

Abstract
Adoptive transfer of T cells can be an effective anti-cancer treatment. However, uncontrolled or unpredictable immediate or persistent toxicities are a source of concern. The ability to conditionally eliminate aberrant cells in vivo therefore is becoming a critical step for the successful translation of this approach to the clinic. We review the evolution of safety systems, focusing on a suicide switch that can be expressed stably and efficiently in human T cells without impairing phenotype, function or antigen specificity. This system is based on the fusion of human caspase 9 to a modified human FK-binding protein, allowing conditional dimerization in the presence of an otherwise bioinert small molecule drug. When exposed to the synthetic dimerizing drug, the inducible caspase 9 (iC9) becomes activated and leads to the rapid apoptosis of cells expressing this construct. We have demonstrated the clinical feasibility and efficacy of this approach after haploidentical hematopoietic stem cell transplant (haplo-HSCT). Here we review the benefits and limitations of the approach.
Introduction
Cellular immunotherapies, including T cells genetically modified to selectively target malignant cells, are a promising cancer treatment as they augment the host immune response [1–4]. Antigen-specific T cells such as virus-specific cytotoxic T lymphocytes can expand in vivo, actively traffic to tumor sites, expand upon exposure to antigens, and persist long term. Moreover, activated T cells can recruit additional and distinct sets of cellular and cytokine-mediated effector mechanisms once antigen is recognized [5–8]. Given the desirable properties of these cell therapies, there has been great interest in adoptively transferring T cells capable of recognizing and destroying human tumors. In clinical trials, T cells modified to recognize specific tumor-associated antigens have produced activity against malignant cells and have led to impressive clinical responses [9–15]. Clinical trials, however, have also shown that antigen-specific T cells can have severe, even fatal, toxicities due to lack of control over their activation, expansion and persistence in vivo. Both on-target (e.g. CD19_CAR-T cells) and off-target toxicities (e.g. using affinity enhanced T cell receptors) have been reported [16–20]. Adverse events following T cell-based therapies may be immediate or delayed, mild or severe, and may persist or worsen over the lifespan of the infused T cells. This ability to persist long-term even in the absence of additional T cell doses distinguishes toxicities associated with adoptive T cell transfer from the pattern of toxicity associated with most small-molecule pharmaceuticals. Thus, how to effectively ablate or control adoptively transferred cells should unwanted effects occur remains a major challenge for cell therapy using conventional antigen-specific T cells.
On-target toxicities may result acutely from excessive cytokine release or from tumor lysis syndrome due to massive activation and proliferation of the infused cells when they encounter tumor [21–24]. On-target but off-tumor toxicities result from damage to normal tissues that share the targeted antigen, for example the hypogammaglobulinemia that follows the prolonged depletion of normal CD19+ B cells by long-lived T cells engineered to express chimeric antigen receptors (CAR) specific for CD19 (CD19-CAR T-cells). In other conditions, cross-reactivity with unrecognized expression of shared/cross-reactive epitopes can lead to severe or fatal neurologic and cardiac toxicities [19,20,25–28]. Thus, there is considerable interest in developing approaches to control the activation, expansion and persistence of infused cells over time. One approach is to use biodegradable cells, in which T lymphocyte transcribed RNA leads to high but transient expression of CAR on T cells [29–31]. In this review, we will instead focus on the ability to conditionally eliminate engineered T cells through the activation of cellular safety switches.
The ideal safety switch has a number of desirable characteristics. The switch should not itself be immunogenic, activation should use an otherwise inert agent that causes no damage to recipient endogenous cells or organs and that does not exacerbate the toxic effects of the cells to be destroyed. The ability for the switch/activating drug to function as a titratable “rheostat” is also desirable, as it would allow very low doses of the drug to be given to control adverse effects without complete abrogation of therapeutic benefit.
Several safety switches have been developed over the past decade but none has every desired characteristic. One set of switches targets surface molecules (CD20, EGFR) [32–37], while a second uses a transgenic enzyme to activate a cytotoxic pro-drug (HSV-thymidine kinase gene + GCV or cytosine deaminase + 5FC) [38–44]. Engineering therapeutic T cells with a peptide-specific antibody-based switch is another strategy to control the activation and expansion of these cells [45].
Evolution of safety switches
Monoclonal antibody-mediated suicide gene
Genetically modifying cells to produce a membrane-expressed protein allows for cell depletion after administration of a specific monoclonal antibody. Human CD20 can be used in this way, inducing cell death by the widely available CD20 mAb, Rituximab. In the presence of Rituximab, up to 90% of CD20 transduced cells are depleted [32–35]. However, normal B cells are also destroyed by this approach and since Rituximab is widely used as a treatment for B cell Non-Hodgkin lymphomas (NHL), the persistence of this antibody in vivo after treatment would lead to inadvertent destruction of any CAR T cells using CD20 as the suicide gene infused to NHL patients after previous Rituximab treatment. An alternative approach uses truncated human epidermal growth factor receptor (EGFR; ErbB-1, HER1 in humans), which is a tyrosine kinase receptor for the ErbB family of growth factor receptors that is not expressed by cells of the hematopoietic and lymphopoietic systems. A truncated EGFR co-expressed with T cells enables cell selection, tracking and ablation in vivo after systemic anti-EGFR monoclonal antibody administration. As yet, we do not know how effective this approach will be in humans [36,37].
Transgenic enzymes and prodrug therapy
Allogeneic hematopoietic stem cell transplantation (allo-HSCT) is the most widely used adoptive immunotherapy to treat cancer and the only curative treatment for some high-risk hematological malignancies. However, the incidence of disease relapse and post-transplant infection in patients receiving an unmanipulated transplant is lower than in patients receiving a T-cell-depleted graft, indicating that mature T cells present in the donor graft can both protect against viral infection and reactivation and produce a graft versus tumor (GVT) effect [46–49]. Investigators have shown that the post-transplant infusion of small numbers of donor T lymphocytes depleted of recipient-reactive T cells can improve immune reconstitution and antiviral immunity in HSCT recipients [50–54]. Engineered T cells with safety switches have been developed to increase the feasibility of infusing higher numbers of donor-derived T cells whilst providing a tool to control the increased risk of acute graft-versus-host disease (GvHD) that would otherwise be associated with any incomplete abrogation of alloreactivity.
