

Abstract
Class I histone deacetylases (HDACs), HDAC1 and HDAC2 often associate together in protein complexes with transcriptional factors such as methyl-CpG-binding protein 2 (MeCP2). Given their high degree of sequence identity, we examined the functional redundancy of HDAC1 and HDAC2 in mature brain. We demonstrate that postnatal forebrain-specific deletion of both HDAC1 and HDAC2 in mice impacts neuronal survival and results in an excessive grooming phenotype caused by dysregulation of the obsessive-compulsive disorder-implicated gene SAP90/PSD-95-associated protein 3 (SAPAP3) in striatum. Moreover, HDAC1- and HDAC2-dependent regulation of SAPAP3 expression requires Mecp2, the gene involved in the pathophysiology of Rett syndrome. We show that postnatal forebrain-specific deletion of Mecp2 causes excessive grooming, which is rescued by restoring striatal Sapap3 expression. Our results provide novel insight into the upstream regulation of SAPAP3, and establish the essential role of striatal HDAC1, HDAC2, and MeCP2 for suppression of repetitive behaviors.
Keywords: MeCP2, behavior, grooming, obsessive-compulsive disorder, Sapap3
Introduction
Histone deacetylases (HDACs) are a class of enzymes that remove acetyl groups from histone tails and promote chromatin remodeling1. The role of individual HDACs in the brain is an active area of investigation with recent data suggesting that the Class I HDAC, HDAC2, is a negative regulator of learning and memory2–6. HDAC2 shares 85% sequence identity with another Class I family member, HDAC1, however loss of HDAC1 in the brain does not impact learning and memory7,8. The target genes of HDAC1 and HDAC2 in brain are unclear and distinguishing their functional roles is complicated by the fact that HDAC1 and HDAC2 can also associate to form a complex with other proteins, such as Methyl-CpG binding protein 2 (MeCP2) to regulate gene expression1,9–11.
MeCP2 is a transcription factor known to play important roles in mediating complex behavior and synaptic function12–16. Loss of function mutations in MECP2 lead to the neurological disorder Rett syndrome17 (RTT), and genomic duplications spanning MECP2 also result in neurological abnormalities with autistic features and behaviors18. RTT patients display a range of phenotypes including repetitive behaviors such as stereotypical hand movements, similar to phenotypes observed in patients with obsessive-compulsive disorder (OCD)19,20. Previous studies have shown that mice lacking MeCP2 recapitulate several of the behavioral aspects of RTT, including social and motor deficits12–14,21,22.
In the present study we were interested in examining whether postnatal deletion of both HDAC1 and HDAC2 results in the phenotypes observed in mice with a brain specific deletion of HDAC2. Rather surprisingly, we identified a functional redundancy between HDAC1 and HDAC2 in neuronal survival that impacted the lifespan of the HDAC1/HDAC2 double knockout mice. We also observed exacerbated grooming behavior that was due to dysregulation of SAPAP3 in the striatum, with a similar phenotype to conditional Mecp2 knockout mice. We were able to rescue the grooming phenotype in conditional Mecp2 knockout mice by expression of SAPAP3 in the striatum suggesting that SAPAP3 is a putative target gene of MeCP2 in association with HDAC1 and HDAC2. Collectively, our data reveal unexpected negative effects of HDAC inhibition in postnatal brain as well as uncover the role of HDAC1, HDAC2, and MeCP2 in regulation of SAPAP3, a gene linked to obsessive-compulsive disorder (OCD).
Results
Postnatal loss of HDAC1 and HDAC2 results in behavioral abnormalities and premature death
To examine the role of HDAC1 and HDAC2 in mature brain, we generated mice with forebrain specific deletions of both genes during postnatal development. Homozygous HDAC1loxP/loxP/HDAC2loxP/loxP mice23 were crossed to calcium/calmodulin-dependent protein kinase II (CaMKII)-Cre93 mice to generate the conditional deletion of Hdac1 and Hdac2 (referred to as cDKO) in forebrain regions during postnatal development13. To determine whether potential phenotypes were due to the loss of both HDAC1 and HDAC2 and not the result of the deletion of either individual HDAC, conditional HDAC1 knockout (HDAC1 cKO) mice as well as conditional HDAC2 knockout (HDAC2 cKO) mice were generated using CaMKII-Cre93. The cDKO mice were born at normal Mendelian ratios and appeared indistinguishable from littermate controls (CTLs) at birth. Immunohistochemistry and western blot analysis of the cDKO mice at 8 weeks of age confirmed the deletion of HDAC1 and HDAC2 in forebrain regions including frontal cortex, hippocampus and striatum with no change in expression in cerebellum (Fig. 1 a–d), consistent with the original characterization of the CaMKII-Cre93 line13. Individual HDAC1 cKO or HDAC2 cKO mice showed a similar pattern of deletion of the gene of interest8. The cDKO mice were indistinguishable in body weight from littermate CTLs for the first few weeks of life, however, at 6 weeks of age the cDKO mice began to lose weight (Fig. 2a) and all cDKO mice died at ~9 weeks of age. Mice with a single copy of either allele did not show alterations in weight nor early postnatal lethality (data not shown). The HDAC1 cKO or HDAC2 cKO mice were also indistinguishable from littermate CTLs in weight and had a normal lifespan (Supplementary Fig. 1a,b). Collectively, these data are consistent with loss of both HDAC1 and HDAC2 in postnatal forebrain regions impacting the viability of the mice.
Figure 1.
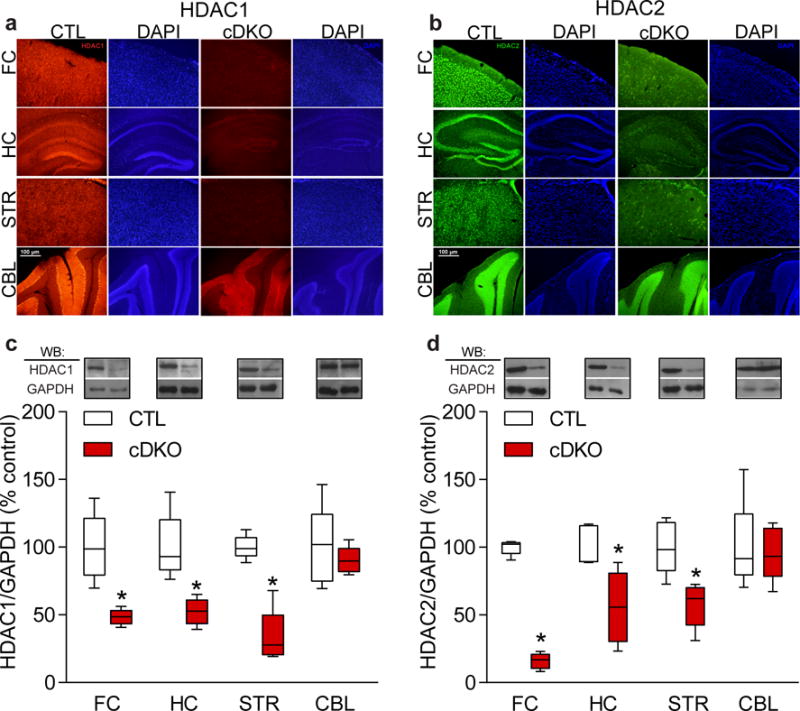
Characterization of conditional HDAC1 and HDAC2 double knockout (cDKO) mice. Fluorescent immunohistochemistry confirmed a selective loss of HDAC1 (a) and HDAC2 (b) in forebrain regions (frontal cortex (FC), striatum (STR), and hippocampus (HC)) but not in cerebellum (CBL) of cDKO mice. Images were captured using an Olympus BX51 epifluorescence microscope and Olympus DP70 software. Scale bar represents 100 μm. Presented are representative images from a cohort of cDKO mice and respective CTLs, and results were replicated in a second cohort of mice. (c,d) Western blot analysis confirmed over 50% reduction of HDAC1 (normalized to GAPDH) (c) and HDAC2 (normalized to GAPDH) (d) in forebrain regions of cDKO mice with no alterations in CBL. Full length blots are presented in Supplementary Fig. 8 (n=5 mice per group for HDAC1; two-tailed t-test; t(8) = 4.577; P = 0.0018 for FC CTL versus cDKO; t(8) = 4.408; P = 0.0037 for HC CTL versus cDKO; t(8) = 6.832; P = 0.0001 for STR CTL versus cDKO; t(8) = 0.6912; P = 0.5090 for CBL CTL versus cDKO; n=5 mice per group for HDAC2; two-tailed t-test; t(8) = 23.06; P < 0.0001 for FC CTL versus cDKO; t(8) = 3.247; P = 0.0118 for HC CTL versus cDKO; t(8) = 3.715; P = 0.0059 for STR CTL versus cDKO; t(8) = 0.8062; P = 0.2536 for CBL CTL versus cDKO). Data are shown as median, 25th and 75th percentile, and min and max value (c–d). *P < 0.05.
Figure 2.
