

Supplemental Digital Content is available in the text.
Keywords: cadherin-4 (CDH4, R-cadherin); gemcitabine (dFdC); genotype–phenotype association; lymphoblastoid cell lines; pancreatic cancer; pharmacogenomics
Abstract
Background
Pancreatic cancer is a rapidly fatal disease with gemcitabine remaining the first-line therapy. We performed a genotype–phenotype association study to identify biomarkers for predicting gemcitabine treatment outcome.
Materials and methods
We selected the top 200 single nucleotide polymorphisms (SNPs) identified from our previous genome-wide association study to associate with overall survival using 400 patients treated with/or without gemcitabine, followed by imputation analysis for regions around the identified SNPs and a replication study using an additional 537 patients by the TaqMan genotyping assay. Functional validation was performed using quantitative reverse transcription-PCR for gemcitabine-induced expression in genotyped lymphoblastoid cell lines and siRNA knockdown for candidate genes in pancreatic cancer cell lines.
Results
Four SNPs in chromosome 1, 3, 9, and 20 showed an interaction with gemcitabine from the discovery cohort of 400 patients (P<0.01). Subsequently, we selected those four genotyped plus four imputed SNPs for SNP×gemcitabine interaction analysis using the secondary validation cohort. Two imputed SNPs in CDH4 and KRT8P35 showed a trend in interaction with gemcitabine treatment. The lymphoblastoid cell lines with the variant sequences showed increased CDH4 expression compared with the wild-type cells after gemcitabine exposure. Knockdown of CDH4 significantly desensitized pancreatic cancer cells to gemcitabine cytotoxicity. The CDH4 SNPs that interacted with treatment are more predictive than prognostic.
Conclusion
We identified SNPs with gemcitabine-dependent effects on overall survival. CDH4 might contribute to variations in gemcitabine response. These results might help us to better predict gemcitabine response in pancreatic cancer.
Introduction
Pancreatic cancer is a rapidly fatal disease with a 5-year survival of around 4–6% 1,2. Current chemotherapy regimens have modest survival benefits. Although newer agents are being tested in pancreatic cancer patients, gemcitabine remains the first-line treatment 3,4. However, the individual response to gemcitabine therapy varies widely 5–7. It is essential to understand how individuals might respond to gemcitabine when treated for pancreatic cancer. Many factors including both tumor and host germline genomic profiles could contribute to the variation in therapeutic response. Many pharmacogenomic studies do not have control populations (patients who were not treated with the drugs of interest); therefore, it is difficult to identify pharmcogenomic predictive biomarkers for a specific drug. In the current studies, we took advantage of two independent patient cohorts in which both gemcitabine-treated patients as well as non-gemcitabine-treated patients were available. These cohorts enabled us to identify not only genetic biomarkers associated with overall survival (OS) but also single nucleotide polymorphism (SNP)–gemcitabine interactions to specifically identify gemcitabine-predictive pharmacogenomic biomarkers. We have previously carried out a genome-wide association study (GWAS) using a cell-based model system, the ‘Human Variation Panel’, to identify genetic markers that were associated with IC50 values of two cytidine analogs: gemcitabine and cytosine arabinoside. This cell model system consists of 300 Epstein–Barr virus-transformed lymphoblastoid cell lines (LCLs) with dense genomic data, including genome-wide SNPs, mRNA expression, and 480 000 CpG methylation sites for each cell line 7,8. This system has been used widely to screen for common germline genetic variants that might be responsible for drug response. We have successfully used this system to both generate a pharmacogenomic hypothesis and to functionally characterize pharmacogenomic signals identified during the clinical GWAS 7–17. In the current study, we selected the top 200 SNPs associated with gemcitabine cytotoxicity (IC50 values) identified during our cell-based GWAS to carry out genotype–phenotype association studies using 400 DNA samples as a discovery cohort and an additional 537 samples as a validation cohort to validate germline genetic variants that might be associated with the patient OS during gemcitabine treatment. The study strategy was designed as shown in Fig. 1.
Fig. 1.

Experimental strategy. Top 200 SNPs associated with gemcitabine cytotoxicity were selected on the basis of our previous GWAS results using the human lymphoblastoid cell line model system, followed by genotyping of DNA samples of pancreatic cancer patients (discovery cohort) from Mayo cohort studies treated with/or without gemcitabine. Imputation analysis was carried out in the regions of ±200 kbp surrounding the top overall survival-associated SNPs. In a replication study, an additional 537 samples (validation cohort) were genotyped for identified candidate SNPs using the TaqMan assay. Top candidate genes were validated by functional assays using gemcitabine and SNP-dependent expression induction, siRNA knockdown, and gemcitabine cytotoxicity assay. GWAS, genome-wide association study; LCL, lymphoblastoid cell line; SNP, single nucleotide polymorphism.
Our findings indicated that four SNPs located in chromosomes 1, 3, 7, and 20, respectively, were identified to be associated with OS (P<0.01) of patients treated with gemcitabine. After imputation of these regions, we selected the top four imputed SNPs plus the four genotyped SNPs for validation in an additional 537 samples using the TaqMan SNP genotyping assay. The replication study showed that two imputed SNPs, rs9637468 located in KRT8P35 and rs4925193 in CDH4, showed a trend of associations with OS during gemcitabine treatment. Further functional validation showed that induction of CDH4 expression was gemcitabine and SNP dependent. Knockdown of CDH4 in three pancreatic cancer cells significantly desensitized cells to gemcitabine treatment, suggesting that the CDH4 gene might contribute to variation in gemcitabine response during pancreatic cancer therapy.
Our genotype–phenotype association analyses, as clinical replication studies of previous cell-based GWAS, followed by functional pharmacogenomics might help to identify and understand novel genetic biomarkers contributing to variations in gemcitabine response and provide us with insights to improve the clinical efficacy of gemcitabine during treatment of pancreatic cancer.
Materials and methods
Drug and cell lines
Gemcitabine was provided by Eli Lilly (Indianapolis, Indiana, USA). Stock solution was dissolved in PBS and stored at −80°C. The human pancreatic cancer AsPC-1, BxPC-3, CFPAC-1, PANC-1, PANC0403, SU86, HupT3, and MIApaca-2 cell lines were obtained from the American Type Culture Collection (Manassas, Virginia, USA) and were cultured in RPMI 1640 medium containing 10% fetal bovine serum.