To improve the safety profile of cellular products after allogeneic HSCT, the herpes simplex virus thymidine kinase (HSV-TK) gene was transferred into donor T cells. HSV-TK enzyme has 1000 times greater affinity for substrates such as gancyclovir (GCV) and acyclovir than host cell thymidine kinase. HSV-TK phosphorylates GCV to the active moiety, which interferes with DNA synthesis, thereby killing dividing cells. Thus, HSV-TK can be used as a suicide gene in the presence of GCV. The first clinical application of this safety switch was its expression in allogeneic donor T cells administered after allo-HSCT to enhance immune recovery. If patients developed acute GvHD, they received GCV as a prodrug. This approach resulted in abrogation of the adverse effects while sparing the anti-viral activity of the infused T cell product [38,39]. Subsequent studies and clinical trials have supported the effectiveness of this approach, which is now in a Phase III clinical trial [55–60].
Although HSV-TK is an effective safety switch for acute GvHD due to transfer of donor T cells after HSCT, it has significant drawbacks that may limit its value as a more broadly used safety gene for other cellular therapies. First, the immunogenicity of HSV-TK can lead to the induction of an immune response to HSV-TK transduced T cells, an effect that will likely be even more common in circumstances when the transduced T cells are administered to more immunocompetent hosts. These immune responses may compromise the persistence of the infused T cells. Secondly, HSV-TK-mediated cell death requires GCV or similar compounds, which are important pharmacological agents for the prophylaxis and treatment of cytomegalovirus infection in immunocompromised hosts. Administration of the prodrug to treat these infections necessarily leads to concomitant and undesired elimination of the transduced cell population. Finally, HSV-TK-mediated killing primarily affects dividing cells and takes several days or even weeks to reach maximum effectiveness, a delay that may be excessive for patients who develop more acute toxicities after treatment with T cell therapies [61–63].
Other investigators have used cytosine deaminase (CD) gene transfer, which converts the antifungal drug 5-fluorocytosine (5-FC) into the cytotoxic 5-fluorouracil (5-FU) [40,41] by catalyzing the hydrolytic deamination of cytosine into uracil and by the further conversion of 5-FU into potent anti-metabolites (5-FdUMP, 5-FdUTP, 5-FUTP) by cellular enzymes. These compounds inhibit thymidylate synthase and the production of RNA and DNA, resulting in cell death. Although these approaches have looked promising in some pre-clinical models, the low efficiency of bacterial CD to convert 5-FC into 5-FU limited the overall therapeutic response. Recently, Ghosh et al. have mutated CD to increase its affinity to 5-FC and reduce the amount of 5-FC required as prodrug [42–44].
Inducible dimerization by small molecules
To improve upon the above approaches by reducing immunogenicity and toxicity while increasing speed and efficacy, we developed an approach that exploited the control of cellular signaling through ligand-mediated dimerization of intracellular proteins. We used cell-permeable synthetic ligands that bind to FK506 binding protein 12 (FKBP12). FKBP12 belongs to the immunophilin family of receptors, a physiological function of which is to bind to and inactivate calcineurin [64–66]. Calcineurin inhibition leads to impaired T-cell receptor signaling and consequent immunosuppression [67]. In order to create a cellular control switch without the unwanted physiological and toxic effects of calcineurin inhibition, Clackson and colleagues redesigned the ligand-FKBP12 interface. They created a specificity-binding pocket in FKBP12 by substituting the bulky phenylalanine with the smaller valine residue (FKBP12-F36V). The redesigned ligand has high affinity and selectivity for FKBP12-F36V and interacts minimally with endogenous FKBP [68]. In 2001, a dimeric form of this ligand, called AP1903, underwent safety testing in healthy volunteers without significant adverse effects [69].
Based on these studies, we devised a safety switch for T cells that exploits dimerization of a modified caspase 9 molecule, a component of the intrinsic (mitochondrial) apoptotic pathway. Under physiological conditions, caspase 9 is activated by the release of cytochrome C from damaged mitochondria. Activated caspase 9 then activates caspase 3, which triggers terminal effector molecules leading to apoptosis (Figure. 1). The optimized inducible caspase 9 molecule (iC9) consists of an FKBP12-F36V domain linked via a flexible Ser-Gly-Gly-Gly-Ser linker to Δcaspase 9, which is caspase 9 without its physiological dimerization domain (caspase activation domain (CARD)). These inducible apoptotic components are followed by a selectable marker, truncated CD19 (ΔCD19), linked by a 2A-like sequence encoding a cleavable peptide. Inducible caspase 9 has low dimerizer-independent basal activity and can be stably expressed in human T cells without impairing their phenotype, function or antigen specificity [70,71]. A single 10nM dose of AP1903, also referred to as chemical inducer of dimerization (CID), induces apoptosis in up to 99% of iC9-transduced cells selected for high expression of the transgenic CD19 marker in vitro and in vivo [70–73]. The killing efficiency is significantly lower in cells with low or intermediate levels of transgene expression. We will discuss below the implications of this decreased killing for multiple rounds of AP1903 treatment in patients [74].
Figure 1. The structure of the iC9 suicide gene.
