

Abstract
Purpose
Hemophilia B, an X-linked disease, manifests with recurrent soft tissue bleeding episodes. Hermansky-Pudlak syndrome, a rare autosomal recessive disorder, is characterized by oculocutaneous albinism and an increased tendency to bleed due to a platelet storage pool defect. We report a novel mutation in HPS6 in a Caucasian man with hemophilia B and oculocutaneous albinism.
Results
The patient was diagnosed with hemophilia B at age 4 months due to recurrent soft tissue bleeding episodes, and he was also diagnosed with Hermansky-Pudlak syndrome at 32 years of age due to unexplained oculocutaneous albinism. His factor IX level was markedly reduced at 13%; whole exome and Sanger sequencing showed the Durham mutation in F9 (NM_000133.3). The diagnosis of Hermansky-Pudlak syndrome subtype 6 was established by demonstrating absence of platelet delta granules on whole mount electron microscopy, an abnormal secondary wave in platelet aggregation studies, and a novel homozygous c.1114 C>T (p.Arg372*) mutation in HPS6 (NM_024747.5) on exome analysis and Sanger sequencing. Clinical phenotyping revealed no evidence of recurrent or unusual infections, interstitial lung disease or pulmonary fibrosis, or neurological disorders. The patient was treated with fresh frozen plasma, recombinant factor IX, and aminocaproic acid. Treatment with desmopressin was added to his regimen after he was diagnosed with Hermansky-Pudlak syndrome. Treatment of bleeding episodes results in effective hemostasis, and the patient has not required platelet or blood product transfusions.
Conclusions
This report highlights the need to consider Hermansky-Pudlak syndrome as an etiology of oculocutaneous albinism even in patients with known hematologic disorders associated with bleeding. Identification of a novel mutation in HPS6 in an individual with hemophilia B shows that, although quite rare, patients may be diagnosed with two independent inherited bleeding disorders. No evidence of lung disease was found in this adult patient with Hermansky-Pudlak syndrome subtype 6.
Keywords: Hermansky-Pudlak syndrome, platelet storage pool disorder, oculocutaneous albinism, Christmas disease, Hemophilia B, Factor IX deficiency
1.1 INTRODUCTION
1.2 Hemophilia B (Christmas disease or Factor IX deficiency) is an X-linked bleeding disorder that is diagnosed by identifying mutations in F9 and low levels of factor IX. Hemophilia B has an estimated incidence of one in 25,000–30,000 male births and has no ethnic or geographic predilection [1]. Deficiency of factor IX predisposes affected individuals to recurrent hemorrhage, especially potentially serious soft tissue bleeding and hemarthroses. Oculocutaneous albinism is not a feature of hemophilia B. Morbidity and mortality are related to life-threatening bleeding episodes, joint damage from recurrent hemarthroses, exposure to blood-borne pathogens, and hypersensitivity reactions to factor IX. Clinical management is aimed at prevention of serious bleeding episodes and their complications by treating patients with virus-inactivated plasma-derived factor concentrates, recombinant factor IX, tranexamic acid, and/or aminocaproic acid [1].
1.3 Hermansky-Pudlak syndrome, a rare autosomal recessive disorder characterized by defective biogenesis of lysosomal-related organelles, is associated with easy bruising and a tendency to bleed due to a platelet storage pool deficiency [2]. Ten genetically distinct subtypes with phenotypic variability have been reported, but they share certain core manifestations, including oculocutaneous albinism and a qualitative platelet disorder [3–5]. The hypopigmentation results from impaired function of melanosomes in melanocytes, while the bleeding occurs due to the absence of platelet dense bodies; both melanosomes and dense bodies are lysosome-related organelles [3]. Patients with certain Hermansky-Pudlak syndrome subtypes (1, 2, and 4) develop a fatal pulmonary fibrosis [6–8]. Hermansky-Pudlak syndrome is diagnosed by demonstration of dense body deficiency on whole mount electron microscopy of platelets or by genetic testing. Platelet dysfunction due to a lack of dense granules confers a life-long bleeding risk, which although mild, can be severe with trauma, surgery, or childbirth. Platelet aggregation studies typically show deficits in the secondary aggregation wave with possible impairment of collagen, adenosine diphosphate, and epinephrine responses. The recommended treatment for the bleeding diathesis is intravenous infusion of desmopressin or transfusion of leukoreduced single-donor platelets for significant bleeding [9].
1.4 Here we report the identification of a novel mutation in HPS6 and clinical phenotyping results in an interesting patient with excessive bleeding, hemophilia B, and oculocutaneous albinism.