DNA samples from pancreatic cancer patients
Patients were enrolled into this study from October 2000 until February 2010 and provided written informed consent for participation in the Mayo Clinic Biospecimen Resource for Pancreas Research. Patients were recruited at the time of their initial visit to the Mayo Clinic for pancreatic cancer. They also provided blood samples in addition to completing demographic/risk factor surveys.
Discovery cohort
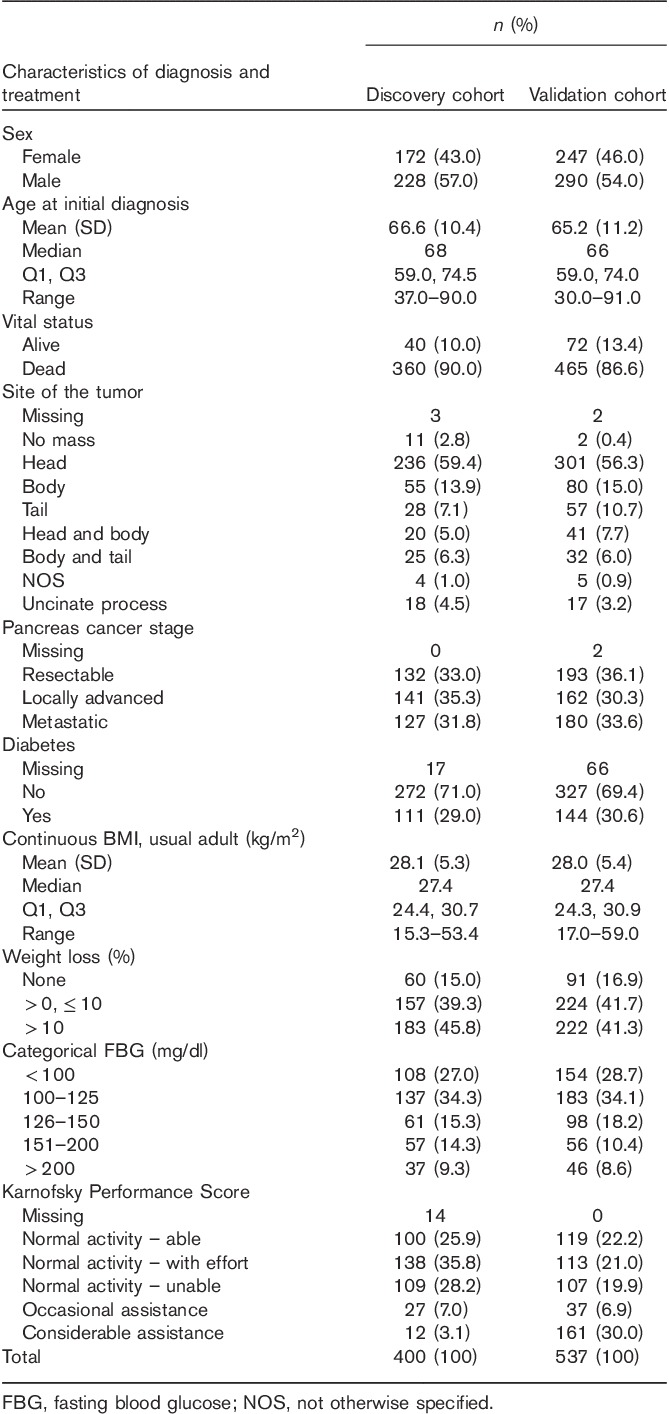
Patients were a subset of the previously published GWA study; all these patients had pancreatic adenocarcinoma and were recruited at Mayo Clinic 18,19. Patients were recruited through the Mayo Clinic Pancreas Cancer Registry (PI. G. Petersen, P50 CA102701) 20. We have obtained extensive clinical and demographic as well as treatment information for these patients, including age, stage, Karnofsky performance score, and use of gemcitabine. A total of 654 individuals with genome-wide SNP data available were included in our discovery cohort. After quality control, 400 individuals were included for the association analysis. Two hundred and fifty-four patients were excluded from the analysis because of various reasons: non-White, survey dropped, had undergone treatment before diagnosis, had a diagnosis of pancreatic cancer before arriving at Mayo, missing survival phenotype, missing genotype, and missing clinical covariate.
Validation cohort
Five hundred and thirty-seven individuals were recruited from the Mayo Clinic Pancreatic Cancer Registry who were not included in the discovery cohort and analyzed as an independent cohort for the validation analysis. The patients were followed up using a mailed survey at 6 months and 1 year in addition to the medical records with documentation of treatment information. Records were abstracted manually using a standardized protocol by a gastrointestinal cancer specialist/medical oncologist blinded to any genotype information 20. Abstracted information included types of chemotherapy and/or radiotherapy, duration of therapy, stage information, and response to treatment and common toxicities. Only patients with complete abstraction information that was sufficient to determine whether they did or did not receive gemcitabine during any part of their treatment were included in the analysis. Survival was calculated as the date of original diagnosis to the date of death or last follow-up.
Genotyping assessment
A total of 537 DNA samples from the validation cohort were selected to perform a genotyping assay for candidate SNPs in the Mayo Clinic Genotyping Shared Resource using the protocol of the TaqMan SNP Genotyping Assay (Applied Biosystems, Carlsbad, California, USA). Specifically, DNA samples were extracted and purified according to the manufacturer’s protocol using a QIAamp tissue kit (Qiagen, Hilden, Germany), followed by estimation of DNA concentration using a NanoDrop ND-1000 Spectrophotometer (Nano Drop Technologies Inc., Wilmington, Delaware, USA). Samples were then diluted to 10 ng/μl. PCRs were carried out in a 5 μl of reaction mix, consisting of 1 μl of genomic DNA template (10 ng), 2.5 μl of 2× TaqMan Universal PCR Master Mix, 0.25 μl of 20× working stock of SNP genotyping assay with specific primers and probes, and 1.25 μl DNAase-free water. PCR conditions consisted of repeated cycles at 95°C for 10 min and 40 cycles at 95°C for 1 min and 60°C for 15 s. Genotype analysis was carried out using the ABI Prism 7700 SDS software (Applied Biosystems). Quality assessment indicated a more than 98% call rate with high concordance among duplicates.