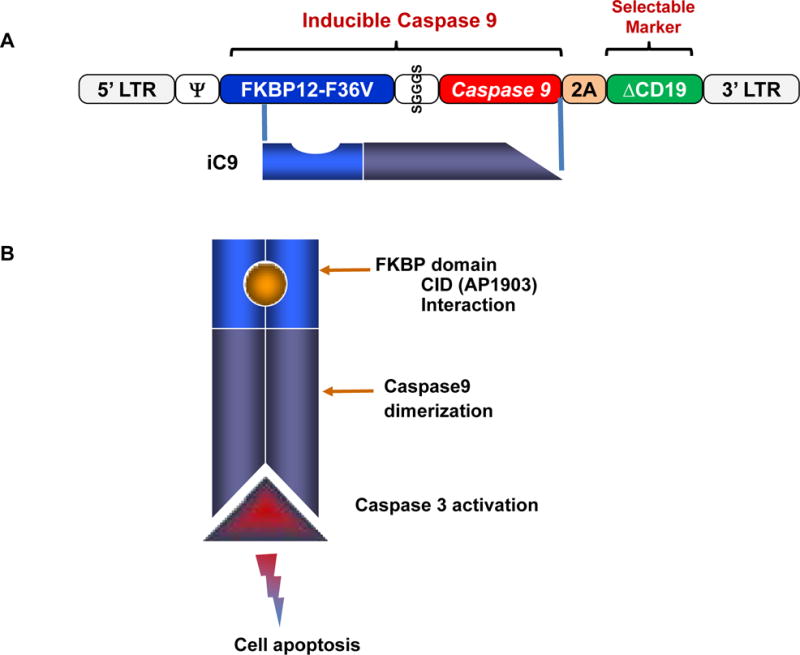
(A) The transgene consists of an inducible caspase 9 (iC9), and a selectable marker, truncated CD19 (ΔCD19), linked by a 2A-like sequence, which encodes a cleavable peptide. iC9 consists of a drug-binding domain (FKBP12-F36V) connected via a short linker (SGGGS) to human caspase 9. (B) Conditional Caspase 9 dimerization is induced in the presence of CID (AP1903), cleaving caspase 3 and leading to cell apoptosis.
Clinical applications of iC9 safety switch post HSCT
Ex vivo allodepletion of donor T cells
As described above (Transgenic enzymes and prodrug therapy), hematopoietic stem cell transplants contain a high frequency of alloreactive T cells. Even when donor and recipient are haploidentical at the HLA loci, the frequency of T cells recognizing the non-shared HLA haplotype can be as high as 10%. To avoid lethal GvHD, it is necessary to extensively deplete T-cells from the donor graft either by positive selection of CD34-positive HSCs or by negative selection of T cells. Although extensive T-cell removal of the graft effectively prevents graft rejection and GvHD, the process also causes prolonged and profound post-transplant immunodeficiency with compromised antiviral immunity, in addition to the increased the risk of relapse as described above. As a consequence, infections remain a significant cause of morbidity and mortality and are a frequent cause of treatment failure after haploidentical transplantation [52–54]. We and others have demonstrated that haploidentical donor T cells can be returned to patients after T cell-depleted allografts with a low incidence of severe GvHD, provided those donor T cells are first depleted of the recipient-reactive component. Preclinical and clinical studies have shown several approaches by which alloreactive T cells can be depleted [51,52,75–77]. Our own studies focused on the use of a CD25 immunotoxin that recognizes the CD25 activation-associated marker expressed on alloreactive donor T cells after in vitro exposure to recipient antigen presenting cells [78,79]. Infusion of these allodepleted T cells improved immune reconstitution and antiviral immunity at doses as low as 3×105 per kg [52,72]. Nonetheless, incorporation of the iC9 safety gene provides the ability to conditionally eliminate these allodepleted T cells in the event of unanticipated GvHD and greatly improves the safety of T-cell addback. We also considered that the feasibility of using iC9 as the sole safety mechanism for haploidentical donor T cell infusions not first subjected to allodepletion.
From an abundance of caution, we started the first clinical trial using the iC9 system on allodepleted haploidentical donor T cells. Patients who had undergone CD34-selected haplo-HSCT were administered escalating doses (1×106–1×107/Kg) of iC9-modified allodepleted T cells from day 30 after transplant. The iC9-T cells expanded and were detected in the peripheral blood as early as 7 days after infusion. Four patients out of ten developed acute GvHD grade 1–2 of the liver and/or skin. When GvHD occurred, >90% of the iC9-T cells were eliminated within 2 hours of dimerizer administration, and GvHD was rapidly (within 24 hours) and permanently resolved. Remarkably, residual iC9-T cells were able to re-expand; these remaining cells contained pathogen-specific precursors and persisted long-term without recurrence of GvHD [80]. Hence, a single dose of AP1903 can rapidly and permanently control of GvHD, and the kinetics of endogenous T-cell reconstitution appeared identical to those observed in patients in whom we did not activate the iC9 transgene.
However, the iC9-T cells infused in this study had already been depleted of alloreactive precursors by ex vivo culture with recipient B cell lymphoblastoid cell lines followed by negative selection of responding (alloreactive) donor T cells. This lengthy and complex process takes about 3 months and so is impractical for patients requiring urgent transplantation, while its complexity makes the process unsuited for scaling to general clinical use.
In vivo allo-depletion of donor T cells
To make the iC9 suicide gene more clinically practicable, we investigated whether iC9 activation alone is sufficient to produce both rapid and long-term control of GvHD caused by alloreplete haploidentical donor T cells in vivo. This new method greatly shortened the manufacturing time to less than 2 weeks. In a subsequent clinical trial, we demonstrated that alloreplete iC9-T cells can engraft, persist at least two years and provide effective antiviral immunity. The iC9-T cells enabled more rapid immune reconstitution than reported after haplo-HSCT without adoptive T cell transfer. Consistent with a previous study in which patients received ex vivo allodepleted iC9-T cells, in this clinical trial the absolute count of endogenous CD3+ T cells was greater than 500 cells per μl at 4 months after iC9-T cell infusion (approximately 5.5 months post-transplantation), while similar T cell counts are reached only between 9–12 months after haplo-HSCT if patients do not receive T lymphocyte add-back [73,80].
Toxicity
In general, the iC9 transgene and dimerizer appear to be non-toxic, although one patient who received multiple doses of the drug (see below: Single versus serial activation) had mild and transient pancytopenia (Grade 2) that was present immediately after each administration of AP1903 and resolved within 72 hrs. This response was not observed in the Phase I/II studies on healthy volunteers or in other treated patients. The mechanism underlying this idiosyncratic reaction is unclear, but it is likely not attributable to direct toxicity to marrow or blood cells since it lasted less than 3 days after each administration of the drug [74].
