apoptosis genes predict response to infliximab therapy in luminal and fistulizing Crohn’s disease. Aliment Pharmacol Ther. 2005;22:613–26. doi: 10.1111/j.1365-2036.2005.02635.x. [DOI] [PubMed] [Google Scholar]
- 166.Hlavaty T, Ferrante M, Henckaerts L, Pierik M, Rutgeerts P, Vermeire S. Predictive model for the outcome of infliximab therapy in Crohn’s disease based on apoptotic pharmacogenetic index and clinical predictors. Inflamm Bowel Dis. 2007;13:372–9. doi: 10.1002/ibd.20024. [DOI] [PubMed] [Google Scholar]
- 167.Arijs I, Li K, Toedter G, et al. Mucosal gene signatures to predict response to infliximab in patients with ulcerative colitis. Gut. 2009;58:1612–9. doi: 10.1136/gut.2009.178665. [DOI] [PubMed] [Google Scholar]
- 168.Arijs I, Quintens R, Van Lommel L, et al. Predictive value of epithelial gene expression profiles for response to infliximab in Crohn’s disease. Inflamm Bowel Dis. 2010;16:2090–8. doi: 10.1002/ibd.21301. [DOI] [PubMed] [Google Scholar]
- 169.Dubinsky MC, Mei L, Friedman M, et al. Genome wide association (GWA) predictors of anti-TNFalpha therapeutic responsiveness in pediatric inflammatory bowel disease. Inflamm Bowel Dis. 2010;16:1357–1366. doi: 10.1002/ibd.21174. [DOI] [PMC free article] [PubMed] [Google Scholar]