Transient transfection and cell proliferation assay
All siRNA pools with a set of four specific siRNAs for an individual candidate gene and a negative nontarget control siRNA pool were purchased from Dharmacon (Chicago, Illinois, USA). Reverse transfection of siRNA was performed in 96-well plates with a mixture of pancreatic cancer cells and 0.2 µl of lipofectamine RNAi-MAX reagent (Invitrogen, Carlsbad, California, USA), as well as 50 nmol/l of siRNA pools. At 24 h, cells were treated with gemcitabine (Eli Lilly, Indianapolis, Indiana, USA) at final concentrations of 0.001, 0.01, 0.1, 1, 10, 100, and 1000 µmol/l for 72 h. MTS cytotoxicity assays were performed using the CellTiter 96 AQueous Non-Radioactive Cell Proliferation Assay kit (Promega Corporation, Madison, Wisconsin, USA), followed by absorbance measurement at 490 nm in a Safire2 microplate reader (Tecan AG, Männedorf, Switzerland).
Real-time quantitative reverse transcription-PCR
Total RNA was isolated from cultured cells using the Quick-RNATM MiniPrep kit (Zymo Research, Orange, California, USA), followed by qRT-PCR with the Power SYBR Green RNA-to-CT 1-Step Kit (Applied Biosystems, Foster City, California, USA) 48 h after transfection. Specifically, primers purchased from QIAGEN (Hilden, Germany) were used to perform qRT-PCR using the Stratagene Mx3005P Real-Time PCR detection system (Agilent Technologies, Santa Clara, California, USA). All experiments were normalized with β-actin as an internal control.
Western blotting assay
Western blotting analysis was carried out with cell lysates. Specifically, 30 μg of protein lysate was subjected to electrophoresis on 12% SDS-PAGE gels. Proteins were transferred to polyvinylidene difluoride membranes. The membranes were then incubated overnight at 4°C with the anti-CDH4 antibody at a 1 : 1000 dilution (Cell Signaling, Boston, Massachusetts, USA), followed by the secondary antibody. Bands were detected with enhanced chemiluminescence (Thermo Scientific, Rockford, Illinois, USA).
Immunohistochemical assay
Formalin-fixed and paraffin-embedded blocks from 45 patients including matched normal surrounding tissues from the same patients were obtained from the Cancer Hospital, Chinese Academy of Medical Sciences. The study was approved by the Human Subjects Research Ethics Committee of the Cancer Hospital as exempt from institutional review board review. For the detection of CDH4, sections were deparaffinized before placing in retrieval solution (citric acid, pH 6.0) for 20 min at 100°C. After cooling for 20 min, slides were quenched with 3% H2O2 for 5 min, before incubating with a 1 : 50 dilution of monoclonal anti-CDH4 antibody (Cell Signaling) for 30 min. Labeling was detected following the manufacturer’s protocol (ZSGB-Bio, Beijing, China). All sections were counterstained with hematoxylin and CDH4 staining was evaluated by two independent pathologists and considered positive on the basis of established criteria 21,22. Score 0 is when there was no specific membrane staining, and positive cases were further classified into 1, 2, and 3 on the basis of the staining intensity of the membrane.
Statistical methods
OS time was calculated since diagnosis. We first used a log-rank test to compare proportional hazards between the discovery and validation cohorts, limiting the risk interval to 2.5 years, and then to test differences in survival, conditional on surviving to 2.5 years. There was no difference in proportional hazards between the two cohorts. Survival was summarized as the median survival and the hazards were essentially proportional up to those times. The 2- and 5-year survival rates were calculated using the Kaplan–Meier method. Comparisons between the discovery and validation sets for continuous variables (age, BMI, etc.) were tested using Wilcoxon rank-sum tests, whereas ordinal categorical variables (Karnofsky Performance Score, etc.) were tested using Armitage trend tests, and finally nonordinal categorical variables were tested using the χ2-test (continuity corrected).
Genotype–phenotype association and interaction with treatment
Kaplan–Meier curves were used to visualize OS. Interactions between the SNP genotype (additive model) and gemcitabine exposure (yes/no) were analyzed (SNP×gemcitabine treatment effect). We also determined the SNP association with OS for patients (overall main effect). These two sets of analyses were completed in a similar manner with both discovery (n=400) and validation cohorts (n=537). Before we carried out the analysis of the effect of SNP on OS, we tested a series of potential covariates including different treatments and other factors (Table 1). As age, stage, Karnofsky Performance Score, and tumor site were shown to be related to survival in previous publications, these factors were included as covariates. Significant ones were adjusted during the association analysis. As the time at which gemcitabine was used varied for individuals, the risk intervals for individuals were divided and we treated different risk intervals as time-varying covariates in the analysis of the OS and SNP–gemcitabine treatment interaction. The association of OS with the interaction of SNPs and gemcitabine was investigated using multivariable Cox proportional hazards regression analysis. The effect of SNPs on gemcitabine treatment was modeled as the interaction of an individual SNP and gemcitabine use (as count of minor allele). To test the pharmacogenomic effect (SNP×gemcitabine interaction), a Wald test was used.
Table 1.
Characteristics of pancreatic cancer patients in the overall survival-association analysis
Imputation analysis
The top significant SNPs associated with OS of those 400 patients from the discovery cohort were selected to explore more functional SNPs in imputation analysis. We imputed the regions of ±200 kbp surrounding those SNPs using MACH 1.0 with 1000 Genome Project, followed by the association of these imputed SNPs with OS 23,24.
Results
Study population
To determine whether any gemcitabine-predictive genetic biomarkers or prognostic biomarkers controlled for treatments in pancreatic cancer patients, we used 400 pancreatic cancer patients treated with (n=218) or without (n=182) gemcitabine for the discovery cohort study and an additional 537 Mayo pancreatic cancer patients treated with (n=296) or without (n=241) gemcitabine with similar demographic and clinical characteristics as a validation cohort to genotype selected SNPs for the association studies. The discovery cohort had already had genome-wide SNP data available as part of the Mayo Pancreatic Cancer Cohort Consortium GWAS project (PanScan Project) 18,19. Specifically, we selected the top 200 SNPs on the basis of the association P-values that were correlated with gemcitabine IC50 values in our LCLs described previously for the association analysis with the OS of those pancreatic cancer patients, as well as interaction analysis with gemcitabine treatment 7,8. We then followed this initial analysis with a validation study using the additional 537 Mayo patient samples to genotype the top candidate SNPs identified from the initial OS association study by the TaqMan SNP Genotyping Assay. The strategy of the experimental design is described in Fig. 1.