Broader application of iC9 gene
As described in the introduction, one setting in which a safety genes would be particularly useful is to control severe on or off-target toxicities from gene modified T cells. Although patients who receive T cell addback after HSCT do not develop the same severe cytokine release syndrome (CRS) as seen after administration of CAR T cells, a milder variant of the disorder is seen with high circulating cytokine levels associated with high fevers and respiratory compromise. One of our patients developed acute GvHD and this became associated with a CRS, as manifested by hyperpyrexia and a high level of circulating cytokines. Through this patient, we were able to determine the potential impact of iC9 activation on CRS. Within 2 hours of AP1903 administration and in the absence of additional therapy the patient’s temperature normalized, skin rash dramatically improved and the elevated plasma cytokine levels declined [73].
We do not yet know whether the iC9 system can be activated in every tissue in which T cell toxicity might be manifest, but there is evidence that AP1903 can effectively deplete iC9-transduced T cells in the CNS as well as in the skin, gut and liver if these organs are affected by GvHD. For instance, one patient developed acute GvHD after VZV meningitis, and was given dimerizing drug with complete response. Comparison of iC9/CD19+ T cells in the CSF before and after dimerizer administration showed a level of depletion equivalent to that seen in peripheral blood.
Thus, iC9 in vivo allodepletion can be achieved by single dose of CID administration, with resolution of associated signs and symptoms of GvHD within 6 to 48 hours. Importantly, there was no recurrence of GvHD associated with the gradual recovery of iC9-T cells following AP1903 administration.
Benefits and limitations
Long-term follow-up revealed that the in vivo persistence of iC9-T cells benefits patients through immediate and sustained protection from major pathogens in the absence of acute or chronic GvHD. This restored immunity is mediated initially by the infused cells themselves, and subsequently by an apparently accelerated reconstitution of endogenous naïve T lymphocytes [80].
Spontaneous dimerizer-independent apoptosis is a potential drawback of iC9, but we have not observed any significant impact on in vivo expansion. Dimerizer-mediated elimination of iC9-T cells requires a minimum expression level for the iC9 transgene. Expression is determined in large part by the site of transgene integration and the level of cell activation. At the time of onset of GvHD, most of the contributing alloreactive T cells will have activated TCR signaling and will therefore express a higher mean level of iC9 than resting cells. As a consequence alloreactive cells contributing to GvHD be even more likely to be eliminated than the bulk T cell population [71,72,74,81]. Nonetheless a small population of alloreactive T cells will likely survive exposure to dimerizing drug, but in clinical studies to date these have been insufficient in number or activity to cause resurgent GvHD. It is possible, however, that a larger treatment series will show occasional failures of elimination and persistence or resurgence of GvHD.
Single vs serial activation
Although a single dose of dimerizing drug can immediately control the symptoms and signs of short-term toxicities induced by adoptively transferred cells, a small number of low expressing residual cells persist, and we observed that these iC9-T cells can subsequently expand and repopulate patients. Although this resurgence does not lead to a recurrence of GvHD, likely because the alloreactive T cells causing GvHD are among the highest expressing population due to their activation state, a resurgence of the transduced population may be undesirable in any setting in which control of the adverse effects requires more sustained T cell ablation. We have shown that under these circumstances repeat administration of CID alone or in combination with other agents may be feasible and safe. After multiple doses of CID, there is additional depletion of T cells, although the percentage of killing is progressively lower with each dose. Nonetheless, up to 85% of iC9-T cells were eliminated from peripheral blood after the third CID treatment in one patient.
Multiple mechanisms likely contribute to the dimerizer resistance of the small population of iC9+ T cells that remain after multiple CID treatments. These factors include proviral integration sites that favor low gene expression, transgene silencing by promoter hypermethylation, high expression of anti-apoptotic proteins in the apoptosis pathway, and sporadic nonsense mutations of the iC9 transgene [74,81–83]. Substituting potent internal promoters and introducing the transgene into lentiviral vectors may help overcome these limitations, as may the use of specific gene editing into consistent sites in the host cell DNA to ensure high levels of constitutive expression. Likely, a variety of these approaches will need to be combined for there to be sustained susceptibility of iC9 in all cells.
Summary
Immunotherapy is one of the most promising approaches for cancer treatment and the iC9 system can rapidly and safely control a broad range of toxicities from adoptively transferred T cells. Our approach exploits a drug that is bio-inert rather than a therapeutically relevant pro-drug, and it may be less immunogenic than the HSV-TK system in immunocompetent recipients. Successful clinical validation of the iC9 safety switch has suggested a number of possible applications in cellular therapies for both hematological diseases and solid tumors.
Highlights.
iC9 safety switch is designed to increase the safety of immunotherapy.
It is based on the fusion of human proteins and non-immunogenicity.
Activation of iC9 can rapidly and safely control a broad range of toxicities.
Serial activation of iC9 safety switch in vivo is feasible and safe.
Acknowledgments
We are thankful Catherine Gillespie for editing the manuscript. The clinical protocols (I.N.D.13813) were supported by NIH-NHLBI grant U54HL08100, and development of the caspase system by P01CA094237 and P50CA126752. The clinical trials also received support from the Clinical Research Center at Texas Children’s Hospital, the Institute for Clinical and Translational Research at Baylor College of Medicine and shared resources of the Dan L Duncan Cancer Center support grant P30CA125123.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Conflict-of-interest disclosure: The Center for Cell and Gene Therapy has a collaborative research agreement with Celgene.