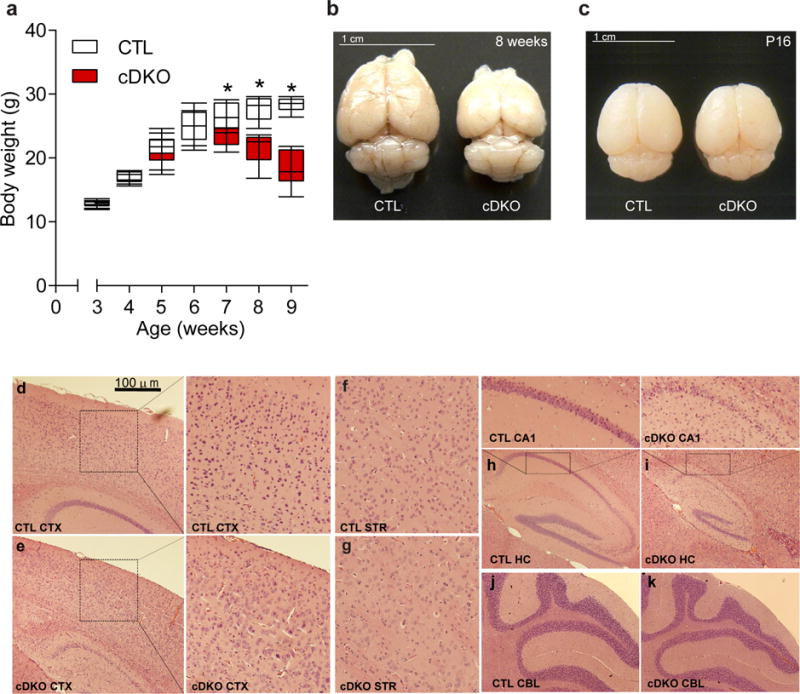
Conditional forebrain deletion of HDAC1 and HDAC2 leads to premature death. (a) cDKO mice significantly lose body weight after 6 weeks of age (n=6 mice per group; two-tailed t-test; t(10) = 0.1132; P = 0.9121 for 3 weeks CTL versus cDKO; two-tailed t-test; t(10) = 0.1838; P = 0.8579 for 4 weeks CTL versus cDKO; two-tailed t-test; t(10) = 0.1854; P = 0.8566 for 5 weeks CTL versus cDKO; two-tailed t-test; t(10) = 0.1417; P = 0.8901 for 6 weeks CTL versus cDKO; two-tailed t-test; t(10) = 2.278; P = 0.0460 for 7 weeks CTL versus cDKO; two-tailed t-test; t(10) = 4.72; P = 0.0008 for 8 weeks CTL versus cDKO; two-tailed t-test; t(10) = 7.922; P < 0.0001 for 9 weeks CTL versus cDKO). (b,c) Representative pictures indicating forebrain volume was smaller in cDKO than in CTLs in 8 week old mice (b) with no changes observed at P16 (c). Images were captured using a Nikon DSLR camera. Scale bar represents 1 cm and results were replicated in > 30 cohorts of mice at 8 weeks of age, and in two cohorts of mice at P16. (d–k) Representative images of Hematoxylin and Eosin (H&E) staining. cDKO mice (e and enlarged) have disrupted cortical lamination compared to CTLs (d and enlarged). Striatal patterns of cDKO mice (g) are indistinguishable from CTLs (f). Overall hippocampal size was dramatically smaller and granule cell layers of CA1 were disrupted in cDKO (i and enlarged), compared to CTLs (h and enlarged). No histological differences were observed in the cerebellum (CBL) between cDKO (k) and CTLs (j). Results were replicated in two additional cohorts of mice. Images were captured using an Olympus BX51 brightfield microscope and Olympus DP70 software. Scale bar represents 100 μm and enlarged images were taken at 40× magnification. Data are shown as median, 25th and 75th percentile, and min and max value (a). *P < 0.05.
Necroscopic analysis of cDKO mice did not reveal peripheral abnormalities (data not shown). However, there was a significant reduction in overall brain size and weight in 8 week old cDKO mice compared to age matched CTLs, which appeared to be due to a decrease in the size of the cortical areas, consistent with the regional deletion of HDAC1 and HDAC2 (Fig. 2b, Supplementary Fig. 1e). The CaMKII-Cre93 line expresses Cre recombinase at postnatal days 10–14, therefore we examined the brain mass of cDKO mice at a time point coinciding with early expression of Cre recombinase13, postnatal day 16, and found no difference compared to CTLs (Fig. 2c), demonstrating that the reduction in brain size occurred after the deletion of HDAC1 and HDAC2. We also observed no differences in brain weight in 8 week old HDAC1 cKO or HDAC2 cKO mice, further supporting that deletion of both HDAC1 and HDAC2 led to the reduction in brain mass (Supplementary Fig. 1c,d). Hematoxylin and eosin (H&E) staining revealed aberrant cellular patterns and layering in cortex (Fig. 2e and enlarged) and hippocampus (Fig. 2i and enlarged) of cDKO mice compared to CTLs (Fig. 2d,h, and enlarged). Rather surprisingly, there were no cell morphology abnormalities observed in the striatum of cDKOs compared to CTLs (Fig. 2f,g) although HDAC1 and HDAC2 expression was significantly reduced in this brain region (Fig. 1a–d), as CaMKII–Cre93 is known to express Cre recombinase in medium spiny neurons of the striatum24. There were also no detectable differences in the cerebellar structure between cDKO and CTL mice, consistent with the forebrain specific deletion (Fig. 2j,k). Terminal deoxynucleotidyl transferase dUTP nick end labeling (TUNEL) revealed increased apoptosis in the cortex and hippocampus, but not in the striatum or cerebellum of cDKOs compared to CTLs consistent with the regional morphological changes (Supplementary Fig. 2), demonstrating a functional redundancy of HDAC1 and HDAC2 on cell survival in cortex and hippocampus.
We examined whether 6 week old cDKO mice, at the time of onset for weight loss, had alterations in locomotor activity or anxiety related behavior. The cDKO mice were significantly hypoactive (Supplementary Fig. 3d and inset) and spent less time in the center of the arena and more time in the periphery in the open field test, suggestive of an increase in anxiety-like behavior (Supplementary Fig. 3e). We also assessed these behaviors in a separate cohort of 3 week old mice, a time point ~1 week after the expression of Cre recombinase, and found that cDKO mice were significantly hypoactive (Supplementary Fig. 3a and inset) with heightened anxiety-related behavior (Supplementary Fig. 3b), showing that loss of both HDAC1 and HDAC2 rapidly contributed to this phenotype.
HDAC1/HDAC2 cDKO mice develop a facial lesion and display excessive grooming similar to OCD-like behaviors
We observed that every cDKO mouse developed a severe lesion on the face and neck regions at ~7 weeks of age (Fig. 3a). The fur lesion could not be attributed to fighting with cage mates as the lesion still appeared in each cDKO mice that was singly housed after weaning (3 weeks of age prior to the appearance of the fur lesion; data not shown), and not in any CTL or HDAC1/2 double heterozygous cage mates, suggesting the lesions were not the result of allogrooming. We also did not observe facial or neck lesions on any of the HDAC1 cKO or HDAC2 cKO mice, demonstrating that the lesion was the result of loss of both HDAC1 and HDAC2. The lesion did not appear to be due to any form of dermatitis, therefore we investigated whether the fur lesion was due to excessive grooming in cDKO mice. We monitored the behavior of 3 and 6 week old cDKO mice for a 30 minute time period (between 8 am – 12 pm) and scored the total amount of time the mouse spent self-grooming. As most studies only score the initial 10 minute period, we assessed this time interval and found the cDKO mice spent approximately twice the amount of time grooming as CTL mice (Fig. 3b; Supplementary Fig. 3c). The phenotype was consistent at the 20 minute, as well as the 30 minute time interval, revealing a significant more than two-fold increase in grooming of cDKO mice compared to CTLs. In contrast, HDAC1 cKO or HDAC2 cKO mice show normal grooming behavior compared to their respective littermate CTLs (Supplementary Fig. 4a,b), demonstrating that the fur lesion as well as the excessive grooming phenotype is due to concurrent loss of HDAC1 and HDAC2.
Figure 3.
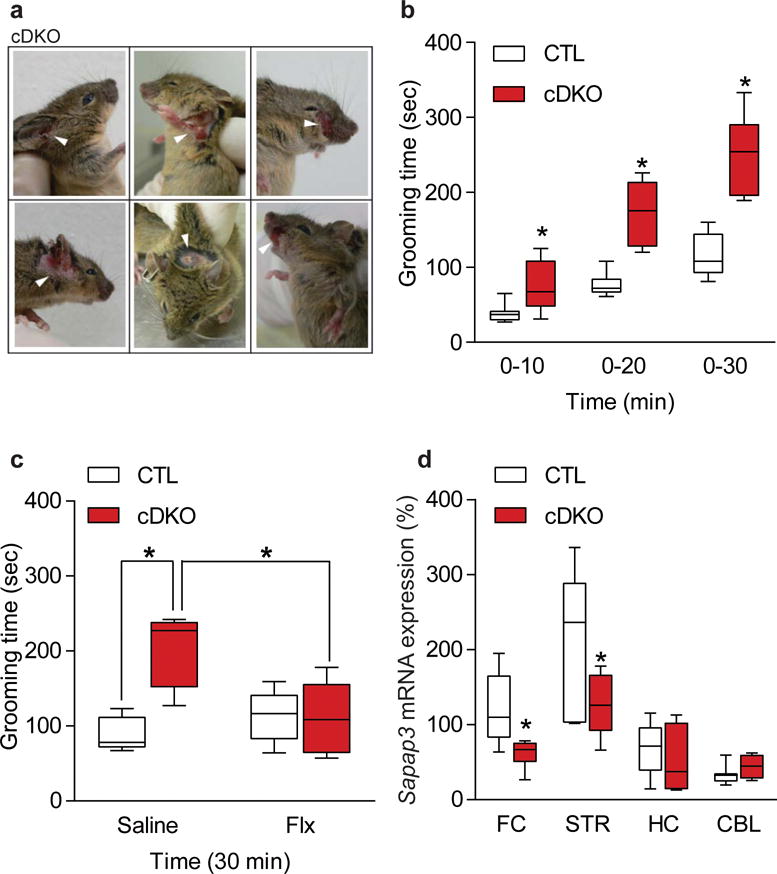
HDAC1 and HDAC2 cDKO mice have increased grooming behavior and dysregulation of SAPAP3. (a) Representative images of the facial lesion which occurs in every cDKO mouse at approximately 7 weeks of age. Results were replicated in every DKO mouse generated in this study. Images were captured using a Nikon DSLR camera. (b) Grooming behavior was assessed by quantifying the total time spent grooming over a 30 minute period. cDKO mice spend significantly more time grooming at 6 weeks of age compared to littermate CTL mice (CTL n=7; cDKO n=6; two-tailed t-test; t(11) = 2.556; P = 0.0267 for 0–10 min CTL versus cDKO; t(11) = 5.129; P = 0.0003 for 0–20 min CTL versus cDKO; t(11) = 5.787; P = 0.0001 for 0–30 min CTL versus cDKO). (c) Fluoxetine (Flx) administration for 21 days attenuated the grooming phenotype in cDKOs to levels comparable to CTLs (CTL Saline n=5; CTL Flx n=6; cDKO Saline n=4; cDKO Flx n=6; two-way ANOVA F(1,17) = 11.1, P = 0.0039 for treatment; Tukey’s post hoc analysis for CTL Saline versus cDKO Saline P = 0.0024, cDKO Saline versus cDKO Flx P = 0.0119). (d) Quantitative-RT (qRT) PCR analysis showed that Sapap3 mRNA levels were significantly down-regulated in the frontal cortex (FC) and striatum (STR) of conditional cDKO mice compared to CTLs, with no change in the hippocampus (HC) and cerebellum (CBL) (FC CTL n=7, cDKO n=6; STR CTL n=6, cDKO=7; HC CTL n=8, cDKO n=7; CBL CTL n=7, cDKO=4; two-tailed t-test; t(11) = 2.769; P = 0.0183 for FC CTL versus cDKO; t(11) = 2.278; P = 0.0437 for STR CTL versus cDKO; t(13) = 0.9407; P = 0.3640 for HC CTL versus cDKO; t(9) = 1.181; P = 0.2677 for CBL CTL versus cDKO). Data are shown as median, 25th and 75th percentile, and min and max value (b–d). *P < 0.05.