A summary of the characteristics of the patients in both cohorts is shown in Table 1. There were no statistically significant differences in sex, age, and diagnosis characteristics or the performance score between the two sets of the patients. The median ages at diagnosis were 68 and 66 years in the two cohorts, respectively. Overall, 90 and 86.6% of patients had died by the time of analysis, reflecting the relatively poor prognosis observed for this type of cancer. As it has been shown that patient age, stage, Karnofsky Performance Score, and tumor site were related to OS in previous publications, these factors were included as covariates in further analysis 18,19.
Genotype–phenotype association of the discovery cohort
We had previously performed a cell-based GWAS using a genetic data-rich cell model system, consisting of 187 human LCLs, and identified top 200 SNPs significantly associated with gemcitabine cytotoxicity (IC50 values), with P-value less than 10−3 for this association study 7. OS time of those 400 patients was calculated since diagnosis 18,19. Of the 200 SNPs selected, none of them was associated significantly with OS of these patients (Supplementary Table 1, Supplemental digital content 1, http://links.lww.com/FPC/B78), suggesting that they might not serve as pancreatic cancer risk factors. However, our findings showed that four SNPs in four genomic regions located in chromosomes 1, 3, 9, and 20, respectively, were strongly associated with OS (P<0.01) of patients when exposed to gemcitabine in the interaction analysis using a Wald test (Table 2 and Supplementary Table 1, Supplemental digital content 1, http://links.lww.com/FPC/B78). The effects of four SNPs on gemcitabine response were in the same direction as that observed in our GWAS analysis using the LCLs, implying that variations in these SNPs might modify the outcome of gemcitabine treatment for pancreatic cancer (Table 2 and Supplementary Table 1, Supplemental digital content 1, http://links.lww.com/FPC/B78). Although these SNPs by gemcitabine interaction were not associated significantly with patient OS after correction for multiple testing (P=1.25E−4), we were aware that these selection criteria were arbitrary, but this would provide us a means to identify more candidates. Follow-up association and functional studies would be useful to narrow down to a number of reasonable genes.
Table 2.
Top genotyped single nucleotide polymorphisms associated with overall survival of 400 pancreatic cancer patients from the discovery cohort in the single nucleotide polymorphism×gemcitabine interaction analysis
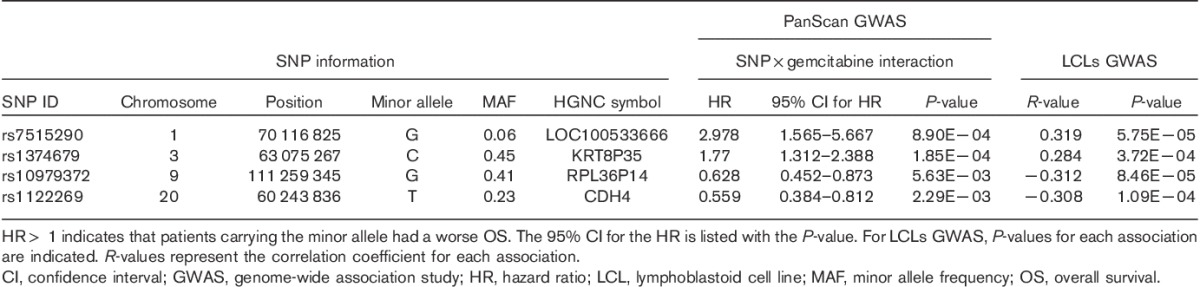
Those four SNPs (rs7515290, rs1374679, rs10979372, and rs1122269) were mapped to four genomic regions containing four HGNC symbols: LOC100533666, KRT8P35, RPL36P14, and CDH4. Similar to other complex diseases, multiple common disease alleles with small effects influence disease risk or drug response in pancreatic cancer. As the four common SNPs [minor allele frequency (MAF)>0.05] showed trends to be associated with gemcitabine IC50 values in LCLs cells and clinical patient outcome in the discovery cohort, we further carried out an imputation analysis including regions around these SNPs. We imputed regions 200 kbp on either side of those four SNPs to explore additional genetic variants around those genomic regions, followed by an SNP–gemcitabine interaction analysis with OS of the discovery cohort patients to determine whether any of the imputed SNPs might have pharmacogenomic effects on gemcitabine during pancreatic cancer treatment (Supplementary Table 2, Supplemental digital content 2, http://links.lww.com/FPC/B79).
Further analysis was carried out to determine whether any of these SNPs may be related to OS after controlling for gemcitabine treatment. None of the SNPs was shown to be a prognostic factor. These results are shown as the main effect of SNP in Supplementary Table 1 (Supplemental digital content 1, http://links.lww.com/FPC/B78).
Genotyping and association analysis of the validation patient cohort
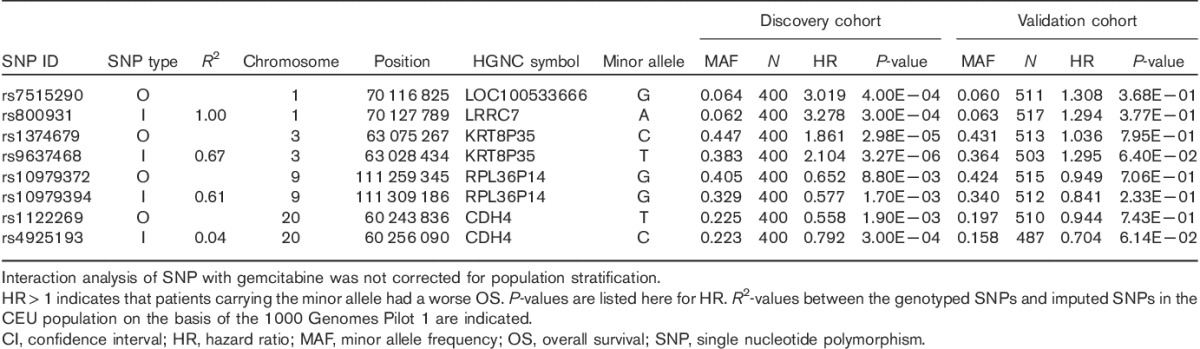
After imputation analysis with the 1000 Genome Project, we selected the four top OS-associated imputed SNPs plus the four genotyped SNPs for validation with 537 additional Mayo samples from a second independent cohort using the TaqMan SNP genotyping assay. In our studies, we sought to validate the finding of gemcitabine-specific SNP effects on OS. We included both individuals with and without gemcitabine treatment for the interaction analysis of these eight SNPs. Two imputed SNPs, rs9637468 located in the downstream of KRT8P35 and rs4925193 in the intron of CDH4, shown trends toward associations with OS of patients from the validation cohort (P=0.064 for rs9637468 and P=0.061 for rs4925193), indicating that they might be genetic biomarkers for the prediction of gemcitabine response during pancreatic cancer therapy, as shown in Table 3. The minor allele of rs9637468 showed an association with worse OS, whereas the rs4925193 minor allele showed an association with better OS. Further functional studies are needed to elucidate the biological implications of these SNPs on gemcitabine response.