References
- 1.Walter EA, Greenberg PD, Gilbert MJ, et al. Reconstitution of cellular immunity against cytomegalovirus in recipients of allogeneic bone marrow by transfer of T-cell clones from the donor. N Engl J Med. 1995;333:1038–1044. doi: 10.1056/NEJM199510193331603. [DOI] [PubMed] [Google Scholar]
- 2.Rooney CM, Smith CA, Ng CY, et al. Infusion of cytotoxic T cells for the prevention and treatment of Epstein-Barr virus-induced lymphoma in allogeneic transplant recipients. Blood. 1998;92:1549–1555. [PubMed] [Google Scholar]
- 3.Dudley ME, Wunderlich JR, Robbins PF, et al. Cancer regression and autoimmunity in patients after clonal repopulation with antitumor lymphocytes. Science. 2002;298:850–854. doi: 10.1126/science.1076514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Gottschalk S, Rooney CM. Adoptive T-Cell Immunotherapy. Curr Top Microbiol Immunol. 2015;391:427–454. doi: 10.1007/978-3-319-22834-1_15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Heslop HE, Slobod KS, Pule MA, et al. Long-term outcome of EBV-specific T-cell infusions to prevent or treat EBV-related lymphoproliferative disease in transplant recipients. Blood. 2010;115(5):925–935. doi: 10.1182/blood-2009-08-239186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Savoldo B, Ramos CA, Liu E, et al. CD28 costimulation improves expansion and persistence of chimeric antigen receptor-modified T cells in lymphoma patients. J Clin Invest. 2011;121(5):1822–1826. doi: 10.1172/JCI46110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Xu Y, Zhang M, Ramos CA, et al. Closely related T-memory stem cells correlate with in vivo expansion of CAR.CD19-T cells and are preserved by IL-7 and IL-15. Blood. 2014;123(24):3750–3759. doi: 10.1182/blood-2014-01-552174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Melenhorst JJ, Castillo P, Hanley PJ, et al. Graft versus leukemia response without graft-versus-host disease elicited by adoptively transferred multivirus-specific T-cells. Mol Ther. 2015;23(1):179–183. doi: 10.1038/mt.2014.192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Louis CU, Savoldo B, Dotti G, et al. Antitumor activity and long-term fate of chimeric antigen receptor-positive T cells in patients with neuroblastoma. Blood. 2011;118(23):6050–6056. doi: 10.1182/blood-2011-05-354449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bollard CM, Gottschalk S, Torrano V, et al. Sustained complete responses in patients with lymphoma receiving autologous cytotoxic T lymphocytes targeting Epstein-Barr virus latent membrane proteins. J Clin Oncol. 2014;32(8):798–808. doi: 10.1200/JCO.2013.51.5304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Porter DL, Levine BL, Kalos M, et al. Chimeric antigen receptor-modified T cells in chronic lymphoid leukemia. N Engl J Med. 2011;365(8):725–733. doi: 10.1056/NEJMoa1103849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Maude SL, Frey N, Shaw PA, et al. Chimeric antigen receptor T cells for sustained remissions in leukemia. N Engl J Med. 2014;371(16):1507–1517. doi: 10.1056/NEJMoa1407222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Garfall AL, Maus MV, Hwang WT, et al. Chimeric Antigen Receptor T Cells against CD19 for Multiple Myeloma. N Engl J Med. 2015;373(11):1040–1047. doi: 10.1056/NEJMoa1504542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ahmed N, Brawley VS, Hegde M, et al. Human Epidermal Growth Factor Receptor 2 (HER2) -Specific Chimeric Antigen Receptor-Modified T Cells for the Immunotherapy of HER2-Positive Sarcoma. J Clin Oncol. 2015;33(15):1688–1696. doi: 10.1200/JCO.2014.58.0225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Cruz CR, Micklethwaite KP, Savoldo B, et al. Infusion of donor-derived CD19-redirected virus-specific T cells for B-cell malignancies relapsed after allogeneic stem cell transplant: a phase 1 study. Blood. 2013;122(17):2965–73. doi: 10.1182/blood-2013-06-506741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Morgan RA, Yang JC, Kitano M, et al. Case report of a serious adverse event following the administration of t cells transduced with a chimeric antigen receptor recognizing erbb2. Mol Ther. 2010;18(4):843–851. doi: 10.1038/mt.2010.24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kochenderfer JN, Dudley ME, Feldman SA, et al. B-cell depletion and remissions of malignancy along with cytokine-associated toxicity in a clinical trial of anti-CD19 chimeric-antigen-receptor-transduced T cells. Blood. 2012;119(12):2709–2720. doi: 10.1182/blood-2011-10-384388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Brentjens RJ, Riviere I, Park JH, et al. Safety and persistence of adoptively transferred autologous CD19-targeted T cells in patients with relapsed or chemotherapy refractory B-cell leukemias. Blood. 2011;118(18):4817–4828. doi: 10.1182/blood-2011-04-348540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Linette GP, Stadtmauer EA, Maus MV, et al. Cardiovascular toxicity and titin cross-reactivity of affinity-enhanced T cells in myeloma and melanoma. Blood. 