2.1 MATERIAL AND METHODS
2.2 Patient Consent and Ethics Approval
The patient and his mother provided written informed consent to protocol 95-HG-0193, “Clinical and Basic Investigations into Hermansky-Pudlak Syndrome,” which was approved by the Institutional Review Board of the National Human Genome Research Institute.
2.3 Pulmonary Function Testing and High-resolution Computed Tomography Scan
Testing was performed at the National Institutes of Health Clinical Center in Bethesda, Maryland. Pulmonary function tests were performed in accordance with guidelines from the American Thoracic Society as described8. High-resolution computed tomography scan images were obtained without intravenous contrast in the prone position as described [10].
2.4 Genetic Sequencing
Genomic DNA was isolated from peripheral blood, and whole exome sequencing was performed by the National Institutes of Health Intramural Sequencing Center and analyzed for mutations as described [11]. To confirm results from whole exome sequencing, DNA was amplified by PCR, and PCR products were sequenced using a genetic analyzer (Applied Biosystems 3130×; Foster City, CA) in accordance with manufacturer’s instructions. The patient’s mother’s genomic DNA was also sequenced and analyzed for mutations in HPS6.
2.5 Platelet Aggregation Testing
Platelet aggregation was measured in 0.105 M sodium citrate-anticoagulated whole blood samples via impedance aggregometry, and platelet dense granule adenosine triphosphate release was measured by chemiluminescence on a Chronolog 700 lumiaggregometry instrument (Havertown, PA). Agonists included collagen at 1 and 5 micrograms/ml, adenosine diphosphate at 10 and 20 micromolar, thrombin at 1 unit/ml, arachidonic acid at 0.5 micromolar, and ristocetin at 0.25 and 1 micrograms/ml. Aggregation and release of adenosine triphosphate from dense granules by epinephrine at 7 and 20 micromolar concentrations were studied in platelet rich plasma using impedance measurements and chemiluminescence.
3.1 RESULTS
3.2 The patient is a 32-year-old Caucasian man with oculocutaneous albinism and hemophilia B, which was diagnosed at four months of age after surgery. Throughout childhood he suffered recurrent, severe bleeding episodes, including hemarthroses and soft tissue bleeding following trauma. There was no history of recurrent or unusual infections, developmental delay, or neurological disease.
3.3 The patient was born full-term to non-consanguineous parents of French and Scottish/Irish descent (Figure 1A); neither parent has albinism. His mother had a history of occasional epistaxis. No information was available on his maternal grandfather. His brother died at seventeen days of life from a cerebral hemorrhage and, thus, was suspected to have hemophilia B; he was not known to have Hermansky-Pudlak syndrome. Two male second cousins once removed have hemophilia B, and a female first cousin once removed, never evaluated for Hermansky-Pudlak syndrome, has albinism.
Figure 1.
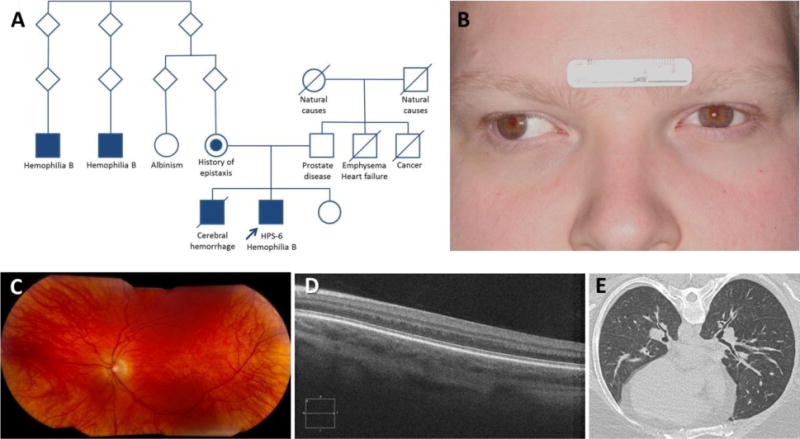
Pedigree and clinical images of patient with Hermansky-Pudlak syndrome-6 and hemophilia B.
(A) Pedigree shows proband (arrow) with Hermansky-Pudlak syndrome-6 (HPS-6) and hemophilia B and his relatives.
(B) Facial photograph reveals a mild pigmentation defect with fair skin and light colored hair and eyes. Exotropia of the right eye is present.
(C) Left eye photograph shows an albinotic poorly pigmented fundus.
(D) Foveal hypoplasia is demonstrated by optical coherence tomography scan.
(E) Representative high-resolution computed tomography scan image of the chest reveals no interstitial lung disease, pulmonary fibrosis, or ground glass opacification.