Table 3.
Results in validation and discovery cohorts for genotyped and imputed single nucleotide polymorphisms selected for the strength of single nucleotide polymorphism×gemcitabine interaction with overall survival in the discovery cohort
Follow-up analysis
Next, we tested the hypothesis of whether these four SNPs located in the downstream of KRT8P35 and the intron of CDH4 might also influence the expression of these two genes in a cis-manner. We carried out an eQTL (expression quantitative trait loci) analysis for these two corresponding genotyped SNPs (rs9637468 and rs4925193) using 15 tumor and eight normal samples from pancreatic cancer patients who were a subset of the samples from the discovery cohort for which we had both genotype and mRNA expression data (Supplementary Table 3A, Supplemental digital content 3, http://links.lww.com/FPC/B80 and 3B, Supplemental digital content 4, http://links.lww.com/FPC/B81). The microarray data for our eQTL analysis were used from the same samples as that in our previous study 25. The KRT8P35 SNPs did not show any cis-relationships in either normal or tumor samples. However, we found that both rs1122269 and rs4925193 showed negative correlations with all three probe sets of CDH4 in a cis-manner, with values of R equal to −0.5 in normal samples (P=0.2) (Supplementary Table 3C, Supplemental digital content 5, http://links.lww.com/FPC/B82), but no cis-associations were found in the tumor tissue samples. The mRNA expression levels of CDH4 (Supplementary Fig. 1A, Supplemental digital content 6, http://links.lww.com/FPC/B83) were associated with OS of those pancreatic cancer patients [hazard ratio (HR)<1 for all the four probe sets of CDH4] (Supplementary Table 3D, Supplemental digital content 7, http://links.lww.com/FPC/B84). The absence of a statistically significant finding could be because of the limited sample size for this analysis. Although the P-values of these probe sets were not statistically significant, they do appear to suggest a protective effect of CDH4 (HR<1 for all four probes). We also tested the trans-association between the two SNPs with gene expression using our pancreatic normal and tumor samples; no statistically significant findings were observed.
To determine the expression level of CDH4 in tumors, we obtained additional sections for 45 pairs of pancreatic ductal adenocarcinoma and their surrounding pancreatic tissue samples from the Cancer Hospital of the Chinese Academy of Medical Sciences to test differences in CDH4 protein expression between tumor and normal tissue. Hematoxylin and eosin-stained sections of adjacent sections of the tissue were prepared to confirm the diagnosis (data not shown). The score levels of CDH4 staining in normal tissues were significantly higher than those in tumor tissues in the 45 pairs of samples (Supplementary Table 4, Supplemental digital content 8, http://links.lww.com/FPC/B85). As shown in two represented cases (Supplementary Fig. 1B, Supplemental digital content 6, http://links.lww.com/FPC/B83), stronger immunostaining of CDH4 was noted in the membranes of normal pancreatic duct epithelium cells, whereas the staining was much weaker or negative in neoplastic epithelium cells as well as in extracellular matrix components. There were 33 (73.3%) pairs of patient samples with positive CDH4 staining in both tumor and normal tissues after quality control of the immunohistochemical (IHC) staining.
Functional validation of candidate genes
We further validated whether those genes containing the OS-associated SNPs might have an influence on gemcitabine cytotoxicity as a result of the contribution to variation in gemcitabine responses during pancreatic cancer therapy. Besides the CDH4 gene, none of the other three genes located in chromosomes 1, 3, and 9 were selected as candidates for functional validation as they are pseudogenes within ±50 kbp sides of the regions of SNPs.
We then determined the effect of different genotypes at the SNP (rs1122269) of the CDH4 gene on the gemcitabine response. Experimentally, we took advantage of our LCLs with GWAS genotyping data for each cell line and selected cells with the individual genotype of interest to determine the effect of the SNP genotype on CDH4 expression in LCLs after gemcitabine exposure. For this, we selected six LCLs with known genotypes for the SNP (rs1122269), exposed cells to various concentrations of gemcitabine, and detected CDH4 mRNA levels in the RT-PCR assay. We found that the cells with the variant sequences showed increased CDH4 expression compared with the wild-type cells after gemcitabine exposure (Fig. 2a).
Fig. 2.

(a) Functional validation in three human pancreatic cancer cell lines. Cells were treated with 30 nmol/l of control siRNA or specific siRNA against CDH4 using three human pancreatic cancer cell lines, followed by the MTS assay after exposure of gemcitabine for 72 h, and knockdown efficiency was confirmed in the quantitative reverse transcription-PCR assay (bar graph). (b) Single nucleotide polymorphism-related differences in CDH4 expression and gemcitabine response in three variant (rs1122269) and three wild-type lymphoblastoid cell lines were detected in quantitative reverse transcription-PCR assay. *P<0.05; **P<0.01.
To further explore the pharmacological effect of CDH4 gene expression on gemcitabine treatment, we performed CDH4 knockdown using a siRNA pool (a set of four specific siRNAs) against CDH4 in human pancreatic cancer cell lines, followed by a gemcitabine cytotoxicity assay. Before CDH4 knockdown, we tested CDH4 protein levels in eight human pancreatic cancer cell lines. As shown in Supplementary Fig. 2 (Supplemental digital content 9, http://links.lww.com/FPC/B86), the protein levels of CDH4 were relatively high enough to be detected in SU86, HupT3, and MIApaca-2 cell clines compared with the others. Therefore, we selected these three lines for further study. Downregulation of CDH4 in all these three lines significantly desensitized cells to gemcitabine treatment, whereas knockdown efficiency was confirmed using real-time qRT-PCR (Fig. 2b).
All the above results implied that genetic variations in CDH4 in pancreatic tumor cells might impact on cellular susceptibility to gemcitabine through an alteration in its gene expression after its treatment. CDH4 has previously been reported as a tumor suppressor in a variety of cancer types. Our study, for the first time, suggested that CDH4 might also be involved in the response to gemcitabine during the treatment of pancreatic cancer.
