

Abstract
Unfrozen archived newborn blood spots (NBS) have been shown to retain sufficient messenger RNA (mRNA) for gene expression profiling. However, the effect of storage time at ambient temperature for NBS samples in relation to the quality of gene expression data is relatively unknown. Here, we evaluated mRNA expression from quantitative real-time PCR (qRT-PCR) and microarray data obtained from NBS samples stored at ambient temperature to determine the effect of storage time on the quality of gene expression. These data were generated in a previous case-control study examining NBS in 53 children with cerebral palsy (CP) and 53 matched controls. NBS sample storage period ranged from 3 to 16 years at ambient temperature. We found persistently low RNA integrity numbers (RIN = 2.3 ± 0.71) and 28S/18S rRNA ratios (∼0) across NBS samples for all storage periods. In both qRT-PCR and microarray data, the expression of three common housekeeping genes—beta cytoskeletal actin (ACTB), glyceraldehyde 3-phosphate dehydrogenase (GAPDH), and peptidylprolyl isomerase A (PPIA)—decreased with increased storage time. Median values of each microarray probe intensity at log2 scale also decreased over time. After eight years of storage, probe intensity values were largely reduced to background intensity levels. Of 21,500 genes tested, 89% significantly decreased in signal intensity, with 13,551, 10,730, and 9,925 genes detected within 5 years, > 5 to < 10 years, and > 10 years of storage, respectively. We also examined the expression of two gender-specific genes (X inactivation-specific transcript, XIST and lysine-specific demethylase 5D, KDM5D) and seven gene sets representing the inflammatory, hypoxic, coagulative, and thyroidal pathways hypothesized to be related to CP risk to determine the effect of storage time on the detection of these biologically relevant genes. We found the gender-specific genes and CP-related gene sets detectable in all storage periods, but exhibited differential expression (between male vs. female or CP vs. control) only within the first six years of storage. We concluded that gene expression data quality deteriorates in unfrozen archived NBS over time and that differential gene expression profiling and analysis is recommended for those NBS samples collected and stored within six years at ambient temperature.
Keywords: mRNA gene expression, quantitative real-time PCR, microarray, newborn blood spots, storage time, ambient temperature
1. Introduction
Newborn blood spotted on filter paper is used throughout the United States and in most industrialized countries for newborn genetic screening. Archives of leftover newborn blood spot (NBS) samples are available in many states [1]. Even after many years of unfrozen storage, mRNA can be extracted from NBS [2–4] and analysis of mRNA phenotype can distinguish individuals with different thalassemia genotypes using archived NBS samples [5]. In our previous studies, we showed that mRNA extracted from unfrozen archived NBS can be applied on microarrays to perform genome-wide gene expression profiling, even though the yield of RNA declined over storage time of NBS samples [6]. We also showed that differential expression could be detected between genders [7] and between cerebral palsy (CP) cases and matched controls [8]. Thus, archived NBS samples can serve as an essential research resource to examine gene expression profiles in the newborn period and possible relation to later child health.
NBS collection differs from most human blood collection methods in that no collecting tube is used, but blood is spotted directly, usually from a heel prick, onto filter paper and dries in minutes. The absence of a liquid environment appears to reduce the activity of ribonucleases and micro RNAs that degrade mRNA, and most mRNA species within the cell are quickly degraded after being transcribed. Some loss of mRNA is likely, especially in older blood spot samples, and this may affect the quality of data such as gene expression profiles obtained from these samples. mRNA degradation is especially possible if blood spots are stored at ambient conditions [9].
A commonly used method for assessing RNA quality is the RNA integrity number (RIN), which ranges from 1 (indicating severe mRNA degradation) to 10 (indicating high quality, largely intact mRNA) [10]. Another common approach for RNA quality assessment is examining the 28S/18S ribosomal RNA (rRNA) ratio. This approach assumes that rRNA quality and quantity reflect that of the underlying mRNA population and a ratio of 2:1 is considered the benchmark for intact RNA [11,12].
Although RIN and 28S/18S rRNA ratios are useful proxies for assessing RNA quality, they are indirect measures of mRNA and do not directly describe the quality and quantity of mRNA. Low scores on these measurements do not necessarily preclude the detection of biologically-relevant, differentially-expressed genes between experimental and control groups.