3.4 Physical examination revealed an obese male with hypopigmented skin, blond hair, and light-colored eyes (Figure 1B). Ophthalmologic examination revealed conjugate nystagmus, exotropia, iris transillumination, foveal hypoplasia with absence of a formed umbo, an albinotic poorly pigmented fundus, reduced visual acuity (20/100 bilaterally), and high myopic astigmatism (Figure 1C). Optical coherence tomography scan confirmed foveal hypoplasia (Figure 1D). His chest was clear to auscultation, and his cardiac examination was normal. There was no lymphadenopathy, organomegaly, or neurological deficit. Bruises in various stages of healing were noted; there were no petechiae or hemarthroses.
3.5 Baseline laboratory tests, including complete blood counts, were normal, and activated partial thromboplastin time was mildly prolonged (Table 1). Pulmonary function tests were normal, and high resolution chest computed tomography scan showed no evidence of interstitial lung disease or pulmonary fibrosis (Figure 1E).
Table 1.
Hematology test results of patient with Hermansky-Pudlak syndrome-6 and hemophilia B
| Laboratory Test | Result | Reference Range |
|---|---|---|
| Platelet count | 177 | 161–347 K/uL |
| Prothrombin time | 12.9 | 11.6 – 15.2 sec |
| aPTT | 44.6 | 25.3 – 37.3 sec |
| Thrombin time | 15.3 | 13.2 – 20.5 sec |
| Fibrinogen | 478 | 177 – 466 mg/dL |
| aPTT mixing study | Immediate - 37.1 1 hour - 36.8 |
25.3 – 37.3 sec |
| Lupus anti-coagulant | Negative | Negative |
| VWF activity | 89 | 52 – 156 IU/dL |
| VWF antigen | 75 | 50 – 197 IU/dL |
| Factor VIII activity | 69 | 41 – 184 IU/dL |
| Factor IX | 13 | 67 – 179 % |
| Factor XI activity | 76 | 74 – 157 % |
| Factor XII activity | 93 | 52 – 172 % |
aPTT = activated partial thromboplastin time
VWF = von Willebrand factor
3.6 Testing was performed to evaluate for hemophilia B and Hermansky-Pudlak syndrome. The factor IX level was 13% (Table 1); whole exome and Sanger sequencing revealed a hemizygous c.316 G>A (p.Gly106Ser) variant (rs137852233) in F9 (Durham mutation) confirming hemophilia B (Figure 2A) [12]. Given his oculocutaneous albinism and easy bruisability, a diagnosis of Hermansky-Pudlak syndrome was considered. Hence, we performed whole mount platelet electron microscopy, which revealed no dense bodies (Figure 2B). Platelet aggregation studies in this patient showed poor responses to adenosine triphosphate and no secondary aggregation wave. His platelets responded to desmopressin, indicating that desmopressin may be effective in prevention or treatment of his bleeding secondary to Hermansky-Pudlak syndrome [13]. Furthermore, to determine the patient’s subtype of Hermansky-Pudlak syndrome, we performed exome analysis and Sanger sequencing, and we identified a novel homozygous c.1114 C>T (p.Arg372*) mutation (rs776754431) in HPS6 (Figure 2C). Consistent with this result, a heterozygous c.1114 C>T mutation in HPS6 was identified in the patient’s mother (Figure 2D). No genetic data are available from his father, and no deletions were identified in the patient’s exome data.
Figure 2.
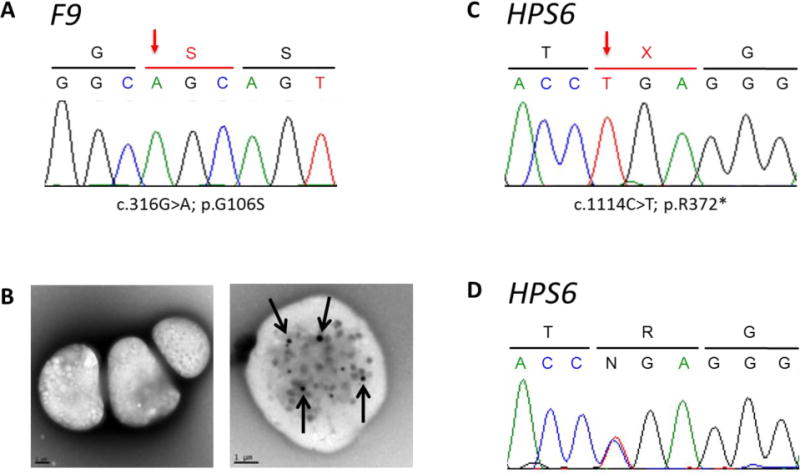
Genetic sequencing and platelet images of patient with Hermansky-Pudlak syndrome-6 and hemophilia B.