Discussion
We previously used the ‘Human Variation Panel’ – a genomic data-rich lymphoblastoid cell line model system – to identify markers that might contribute toward variation in response to gemcitabine 7,8. With the cell-based GWAS data, we had identified the top 200 most significant SNPs associated with gemcitabine cytotoxicity in the LCLs as an index of the drug response 7. In the present study, we selected these 200 SNPs and carried out a genotype–phenotype association study using information and samples obtained on our Mayo pancreatic patients. We found that four SNPs in the genomic region of chromosomes 1, 3, 9, and 20 were associated with gemcitabine-specific effects on OS (P<0.01). This was consistent with the observation from our previous cell-based GWAS results (as shown in Table 2).
Furthermore, we also tested the effect of gemcitabine treatment by the genotypes of those four top OS-associated SNPs using the samples from the discovery cohort patients. As shown in Supplementary Table 5 (Supplemental digital content 10, http://links.lww.com/FPC/B87), the results implied that the SNPs in KRT8P35 and CDH4 might be predictive markers for gemcitabine response. Individuals with more common alleles for SNP (rs1374679) in KRT8P35 had better OS. The SNP (rs1122269) in the CDH4 gene showed the opposite trend, with the minor alleles associated with better outcomes. We had also carried out a Kaplan–Meier analysis to test a gemcitabine-specific SNP effect using samples from patients treated with or without gemcitabine (Supplementary Fig. 3, Supplemental digital content 11, http://links.lww.com/FPC/B88). These Kaplan–Meier plots indicated that variants in the genomic regions might be predictive markers for the gemcitabine treatment in pancreatic cancer. Kaplan–Meier curves for the other two SNPs (rs7515290 and rs10979372) were also shown to detect the effect of gemcitabine treatment (Supplementary Fig. 3, Supplemental digital content 11, http://links.lww.com/FPC/B88).
We then carried out an imputation analysis to determine whether additional SNPs might predict gemcitabine response. Two imputed SNPs, rs9637468 located downstream of KRT8P35 and rs4925193 in the intron of CDH4, were associated with gemcitabine-specific effects on OS from both the PanScan discovery and validation cohorts, indicating that these two SNPs might be genetic biomarkers to predict gemcitabine response. More functional studies would be required to elucidate the mechanism by which these variants impact on drug responses of gemcitabine during the treatment of pancreatic cancer.
In this study, to investigate the pharmacogenomic effects of these potential genetic markers, we carried out an eQTL (expression quantitative trait loci) analysis to determine whether they might transcriptionally regulate nearby gene expression in either a cis-manner or a trans-manner. The rs9637468 did not show any cis-relationships in either normal or tumor samples, but we did observe trans-associations (P<10−4) with the expression of other genes in both normal and tumor samples (Supplementary Table 3A, Supplemental digital content 3, http://links.lww.com/FPC/B80 and 3B, Supplemental digital content 4, http://links.lww.com/FPC/B81). This indicated that rs9637468 might impact on the expression and/or the function of other unknown genes or genomic regions, further contributing to the individual chemosensitivity of this variant carrier when treated with gemcitabine. However, in our eQTL analysis, the two SNPs (rs1122269 and rs4925193) of CDH4 showed negative cis-correlations with the expression of CDH4 mRNA for all three of its probe sets in the normal tissue samples, although the P-values were not statistically significant (Supplementary Table 3C, Supplemental digital content 5, http://links.lww.com/FPC/B82). We also found that the mRNA expression levels of CDH4 were associated with the OS of these pancreatic cancer patients (HR<1 for all the four probe sets of CDH4) (Supplementary Table 3D, Supplemental digital content 7, http://links.lww.com/FPC/B84). We found that both mRNA and protein expression levels were lower in pancreatic tumor tissues compared with that in normal tissue samples using microarray and IHC staining assays (Supplementary Fig. 1A and 1B, Supplemental digital content 6, http://links.lww.com/FPC/B83), suggesting the tumor-suppressive function of CDH4 in pancreatic cancer. More importantly, induction of CDH4 expression was gemcitabine dependent and SNP (rs1122269 and rs4925193) was genotype dependent in LCLs (Fig. 2a). Further functional study in the three pancreatic cancer cell lines showed that the decrease in CDH4 expression using siRNA knockdown desensitized cells to gemcitabine cytotoxicity (Fig. 2b). These results implied that, if a pancreatic cancer patient carried the CDH4 variant (rs1122269), its gene expression in cancer cells would be upregulated by gemcitabine exposure, leading to more sensitivity to the drug. Therefore, our functional studies suggested that the SNP (rs1122269) of CDH4 might serve as a predictive biomarker for treatment outcomes of gemcitabine in pancreatic cancer patients.
The CDH4 gene encodes a protein, cadherin-4, in humans, also called R-cadherin, which belongs to the cadherin superfamily 26,27. The encoded protein is a calcium-dependent cell–cell adhesion glycoprotein composed of five extracellular cadherin repeats, a transmembrane region, and a highly conserved cytoplasmic tail 28–30. This cadherin is believed to play an important role during neuronal outgrowth and clinical neurological disease, as well as muscle and pancreas development 31–33. A previous study showed that inappropriate expression of R-cadherin (CDH4) in tumor cells results in decreased expression of endogenous cadherins (cadherin switching) and sustained signaling through Rho GTPases. CDH4 has been reported to contribute to tumorigenesis, invasiveness, and metastasis as a tumor suppressor in colorectal cancer, gastricointestinal cancer, ovarian cancer, chondrosarcoma, and nasopharyngeal carcinoma 26,27,34–37. Together with the results from the present genotype–phenotype association and follow-up functional studies, this indicated that CDH4 might play an important role in the response of gemcitabine and also act as a tumor suppressor in human pancreatic cancer as reported by previous studies. It was the first time that we identified the CDH4 gene associated with individual response after gemcitabine exposure during pancreatic cancer therapy. This suggested that CDH4 might also serve as a useful predictive/prognostic biomarker for individualized medicine, as well as a novel target for antineoplastic drug development. More functional characterization of polymorphisms in CDH4 and the gene itself would be required to detect the impact on gemcitabine treatment outcomes.
