

Abstract
The treatment of hepatitis C virus (HCV) infection has been revolutionized in recent years by the development of direct-acting antiviral regimens that do not contain peginterferon (pegIFN) and/or ribavirin (RBV). While direct-acting antiviral-based regimens have been shown to be greatly superior to pegIFN/RBV-based regimens in terms of efficacy and safety, they have a greater susceptibility to drug–drug interactions (DDIs). Daclatasvir (DCV)—the benchmark pangenotypic nonstructural protein 5A inhibitor—has been shown to be efficacious and generally well tolerated in partnership with other HCV direct-acting antivirals, including sofosbuvir, asunaprevir (ASV), and ASV plus beclabuvir. DCV may be the object of a DDI via the induction or inhibition of cytochrome P450 (CYP) 3A4 and/or P-glycoprotein (P-gp) by the concomitant medication, or the precipitant of a DDI via DCV-based induction/inhibition of CYP 3A4 or inhibition of P-gp, organic anion transporting polypeptide 1B1/B3, and/or breast cancer resistance protein. This article presents an overview of the drug interaction studies conducted during the clinical development of DCV, the findings of these studies that led to the guidance on concomitant medication use and dosage along with any required DCV dose modifications, and the use of the known metabolic pathway of DCV to guide concomitant dosing where direct drug–drug studies have not been conducted. The robust characterization of the DCV clinical pharmacology program has demonstrated that DCV has few or no clinically relevant DDIs with medications with which it is likely to be co-administered, and the majority of DDIs that do occur can be predicted and easily managed.
Funding: Bristol-Myers Squibb.
Electronic supplementary material
The online version of this article (doi:10.1007/s12325-016-0407-5) contains supplementary material, which is available to authorized users.
Keywords: Concomitant medications, Daclatasvir, Drug–drug interactions, Hepatitis C virus, Infectious diseases
Introduction
The development of direct-acting antiviral agents (DAAs) represents a major evolution in the treatment of hepatitis C virus (HCV) infection in both efficacy and safety compared with peginterferon (pegIFN) in combination with ribavirin (RBV) [1].
While the more recently developed DAAs have a lower potential for drug–drug interactions (DDIs) relative to the first-generation HCV NS3 protease inhibitors, the majority of new DAAs have some interaction with cytochrome P450 (CYP) enzymes, e.g., CYP3A4, and/or transporters such as P-glycoprotein (P-gp) [2]. In addition, some DAAs display pH-dependent solubility and may therefore be susceptible to DDIs in the presence of gastric acid-reducing agents. Moreover, DAA-only regimens require combination therapy of two or more DAAs with differing mechanisms of action thereby increasing the DDI considerations, particularly in the presence of concomitant medications.
DDIs that result in reduced exposure of HCV nonstructural protein 5A (NS5A) inhibitors (e.g., daclatasvir [DCV]) are particularly relevant in virologic terms due to the likelihood of the selection and persistence of resistance-associated variants if treatment failure occurs [3].
DCV—the benchmark pangenotypic NS5A inhibitor approved in the US, Europe, Japan, and multiple nations across Latin America, the Middle East, and Asia Pacific—has been shown to be highly effective and generally well tolerated in phase 3 studies with other HCV DAAs, including sofosbuvir (SOF), asunaprevir (ASV), and ASV with beclabuvir (BCV) [4–10]. Furthermore, DCV has been shown to have a low potential for DDIs. Simple dose adjustments can mitigate the majority of the few DDIs that may occur when DCV is co-administered with concomitant medications.
The DDI profile of DCV, both as a precipitant and object of the interaction, has been assessed in a comprehensive clinical pharmacology program with widely accepted probe substrates, inhibitors and inducers of CYP enzymes, and human drug transporters. In addition, DDIs between DCV and first-line antiretrovirals, immunosuppressants, treatments for opioid dependence, gastric acid-reducing agents, and other DAAs and common concomitant medications have also been assessed.