(A) F9 chromatogram showing a c.316G>A; p.G106S mutation (arrow).
(B) Representative photomicrograph of platelets imaged by whole mount transmission electron microscopy reveals an absence of dense bodies in this patient with HPS (left panel). Imaging of a normal platelet demonstrates dense bodies (arrows) (right panel).
(C) HPS6 chromatogram revealing a c.1114 C>T; p.Arg372* mutation (arrow) in the patient.
(D) HPS6 chromatogram revealing a heterozygous c.1114 C>T mutation in the patient’s mother.
3.7 The patient was treated with fresh frozen plasma infusions, recombinant factor IX, and aminocaproic acid for hemophilia B and with desmopressin for Hermansky-Pudlak syndrome. Treatment of bleeding episodes results in effective hemostasis, and the patient has never received a platelet or blood transfusion.
4.1 DISCUSSION
4.2 We report a hemizygous mutation in F9, located on chromosome Xq27.1–q27.2, and a novel homozygous mutation in HPS6, located on chromosome 10q24.32, in a 32-year-old man with bleeding and oculocutaneous albinism. Taken together, the patient’s clinical findings and molecular studies indicate that he has both hemophilia B and Hermansky-Pudlak syndrome-6. Consistent with these diagnoses, factor IX levels were low, platelet aggregation was deficient, and platelet dense bodies were absent.
4.3 Patients with Hermansky-Pudlak syndrome-6 typically exhibit a relatively mild phenotype with minor degrees of hypopigmentation and visual acuity deficits. In addition, patients with Hermansky-Pudlak syndrome-6 have not been reported to develop pulmonary fibrosis, which is a leading cause of mortality in patients with certain Hermansky-Pudlak syndrome subtypes (1, 2, and 4) [6–8]. Given the high penetrance of pulmonary fibrosis in 3 of 10 subtypes of Hermansky-Pudlak syndrome, identifying a patient’s genotype has important prognostic implications. Our patient, whose evaluation shows an absence of pulmonary fibrosis and whose phenotype is similar to other cases of Hermansky-Pudlak syndrome-6, had a homozygous c.1114 C>T (p.Arg372*) mutation in HPS6, which has not been reported [14].
4.4 Hemophilia B is due to mutations in F9, and more than a thousand sequence alterations in F9 have been identified ranging from single point mutations to complete deletions [1]. Genotype-phenotype associations have been reported, with some severe F9 mutations resulting in much lower levels of factor IX activity compared to other mutations. Our patient has a hemizygous c.316 G>A (p.Gly106Ser) variant in a highly conserved glycine residue of F9, known as the Durham mutation, which is associated with a mild phenotype [12].
5.1 CONCLUSIONS
In summary, we report an unusual patient with two inherited bleeding diatheses affecting primary and secondary hemostasis. Although Hermansky-Pudlak syndrome has been associated with low von Willebrand factor antigen levels, this is the first reported case of a patient with Hermansky-Pudlak syndrome and hemophilia B [15]. The clinical management of these disorders differs, so that diagnosing both Hermansky-Pudlak syndrome and hemophilia B enables this patient to receive effective prophylaxis and treatment for bleeding. Furthermore, identification of a mutation in HPS6 in this patient is clinically relevant, because his prognosis is generally favorable compared to patients with mutations in Hermansky-Pudlak syndrome genes that are associated with pulmonary fibrosis.
Highlights.
Two independent inherited bleeding disorders can occur in the same patient.
Whole exome and Sanger sequencing identify a novel mutation in HPS6.
Hermansky-Pudlak syndrome subtype 6 is diagnosed in an adult with hemophilia B.
Acknowledgments
We thank our patients who participate in our research program.
