

Abstract
Congenital absence of nose (Arhinia) is extremely rare. A male baby was born at term via uncomplicated vaginal delivery and presented with complete arhinia, bilateral microphthalmia, lower eyelid coloboma and feeding difficulty. Reconstructive surgery was postponed until preschool age. On follow up at 1 year of age baby is feeding liquid and semisolid food and growing well.
Keywords: Arhinia, Microphthalmia, Coloboma, Congenital anomaly
Introduction
Congenital absence of nose is an extremely rare congenital malformation with only 45 cases reported since 1931 [1]. Arhinia causes severe breathing difficulty and feeding problem in neonates. Other congenital anomalies such as eyes, midline defects and central nervous system anomalies may also be associated. The etiology largely remains unknown.
Case Report
A boy baby was born at 38 weeks of gestation via uncomplicated vaginal delivery from a 28 years old mother with uneventful antenatal history. Ultrasound done at 30 and 37 weeks of gestation were normal. There was no history of consanguinity, no family history of any congenital malformation and no history of any drug intake during pregnancy. Previous one pregnancy was normal healthy boy 7 years back. Baby had birth weight of 2444 g (10–25th percentile), height 47 cm (25th percentile) and head circumference 32 cm (10–25th percentile). On physical examination baby had absent nose, high arched palate, hypertelorism, bilateral microphthalmia with rudimentary eyeballs, absent light and red reflex and bilateral lower eyelid coloboma (Fig. 1). Baby didn’t have respiratory difficulty but had feeding difficulty. His chest X-ray, cranial ultrasound, echocardiography and CT scan of brain was normal. CT scan of paranasal sinuses (Fig. 2) revealed absent nasal cavity, poorly developed nasal septum, bilateral small maxillary antrum, non-fusion of both zygomatic bones, presence of both eyeballs and lens and absence of cribriform plate. Feeding through Oro-gastric tube was started on day 1. Baby had cyanosis during oral feeding so baby was discharged on oro-gastric tube feeding with proper advice due to insistence of parents. On follow up at 1 year of age baby is able to feed by paladai without any respiratory difficulty. Karyotype of the baby was normal 46 XY.
Fig. 1.
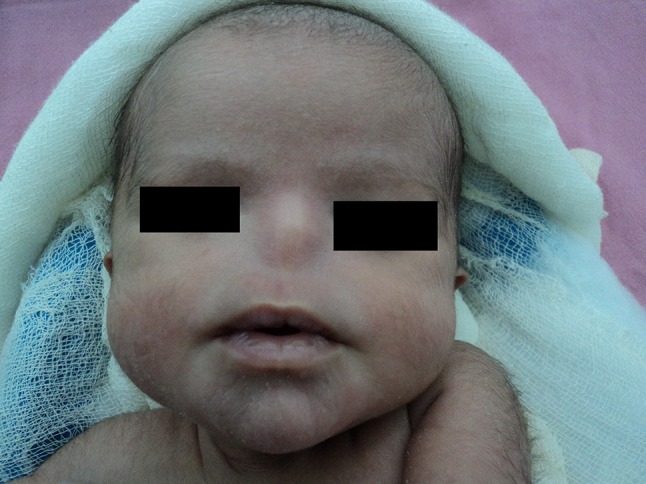
Complete absence of nose
Fig. 2.
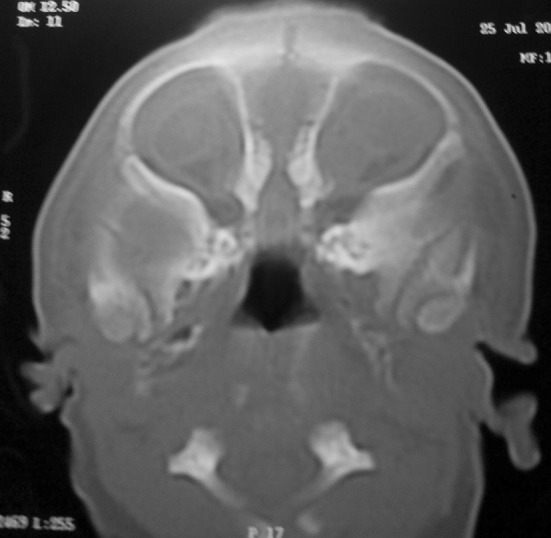
CT scan of paranasal sinus shows: absent nasal cavity
Discussion
Congenital arhinia is the absence of external nose, nasal cavity and olfactory apparatus. The pathogenesis of congenital arhinia is not well understood. The nose forms between 3rd and 10th weeks of gestation. At 24 days of life there are five prominences which surround stomoedeum: paired maxillary and mandibular processes and midline frontonasal prominence [2]. By the end of 4th week nasal placodes develop on both sides of frontonasal process. Cells in nasal placodes are derived from neural crest. Mesenchymal proliferation at the edges of nasal placodes lead to horseshoe shaped elevation of margins called medial and lateral nasal prominences. The deep groove in which nasal placodes are lying are called nasal pits which continue to deepen and form nasal sacs. By 7th week medial nasal prominences fuses in midline and maxillary processes fuse with medial nasal prominences. This fusion is complete at 10th weeks such that medial nasal prominences form philtrum and maxillary prominences form the rest of the upper lip. The frontonasal prominence forms bridge of nose, medial nasal prominence forms crest and tip and lateral nasal prominence forms alar nasi. By 7th week proliferation of epithelial cells form an epithelial plug which fills nasal cavity. This epithelial plug dissolves during 13–15th week [3] following which open nasal cavity forms by 16th week. During 6th week ectodermal cells of nasal placodes differentiate to form primary sensory neurons. These cells develop axons and by 7th weeks these axons contact olfactory bulbs which contain secondary neurons.