Chronic administration of fluoxetine attenuates excessive grooming
Increased grooming in mice has been suggested to model aspects of obsessive-compulsive disorder (OCD), with previous data establishing that the serotonin selective reuptake inhibitor (SSRI), fluoxetine, can alleviate excessive grooming in mice25,26 similar to the attenuation of symptoms in individuals with OCD27–29. We treated 3 week old cDKO and littermate CTLs with fluoxetine (intraperitoneal injections, 10 mg kg−1/day) and examined whether the excessive grooming behavior in cDKO mice was responsive to the drug treatment. We found that one week of fluoxetine treatment did not attenuate the increased grooming behavior of the cDKO mice (Supplementary Fig. 4c). However, three weeks of fluoxetine administration significantly reduced the amount of time spent grooming comparable to a level seen in CTLs (Fig. 3c), recapitulating clinical data that SSRIs take several weeks to reach efficacy30.
HDAC1/HDAC2 cDKO mice have altered Sapap3 expression
The gene encoding post-synaptic scaffolding protein SAP90/PSD-95-associated protein 3 (SAPAP3) is linked to obsessive-compulsive and grooming disorders in humans31,32. Mice lacking Sapap3 display an excessive grooming phenotype resulting in a facial lesion25, similar to the cDKO mice. We therefore assessed whether Sapap3 expression was altered by the loss of HDAC1 and HDAC2. Quantitative-RT (qRT) PCR analysis revealed a significant decrease in Sapap3 expression in frontal cortex and striatum, with no alterations in hippocampus and cerebellum (Fig. 3d), in cDKO mice. This result is in agreement with previous work showing that the grooming phenotype of the Sapap3 knockout mice is mediated through the corticostriatal pathway25. Sapap3 expression was not altered in HDAC1 cKO or HDAC2 cKO mice consistent with the premise that loss of both HDAC1 and HDAC2 was necessary to dysregulate its expression (Supplementary Fig. 4d,e). We also examined the expression of Slitrk5, another gene that has been linked to excessive grooming behavior in mice26, and found no change in expression in the striatum or cortex, showing specificity for the altered Sapap3 expression in the cDKO mice (Supplementary Fig. 4f).
Striatal specific deletion of HDAC1/HDAC2 recapitulates excessive grooming seen in cDKO mice
We next examined whether deletion of HDAC1 and HDAC2 selectively in the striatum is sufficient to disrupt Sapap3 expression and elicit the excessive grooming behavior. We used stereotaxic methods to bilaterally inject into the dorsal striatum of adult Hdac1loxP/loxP/Hdac2loxP/loxP mice an adeno-associated viral (AAV) vector expressing Green Fluorescent Protein tagged to Cre recombinase (GFP-Cre) or GFP alone as a control (Fig. 4a). Mice were behaviorally tested three weeks after surgery, a time point sufficient for Cre mediated recombination with AAV-GFP-Cre33 then sacrificed to confirm viral placements using laser microscopy (Fig. 4b), with off-target injected animals eliminated from further analysis. qRT-PCR showed an approximate 50% reduction in HDAC1 and HDAC2 expression in dorsal striatum of AAV-GFP-Cre compared to AAV-GFP injected mice (Fig. 4c). The striatum specific deletion of HDAC1 and HDAC2 did not result in any obvious structural alterations or cell death within the striatum (Supplemental Fig. 5d), and did not impact lifespan, weight, locomotor activity, anxiety-like behavior, or motor coordination compared to AAV-GFP injected mice (Supplementary Fig. 5a–c). However, deletion of HDAC1 and HDAC2 selectively in the striatum resulted in a significant increase in the time spent grooming (Fig. 4d) with a significant decrease in Sapap3 expression in the striatum compared to AAV-GFP injected mice (Fig. 4e) similar to data from the cDKO mice.
Figure 4.
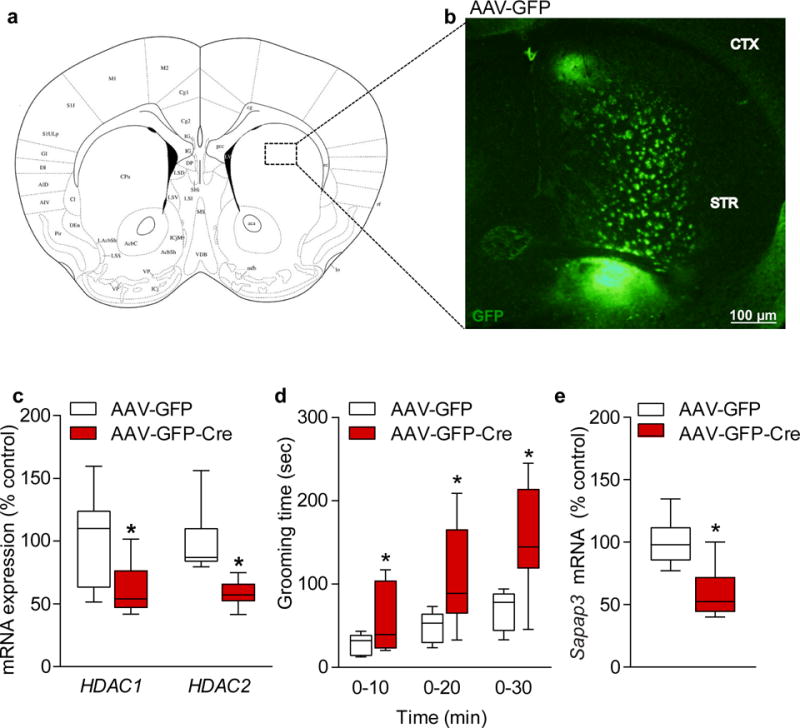
Striatal specific deletion of HDAC1 and HDAC2 recapitulates the excessive grooming phenotype. (a) Representative diagram of the approximate virus injection site in the dorsal striatum of Hdac1loxP/loxP/Hdac2loxP/loxP mice. (b) Striatal sections containing GFP epifluorescence were laser microdissected and subjected to quantitative-RT (qRT) PCR for quantitation of Hdac1 and Hdac2 mRNA levels. Presented is a representative coronal section indicating the GFP infected neurons at the injection site. Images were captured using an Olympus BX51 epifluorescence microscope and Olympus DP70 software. Results were replicated in seven additional mice. Scale bar represents 100 μm. (c) Quantitative-RT (qRT) PCR confirmed a significant knockdown of approximately 50% of both of Hdac1 and Hdac2 mRNA levels in the striatum of Hdac1loxP/loxP/Hdac2loxP/loxP mice which received AAV-GFP-Cre, in comparison to mice which received AAV-GFP (HDAC1 AAV-GFP n=7; AAV-GFP-Cre n=6; two-tailed t-test; t(11) = 2.211; P = 0.0491 for AAV-GFP versus AAV-GFP-Cre; HDAC2 AAV-GFP n=7; AAV-GFP-Cre n=8; two-tailed t-test; t(13) = 4.084; P = 0.0013 for AAV-GFP versus AAV-GFP-Cre). (d) Hdac1loxP/loxP/Hdac2loxP/loxP mice which received AAV-GFP-Cre spent significantly more time grooming compared to Hdac1loxP/loxP/Hdac2loxP/loxP mice injected with AAV-GFP, recapitulating a similar phenotype observed in the cDKO mice (AAV-GFP n=9; AAV-GFP-Cre n=8; two-tailed t-test; t(15) = 2.134; P = 0.0497 for 0–10 min AAV-GFP versus AAV-GFP-Cre; t(15) = 2.863; P = 0.0119 for 0–20 min AAV-GFP versus AAV-GFP-Cre; t(15) = 3.709; P = 0.0021 for 0–30 min AAV-GFP versus AAV-GFP-Cre). (e) Striatal mRNA expression of Sapap3 in the Hdac1loxP/loxP/Hdac2loxP/loxP mice injected with AAV-GFP-Cre was significantly reduced compared to those with AAV-GFP (AAV-GFP n=6; AAV-GFP-Cre n=6; two-tailed t-test; t(10) = 3.425; P = 0.0065 for AAV-GFP versus AAV-GFP-Cre). Data are shown as median, 25th and 75th percentile, and min and max value (c–e). *P < 0.05.