2013;122(6):863–871. doi: 10.1182/blood-2013-03-490565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Cameron BJ, Gerry AB, Dukes J, et al. Identification of a Titin-derived HLA-A1-presented peptide as a cross-reactive target for engineered MAGE A3-directed T cells. Sci Transl Med. 2013;5(197):197ra103. doi: 10.1126/scitranslmed.3006034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kalos M, Levine BL, Porter DL, et al. T cells with chimeric antigen receptors have potent antitumor effects and can establish memory in patients with advanced leukemia. Sci Transl Med. 2011;3:95ra73. doi: 10.1126/scitranslmed.3002842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Grupp SA, Kalos M, Barrett D, et al. Chimeric antigen receptor-modified T cells for acute lymphoid leukemia. New Engl J Med. 2013;368(16):1509–1518. doi: 10.1056/NEJMoa1215134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lee DW, Kochenderfer JN, Stetler-Stevenson M, et al. T cells expressing CD19 chimeric antigen receptors for acute lymphoblastic leukaemia in children and young adults: a phase 1 dose-escalation trial. Lancet. 2015;385(9967):517–528. doi: 10.1016/S0140-6736(14)61403-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Teachey DT, Lacey SF, Shaw PA, et al. Identification of Predictive Biomarkers for Cytokine Release Syndrome after Chimeric Antigen Receptor T cell Therapy for Acute Lymphoblastic Leukemia. Cancer Discov. 2016 Apr 13; doi: 10.1158/2159-8290.CD-16-0040. pii: CD-16-0040 [Epub ahead of print] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Brentjens RJ, Davila ML, Riviere I, et al. CD19-targeted T cells rapidly induce molecular remissions in adults with chemotherapy-refractory acute lymphoblastic leukemia. Sci Transl Med. 2013;5:177ra138. doi: 10.1126/scitranslmed.3005930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kochenderfer JN, Wilson WH, Janik JE, et al. Eradication of B-lineage cells and regression of lymphoma in a patient treated with autologous T cells genetically engineered to recognize CD19. Blood. 2010;116(20):4099–4102. doi: 10.1182/blood-2010-04-281931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Davila ML, Riviere I, Wang X, et al. Efficacy and toxicity management of 19-28z CAR T cell therapy in B cell acute lymphoblastic leukemia. Sci Transl Med. 2014;6(224):224ra225. doi: 10.1126/scitranslmed.3008226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Fedorov VD, Themeli M, Sadelain M. PD-1- and CTLA-4-based inhibitory chimeric antigen receptors (iCARs) divert off-target immunotherapy responses. Sci Transl Med. 2013;5(215):215ra172. doi: 10.1126/scitranslmed.3006597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Zhao Y, Moon E, Carpenito C, Paulos CM, Liu X. Multiple injections of electroporated autologous T cells expressing a chimeric antigen receptor mediate regression of human disseminated tumor. Cancer Res. 2010;70(22):9053–9061. doi: 10.1158/0008-5472.CAN-10-2880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Maus MV, Haas AR, Beatty GL, et al. T cells expressing chimeric antigen receptors can cause anaphylaxis in humans. Cancer Immunol Res. 2013;1(1):26–31. doi: 10.1158/2326-6066.CIR-13-0006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Beatty GL, Haas AR, Maus MV, et al. Mesothelin-specific chimeric antigen receptor mRNA-engineered T cells induce anti-tumor activity in solid malignancies. Cancer Immunol Res. 2014;2(2):112–120. doi: 10.1158/2326-6066.CIR-13-0170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Serafini M, Manganini M, Borleri G, et al. Characterization of CD20-transduced T lymphocytes as an alternative suicide gene therapy approach for the treatment of graft-versus-host disease. Hum Gene Ther. 2004;15(1):63–76. doi: 10.1089/10430340460732463. [DOI] [PubMed] [Google Scholar]
- 33.Griffioen M, van Egmond EH, Kester MG, Willemze R, Falkenburg JH, Heemskerk MH. Retroviral transfer of human CD20 as a suicide gene for adoptive t-cell therapy. Haematologica. 2009;94(9):1316–1320. doi: 10.3324/haematol.2008.001677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Introna M, Barbui AM, Bambacioni F, et al. Genetic modification of human T cells with CD20: A strategy to purify and lyse transduced cells with anti-CD20 antibodies. Human Gene Ther. 2000;11(4):611–620. doi: 10.1089/10430340050015798. [DOI] [PubMed] [Google Scholar]
- 35.Philip B, Kokalaki E, Mekkaoui L, et al. A highly compact epitope-based marker/suicide gene for easier and safer T-cell therapy. Blood. 2014;124(8):1277–1287. doi: 10.1182/blood-2014-01-545020. [DOI] [PubMed] [Google Scholar]
- 36.Wang X, Chang WC, Wong CW, et al. A transgene-encoded cell surface polypeptide for selection, in vivo tracking, and ablation of engineered cells. Blood. 2011;118(5):1255–1263. doi: 10.1182/blood-2011-02-337360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Jensen MC, Riddell SR. Designing chimeric antigen receptors to effectively and safely target tumors. Curr Opin Immunol. 2015;33:9–15. doi: 10.1016/j.coi.2015.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Bonini C, Ferrari G, Verzeletti S, et al. HSV-TK gene transfer into donor lymphocytes for control of allogeneic graft-versus-leukemia. Science. 1997;276(5319):1719–1724. doi: 10.1126/science.276.5319.1719. [DOI] [PubMed] [Google Scholar]
- 39.Tiberghien P, Ferrand C, Lioure B, et al. Administration of herpes simplex-thymidine kinase-expressing donor T cells with a T-cell-depleted allogeneic marrow graft. Blood. 2001;97(1):63–72. doi: 10.1182/blood.v97.1.63. [DOI] [PubMed] [Google Scholar]
- 40.Haack K, Moebius U, Knebel Doeberitz MV, et al. Detection of cytosine deaminase in genetically modified tumor cells by specific antibodies. Hum Gene Ther. 1997;8(11):1395–1401. doi: 10.1089/hum.1997.8.11-1395. [DOI] [PubMed] [Google Scholar]
- 41.Tiraby M, Cazaux C, Baron M, Drocourt D, Reynes JP, Tiraby G. Concomitant expression of E. coli cytosine deaminase and uracil phosphoribosyltransferase improves the cytotoxicity of 5-fluorocytosine. FEMS Microbiol Lett. 1998;167(1):41–49. doi: 10.1111/j.1574-6968.1998.tb13205.x. [DOI] [PubMed] [Google Scholar]