In this study, we addressed two topics related to assessing the quality of mRNA microarray data obtained from unfrozen archived NBS samples. First, we evaluated whether mRNA microarray data quality reflects the yield of mRNA or level of mRNA degradation with respect to NBS storage period. Second, we assessed the detectability of differentially-expressed genes between genders and CP vs. controls from mRNA microarray data, also based on NBS storage period. Our study will provide the optimal cutoff for a recommended ambient storage period of NBS samples to maximize the benefit of using these samples for microarray gene expression profiling studies.
2. Materials and methods
2.1. Study subjects
Our mRNA microarray dataset was acquired from unfrozen archived NBS of 53 singleton CP cases and 53 matched controls (similar year of birth, sex and gestational age) included in our previous case-control study to investigate the molecular etiology of CP [8]. Among the 106 study subjects, 31 were females and 75 were males, with an NBS storage period ranging from 2.9 – 16 years. In 2009, Michigan Department of Community Health (MDCH) began storing all newly collected NBS at −20 °C. However, our study subjects were born within 1994 – 2007 and NBS samples were retrieved from the archive in 2008 (stored at ambient temperature without climate control). These NBS samples were retrieved from MDCH upon receipt of written informed consent from parents or guardians. This project was approved by the institutional review boards of all participating institutions.
2.2. RNA isolation, microarray and qRT-PCR assays
Total RNA was extracted from three 3 mm punches of NBS using the illustra RNAspin Mini RNA kit (GE Healthcare). Due to large sample size, all spots were not extracted at the same time but in batches of 24. Extracted mRNA was treated the same way for all spots prior to analysis. To minimize variables, all assays were performed by a single technician and extracted mRNA was immediately stored at −80 °C until the next procedure. The Agilent Whole Human Genome Gene Expression 8 × 60 K Microarray platform was used for microarray assays. Quantitative real-time PCR (qRT-PCR) was performed using Applied Biosystems 7500 Fast Real-Time PCR System. Details regarding sample processing, RNA isolation, microarray and qRT-PCR techniques were published in our previous study [8].
2.3. Assessment of mRNA microarray data quality
We examined the following four features to assess mRNA microarray data:
Distribution of RIN and 28S/18S rRNA ratios across storage periods of NBS samples.
Overall distribution of raw fluorescent probe intensity data across storage periods of NBS samples, illustrated by brightness of raw digital images as well as density and scatter plots of log2 signal intensity.
Linear regression slopes of log2 signal intensity across storage periods of NBS using raw aggregated gene expression data for all genes available in the microarrays.
Number of aggregated genes being filtered and number of remaining genes after filtering, in correspondence with storage period of NBS samples.
We employed the commonly used approach for filtering unqualified spots of microarray data by Paterson et al. in which probe intensity was removed when the gProcessed signal was less than two times the gProcessed signal error [13]. We applied quantile normalization on filtered microarray data with a slight modification, which was stratifying the storage period of NBS samples. Probe intensity data was then aggregated to a gene level using the average values of log2 signal intensity data for each gene with multiple probes. R limma package [14] was used for microarray data processing.
2.4. Assessment of housekeeping gene expression based on NBS storage period
To further evaluate the pattern of mRNA expression signals over storage period of NBS samples, we examined the signal intensities of three commonly used housekeeping genes—peptidylprolyl isomerase A (PPIA), beta cytoskeletal actin (ACTB), and glyceraldehyde 3-phosphate dehydrogenase (GAPDH)—in microarray and qRT-PCR data.
2.5. Evaluation on the power of detecting differentially-expressed genes in NBS samples stored across 3 – 16 years at ambient temperature
We also assessed the impact of storage period on the power of detecting differentially-expressed genes in NBS samples. First, we examined the effect of storage period on detecting two sex-linked genes—X inactivation-specific transcript, XIST and lysine-specific demethylase 5D, KDM5D—that are known to be differentially-regulated between male and female samples [7]. A linear model for microarray data implemented in the R limma package [14] was used to analyze differential expression of these two genes. Second, we examined the effect of storage period on detecting seven gene sets—four empirical and three canonical gene sets representing the inflammatory, hypoxic, coagulative, and thyroidal pathways—hypothesized to be differentially-expressed in newborns who later developed CP or without CP. GAGE (generally applicable gene set enrichment for pathway analysis) [15] was applied for gene set statistical analysis, as described in our previous study [8]. GAGE t-statistics of each matched case-control pair were standardized by converting to the equivalent z-statistics for empirical inflammatory, hypoxic, and thyroidal gene sets, using Stouffer's method [16], which showed significant differential expression between CP and controls in our previous study. The absolute values of these converted z-statistics were then plotted against storage period of NBS samples for each case-control pair. All analyses were performed using R version 2.13.2.