Postnatal conditional deletion of Mecp2 recapitulates OCD-like phenotype of cDKO mice
Our data so far demonstrate that loss of both HDAC1 and HDAC2 in the striatum down-regulates Sapap3 expression and results in an excessive grooming phenotype. HDAC1 and HDAC2 do not bind DNA directly1,9,34 but rather participate in protein complexes to impact gene transcription. Methyl-CpG-binding protein 2 (MeCP2) is a transcription factor that has been shown to interact with HDAC1 and HDAC2 in a co-repressor complex and regulate gene expression10,11. We confirmed that this interaction also occurs in the striatum by crossing Mecp22loxP/+ mice with CaMKII-Cre93 mice to generate conditional Mecp2 knockout mice (Mecp2 cKO), and performing immunoprecipitation (IP) with an antibody against HDAC2 on striatal samples from CTL and Mecp2 cKO mice (Supplementary Fig. 6). The function of MeCP2 in the central nervous system is complex, as it has been shown to activate and repress transcription11,35,36. To investigate whether MeCP2 binds the Sapap3 promoter, we performed chromatin immunoprecipitation (ChIP) from the striatum of MeCP2 cKO mice and CTLs using an antibody to MeCP2. QPCR analysis of the ChIP DNA revealed specific MeCP2 binding to the Sapap3 promoter region (as indicated by the H3K4me3 promoter mark) ~600 bp upstream of the transcriptional start site (P600), adjacent to a CpG island (Fig. 5 a,b). HDACs are not known to bind DNA directly1,9,34, suggesting that MeCP2 is mediating the effects of HDAC1 and HDAC2 on SAPAP3 function. Consistent with our results, previous work has shown that mice with loss of Mecp2 in inhibitory forebrain neurons display an excessive grooming phenotype22. In our experiments, the CaMKII-Cre93 line used to generate the Mecp2 cKO mice expresses Cre recombinase in excitatory neurons in broad forebrain regions as well as inhibitory medium spiny neurons in the striatum24. We found that Mecp2 cKO mice also show a significant increase in time spent grooming (Fig. 5c) with a significant decrease in Sapap3 expression in the striatum (Fig. 5d) compared to littermate CTLs.
Figure 5.
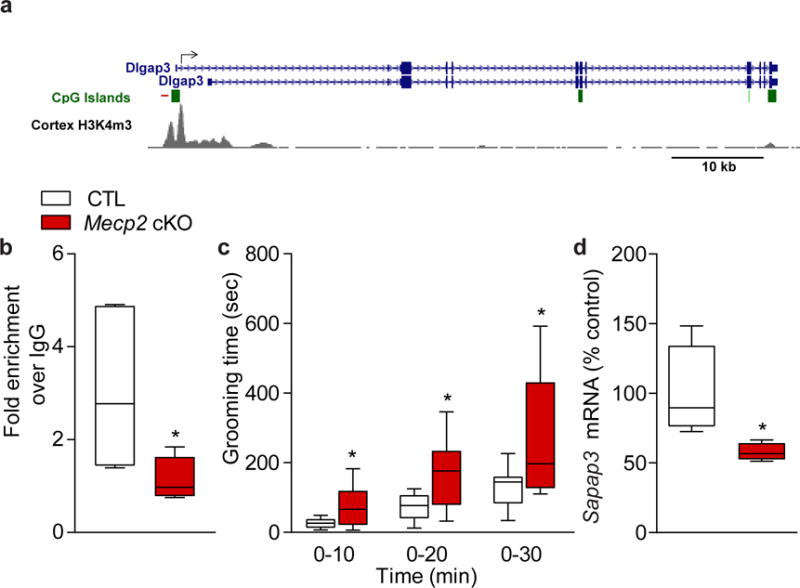
Loss of Mecp2 leads to excessive grooming and dysregulation of SAPAP3. (a) The Sapap3 (Dlgap3) promoter region showing the location of ChIP QPCR primers (red line) relative to the transcription start site (arrow) and the adjacent CpG island (green rectangle). The H3K4me3 promoter mark is based on mouse cortex ChIP-seq data from the LICR Histone track of the University of California Santa Cruz genome browser (http://genome.ucsc.edu), NCBI37/mm9 mouse genome build37. (b) MeCP2 binds to the promoter region of Sapap3. ChIP with antibody to MeCP2 shows that it binds to the promoter region of Sapap3. QPCR data are normalized to input and plotted as fold enrichment over IgG (n=5 for all groups; two-tailed t-test; t(8) = 2.404; P = 0.0429 for CTL versus cKO). (c) Conditional Mecp2 cKO mice (cKO) spend significantly more time grooming compared to littermate CTL mice (CTL n=15; Mecp2 cKO n=12; two-tailed t-test; t(25) = 3.133; P = 0.0044 for 0–10 min CTL versus Mecp2 cKO; t(25) = 3.62; P = 0.0013 for 0–20 min CTL versus Mecp2 cKO; t(25) = 3.036; P = 0.0055 for 0–30 min CTL versus Mecp2 cKO). (d) Sapap3 mRNA expression in the striatum of Mecp2 cKO mice is significantly reduced compared to CTLs (n=4 mice per group; two-tailed t-test; t(6) = 2.487; P = 0.0474 for CTL versus Mecp2 cKO). Data are shown as median, 25th and 75th percentile, and min and max value (b–d). *P < 0.05.
Restoring Sapap3 in striatum of Mecp2 cKO mice rescues excessive grooming phenotype
To establish a direct link between MeCP2 and SAPAP3 in mediating the grooming phenotype, we generated adeno-associated DJ serotype viruses expressing either GFP only (control) or an HA-tagged SAPAP3 (AAV-SAPAP3), and stereotaxically injected them bilaterally into the dorsal striatum of adult Mecp2 cKO and CTL mice to test whether this would rescue the grooming phenotype (Fig. 6a). We confirmed the localized expression of SAPAP3 to the dorsal striatum by immunohistochemistry (Fig. 6b). We also corroborated the rescue of Sapap3 expression in the striatum of Mecp2 cKO mice injected with the AAV-SAPAP3 virus by qRT-PCR analysis (Fig. 6c). We found that AAV-mediated expression of SAPAP3 in the striatum of the Mecp2 cKO mice rescued the excessive grooming behavior to a level comparable to CTL mice injected with GFP alone (Fig. 6d), indicating that this deficit was the result of decreased expression of Sapap3. The AAV expression of Sapap3 in the dorsal striatum did not alter locomotor activity, and in contrast to the reversal of the grooming phenotype, the previously reported impaired performance of the Mecp2 cKO mice on the rotarod14 was still present (Supplemental Fig. 7a,b), suggesting this deficit is mediated through a different mechanism.
Figure 6.
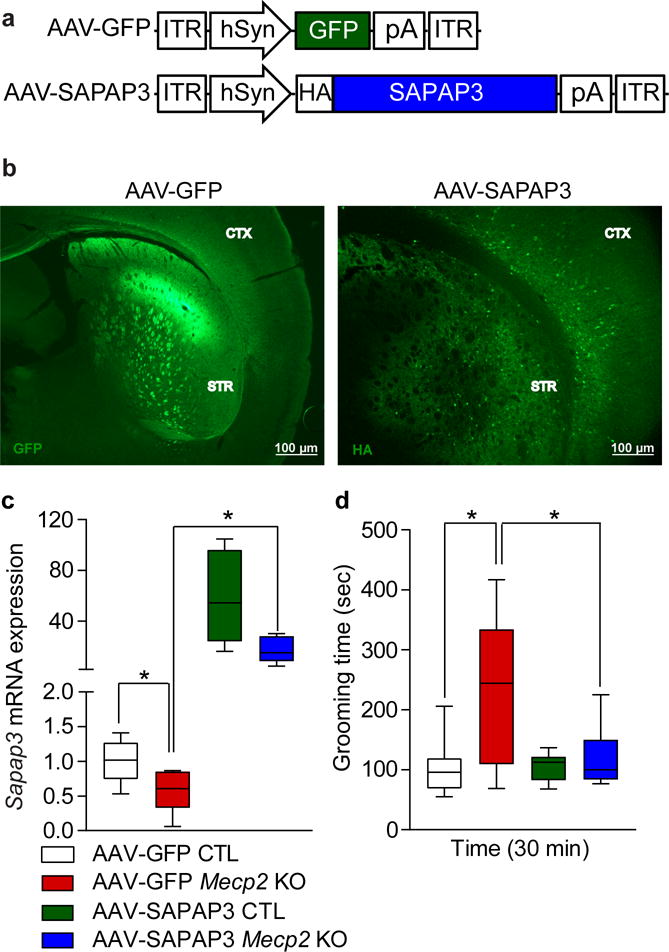
Restoring MeCP2 in the striatum rescues grooming behavior in Mecp2 cKO mice. (a) Diagram representing the map of the AAV-GFP and AAV-SAPAP3 constructs. (b) Representative images of approximate virus injection site in the dorsal striatum for Sapap3 rescue experiments. Coronal sections were immunostained using anti-GFP (left) and anti-HA (right) to visualize infectivity. Results were replicated in a second cohort of mice. Images were captured on a Nikon ECLIPSE 80i Upright microscope using NIS-element software. Scale bar represents 100 μm. (c) Striatal mRNA expression of Sapap3 in Mecp2 cKO animals injected with AAV-GFP is significantly reduced by approximately 50% compared to CTL mice. Mecp2 cKO mice injected with AAV-SAPAP3 show rescued expression of Sapap3 compared to Mecp2 cKO mice injected with AAV-GFP virus (AAV-GFP CTL n=9; AAV-GFP Mecp2 cKO n=6; AAV-SAPAP3 CTL n=8; AAV-SAPAP3 Mecp2 cKO n=5; two-way ANOVA F(1,24) = 7.752, P = 0.0103 for interaction; Holm-Sidak post hoc analysis for multiple comparisons for AAV-GFP CTL versus AAV-GFP Mecp2 cKO P = 0.0162, AAV-GFP Mecp2 cKO versus AAV-SAPAP3 Mecp2 cKO P = 0.0027). (d) Mecp2 cKO mice injected with AAV-GFP spend significantly more time grooming compared to CTL mice injected with AAV-GFP. Excessive grooming is alleviated in Mecp2 cKO mice injected with AAV-SAPAP3 to levels comparable to AAV-GFP CTLs (AAV-GFP CTL n=10; AAV-GFP Mecp2 cKO n=8; AAV-SAPAP3 CTL n=10; AAV-SAPAP3 Mecp2 cKO n=7; two-way ANOVA F(1,31) = 0.0190, P = 0.0190 for interaction; Tukey’s post hoc analysis for AAV-GFP CTL versus AAV-GFP Mecp2 cKO P = 0.0017, AAV-GFP Mecp2 cKO versus AAV-SAPAP3 Mecp2 cKO P = 0.0158). Data are shown as median, 25th and 75th percentile, and min and max value (c–d). *P < 0.05.