In addition, we have identified several SNPs located in the other three pseudogenes that were associated with outcomes depending on gemcitabine exposure. Although these SNPs did not appear to be associated with cis-expression using clinical tissue samples, it is possible that the genotypes have a trans-effect on other genes. Other mechanisms such as long-chain noncoding RNAs and microRNA might also be affected by these SNPs, which in turn regulate genes/pathways contributing toward gemcitabine response in pancreatic cancer.
Certainly, there were some limitations in our studies. First, the imputation analysis for the SNP (rs7515290) with low MAF (MAF=0.06) might lead to a type I error and a false-positive result. Also, there might be confounding factors affecting the pharmacogenetics effects of both genotyped and imputed SNPs (rs7515290 and rsrs800931) in the analysis. More follow-up validation studies using additional cohorts of clinical samples or functional characterization experiments will be necessary in future. Second, our cohorts in the studies were not randomized prospective studies. As an observational study, the population is probably more representative of the pancreatic cancer patients seen in academic medical centers. However, the patients selected in the study were only accessible to us with gemcitabine-related treatment information. Additional randomized studies will be required to assess the clinical utility of risk stratification. There were also some epidemiologic risk factors that might confound with the pharmacological effect of genetic markers, such as adiposity, smoking, diabetes, and family history already known for pancreatic cancer.
Taken together, our cell line model system has proven to be a powerful tool both for the identification of pharmacogenomic hypotheses and for the pursuit of hypotheses from the clinical GWAS 7,9,12,14,25. Our findings from the translational research of cell-based GWAS results into clinical validation by genotype–phenotype association analysis, and experimentally functional studies, would help to identify genetic biomarkers contributing to individual variability in gemcitabine response during the treatment of pancreatic cancer and help use those predictive/prognostic biomarkers to select responsive patient subpopulations for gemcitabine therapy and improve its treatment outcome.
Supplementary Material
Supplemental digital content is available for this article. Direct URL citations appear in the printed text and are provided in the HTML and PDF versions of this article on the journal's website (www.pharmacogeneticsandgenomics.com).
Acknowledgements
The microarray data and SNP data from the LCL-based GWAS study have been submitted to the NCBI Gene Expression Omnibus databases (http://www.ncbi.nlm.nih.gov/geo) under SuperSeries accession no. GSE24277.
The SNP data from the Whole Genome Scan for Pancreatic Cancer Risk in the Pancreatic Cancer Cohort Consortium and Pancreatic Cancer Case-Control Consortium (PanScan) have been deposited in the NCBI dbGaP datasets (http://www.ncbi.nlm.nih.gov/gap) under dbGaP authorized accession no. phs000206.v5.p3, along with the weblink of genotype/phenotype interaction studies (http://epi.grants.cancer.gov/PanScan/).
The microarray data from the PanScan study to perform our eQTL analysis have been deposited in the NCBI GEO databases (http://www.ncbi.nlm.nih.gov/geo) with Series accession no. GSE16515.
Financial support: The basic scientific research Project of the Central Public Welfare Foundation for Scientific Research, IMBF201401 (L.L.), The Peking Union Medical College (PUMC) Youth Fund and Fundamental Research Funds for the Central Universities Foundation, 3332015166 (L.L.), The Gerstner Family Career Development Award in Individualized Medicine at Mayo Clinic (L.L.), and National Cancer Institute R01 Grant, CA196648 (L.W.).
Conflicts of interest
There are no conflicts of interest.
Footnotes
Liang Li and Jian-Wei Zhang contributed equally to the writing of this article.
References
- 1.Li D, Xie K, Wolff R, Abbruzzese JL. Pancreatic cancer. Lancet 2004; 363:1049–1057. [DOI] [PubMed] [Google Scholar]
- 2.Maitra A, Hruban RH. Pancreatic cancer. Annu Rev Pathol 2008; 3:157–188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Jaffee EM, Hruban RH, Canto M, Kern SE. Focus on pancreas cancer. Cancer Cell 2002; 2:25–28. [DOI] [PubMed] [Google Scholar]
- 4.Middleton G, Silcocks P, Cox T, Valle J, Wadsley J, Propper D, et al. Gemcitabine and capecitabine with or without telomerase peptide vaccine GV1001 in patients with locally advanced or metastatic pancreatic cancer (TeloVac): an open-label, randomised, phase 3 trial. Lancet Oncol 2014; 15:829–840. [DOI] [PubMed] [Google Scholar]
- 5.Kocabas NA, Aksoy P, Pelleymounter LL, Moon I, Ryu JS, Gilbert JA, et al. Gemcitabine pharmacogenomics: deoxycytidine kinase and cytidylate kinase gene resequencing and functional genomics. Drug Metab Dispos 2008; 36:1951–1959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hertel LW, Boder GB, Kroin JS, Rinzel SM, Poore GA, et al. Evaluation of the antitumor activity of gemcitabine (2’,2’-difluoro-2’-deoxycytidine). Cancer Res 1990; 50:4417–4422. [PubMed] [Google Scholar]
- 7.Li L, Fridley BL, Kalari K, Jenkins G, Batzler A, Weinshilboum RM, Wang L. Gemcitabine and arabinosylcytosin pharmacogenomics: genome-wide association and drug response biomarkers. PloS one 2009; 4:e7765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Li L, Fridley B, Kalari K, Jenkins G, Batzler A, Safgren S, et al. Gemcitabine and cytosine arabinoside cytotoxicity: association with lymphoblastoid cell expression. Cancer Res 2008; 68:7050–7058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Niu N, Qin Y, Fridley BL, Hou J, Kalari KR, Zhu M, et al. Radiation pharmacogenomics: a genome-wide association approach to identify radiation response biomarkers using human lymphoblastoid cell lines. Genome Res 2010; 20:1482–1492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Huang RS, Duan S, Bleibel WK, Kistner EO, Zhang W, Clark TA, et al. A genome-wide approach to identify genetic variants that contribute to etoposide-induced cytotoxicity. Proc Natl Acad Sci USA 2007; 104:9758–9763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Huang RS, Duan S, Kistner EO, Hartford CM, Dolan ME. Genetic variants associated with carboplatin-induced cytotoxicity in cell lines derived from Africans. Mol Cancer Ther 2008; 