3. Results
3.1. mRNA microarray and qRT-PCR data quality in relation with storage period of NBS samples
3.1.1. Variation in RIN and 28S/18S rRNA ratio
Average RIN in 106 NBS samples was 2.3 ± 0.71 and RIN values did not vary significantly over storage period of 3 – 16 years at ambient temperature (Figure 1a). The overall 28S/18S rRNA ratio was zero (presumably reflecting the absence of 28S rRNA peaks) and similar with RIN, the ratio showed no time-related trend (data not shown).
Figure 1.
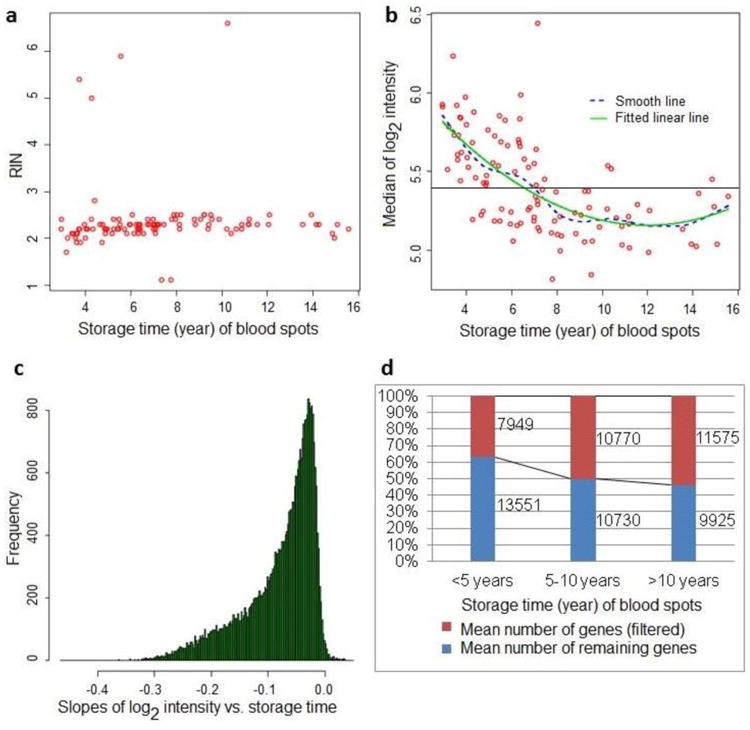
RNA and gene expression microarray data quality in NBS samples stored for 3 to 16 years at ambient temperature. a. Distribution of RIN (RNA integrity number) across storage time (year) of blood spots; b. Median values of log2 probe intensity in microarray data acquired from NBS samples stored for 3 to 16 years (background intensity levels are below the horizontal line); c. Linear regression slopes of log2 signal intensity for all genes plotted against storage period of NBS after filtering and aggregated to a gene level; d. Number of genes being filtered and number of remaining genes after filtering by NBS storage periods.
3.1.2. Fluorescent probe intensity of raw microarray data
Direct visual inspection of the raw digital image of microarray is part of the quality control procedure to ensure that overall fluorescent probe intensity of mRNA is not affected by image artifacts. In general, visual inspection showed “brighter” microarray images in newer NBS samples compared with older samples (Figure S1). This observation is confirmed with density plots of log2-transformed raw (non-normalized) probe intensity data showing higher signal intensities in NBS with shorter storage periods (Figure S2). The median values of log2 probe intensity/unfiltered gene expression declined significantly and linearly over time (p < 0.0001). The average median values of log2 signal intensity in raw microarray data before filtering declined from 5.8 in NBS samples within 5 years of storage to 5.2 in NBS samples at 5 – 10 years of storage, with little or no further decline after 10 years of storage at ambient temperature. In addition, median values of log2 signal intensity were reduced to background intensity levels for the majority of NBS samples older than 8 years of storage (Figure 1b).
After raw probe signal was aggregated to a gene level, log2 signal intensity of each gene was regressed against the duration of storage. Detection of hemoglobin subunit alpha 2 (HBA2) and hemoglobin subunit beta (HBB) genes declined significantly over time with linear regression slopes of −0.18 (p = 0.0003) and −0.25 (p < 0.0001), respectively (Figure S3). Since these NBS samples were processed without globin depletion, globins remain the most abundant genes, with log2 signal intensity > 12 even in NBS samples stored for more than 10 years at ambient temperature.