- 42.Kievit E, Bershad E, Ng E, et al. Superiority of yeast over bacterial cytosine deaminase for enzyme/prodrug gene therapy in colon cancer xenografts. Cancer Res. 1999;59:1417–1421. [PubMed] [Google Scholar]
- 43.Pandha HS, Martin LA, Rigg A, et al. Genetic prodrug activation therapy for breast cancer: A phase I clinical trial of erbB-2-directed suicide gene expression. J Clin Oncol. 1999;17(7):2180–2189. doi: 10.1200/JCO.1999.17.7.2180. [DOI] [PubMed] [Google Scholar]
- 44.Raza A, Kohila V, Ghosh SS. Redesigned Escherichia coli cytosine deaminase: a new facet of suicide gene therapy. J Gene Med. 2015;17(6–7):132–139. doi: 10.1002/jgm.2831. [DOI] [PubMed] [Google Scholar]
- 45.Rodgers DT, Mazagova M, Hampton EN, et al. Switch-mediated activation and retargeting of CAR-T cells for B-cell malignancies. Proc Natl Acad Sci U S A. 2016;113(4):E459–468. doi: 10.1073/pnas.1524155113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Ballen KK, Koreth J, Chen YB, Dey BR, Spitzer TR. Selection of optimal alternative graft source: mismatched unrelated donor, umbilical cord blood, or haploidentical transplant. Blood. 2012;119(9):1972–1980. doi: 10.1182/blood-2011-11-354563. [DOI] [PubMed] [Google Scholar]
- 47.Palma J, Salas L, Carrión F, et al. Haploidentical stem cell transplantation for children with high-risk leukemia. Pediatr Blood Cancer. 2012;59(5):895–901. doi: 10.1002/pbc.24022. [DOI] [PubMed] [Google Scholar]
- 48.Aversa F, Terenzi A, Tabilio A, et al. Full haplotype-mismatched hematopoietic stem-cell transplantation: a phase II study in patients with acute leukemia at high risk of relapse. J Clin Oncol. 2005;23(15):3447–3454. doi: 10.1200/JCO.2005.09.117. [DOI] [PubMed] [Google Scholar]
- 49.Bethge WA, Haegele M, Faul C, et al. Haploidentical allogeneic hematopoietic cell transplantation in adults with reduced-intensity conditioning and CD3/CD19 depletion: fast engraftment and low toxicity. Exp Hematol. 2006;34(12):1746–1752. doi: 10.1016/j.exphem.2006.08.009. [DOI] [PubMed] [Google Scholar]
- 50.Andre-Schmutz I, Le Deist F, Hacein-Bey-Abina S, et al. Immune reconstitution without graft-versus-host disease after haemopoietic stem-cell transplantation: a phase 1/2 study. Lancet. 2002;360(9327):130–137. doi: 10.1016/S0140-6736(02)09413-8. [DOI] [PubMed] [Google Scholar]
- 51.Amrolia PJ, Muccioli-Casadei G, Yvon E, et al. Selective depletion of donor alloreactive T cells without loss of antiviral or anti-leukemic responses. Blood. 2003;102(6):2292–2299. doi: 10.1182/blood-2002-11-3516. [DOI] [PubMed] [Google Scholar]
- 52.Amrolia PJ, Muccioli-Casadei G, Huls H, et al. Adoptive immunotherapy with allodepleted donor T-cells improves immune reconstitution after haploidentical stem cell transplantation. Blood. 2006;108(6):1797–1808. doi: 10.1182/blood-2006-02-001909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Perruccio K, Tosti A, Burchielli E, et al. Transferring functional immune responses to pathogens after haploidentical hematopoietic transplantation. Blood. 2005;106(13):4397–4406. doi: 10.1182/blood-2005-05-1775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Leen AM, Christin A, Myers GD, et al. Cytotoxic T lymphocyte therapy with donor T cells prevents and treats adenovirus and Epstein-Barr virus infections after haploidentical and matched unrelated stem cell transplantation. Blood. 2009;114(19):4283–4292. doi: 10.1182/blood-2009-07-232454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Ciceri F, Bonini C, Marktel S, et al. Antitumor effects of HSV-TK–engineered donor lymphocytes after allogeneic stem-cell transplantation. Blood. 2007;109(11):4698–4707. doi: 10.1182/blood-2006-05-023416. [DOI] [PubMed] [Google Scholar]
- 56.Ciceri F, Bonini C, Stanghellini MT, et al. Infusion of suicide-gene-engineered donor lymphocytes after family haploidentical haemopoietic stem-cell transplantation for leukaemia (the TK007 trial): a non-randomised phase I–II study. Lancet Oncol. 2009;10(5):489–500. doi: 10.1016/S1470-2045(09)70074-9. [DOI] [PubMed] [Google Scholar]
- 57.Sangro B, Mazzolini G, Ruiz M, et al. A phase I clinical trial of thymidine kinase-based gene therapy in advanced hepatocellular carcinoma. Cancer Gene Ther. 2010;17(12):837–843. doi: 10.1038/cgt.2010.40. [DOI] [PubMed] [Google Scholar]
- 58.Nasu Y, Saika T, Ebara S, et al. Suicide gene therapy with adenoviral delivery of HSV-tK gene for patients with local recurrence of prostate cancer after hormonal therapy. Mol Ther. 2007;15(4):834–840. doi: 10.1038/sj.mt.6300096. [DOI] [PubMed] [Google Scholar]
- 59.Colombo F, Barzon L, Franchin E, et al. Combined HSV-TK/IL-2 gene therapy in patients with recurrent glioblastoma multiforme: biological and clinical results. Cancer Gene Ther. 2005;12(10):835–848. doi: 10.1038/sj.cgt.7700851. [DOI] [PubMed] [Google Scholar]
- 60.Van der Linden RR, Haagmans BL, Mongiat-Artus P, et al. Virus specific immune responses after human neoadjuvant adenovirus-mediated suicide gene therapy for prostate cancer. Eur Urol. 2005;48(1):153–161. doi: 10.1016/j.eururo.2005.02.013. [DOI] [PubMed] [Google Scholar]
- 61.Traversari C, Marktel S, Magnani Z, et al. The potential immunogenicity of the TK suicide gene does not prevent full clinical benefit associated with the use of TK-transduced donor lymphocytes in HSCT for hematologic malignancies. Blood. 2007;109(11):4708–15. doi: 10.1182/blood-2006-04-015230. [DOI] [PubMed] [Google Scholar]
- 62.Garin MI, Garrett E, Tiberghien P, et al. Molecular mechanism for ganciclovir resistance in human T lymphocytes transduced with retroviral vectors carrying the herpes simplex virus thymidine kinase gene. Blood. 2001;97:122–9. doi: 10.1182/blood.v97.1.122. [DOI] [PubMed] [Google Scholar]