7:3038–3046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Im HK, Gamazon ER, Stark AL, Huang RS, Cox NJ, Dolan ME. Mixed effects modeling of proliferation rates in cell-based models: consequence for pharmacogenomics and cancer. PLoS Genet 2012; 8:e1002525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wang L, McLeod HL, Weinshilboum RM. Genomics and drug response. N Engl J Med, 364:1144–1153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ingle JN, Schaid DJ, Goss PE, Liu M, Mushiroda T, Chapman JA, et al. Genome-wide associations and functional genomic studies of musculoskeletal adverse events in women receiving aromatase inhibitors. J Clin Oncol 2010; 28:4674–4682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Liu M, Ingle JN, Fridley BL, Buzdar AU, Robson ME, Kubo M, et al. TSPYL5 SNPs: association with plasma estradiol concentrations and aromatase expression. Mol Endocrinol 2013; 27:657–670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Liu M, Wang L, Bongartz T, Hawse JR, Markovic SN, Schaid DJ, et al. Aromatase inhibitors, estrogens and musculoskeletal pain: estrogen-dependent T-cell leukemia 1A (TCL1A) gene-mediated regulation of cytokine expression. Breast Cancer Res 2012; 14:R41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Manco L, Ribeiro ML. Gene symbol: NT5C3. Disease: pyrimidine 5’-nucleotidase (P5’N) deficiency. Hum Genet 2006; 119:673–674. [PubMed] [Google Scholar]
- 18.Petersen GM, Amundadottir L, Fuchs CS, Kraft P, Stolzenberg-Solomon RZ, Jacobs KB, et al. A genome-wide association study identifies pancreatic cancer susceptibility loci on chromosomes 13q22.1, 1q32.1 and 5p15.33. Nat Genet 2010; 42:224–228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Amundadottir L, Kraft P, Stolzenberg-Solomon RZ, Fuchs CS, Petersen GM, Arslan AA, et al. Genome-wide association study identifies variants in the ABO locus associated with susceptibility to pancreatic cancer. Nat Genet 2009; 41:986–990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.McWilliams RR, Matsumoto ME, Burch PA, Kim GP, Halfdanarson TR, de Andrade M, et al. Obesity adversely affects survival in pancreatic cancer patients. Cancer 2010; 116:5054–5062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Shia SJ, Klimstra DS, Li AR, Qin J, Saltz L, Teruya-Feldstein J, et al. Epidermal growth factor receptor expression and gene amplification in colorectal carcinoma: an immunohistochemical and chromogenic in situ hybridization study. Mod Pathol 2005; 18:1350–1356. [DOI] [PubMed] [Google Scholar]
- 22.Hermano HE, Meirovitz A, Meir K, Nussbaum G, Appelbaum L, Peretz T, Elkin M. Macrophage polarization in pancreatic carcinoma: role of heparanase enzyme. J Natl Cancer Inst 2014; 106:dju332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Li L, Fridley BL, Kalari K, Niu N, Jenkins G, Batzler A, et al. Discovery of genetic biomarkers contributing to variation in drug response of cytidine analogues using human lymphoblastoid cell lines. BMC Genomics 2014; 15:93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Browning SR, Browning BL. Rapid and accurate haplotype phasing and missing-data inference for whole-genome association studies by use of localized haplotype clustering. Am J Hum Genet 2007; 81:1084–1097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Pei H, Li L, Fridley BL, Jenkins GD, Kalari KR, Lingle W, et al. FKBP51 affects cancer cell response to chemotherapy by negatively regulating Akt. Cancer cell 2009; 16:259–266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Bonacci TM, Hirsch DS, Shen Y, Dokmanovic M, Wu WJ. Small GTPase Rho regulates R-cadherin through Dia1/profilin-1. Cell Signal 2012; 24:2102–2110. [DOI] [PubMed] [Google Scholar]
- 27.Miotto E, Sabbioni S, Veronese A, Calin GA, Gullini S, Liboni A, et al. Frequent aberrant methylation of the CDH4 gene promoter in human colorectal and gastric cancer. Cancer Res 2004; 64:8156–8159. [DOI] [PubMed] [Google Scholar]
- 28.Shan WS, Tanaka H, Phillips GR, Arndt K, Yoshida M, Colman DR, Shapiro L. Functional cis-heterodimers of N- and R-cadherins. J Cell Biol 2000; 148:579–590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Sano K, Tanihara H, Heimark RL, Obata S, Davidson M, St, John T, et al. Protocadherins: a large family of cadherin-related molecules in central nervous system. EMBO J 1993; 12:2249–2256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Tanihara H, Sano K, Heimark RL, St, John T, Suzuki S. Cloning of five human cadherins clarifies characteristic features of cadherin extracellular domain and provides further evidence for two structurally different types of cadherin. Cell Adhes Commun 1994; 2:15–26. [DOI] [PubMed] [Google Scholar]
- 31.Rosenberg P, Esni F, Sjodin A, Larue L, Carlsson L, Gullberg D, et al. A potential role of R-cadherin in striated muscle formation. Dev Biol 1997; 187:55–70. [DOI] [PubMed] [Google Scholar]
- 32.Dahl U, Sjodin A, Larue L, Radice GL, Cajander S, Takeichi M, et al. Genetic dissection of cadherin function during nephrogenesis. Mol Cell Biol 2002; 22:1474–1487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Sjodin A, Dahl U, Semb H. Mouse R-cadherin: expression during the organogenesis of pancreas and gastrointestinal tract. Exp Cell Res 1995; 221:413–425. [DOI] [PubMed] [Google Scholar]
- 34.Johnson E, Theisen CS, Johnson KR, Wheelock MJ. R-cadherin influences cell motility via Rho family GTPases. J Biol Chem 2004; 279:31041–31049. [DOI] [PubMed] [Google Scholar]
- 35.Agiostratidou G, Li M, Suyama K, Badano I, Keren R, Chung S, et al. Loss of retinal cadherin facilitates mammary tumor progression and metastasis. Cancer Res 2009; 69:5030–5038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Du C, Huang T, Sun D, Mo Y, Feng H, Zhou X, et al. CDH4 as a novel putative tumor suppressor gene epigenetically silenced by promoter hypermethylation in nasopharyngeal carcinoma. Cancer Lett 2011; 309:54–61. [DOI] [PubMed] [Google Scholar]
- 37.Maeda M, Johnson E, Mandal SH, Lawson KR, Keim SA, Svoboda RA, et al. Expression of inappropriate cadherins by epithelial tumor cells promotes endocytosis and degradation of E-cadherin via competition for p120(ctn). Oncogene 2006; 25:4595–4604. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental digital content is available for this article. Direct URL citations appear in the printed text and are provided in the HTML and PDF versions of this article on the journal's website (www.pharmacogeneticsandgenomics.com).