After filtering, log2 signal intensity/gene expression of the majority of genes declined over storage time. The linear regression slopes were negative for the majority of genes (median slope = −0.06), with −0.37 as the most negative slope (Figure 1c). Of 21,500 genes tested, 89% significantly declined with storage time at ambient temperature (negative slope p < 0.05).
3.1.3. Number of detected genes in various NBS storage periods
We examined the number of genes in the microarray data that showed signal intensity either above or within the background intensity level. After filtering, the number of genes within the background level increased linearly with storage time (p < 0.0001), with approximately 35%, 50% and 55% of genes within background intensity for storage up to 5 years, 5 – 10 years, and more than 10 years, respectively. For the same storage periods, of 21,500 genes available in the arrays, the mean number of genes above background intensity was 13,551, 10,730 and 9,925, respectively (Figure 1d).
3.1.4. Expression of housekeeping genes in microarray and qPCR data
We found log2 signal intensity of ACTB, PPIA, and GAPDH to be significantly decreased over time in the microarray data (linear regression slopes = −0.23, −0.15, and −0.14, respectively; p < 0.0001 for all) (Figure 2 upper panel). In addition, the mean qRT-PCR cycle threshold (i.e. number of PCR cycles required for signal detection; cycle threshold is inversely proportional to the relative expression in the gene of interest) was found to be significantly increased over NBS sample storage time (Figure 2 lower panel).
Figure 2.
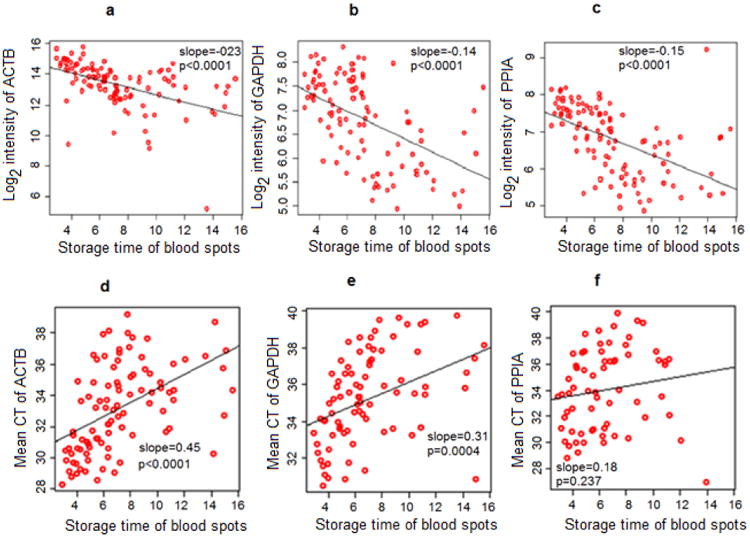
Expression of three commonly used housekeeping genes over NBS sample storage time (in year) from microarray and qRT-PCR data. Upper panel (microarray gene expression data): a. ACTB gene expression; b. GAPDH gene expression; c. PPIA gene expression. Lower panel (qRT-PCR data): d. Mean CT value of ACTB; e. Mean CT value of GAPDH; f. Mean CT value of PPIA. qRT-PCR: quantitative real time polymerase chain reaction; ACTB: beta cytoskeletal actin; GAPDH: glyceraldehyde 3-phosphate dehydrogenase; PPIA: peptidylprolyl isomerase A; CT: cycle threshold.
3.2. Detecting differential gene expression in NBS stored at ambient temperature over time
3.2.1. Detecting differential expression of gender-specific genes over time
Previously, we showed that gender-specific genes can be detected and differentially-expressed in NBS samples [7]. Here, we evaluated XIST and KDM5D gene expression changes in NBS samples over time. XIST is expressed exclusively in female samples as part of the X chromosome inactivation process, and thus, XIST expression in female samples is expected to be distinctly higher than that of male samples. From our NBS data, we found XIST gene expression at 41th percentile of transcription levels in male samples and 70th percentile in female samples. The mean log2 signal intensity of XIST in female NBS samples was significantly higher than that of male NBS samples (p < 0.0001). Log2 signal intensity of XIST significantly decreased over storage time in both female (slope = −0.14, p = 0.0012) and male (slope = −0.02, p = 0.0226) samples. However, signal intensity values declined more dramatically in female than male samples (Figure 3a). Log2 signal intensity of XIST in female NBS samples started to overlap with those in male samples > 6 years of storage. Differences in mean log2 signal intensity (Δ mean) between female vs. male were 1.58 and 1.05 for ≤ 6 years of storage and > 6 years of storage, respectively (Figure 3a).