This study was supported in part by the Intramural Research Programs of the National Human Genome Research Institute, the Clinical Center, the National Institute of Biomedical Imaging and Bioengineering, the National Eye Institute, and the Office of the Director, National Institutes of Health; HL119503 (LRY); and HL127672 (LRY). The Rare Lung Diseases Consortium (U54 HL127672) is part of Rare Diseases Clinical Research Network (RDCRN), an initiative of the Office of Rare Diseases Research, National Center for Advancing Translational Sciences. This consortium is funded through collaboration between NCATS and the National Heart, Lung, and Blood Institute.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Escobar MA, Key NS. In: Williams Hematology. Kaushansky K, Lichtman M, Prchal J, Levi MM, Press O, Burns L, Caligiuri M, editors. McGraw-Hill; New York, NY, USA: 2016. Chapter 123. [Google Scholar]
- 2.Gahl WA, Brantly M, Kaiser-Kupfer MI, Iwata F, Hazelwood S, Shotelersuk V, et al. Genetic defects and clinical characteristics of patients with a form of oculocutaneous albinism (Hermansky-Pudlak syndrome) N Engl J Med. 1998;338:1258–1264. doi: 10.1056/NEJM199804303381803. [DOI] [PubMed] [Google Scholar]
- 3.Huizing M, Helip-Wooley A, Westbroek W, Gunay-Aygun M, Gahl WA. Disorders of lysosome-related organelle biogenesis: clinical and molecular genetics. Annu Rev Genomics Hum Genet. 2008;9:359–386. doi: 10.1146/annurev.genom.9.081307.164303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Cullinane AR, Curry JA, Carmona-Rivera C, Summers CG, Ciccone C, Cardillo ND, et al. A BLOC-1 mutation screen reveals that PLDN is mutated in Hermansky-Pudlak syndrome type 9. Am J Hum Genet. 2011;88:778–787. doi: 10.1016/j.ajhg.2011.05.009. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 5.Ammann S, Schulz A, Krägeloh-Mann I, Dieckmann NM, Niethammer K, Fuchs S, et al. Mutations in AP3D1 associated with immunodeficiency and seizures define a new type of Hermansky-Pudlak syndrome. Blood. 2016;127:997–1006. doi: 10.1182/blood-2015-09-671636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Brantly M, Avila NA, Shotelersuk V, Lucero C, Huizing M, Gahl WA. Pulmonary function and high-resolution CT findings in patients with an inherited form of pulmonary fibrosis, Hermansky-Pudlak syndrome, due to mutations in HPS-1. Chest. 2000;117:129–136. doi: 10.1378/chest.117.1.129. [DOI] [PubMed] [Google Scholar]
- 7.Anderson PD, Huizing M, Claassen DA, White J, Gahl WA. Hermansky-Pudlak syndrome type 4 (HPS-4): clinical and molecular characteristics. Hum Genet. 2003;113:10–17. doi: 10.1007/s00439-003-0933-5. [DOI] [PubMed] [Google Scholar]
- 8.Gochuico BR, Huizing M, Golas GA, Scher CD, Tsokos M, Denver SD, et al. Interstitial lung disease and pulmonary fibrosis in Hermansky-Pudlak syndrome-2, an AP-3 complex disease. Mol Med. 2012;18:56–64. doi: 10.2119/molmed.2011.00198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Seward SL, Jr, Gahl WA. Hermansky-Pudlak syndrome: health care throughout life. Pediatrics. 2013;132:153–160. doi: 10.1542/peds.2012-4003. [DOI] [PubMed] [Google Scholar]
- 10.Cullinane AR, Yeager C, Dorward H, Carmona-Rivera C, Wu HP, Moss J, et al. Dysregulation of Galectin-3: Implications for Hermansky-Pudlak Syndrome Pulmonary Fibrosis. Am J Respir Cell Mol Biol. 2014;50:605–613. doi: 10.1165/rcmb.2013-0025OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Stephen J, Vilboux T, Haberman Y, Pri-Chen H, Pode-Shakked B, Mazaheri S, et al. Congenital protein losing enteropathy: an inborn error of lipid metabolism due to DGAT1 mutations. Eur J Hum Genet. 2016;24:1268–1273. doi: 10.1038/ejhg.2016.5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Denton PH, Fowlkes DM, Lord ST, Reisne HM. Hemophilia B Durham: a mutation in the first EGF-like domain of factor IX that is characterized by polymerase chain reaction. Blood. 1998;72:1407–1411. [PubMed] [Google Scholar]
- 13.Wijermans PW, van Dorp DB. Hermansky-Pudlak syndrome: Correction of bleeding time by 1-desamino-8D-arginine vasopressin. Am J Hematol. 1989;30:154–157. doi: 10.1002/ajh.2830300307. [DOI] [PubMed] [Google Scholar]
- 14.Huizing M, Pederson B, Hess RA, Griffin A, Helip-Wooley A, Westbroek W, et al. Clinical and cellular characterisation of Hermansky-Pudlak syndrome type 6. J Med Genet. 2009;46:803–810. doi: 10.1136/jmg.2008.065961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Witkop CJ, Jr, Bowie EJ, Krumwiede MD, Swanson JL, Plumhoff EA, White JG. Synergistic effect of storage pool deficient platelets and low plasma von Willebrand factor on the severity of the hemorrhagic diathesis in Hermansky-Pudlak syndrome. Am J Hematol. 1993;44:256–259. doi: 10.1002/ajh.2830440407. [DOI] [PubMed] [Google Scholar]