- 63.Marin V, Cribioli E, Philip B, et al. Comparison of different suicide-gene strategies for the safety improvement of genetically manipulated T cells. Hum Gene Ther Methods. 2012;23(6):376–86. doi: 10.1089/hgtb.2012.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Spencer DM, Wandless TJ, Schreiber SL, Crabtree GR. Controlling signal transduction with synthetic ligands. Science. 1993;262(5136):1019–1024. doi: 10.1126/science.7694365. [DOI] [PubMed] [Google Scholar]
- 65.Fan L, Freeman KW, Khan T, Pham E, Spencer DM. Improved artificial death switches based on caspases and FADD. Hum Gene Ther. 1999;10(14):2273–2285. doi: 10.1089/10430349950016924. [DOI] [PubMed] [Google Scholar]
- 66.Schreiber SL. Chemistry and biology of the immunophilins and their immunosuppressive ligands. Science. 1991;251(4991):283–287. doi: 10.1126/science.1702904. [DOI] [PubMed] [Google Scholar]
- 67.Clipstone NA, Crabtree GR. Identification of calcineurin as a key signaling enzyme in T-lymphocyte activation. Nature. 1992;357(6380):695–697. doi: 10.1038/357695a0. [DOI] [PubMed] [Google Scholar]
- 68.Clackson T, Yang W, Rozamus LW, et al. Redesigning an FKBP-ligand interface to generate chemical dimerizers with novel specificity. Proc Natl Acad Sci U S A. 1998;95(18):10437–10442. doi: 10.1073/pnas.95.18.10437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Iuliucci JD, Oliver SD, Morley S, et al. Intravenous safety and pharmacokinetics of a novel dimerizer drug, AP1903, in healthy volunteers. J Clin Pharmacol. 2001;41(8):870–879. doi: 10.1177/00912700122010771. [DOI] [PubMed] [Google Scholar]
- 70.Straathof KC, Pulè MA, Yotnda P, et al. An inducible caspase 9 safety switch for T-cell therapy. Blood. 2005;105(11):4247–4254. doi: 10.1182/blood-2004-11-4564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Tey SK, Dotti G, Rooney CM, Heslop HE, Brenner MK. Inducible caspase 9 suicide gene to improve the safety of allodepleted T cells after haploidentical stem cell transplantation. Biol Blood Marrow Transplant. 2007;13(8):913–924. doi: 10.1016/j.bbmt.2007.04.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Di Stasi A, Tey SK, Dotti G, et al. Inducible apoptosis as a safety switch for adoptive cell therapy. N Engl J Med. 2011;365(18):1673–1683. doi: 10.1056/NEJMoa1106152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Zhou X, Dotti G, Krance RA, et al. Inducible caspase-9 suicide gene controls adverse effects from alloreplete T cells after haploidentical stem cell transplantation. Blood. 2015;125(26):4103–4113. doi: 10.1182/blood-2015-02-628354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Zhou X, Naik S, Dakhova O, et al. Serial Activation of the Inducible Caspase 9 Safety Switch after Human Stem Cell Transplantation. Mol Ther. 2016;24(4):823–831. doi: 10.1038/mt.2015.234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Solomon SR, Tran T, Carter CS, et al. Optimized clinical-scale culture conditions for ex vivo selective depletion of host-reactive donor lymphocytes: a strategy for GvHD prophylaxis in allogeneic PBSC transplantation. Cytotherapy. 2002;4(5):395–406. doi: 10.1080/146532402320775982. [DOI] [PubMed] [Google Scholar]
- 76.Szabolcs P, Park KD, Marti L, et al. Superior depletion of alloreactive T cells from peripheral blood stem cell and umbilical cord blood grafts by the combined use of trimetrexate and interleukin-2 immunotoxin. Biol Blood Marrow Transplant. 2004;10(11):772–783. doi: 10.1016/j.bbmt.2004.07.005. [DOI] [PubMed] [Google Scholar]
- 77.Vaclavkova P, Cao Y, Wu LK, Michalek J, Vitetta ES. A comparison of an anti-CD25 immunotoxin, Ontak and anti-CD25 microbeads for their ability to deplete alloreactive T cells in vitro. Bone Marrow Transplant. 2006;37(6):559–567. doi: 10.1038/sj.bmt.1705286. [DOI] [PubMed] [Google Scholar]
- 78.Montagna D, Yvon E, Calcaterra V, et al. Depletion of alloreactive T cells by a specific anti-interleukin-2 receptor p55 chain immunotoxin does not impair in vitro antileukemia and antiviral activity. Blood. 1999;93(10):3550–3557. [PubMed] [Google Scholar]
- 79.André-Schmutz I, Le Deist F, Hacein-Bey-Abina S, et al. Immune reconstitution without graft-versus-host disease after haemopoietic stem-cell transplantation: a phase 1/2 study. Lancet. 2002;360(9327):130–137. doi: 10.1016/S0140-6736(02)09413-8. [DOI] [PubMed] [Google Scholar]
- 80.Zhou X, Di Stasi A, Tey SK, et al. Long-term outcome after haploidentical stem cell transplant and infusion of T cells expressing the inducible caspase 9 safety transgene. Blood. 2014;123(25):3895–3905. doi: 10.1182/blood-2014-01-551671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Chang E, Liu H, West JA, et al. (2015). Clonal Dynamics in vivo of Virus Integration Sites of T cells expressing a safety switch. Mol Ther. 2016;24(4):736–745. doi: 10.1038/mt.2015.217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Yagyu S, Hoyos V, Del Bufalo F, Brenner MK. Multiple mechanisms determine the sensitivity of human-induced pluripotent stem cells to the inducible caspase-9 safety switch. Mol Ther. 2016 doi: 10.1038/mtm.2016.3. In press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Barese CN, Felizardo TC, Sellers SE, et al. Regulated Apoptosis of Genetically Modified Hematopoietic Stem and Progenitor Cells Via an Inducible Caspase-9 Suicide Gene in Rhesus Macaques. Stem Cells. 2015;33(1):91–100. doi: 10.1002/stem.1869. [DOI] [PMC free article] [PubMed] [Google Scholar]