Figure 3.
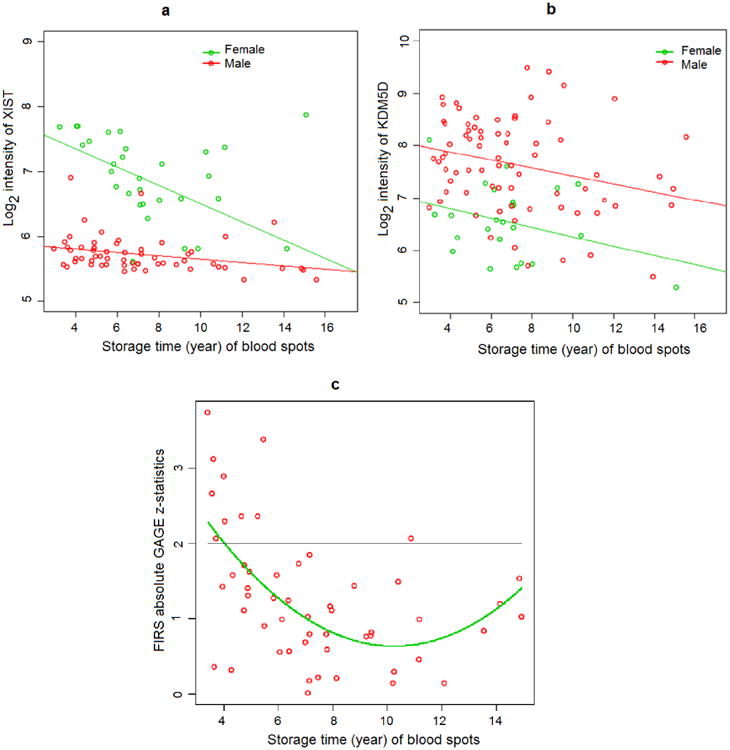
Effect of NBS sample storage time (in year) on detecting differential gene expression in microarray. a. Signal intensities of XIST in male and female NBS samples; b. Signal intensities of KDM5D in male and female NBS samples; c. Absolute values of GAGE z-statistics for the FIRS gene set in matched case-control pairs of NBS samples. XIST: X inactivation-specific transcript; KDM5D: lysine-specific demethylase 5D; FIRS: fetal inflammatory response syndrome; GAGE: generally applicable gene set enrichment for pathway analysis.
KDM5D is mapped on the Y chromosome and gene expression of KDM5D is detected in all male tissues. Thus, the expression signal of KDM5D is expected to be higher in male than female samples. From our NBS data, we found KDM5D gene expression at the 80th percentile of transcription levels in male samples and 68th percentile in female samples. Log2 signal intensity of KDM5D significantly decreased over time in male (slope = −0.08, p = 0.0205) but not female NBS samples (slope= −0.09, p = 0.115), probably due to a smaller female sample size (Figure 3b).
3.2.2. Detecting differential expression of gene sets over time
In our previous study, we hypothesized that gene sets representing hypoxia, coagulation, thyroid hormone function, and inflammation pathways would be dysregulated in infants who later developed CP [8]; fetal inflammatory response syndrome (FIRS) gene set [17] showed the greatest differential expression between CP and healthy controls. Here, by comparing cases to controls, we found that absolute values in z-statistics declined substantially over time (slope = −0.72, p < 0.001) and the majority of significant differences (absolute values in z-statistics > 1.96) was found in NBS samples < 6 years of storage (Figure 3c). The Chi-square test for global significance in z-statistics between case-control pairs for the FIRS gene set was statistically significant in NBS samples ≤ 6 years of storage (p < 0.0001) and not significant in NBS samples > 6 years of storage (p = 0.43). On the other hand, empirical hypoxia and thyroidal gene sets—two other gene sets that were significantly up-regulated in preterm-born CP compared to healthy controls—did not show any significant difference in z-statistics over NBS storage time. After filtering, the number of genes detected in our seven gene sets that reflect four pathophysiological pathways to CP also declined significantly and linearly with storage duration (p < 0.0001) (Figure 4).