Discussion
Our results reveal that postnatal deletion of both HDAC1 and HDAC2 in the brain results in several adverse effects, including neuronal apoptosis in cortical and hippocampal regions as well as early postnatal lethality, demonstrating functionally redundant roles in neuronal survival past embryogenesis. Given the recent efforts in the development of HDAC2 inhibitors for the treatment of neurodegeneration and cognitive enhancement, our results suggest caution with compounds targeting both HDAC1 and HDAC2, and emphasize the importance of subtype specific inhibitors. We also observed that the concurrent loss of HDAC1 and HDAC2 results in an increase in striatum-dependent repetitive behaviors and dysregulation of Sapap3 expression, a gene linked to obsessive-compulsive and grooming disorders in humans31,32. We were able to demonstrate that the selective deletion of HDAC1 and HDAC2 in the dorsal striatum results in altered Sapap3 expression as well as increased repetitive grooming similar to that observed in cDKO mice. Mecp2, the gene that causes Rett syndrome and various neurodevelopmental abnormalities, has been shown to interact with HDAC1 and HDAC2 in a protein complex14,15. We find that MeCP2 binds to the Sapap3 promoter and that deletion of Mecp2 in broad forebrain regions of the mouse, similar to the loss of Mecp2 in inhibitory neurons22, results in increased repetitive behaviors as well as decreased Sapap3 expression in the dorsal striatum. We then show that the grooming phenotype in the Mecp2 cKO mice can be rescued by reintroducing Sapap3 expression in the dorsal striatum. Our results showing that MeCP2 binds to the Sapap3 promoter, that loss of Mecp2 attenuates Sapap3 expression, and that restoring Sapap3 in the striatum rescues the grooming phenotype, suggests that the repetitive behaviors observed in Rett syndrome may be due to dysregulation of SAPAP3 in the striatum. Collectively, these data suggest that HDAC1 and HDAC2 in concert with MeCP2 within the striatum regulate repetitive behaviors through transcription dependent processes, and provide novel insight into the neurobiological mechanisms underlying repetitive behaviors.
Methods Summary
Animals were housed and maintained as previously described8. Molecular studies consisted of western blot analysis, quantitative PCR, or chromatin immunoprecipitation on tissue lysates from frontal cortex, hippocampus, striatum, or cerebellum. Histological analysis consisted of immunohistochemistry, terminal deoxynucleotidyltransferase-mediated UTP end labeling, and Hematoxylin and Eosin staining and performed as previously described8,38. Behavioral studies and stereotaxic surgeries were performed using male mice as previously described8,21. Obsessive-compulsive-like behavior was assessed by scoring grooming behavior. Briefly, mice were placed in a home cage environment and total time spent grooming was measured for 30 minutes. All drugs were administered via intraperitoneal injection. All experiments were performed and analyzed blind to test variable. Sample sizes were estimated based on our previous experience performing similar experiments.
Mice
The conditional Hdac1 knockout (Hdac1 cKO), Hdac2 knockout (Hdac2 cKO) and Hdac1 and Hdac2 double knockout (cDKO) mice were generated by breeding transgenic mice expressing Cre recombinase under the control of the calcium/calmodulin-dependent kinase II promoter (CaMKII-Cre93 line) with Hdac1loxP/loxP, Hdac2loxP/loxP, or Hdac1loxP/loxP/Hdac2loxP/loxP mice23,38. Previous work has demonstrated that the CaMKII-Cre93 mice express Cre recombinase at postnatal days 10–14 selectively in forebrain neurons13. The Hdac1loxP/loxP, Hdac2loxP/loxP, and the CaMKII-Cre93 lines were on a mixed 129/BALBC background and backcrossed to a C57BL/6 line for at least 10 generations. The Hdac1 cKO, Hdac2 cKO, and cDKO mice were genotyped using PCR analysis from genomic DNA isolated from tails as previously described23. Littermates not carrying the Cre recombinase transgene regardless of loxP alleles were used as control mice in all experiments. Conditional Mecp2 knockout (Mecp2 cKO) mice are previously reported14. Mice were maintained on a 12 hour light/dark cycle with ad libitum access to food and water. Mice were housed 3–5 mice per cage, with the exception of when mice were singly housed for analyzing the lesion in cDKO mice. All animal protocols were approved by the Institutional Animal Care and Use Committee at The University of Texas Southwestern Medical Center.
Drug injections
All injections were delivered intraperitoneally on naïve male mice. The cDKO mice and control littermates received a once daily injection of either 0.9% saline or fluoxetine (Eli Lilly) at a concentration of 10 mg kg−1 in 0.9% saline, during the morning hours of the day (8am–12pm) for 7 or 21 days.
Protein quantification
Brain regions were dissected out and homogenized in a lysis buffer containing 25 mM HEPES, pH 7.9, 150 mM NaCl, 1 mM PMSF, 20 mM NaF, 1 mM DTT, 0.1% NP40, and proteinase inhibitor cocktails (Sigma), and spun down to isolate the lysate. Protein concentrations were determined by Bradford assays and 20 μg of the protein was loaded on 10% SDS-PAGE gels, electrophoresed, transferred to nitrocellulose membranes, then blocked with 5% non-fat milk prior to overnight incubation with primary antibodies. Dilutions of primary antibodies were 1:2000 for both HDAC1 (Abcam, ab19845)8 and HDAC2 antibodies (Abcam, ab32117)8, 1:1000 for MeCP2 antibody (Thermofisher, PA1-888)39, and 1:50,000 for glyceraldehyde-3-phosphate dehydrogenase (GAPDH) antibody (Cell Signaling Technology, 2118S)8. The following day membranes were washed, then incubated with peroxidase-labeled anti-rabbit secondary antibody (Vector, P1-1000) at 1:5000 for HDAC1, HDAC2, and MeCP2, and 1:10,000 for GAPDH. Protein bands were detected using enhanced chemiluminescense (ECL) and exposed to film. Immunoreactivity was quantitated by the NIH image J analysis software. HDAC1 or HDAC2 was normalized to GAPDH bands. For HDAC1 and HDAC2 westerns, following the transfer, each membrane was cut below the 50 KDa marker in order to individually probe for either HDAC1 or HDAC2, and GAPDH on the same blot. Separate gels were run for FC, HC, STR, and CBL and all blots were processed in parallel. For western blot following immunoprecipitation, primary antibodies were 1:1000 for MeCP2 (Thermofisher, PA1-888) and 1:6000 for Actin (Abcam, ab6276)40. Secondary antibodies were 1:5000 for IRDye® 680RD donkey anti-mouse (LI-COR, 926-68072) and 1:5000 for IRDye® 800CW donkey anti-rabbit (LI-COR, 926-32213). Blots were imaged using a LICOR Odyssey® CLx Imaging System (LI-COR).
Immunohistochemistry
Mice were transcardially perfused with 4% paraformaldehyde (PFA) in 0.1 M phosphate-buffered saline (PBS) and brains were removed from the skull. Following post-fixation in 4% PFA overnight, the brains were cryo-protected in 30% sucrose in 0.1M PBS prior to sectioning on a freezing microtome. The brains were coronally sectioned at 30 μm and subjected to immunohistochemistry. Briefly, free floating sections were incubated overnight in primary antibody solution composed of 3% normal goat serum, and 0.3% Triton X-100 in PBS. Dilutions for the primary antibodies were 1:250 for rabbit anti-HDAC1 (Abcam, ab19845)8, 1:2000 for rabbit anti-HDAC2 (Abcam, ab32117)8, 1:1600 for anti-HA (Cell Signaling, 3724S)41, and 1:200 for anti-GFP (Cell Signaling, 2956S). For HDAC1 staining, the sections were treated in 10 mM citric acid (pH 6) for 15 minutes at 95 °C for antigen retrieval prior to the primary antibody incubation. Immunoreactivity was visualized by secondary antibodies conjugated with either Alexa Fluor 594 (HDAC1, Invitrogen, A11012)8 or Alexa Flour 488 (HDAC2, HA, GFP, Invitrogen, A11008)42. The sections were incubated at a 1:200 dilution at room temperature for 2 hours, counter-stained with 4′,6-diamidino-2-phenylindole (DAPI), and then mounted on superfrost plus slides in Vectashield mounting media (Vector Laboratories).
Hematoxylin and Eosin (H&E) Staining
H&E staining was carried out as previously described38. Briefly, paraffin sections made from formalin fixed tissue were affixed to microscope slides through sequential room temperature and heated air-drying. Dried sections were deparaffinized and stained with hematoxylin, then destained using 70% ethanol. Sections were then stained with eosin, destained, and dehydrated in ascending ethanol solutions. Sections were rinsed in xylene then cover slipped with synthetic mounting media.
TUNEL
Terminal deoxynucleotidyltransferase-mediated UTP end labeling (TUNEL) staining for apoptotic cells was done according to the manufacturer’s protocol (Promega DeadEnd Fluorometric TUNEL System, Madison, WI). Apoptotic cells were labeled with fluorescein and the sections were counterstained with propidium iodide.