In arhinia there is failure of development of nasal placode on one or both side leading to failure of growth of medial and lateral nasal prominences. This also secondarily leads to failure of development of olfactory bulb and tract. Failure of absorption of nasal epithelial plug during 13–15th weeks of gestation, premature fusion of medial nasal prominences and migration defect of neural crest cells have also been proposed in the pathogenesis of arhinia [4].
Most of the cases are sporadic but familial cases have been reported. Significant antenatal history is not found and karyotype is normal in most cases and no sex predilection is found. McGlone reviewed 27 cases until 2003 [5]. Zhang et al. [1] reported 44th case and Ali [6] reported congenital arhinia with lacrimal mucocele. Arhinia may be associated with other anomalies like absence of olfactory bulbs and nerves, absent paranasal sinuses, high arched or cleft palate, eye anomalies, low set ears and CNS abnormalities [7]. Bosma reported a case of arhinia, choanal atresia, microphthalmia, cleft palate, inguinal hernias, hypogonadotropic hypogonadism with cryptorchidism and normal intelligence [8]. Abnormality in chromosome 9 [7] was reported in two cases and reciprocal translocation between chromosome 3 and 12 was reported in one case [9]. Antenatal diagnosis of arhinia has been reported.
Arhinia leads to airway obstruction and feeding difficulty. In our patient there was no breathing difficulty but baby had feeding problem. Management of congenital arhinia mainly involves management of airways and feeding difficulty. Neonates are obligate nasal breathers. Immediate management includes oral airway, tracheostomy or surgical creation of nasal airway in case of life threatening respiratory distress in neonatal period. Surgical reconstruction needs multidisciplinary approach involving neonatologist, pediatric, plastic, and craniofacial surgeons. Surgical reconstruction is preferably delayed until preschool age because facial development is almost complete till then [10]. Parents also need psychological support. Baby learns mouth breathing early. Feeding difficulty can be managed with oro-gastric tube feeding.
Conclusion
Congenital arhinia is a rare anomaly and needs multidisciplinary approach. Airway and feeding management is mainstay of treatment. Reconstructive surgery can be delayed until preschool age.
Author’s Contribution
Dr. Uttam Mondal: Diagnosis, work up and management of the case, Dr. Rameshwar Prasad: management and follow up of the case, review of literature and preparation of final manuscript.
Compliance with Ethical Standards
Conflict of interest
Dr. Uttam Mondal declares that he has no conflict of interest. Dr. Rameshwar Prasad declares that he has no conflict of interest.
Ethical Approval
This article does not contain any studies with human participants or animals performed by any of the authors.
Informed Consent
Informed consent was obtained from all individual participants included in the study.
Contributor Information
Uttam Mondal, Email: mondaluttam9@gmail.com.
Rameshwar Prasad, Email: drrameshwarprasad@hotmail.com.
References
- 1.Zhang MM, Hu YH, He W, Hu KK. Congenital arhinia: a rare case. Am J Case Rep. 2014;15:115–118. doi: 10.12659/AJCR.890072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Som PM, Naidich TP. Illustrated review of the embryology and development of the facial region, part 1: early face and lateral nasal cavities. Am J Neuroradiol. 2013;34(12):2233–2240. doi: 10.3174/ajnr.A3415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Nishimura Y. Embryological study of nasal cavity development in human embryos with reference to congenital nostril atresia. Acta Anat (Basel) 1993;147(3):140–144. doi: 10.1159/000147494. [DOI] [PubMed] [Google Scholar]
- 4.Olsen ØE, Gjelland K, Reigstad H, Rosendahl K. Congenital absence of the nose: a case report and literature review. Pediatr Radiol. 2001;31(4):225–232. doi: 10.1007/s002470000419. [DOI] [PubMed] [Google Scholar]
- 5.McGlone L. Congenital arhinia. J Paediatr Child Health. 2003;39(6):474–476. doi: 10.1046/j.1440-1754.2003.00193.x. [DOI] [PubMed] [Google Scholar]
- 6.Ali MJ. Bilateral lacrimal mucoceles in a setting of congenital arhinia. Ophthal Plast Reconstr Surg. 2014;30(6):e167. doi: 10.1097/IOP.0000000000000284. [DOI] [PubMed] [Google Scholar]
- 7.Cohen D, Goitein KJ. Arhinia revisited. Rhinology. 1987;25(4):237–244. [PubMed] [Google Scholar]
- 8.Bosma JF, Henkin RI, Christiansen RL, Herdt JR. Hypoplasia of the nose and eyes, hyposmia, hypogeusia, and hypogonadotrophic hypogonadism in two males. J Craniofac Genet Dev Biol. 1981;1(2):153–184. [PubMed] [Google Scholar]
- 9.Hou J-W. Congenital arhinia with de novo reciprocal translocation, t(3; 12)(q13.2; p11.2) Am J Med Genet Part A. 2004;130:200–203. doi: 10.1002/ajmg.a.30268. [DOI] [PubMed] [Google Scholar]
- 10.Weinberg A, Neuman A, Benmeir P, Lusthaus S, Wexler MR. A rare case of arhinia with severe airway obstruction: case report and review of the literature. Plast Reconstr Surg. 1993;91(1):146–149. doi: 10.1097/00006534-199301000-00024. [DOI] [PubMed] [Google Scholar]