Figure 4.
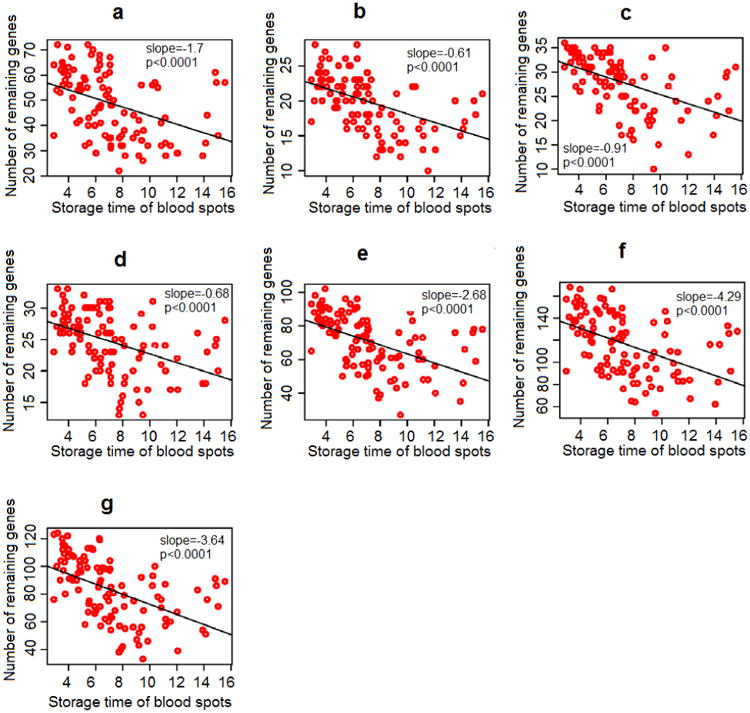
Number of genes after filtering in 7 gene sets across storage period of 3 to 16 years. a. Canonical coagulation gene set; b. Canonical inflammation gene set; c. Empirical inflammation (or fetal inflammatory response syndrome (FIRS)) gene set; d. Canonical hypoxia gene set; e. Empirical hypoxia gene set; f. Empirical thyroidal gene set; g. Canonical thyroidal gene set.
4. Discussion
Zero 28S/18S ratio in the majority of NBS samples was primarily due to the absence of 28S peak data. 28S/18S ratio is an indirect measurement of mRNA degradation since it is based on the availability of rRNA. Although rRNA accounts for more than 80% of RNA in cells, it is unclear how rRNA reflects the quality of an underlying mRNA population within NBS samples. Some studies suggest that the correlation between 28S/18S ratio and mRNA integrity is weak [18,19]. Also, the consistently low RIN scores suggested mRNA degradation occurred in NBS samples stored between 3 to 16 years at ambient temperature. While RIN may describe the relative quality of mRNA, it cannot by itself reveal how mRNA degradation affects the detection of gene expression in microarray or qRT-PCR technique. Here, we demonstrated that low 28S/18S ratio and RIN scores did not necessarily preclude detection of mRNA species, as it was possible to detect mRNA gene expression in NBS samples up to 16 years of storage at ambient temperature.
Microarray data acquired from archived NBS samples stored at ambient temperature showed a consistent and nearly linear decline of mRNA expression signal with increased storage periods. After approximately 8 years of storage, median values of log2 probe intensity decreased to background intensity levels for most of the samples. Expression for the majority of genes also reduced significantly over time. This inclination was not an artifact from microarray processes as the expression of three common housekeeping genes (ACTB, PPIA and GAPDH) was also reduced over time when validated with qRT-PCR. Since globins, the most abundant proteins, were not removed from our samples, we also investigated their gene expression changes over time. Although the gene expression of HBA2 and HBB declined significantly over time, their log2 signal intensity remained high (> 12) after 10 years of storage. On the other hand, genes with lower abundance exhibited reduced signal intensities to that of background levels after 10 years.