Vector construction and AAV preparation
A lentiviral vector containing the initial eGFP-SAPAP3 construct was received from the lab of Dr. Guoping Feng. Two AAV vectors were constructed from this plasmid. The eGFP and SAPAP3 were PCR amplified independently and cloned using standard methods into an expression cassette containing the human Synapsin promoter and Growth Hormone polyadenylation signal surrounded by AAV-2 inverted tandem repeats. A Human influenza hemagglutinin (HA) tag was attached to the N-terminus of SAPAP3 during PCR amplification prior to cloning. Primers used were: GFP.EcoRV.R, 5′-ATA TGA TAT CTC ACT TGT ACA GCT CGT CCA TGC CG-3′; GFPSAPAP3.SpeI.F, 5′-ATA TAC TAG TAC CAA TGG TGA GCA AGG GCG AGG AGC-3′; SpeI.HA-SAPAP3F, 5′-ATA TAC TAG TAC CAA TGT ACC CAT ACG ATG TTC CAG ATT ACG CTG GCG GCT CCG GAG GAA GGG GTT ACC ATG G-3′; SAPAP3.EcoRV.R, 5′-ATA TGA TAT CTC ACA GCC TGG TCT GGG CCT CG-3′. pAAV-GFP or pAAV-HA-SAPAP3 were cotransfected with pAAV-DJ and pHelper into HEK-293 cells. 72 hours after transfection, cells were harvested, lysed by freeze-thaw, and incubated with Benzonase (Sigma) at 37 °C. To isolate AAV vector, iodixanol gradient was performed. Cell lysate was transferred to iodixanol gradient in quick-seal centrifugation tube and centrifuged at 48,000 rpm in a Beckman Type 70Ti rotor at 18 °C for 2 hours and 10 minutes. After centrifugation, 40% gradient fraction was collected. Collected fraction was applied onto Amicon Ultra 100K (Millipore), washed with PBS (−) and concentrated. Viral titers were determined by RT-PCR using primers for HGpolyA, and are 1.27×1012 GC ml−1 for AAV-GFP and 4.9×1012 GC ml−1 for AAV-SAPAP3.
Adeno-associated virus injection
The adeno-associated virus-green fluorescent protein (AAV-GFP) or adeno-associated virus expressing Cre recombinase tagged with GFP (AAV-GFP-Cre) were obtained from Penn Vector Core; AAV-GFP and AAV-GFP-Cre are AAV1.CMV.PI.EGFP.WPRE.bGH (viral titer of 3.18×1013 GC ml−1) and AAV1.CMV.HI.GFP-Cre.SV40 (viral titer of 7.88×1012 GC ml−1), respectively. Previous work demonstrated that GFP-tagged Cre recombinase possesses normal enzymatic activity21,33. To inject the AAV-GFP, AAV-GFP-Cre or AAV-SAPAP3 in striatum, 3–5 month old naïve Hdac1loxP/loxP/Hdac2loxP/loxP male mice were subjected to stereotaxic surgery as previously described43. Briefly, anesthetized animals placed into the stereotaxic frame received 1 μl of AAV-GFP or AAV-GFP-Cre, or 0.5 μl of AAV-SAPAP3 bilaterally with the coordinates relative to Bregma at anteroposterior +1.2 mm, lateral +2.5 mm, and dorsoventral −3.0 mm with a 10° angle. Mice were allowed to recover for three weeks before the commencement of behavioral testing.
RNA extraction and quantitative-RT (qRT) PCR
To determine relative expression of Hdac1 and HDAC2 mRNA after stereotaxic AAV injection, we performed qRT-PCR as described previously21. Briefly, the animals were sacrificed by rapid decapitation and the brains were sectioned at 14 μm. The dorsal striatum expressing GFP or Cre-GFP was laser microdissected from each section using AS LMD (Leica) system. Eight sections were pooled to extract RNA using PicoPure RNA isolation kit (Arcturus). Each section was 140 μm apart, thus encompassing the majority of the AAV infusion site in the striatum. Conditions for construction of complementary DNA (cDNA) were described earlier21. Using cDNA as a template, transcripts for Hdac1, HDAC2, Sapap3, and Gapdh were amplified using Power SYBR Green PCR master mix (Applied Biosystems) in a 7500 Real-Time PCR system (Applied Biosystems). The thermal cycling conditions for PCR amplification consisted of 1 cycle of 50 °C for 2 m and 95 °C for 10 m, 40 cycles of 95 °C for 15 s, 60 °C for 60 s, and 1 dissociation cycle of 95 °C for 15 s, 60 °C for 60 s, 95 °C for 15 s, and 60 °C for 15 s. Primers are 5′-agg gca cca aga gga aag tct gtt-3′ and 5′-gca gca aat tgt gag tca tgc gga-3′ for Hdac1; 5′-GCG TAC AGT CAA GGA GGC GG-3′ and 5′-GCT TCA TGG GAT GAC CCT GGC-3′ for Hdac2; 5′-AGC AGT ACC TTC CCC AGG AT-3′ and 5′-AAA CTG GTC CAG GAG TGT GG-3′ for Sapap3; 5′-AGG TCG GTG TGA ACG GAT TTG-3′ and 5′-TGT AGA CCA TGT AGT TGA GGT CA-3′ for Gapdh. Primers for Sapap3 following viral injection with AAV-SAPAP3 are 5′-GTG GAC ACA GCC AGG ATC AAC-3′ and 5′-GCT GCT GCT GGT TAA ACT CTT CGC-3′.
For Sapap3 expression analysis in Hdac1 cKO, Hdac2 cKO, cDKO, and Mecp2 cKO mice, the following brain regions were dissected: FC, HC, STR, and CBL, and total RNA was extracted using Trizol reagent (Ambion) according to the manufacturer’s instructions. Single stranded cDNA was synthesized by treating extracted RNA with random primers and SuperScript Reverse Transcriptase III (Invitrogen). Amplification was performed using Power SYBR Green PCR master mix in a 7500 Real Time PCR detection System (Applied Biosystems).
Immunoprecipitation (IP)
Mouse striata were dissected and homogenized in 120 μl of cold lysis buffer containing 20 mM Tris pH 7.5, 100 mM NaCl, 1 mM EDTA, 1mM PMSF, 0.5% NP40, and protease and phosphatase inhibitor cocktails (1 tablet each of PhosSTOP and cOmplete, Sigma), incubated for 30 minutes at 4 °C on a rotator, then spun down to isolate the lysate. 20 μl of lysate was saved at −20 °C as input material for protein quantification. The remaining lysate was incubated with 880 μl of cold lysis buffer and ~12 μg of anti-HDAC2 (Abcam, ab7029)44 overnight at 4 °C on a rotator for IP. Recombinant Protein G Agarose beads (Invitrogen) were washed twice with 1X PBS and blocked overnight in 1% BSA at 4 °C on a rotator. The following day, the beads were washed twice with lysis buffer and added to the lysate samples, and rotated for 2 hours at 4 °C. Immunoprecipitates were washed four times with cold lysis buffer, resuspended in 25 μl cold lysis buffer and 20 μl of Laemmli sample buffer (BioRad), boiled and analyzed by western blot.
Chromatin immunoprecipitation (ChIP)
ChIP was performed as previously described45 with modifications. Briefly, a pool of three dissected mouse striata were homogenized in crosslinking-buffer (0.1 M NaCl, 1 mM EDTA, 0.5 mM EGTA, 25 mM Hepes-KOH, pH 8.0) containing 1% paraformaldehyde. Cross-linking was quenched by adding glycine (final concentration is 125 mM) for 5 minutes after a 10 minute incubation at RT). Cells were then rinsed three times in ice-cold PBS containing complete protease inhibitor cocktail tablets (Roche Diagnostics) and lysed by Buffer I (50 mM Hepes-KOH, pH 7.5, 140 mM NaCl, 1 mM EDTA, pH 8.0, 10 % Glycerol, 0.5 % NP-40, complete protease inhibitor cocktail) for 10 minutes at 4 °C. Nuclei were then pelleted by centrifugation at 2000 g for 10 minutes at 4 °C. The isolated nuclei were rinsed with IP buffer (10 mM Tris-HCl, pH 8.0, 300 mM NaCl, 1 mM EDTA, pH 8.0, 0.5 mM EGTA, pH 8.0, 1% Triton X-100, 0.1% deoxycholic acid (sodium salt) and complete protease inhibitor cocktail). Samples were fragmented by sonication in IP buffer. The sizes of DNA fragments range from 200 bp to 2 kb. Insoluble material was removed by centrifugation at 12,000 g for 10 min at 4 °C. The supernatant was transferred to a new tube and the final volume of the resulting nuclear lysate was adjusted to 1 ml by adding additional IP buffer. 1/20 volume of the ChIP sample (50 μl from 1 ml lysate) was saved as input material. The rest of the lysate was incubated with the indicated antibody (MeCP2 (Millipore, MABE328)46 or IgG (Millipore, NI04-100UG), 2 μg)46 overnight at 4 °C for immunoprecipitation.
The next day, 20 μl of pre-rinsed Protein A/G PLUS Agarose (Santa Cruz Biotechnology) was added to each ChIP reaction and further incubated for 2 hours at 4 °C. The beads bound by immune-complexes were pelleted and washed twice with each of the following buffers: low salt buffer (0.1% SDS, 1% Triton X-100, 2 mM EDTA, 20 mM Tris-HCl, pH 8.1, 150 mM NaCl), high salt buffer (0.1% SDS, 1% Triton X-100, 2 mM EDTA, 20 mM Tris-HCl, pH 8.1, 500 mM NaCl), and LiCl buffer (0.25M LiCl, 1% IGEPAL CA630, 1% deoxycholic acid (sodium salt), 1mM EDTA, 10mM Tris, pH 8.1). In each wash, the beads were incubated with wash buffer for 10 minutes at 4 °C. The washed beads were then rinsed once with 1x TE (10 mM Tris-HCl, pH 8.0, 1 mM EDTA). The immunoprecipitated material was eluted from the beads twice by adding 150 μl of elution buffer (10 mM Tris-HCl, pH 8.0, 1 mM EDTA, pH 8.0, 1% SDS) to each ChIP reaction and incubating the sample at 65 °C for 10 min with brief vortexing every 2 minutes. 250 μl of elution buffer was also added to the saved input material (50 μl) and this sample was processed together with the ChIP samples. The eluates were combined and crosslinking was reversed by incubation at 65 °C for ~4–5 hours. To each eluate (300 μl), 5μl of diluted (1:50, 2 mg ml−1) RNaseA (Qiagen) was added and incubated for 1 hour at 37 °C. Then 7 μl of Proteinase K (20 mg ml−1; New England Biolabs) was added and incubated for 2 hours at 50 °C. The immunoprecipitated genomic DNA fragments were extracted with Phenol/Chloroform (Invitrogen). The extracted DNA fragments were then purified using the QIAquick PCR purification kit (Qiagen) and DNA fragments were eluted in 60 μl of 10 mM Tris-HCl, pH 8.5.
ChIP quantitative PCR (QPCR)
For QPCR using ChIP samples, 1 μl of ChIP DNA was used as a direct template and amplification was performed using Power SYBR Green PCR master mix in a 7500 Real Time PCR detection System (Applied Biosystems). Primer sequences for the Sapap3 P600 region are as follows: 5′-GGG ACT AGT GCG GAG AA-3′ and 5′- TCT TAG GCT CCT GTC CTT AG -3′. For the data analysis, a ΔCt value was obtained by subtracting the ΔCt value of input DNA. A ΔΔCt value was then calculated by subtracting the ΔCt value for IgG. The data are presented as fold enrichment over IgG binding.
Behavioral Overview
For all behavioral testing, experimental animals were male mice, and age matched littermates were used as CTLs. Testing groups for behavioral cohorts were balanced by age and genotype, and randomization of experimental groups was not performed. Mice were naïve prior to the start of the initial behavioral test within each series of experiments. All experiments were conducted during the light cycle and scored by an observer blind to genotypes and group assignments. Mice were habituated to testing facilities 1 hour prior to behavioral assessment. For all experiments, error bars represent either mean ± s.e.m for dot plots or median, 25th and 75th percentile and min and max value of the data set for box-and-whisker plots, and significance was *P <0.05. Mice were tested in cohorts for various behavioral tasks. Distinct cohorts of HDAC1 cKO, HDAC2 cKO, cDKO, and respective CTL mice were used to assess body weight and brain weight. Distinct cohorts of cDKO mice and CTLs were used to test locomotor activity and open field, and separate cohorts were used to test 6 week and 3 week old mice in each task. A distinct cohort of cDKO and CTL mice was used to score grooming behavior at 6 weeks old, and a separate cohort of cDKO and CTL mice was used to score grooming at 3 weeks old. Separate cohorts of HDAC1 cKO, HDAC2 cKO, and respective CTL mice were used to test grooming behavior. A distinct cohort of cDKOs and CTLs was used to test grooming behavior following one week of fluoxetine treatment, and the same cohort was scored for grooming behavior following 3 weeks of fluoxetine treatment. A distinct cohort of Hdac1loxP/loxP/Hdac2loxP/loxP mice was used for stereotaxic injections with AAV-GFP and AAV-GFP-Cre and after recovery this same cohort of mice was tested in multiple behavioral tasks in the following order; locomotor activity, open field, grooming, and rotarod. A distinct cohort of mice was used to assess grooming in Mecp2 cKO and CTL mice. For rescue experiments using AAV-GFP and AAV-SAPAP3, a distinct cohort of Mecp2 cKO and CTL mice was used for stereotaxic injections, and following recovery this same cohort of mice was tested in multiple behavioral tasks in the following order; locomotor activity, grooming, and rotarod.
Locomotor activity
Mice were placed individually in a standard mouse home cage (18 cm × 28 cm) with fresh bedding, and activity was monitored over 2 hours by five horizontal photobeams linked to data acquisition software (Photobeam Activity System, San Diego Instruments, San Diego, CA). Ambulatory activity was measured by counting the number of consecutive beam breaks in 5-minute increments.
Open field
Mice were placed in the periphery of a novel open field environment (44 cm × 44 cm, walls 30 cm high) in a dimly lit room and allowed to explore for 10 min. The animals were monitored from above by a video camera connected to a computer running video tracking software (Ethovision 3.0, Noldus, Leesburg, Virginia) to determine the total time spent in the periphery (5 cm from the walls), center (14 cm × 14 cm), and complete center (34 cm × 34cm). The amount of locomotor activity was also determined and in all experiments was consistent with locomotor activity data using standard mouse home cages. The open field arenas were cleaned between mice.
Rotarod
Each mouse was placed on the rotarod (IITC Life Science), which accelerated from 0 to 45 rpm over the course of 60 seconds. Each session ended when the mouse fell off the rod and the total time spent on the rotarod before falling was measured. The mouse was returned to its original cage for 1 hour. The test was repeated for a total of 8 trials over the course of 2 days (4 trials per day).
Grooming
Grooming behavior was assessed as previously described with modifications25. Mice were placed individually into a fresh mouse cage and allowed to move freely for 30 minutes. Test sessions were recorded under red light by a video camera directly in front of the mice located inside the testing room. An observer blind to group and genotype analyzed the video tape and scored self-grooming of any part of the body including the face, head, ears, and full-body grooming. The total amount of time spent grooming (duration) was measured at 10 minute intervals. Continuous grooming for greater than one second was recorded as a grooming bout, and sessions separated by two or more seconds constituted a new bout (modified from Welch et al.).
Statistical analysis
Data are plotted either dot plots with error bars representing mean ± s.e.m, or box-and-whisker plots with interquartile ranges represented as the following: whiskers (error bars) are min and max of data and box is 25th, median, and 75th percentile as calculated in Prism GraphPad software. Statistical differences were calculated in Prism GraphPad software using unpaired, two-tailed t-test when comparing two groups, or one-way or two-way ANOVA with multiple comparisons when appropriate for comparing three or more groups. Tukey, Bonferroni, or Holm-Sidak post hoc tests were used following one-way or two-way ANOVA as appropriate. Statistical significance was defined as *P < 0.05. For all experiments requiring statistical analysis, the statistical test used, exact P values, sample size (n), t values, ANOVA F values, and degrees of freedom for each experiment is indicated in the figure legend. No statistical methods were used to pre-determine sample sizes, however sample sizes were estimated based on similar experiments reported in previous publications from our lab8,39,43,47. Data distribution was assumed to be normal with similar variance between groups, however this was not formally tested. The Grubbs test was used when appropriate to identify and remove significant outliers.
A supplementary methods checklist is available online.
Supplementary Material
Acknowledgments
This work was supported by National Institute of Health grant MH081060 (LMM) and MH66198 (ETK). The authors thank E. Olson (UT Southwestern Medical Center) for generously providing Hdac1loxP/loxP/Hdac2loxP/loxP, Hdac1loxP/loxP, and Hdac2loxP/loxP mice, J. Richardson for performing the necropsy of the mice, and G. Feng (Duke University Medical Center) for the initial vector construct of Sapap3. The authors are grateful to E. Olson, R. Bassel-Duby, W. Xu, and TK. Kim for helpful comments and suggestions. We also thank Brent Trauterman for his excellent technical assistance. L. Monteggia holds the Ginny and John Eulich Professorship in Autism Spectrum Disorders at UT Southwestern.
Footnotes
Competing financial interests
L. Monteggia is on the scientific advisory board for Rodin Therapeutics.
Author Contributions
M. Mahgoub and M. Adachi performed the behavioral experiments. M. Mahgoub, M. Adachi, K. Suzuki, M. Chahrour, and X. Liu, contributed to the molecular experiments. M. Mahgoub performed the statistical analyses. M. Mahgoub made the figures and wrote the corresponding sections of the paper. L. Monteggia and E. Kavalali designed the study, supervised the experiments and wrote the paper.
Data availability
The data that support the findings of this study are available from the corresponding author upon request.