In older NBS samples with overall signal intensities that are lower compared to newer samples, one would presume that the power to detect differential expression of biologically-relevant genes will be reduced. In this study, differential expression of two gender-specific genes, XIST and KDM5D, could be distinctly detected in NBS samples stored up to 6 years at ambient temperature. Similarly, the FIRS gene set which was the most differentially-expressed between CP and healthy controls was found to be statistically significant only in NBS samples ≤ 6 years of storage. However, the empirical hypoxia and thyroidal gene sets, two other gene sets that were significantly up-regulated in CP, did not show a significant difference in z-statistics over the same NBS storage periods. This may be due to a smaller number of genes in the FIRS gene set (36 genes) compared with the empirical hypoxia and thyroidal gene sets (n = 127 and n = 140, respectively). Further study is needed to investigate the power of detection over time in relation to the number of genes in our gene set analysis. We showed that although mRNA can still be recovered from older blood spots, the power to detect differentially-expressed biologically-relevant genes may be limited to NBS samples stored within 6 years at ambient temperature.
In Michigan, NBS samples were archived at ambient temperature from 1987 to 2008. Since 2009, NBS samples have been stored at a controlled temperature storage of −20 °C and those prior to 2009 have since been moved to −20 °C storage. We have previously shown that storing NBS samples at −20 °C significantly improves the number of genes detected in the microarray platform [9]. For NBS samples stored at −20 °C, microarray gene expression data did not change significantly between samples collected 10 years apart [20]. Therefore, whenever possible, NBS samples are recommended for archival storage at −20 °C to maximize the retrieval of gene expression data.
5. Conclusions
In summary, gene expression data can be generated from mRNA of NBS samples stored for 3 – 16 years at ambient temperature. However, median values of log2 probe intensity in raw gene expression microarray data decreased over time, with values reduced to background intensity levels for the majority of NBS samples older than 8 years of storage. After filtering, the number of genes detected above background intensity decreased over time, with less than 10,000 genes detected in NBS samples > 10 years of storage. Expression of housekeeping genes also decreased over time. Biologically-relevant genes are significantly differentiated in NBS samples < 6 years of storage. Thus, to study differential gene expression using microarray, NBS samples stored within 6 years at ambient temperature are recommended.
Supplementary Material
Acknowledgments
We would like to thank the study subjects and the OWL (Origin, Wellness and Life History of Cerebral Palsy) team for providing the qRT-PCR and microarray data. We would also like to thank Kyle Furge for his input on the analysis of microarray data. This work was supported by the National Institutes of Health [grant number R01 NS055101]. NTH was partially funded by the Vietnam Education Foundation.
Footnotes
Conflict of interest: The authors state that there is no conflict of interest.
Authors' contributions: Conceived and designed the experiments: NTH, SKK and NP. Performed the experiments: SKK, JVB and JHR. Obtained and interpreted the data: NTH, SKK, JVB, JHR and NP. Wrote the manuscript: NTH, NP and SKK.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Olney RS, Moore CA, Ojodu JA, Lou Lindegren M, Hannon WH. Storage and use of residual dried blood spots from state newborn screening programs. J Pediatr. 2006;148:618–622. doi: 10.1016/j.jpeds.2005.12.053. [DOI] [PubMed] [Google Scholar]
- 2.Karlsson H, Guthenberg C, von Döbeln U, Kristenssson K. Extraction of RNA from dried blood on filter papers after long-term storage. [accessed June 9, 2016];Clin Chem. 2003 49:979–81. doi: 10.1373/49.6.979. http://www.ncbi.nlm.nih.gov/pubmed/12766003. [DOI] [PubMed] [Google Scholar]
- 3.Gauffin F, Nordgren A, Barbany G, Gustafsson B, Karlsson H. Quantitation of RNA decay in dried blood spots during 20 years of storage. Clin Chem Lab Med. 2009;47:1467–1469. doi: 10.1515/CCLM.2009.351. [DOI] [PubMed] [Google Scholar]