References
- 1.Haberland M, Montgomery RL, Olson EN. The many roles of histone deacetylases in development and physiology: implications for disease and therapy. Nature reviews Genetics. 2009;10:32–42. doi: 10.1038/nrg2485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Levenson JM, et al. Regulation of histone acetylation during memory formation in the hippocampus. The Journal of biological chemistry. 2004;279:40545–40559. doi: 10.1074/jbc.M402229200. [DOI] [PubMed] [Google Scholar]
- 3.Fischer A, Sananbenesi F, Wang X, Dobbin M, Tsai LH. Recovery of learning and memory is associated with chromatin remodelling. Nature. 2007;447:178–182. doi: 10.1038/nature05772. [DOI] [PubMed] [Google Scholar]
- 4.Vecsey CG, et al. Histone deacetylase inhibitors enhance memory and synaptic plasticity via CREB:CBP-dependent transcriptional activation. The Journal of neuroscience: the official journal of the Society for Neuroscience. 2007;27:6128–6140. doi: 10.1523/JNEUROSCI.0296-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Barrett RM, Wood MA. Beyond transcription factors: the role of chromatin modifying enzymes in regulating transcription required for memory. Learn Mem. 2008;15:460–467. doi: 10.1101/lm.917508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Morris MJ, Karra AS, Monteggia LM. Histone deacetylases govern cellular mechanisms underlying behavioral and synaptic plasticity in the developing and adult brain. Behav Pharmacol. 2010;21:409–419. doi: 10.1097/FBP.0b013e32833c20c0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Guan JS, et al. HDAC2 negatively regulates memory formation and synaptic plasticity. Nature. 2009;459:55–60. doi: 10.1038/nature07925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Morris MJ, Mahgoub M, Na ES, Pranav H, Monteggia LM. Loss of histone deacetylase 2 improves working memory and accelerates extinction learning. The Journal of neuroscience: the official journal of the Society for Neuroscience. 2013;33:6401–6411. doi: 10.1523/JNEUROSCI.1001-12.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Shahbazian MD, Grunstein M. Functions of site-specific histone acetylation and deacetylation. Annu Rev Biochem. 2007;76:75–100. doi: 10.1146/annurev.biochem.76.052705.162114. [DOI] [PubMed] [Google Scholar]
- 10.Jones PL, et al. Methylated DNA and MeCP2 recruit histone deacetylase to repress transcription. Nature genetics. 1998;19:187–191. doi: 10.1038/561. [DOI] [PubMed] [Google Scholar]
- 11.Nan X, et al. Transcriptional repression by the methyl-CpG-binding protein MeCP2 involves a histone deacetylase complex. Nature. 1998;393:386–389. doi: 10.1038/30764. [DOI] [PubMed] [Google Scholar]
- 12.Guy J, Hendrich B, Holmes M, Martin JE, Bird A. A mouse Mecp2-null mutation causes neurological symptoms that mimic Rett syndrome. Nature genetics. 2001;27:322–326. doi: 10.1038/85899. [DOI] [PubMed] [Google Scholar]
- 13.Chen RZ, Akbarian S, Tudor M, Jaenisch R. Deficiency of methyl-CpG binding protein-2 in CNS neurons results in a Rett-like phenotype in mice. Nature genetics. 2001;27:327–331. doi: 10.1038/85906. [DOI] [PubMed] [Google Scholar]
- 14.Gemelli T, et al. Postnatal loss of methyl-CpG binding protein 2 in the forebrain is sufficient to mediate behavioral aspects of Rett syndrome in mice. Biological psychiatry. 2006;59:468–476. doi: 10.1016/j.biopsych.2005.07.025. [DOI] [PubMed] [Google Scholar]
- 15.Nelson ED, Kavalali ET, Monteggia LM. MeCP2-dependent transcriptional repression regulates excitatory neurotransmission. Curr Biol. 2006;16:710–716. doi: 10.1016/j.cub.2006.02.062. [DOI] [PubMed] [Google Scholar]
- 16.Chao HT, Zoghbi HY, Rosenmund C. MeCP2 controls excitatory synaptic strength by regulating glutamatergic synapse number. Neuron. 2007;56:58–65. doi: 10.1016/j.neuron.2007.08.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Amir RE, et al. Rett syndrome is caused by mutations in X-linked MECP2, encoding methyl-CpG-binding protein 2. Nature genetics. 1999;23:185–188. doi: 10.1038/13810. [DOI] [PubMed] [Google Scholar]
- 18.Moretti P, Zoghbi HY. MeCP2 dysfunction in Rett syndrome and related disorders. Curr Opin Genet Dev. 2006;16:276–281. doi: 10.1016/j.gde.2006.04.009. [DOI] [PubMed] [Google Scholar]
- 19.Hagberg B, Aicardi J, Dias K, Ramos O. A progressive syndrome of autism, dementia, ataxia, and loss of purposeful hand use in girls: Rett’s syndrome: report of 35 cases. Ann Neurol. 1983;14:471–479. doi: 10.1002/ana.410140412. [DOI] [PubMed] [Google Scholar]
- 20.Zoghbi H. Genetic aspects of Rett syndrome. J Child Neurol. 1988;3(Suppl):S76–78. doi: 10.1177/0883073888003001s15. [DOI] [PubMed] [Google Scholar]
- 21.Adachi M, Autry AE, Covington HE, 3rd, Monteggia LM. MeCP2-mediated transcription repression in the basolateral amygdala may underlie heightened anxiety in a mouse model of Rett syndrome. The Journal of neuroscience: the official journal of the Society for Neuroscience. 2009;29:4218–4227. doi: 10.1523/JNEUROSCI.4225-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Chao HT, et al. Dysfunction in GABA signalling mediates autism-like stereotypies and Rett syndrome phenotypes. Nature. 2010;468:263–269. doi: 10.1038/nature09582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Montgomery RL, et al. Histone deacetylases 1 and 2 redundantly regulate cardiac morphogenesis, growth, and contractility. Genes & development. 2007;21:1790–1802. doi: 10.1101/gad.1563807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Fan G, et al. DNA hypomethylation perturbs the function and survival of CNS neurons in postnatal animals. The Journal of neuroscience: the official journal of the Society for Neuroscience. 2001;21:788–797. doi: 10.1523/JNEUROSCI.21-03-00788.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Welch JM, et al. Cortico-striatal synaptic defects and OCD-like behaviours in Sapap3-mutant mice. Nature. 2007;448:894–900. doi: 10.1038/nature06104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Shmelkov SV, et al. Slitrk5 deficiency impairs corticostriatal circuitry and leads to obsessive-compulsive-like behaviors in mice. Nat Med. 2010;16:598–602. doi: 10.1038/nm.2125. 591p following 602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.McDougle CJ. Update on pharmacologic management of OCD: agents and augmentation. J Clin Psychiatry. 1997;58(Suppl 12):11–17. [PubMed] [Google Scholar]
- 28.Rapoport JL, Inoff-Germain G. Treatment of obsessive-compulsive disorder in children and adolescents. J Child Psychol Psychiatry. 2000;41:419–431. [PubMed] [Google Scholar]
- 29.Vythilingum B, Cartwright C, Hollander E. Pharmacotherapy of obsessive-compulsive disorder: experience with the selective serotonin reuptake inhibitors. Int Clin Psychopharmacol. 2000;15(Suppl 2):S7–13. doi: 10.1097/00004850-200008002-00003. [DOI] [PubMed] [Google Scholar]
- 30.Charney DS, Nestler EJ, Sklar P, Buxbaum JD. Neurobiology of mental illness. Oxford University Press; 2013. [Google Scholar]
- 31.Bienvenu OJ, et al. Sapap3 and pathological grooming in humans: Results from the OCD collaborative genetics study. American journal of medical genetics. Part B, Neuropsychiatric genetics: the official publication of the International Society of Psychiatric Genetics. 2009;150B:710–720. doi: 10.1002/ajmg.b.30897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Zuchner S, et al. Multiple rare SAPAP3 missense variants in trichotillomania and OCD. Molecular psychiatry. 2009;14:6–9. doi: 10.1038/mp.2008.83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Berton O, et al. Essential role of BDNF in the mesolimbic dopamine pathway in social defeat stress. Science. 2006;311:864–868. doi: 10.1126/science.1120972. [DOI] [PubMed] [Google Scholar]
- 34.Grunstein M. Histone acetylation in chromatin structure and transcription. Nature. 1997;389:349–352. doi: 10.1038/38664. [DOI] [PubMed] [Google Scholar]
- 35.Chahrour M, et al. MeCP2, a key contributor to neurological disease, activates and represses transcription. Science. 2008;320:1224–1229. doi: 10.1126/science.1153252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Yasui DH, et al. Integrated epigenomic analyses of neuronal MeCP2 reveal a role for long-range interaction with active genes. Proceedings of the National Academy of Sciences of the United States of America. 2007;104:19416–19421. doi: 10.1073/pnas.0707442104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kent WJ, et al. The human genome browser at UCSC. Genome Res. 2002;12:996–1006. doi: 10.1101/gr.229102.. Article published online before print in May 2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Montgomery RL, Hsieh J, Barbosa AC, Richardson JA, Olson EN. Histone deacetylases 1 and 2 control the progression of neural precursors to neurons during brain development. Proceedings of the National Academy of Sciences of the United States of America. 2009;106:7876–7881. doi: 10.1073/pnas.0902750106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Na ES, et al. A mouse model for MeCP2 duplication syndrome: MeCP2 overexpression impairs learning and memory and synaptic transmission. The Journal of neuroscience: the official journal of the Society for Neuroscience. 2012;32:3109–3117. doi: 10.1523/JNEUROSCI.6000-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Adeosun SO, et al. Cognitive deficits and disruption of neurogenesis in a mouse model of apolipoprotein E4 domain interaction. The Journal of biological chemistry. 2014;289:2946–2959. doi: 10.1074/jbc.M113.497909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Liu X, et al. Interleukin 1 type 1 receptor restore: a genetic mouse model for studying interleukin 1 receptor-mediated effects in specific cell types. The Journal of neuroscience: the official journal of the Society for Neuroscience. 2015;35:2860–2870. doi: 10.1523/JNEUROSCI.3199-14.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Fremont R, Tewari A, Khodakhah K. Aberrant Purkinje cell activity is the cause of dystonia in a shRNA-based mouse model of Rapid Onset Dystonia-Parkinsonism. Neurobiol Dis. 2015;82:200–212. doi: 10.1016/j.nbd.2015.06.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Adachi M, Barrot M, Autry AE, Theobald D, Monteggia LM. Selective loss of brain-derived neurotrophic factor in the dentate gyrus attenuates antidepressant efficacy. Biological psychiatry. 2008;63:642–649. doi: 10.1016/j.biopsych.2007.09.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Potts RC, et al. CHD5, a brain-specific paralog of Mi2 chromatin remodeling enzymes, regulates expression of neuronal genes. PLoS One. 2011;6:e24515. doi: 10.1371/journal.pone.0024515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Flavell SW, et al. Genome-wide analysis of MEF2 transcriptional program reveals synaptic target genes and neuronal activity-dependent polyadenylation site selection. Neuron. 2008;60:1022–1038. doi: 10.1016/j.neuron.2008.11.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Li W, Calfa G, Larimore J, Pozzo-Miller L. Activity-dependent BDNF release and TRPC signaling is impaired in hippocampal neurons of Mecp2 mutant mice. Proceedings of the National Academy of Sciences of the United States of America. 2012;109:17087–17092. doi: 10.1073/pnas.1205271109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Autry AE, et al. NMDA receptor blockade at rest triggers rapid behavioural antidepressant responses. Nature. 2011;475:91–95. doi: 10.1038/nature10130. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.