- 4.Matsubara Y, Ikeda H, Endo H, Narisawa K. Dried blood spot on filter paper as a source of mRNA. [accessed June 9, 2016];Nucleic Acids Res. 1992 20:1998. doi: 10.1093/nar/20.8.1998. http://www.ncbi.nlm.nih.gov/pubmed/1579508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Zhang YH, McCabe ER. RNA analysis from newborn screening dried blood specimens. [accessed June 9, 2016];Hum Genet. 1992 89:311–4. doi: 10.1007/BF00220548. http://www.ncbi.nlm.nih.gov/pubmed/1351035. [DOI] [PubMed] [Google Scholar]
- 6.Haak PT, Busik JV, Kort EJ, Tikhonenko M, Paneth N, Resau JH. Archived unfrozen neonatal blood spots are amenable to quantitative gene expression analysis. Neonatology. 2009;95:210–216. doi: 10.1159/000155652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Resau JH, Ho NT, Dykema K, Faber MS, Busik JV, Nickolov RZ, Furge KA, Paneth N, Jewell S, Khoo SK. Evaluation of sex-specific gene expression in archived dried blood spots (DBS) Int J Mol Sci. 2012;13:9599–608. doi: 10.3390/ijms13089599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ho NT, Furge K, Fu W, Busik J, Khoo SK, Lu Q, Lenski M, Wirth J, Hurvitz E, Dodge N, Resau J, Paneth N. Gene expression in archived newborn blood spots distinguishes infants who will later develop cerebral palsy from matched controls. Pediatr Res. 2013;73:450–6. doi: 10.1038/pr.2012.200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wei C, Lu Q, Khoo SK, Lenski M, Fichorova RN, Leviton A, Paneth N. Comparison of frozen and unfrozen blood spots for gene expression studies. J Pediatr. 2014;164 doi: 10.1016/j.jpeds.2013.09.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Schroeder A, Mueller O, Stocker S, Salowsky R, Leiber M, Gassmann M, Lightfoot S, Menzel W, Granzow M, Ragg T. The RIN: an RNA integrity number for assigning integrity values to RNA measurements. BMC Mol Biol. 2006;7:3. doi: 10.1186/1471-2199-7-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sambrook J, Russel DW. Molecular Cloning: A Laboratory Manual. 3rd. Cold Spring Harb. Lab. Press; Cold Spring Harb. NY: 2001. [Google Scholar]
- 12.Van de Goor TA. The principle and promise of Labchip technology. PharmaGenomics. 2003:16–18. [Google Scholar]
- 13.Patterson Ta, Lobenhofer EK, Fulmer-Smentek SB, Collins PJ, Chu TM, Bao W, Fang H, Kawasaki ES, Hager J, Tikhonova IR, Walker SJ, Zhang L, Hurban P, de Longueville F, Fuscoe JC, Tong W, Shi L, Wolfinger RD. Performance comparison of one-color and two-color platforms within the MicroArray Quality Control (MAQC) project. Nat Biotechnol. 2006;24:1140–1150. doi: 10.1038/nbt1242. [DOI] [PubMed] [Google Scholar]
- 14.Smith GK. limma: Linear Models for Microarray Data. Bioinforma Comput Biol Solut Using R Bioconductor. 2005:397–420. doi:citeulike-article-id:5722720. [Google Scholar]
- 15.Luo W, Friedman MS, Shedden K, Hankenson KD, Woolf PJ. GAGE: generally applicable gene set enrichment for pathway analysis. BMC Bioinformatics. 2009;10:161. doi: 10.1186/1471-2105-10-161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Riley JW, Stouffer SA, Suchman EA, Devinney LC, Star SA, Williams RM. The American Soldier: Adjustment During Army Life. Am Sociol Rev. 1949;14:557. doi: 10.2307/2087216. [DOI] [Google Scholar]
- 17.Madsen-Bouterse SA, Romero R, Tarca AL, Kusanovic JP, Espinoza J, Kim CJ, Kim JS, Edwin SS, Gomez R, Draghici S. The transcriptome of the fetal inflammatory response syndrome. Am J Reprod Immunol. 2010;63:73–92. doi: 10.1111/j.1600-0897.2009.00791.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Miller CL, Diglisic S, Leister F, Webster M, Yolken RH. Evaluating RNA status for RT-PCR in extracts of postmortem human brain tissue. Biotechniques. 2004;36:628–633. doi: 10.2144/04364ST03. [DOI] [PubMed] [Google Scholar]
- 19.Imbeaud S, Graudens E, Boulanger V, Barlet X, Zaborski P, Eveno E, Mueller O, Schroeder A, Auffray C. Towards standardization of RNA quality assessment using user-independent classifiers of microcapillary electrophoresis traces. Nucleic Acids Res. 2005;33:1–12. doi: 10.1093/nar/gni054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Grauholm J, Khoo SK, Nickolov RZ, Poulsen JB, Bækvad-Hansen M, Hansen CS, Hougaard DM, Hollegaard MV. Gene expression profiling of archived dried blood spot samples from the Danish Neonatal Screening Biobank. Mol Genet Metab. 2015;116:119–124. doi: 10.1016/j.ymgme.2015.06.011. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.