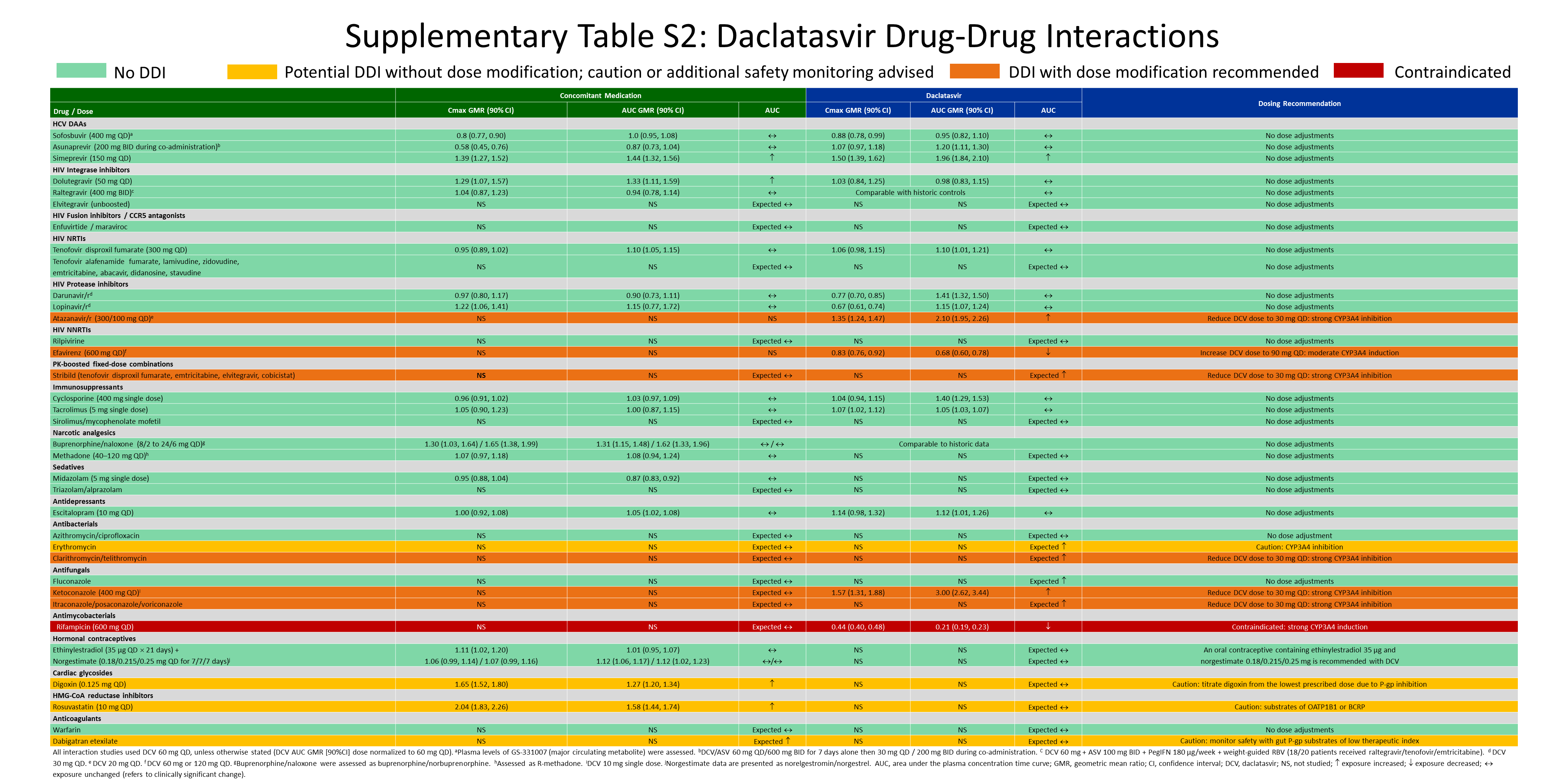
This review describes DDIs between DCV and common concomitant medications. An overview, where conducted, of the DDI study designs, patient populations, and dosing schedules is presented in the supplementary material, Table S1; a desktop summary of DCV DDIs (determined in clinical studies and predicted) is presented in Table S2 in the supplementary material, whereas individual DDIs are discussed below.
The DDI studies used standard statistical criteria and effects were assessed by geometric mean ratios (GMRs) of maximal concentration (C max), area under concentration–time curve (AUC), and minimal concentration, where applicable. All procedures followed were in accordance with the ethical standards of the responsible committee on human experimentation (institutional and national) and with the Helsinki Declaration of 1964, as revised in 2013. Informed consent was obtained from all patients included in the studies. Where possible, data from observed DDI studies were extrapolated to provide guidance for other drugs via similar mechanisms of interaction.
The Pharmacokinetic Basis of DCV DDIs
A DDI is present when one drug (the precipitant) alters the absorption, distribution, metabolism, or excretion of another drug (the object or victim) or a combination of these processes. For DCV, observations during in vitro and absorption, distribution, metabolism, and excretion studies in healthy subjects demonstrated that DCV undergoes minimal, primarily CYP3A4-mediated metabolism, and is excreted in the feces mostly as the parent drug [11]. It was further shown that DCV is a time-dependent inhibitor of CYP3A4 in vitro, a weak inducer of CYP3A4 in vivo, as well as a substrate and inhibitor of P-gp, organic anion transporting polypeptide (OATP), and breast cancer resistance protein (BCRP; [data on file]). These data suggest that DCV may be the object of a DDI via the induction or inhibition of CYP3A4 and/or P-gp by the concomitant medication, or the precipitant of a DDI via DCV-based induction/inhibition of CYP3A4 or via inhibition of P-gp, OATP1B1/B3, and/or BCRP.
Studies in healthy human subjects were conducted to assess the magnitude of potential DDIs where DCV would be the object or precipitant of an interaction. DDI studies were conducted with representative drugs from therapeutic classes likely to be co-administered with DCV, e.g., antiretrovirals, immunosuppressants, antidepressants, and hormonal contraceptives. The magnitude of DDIs where DCV would be the object of the interaction were investigated using a widely recognized strong CYP3A4 inducer (rifampin [Study AI444-012; data on file]) and known CYP3A4 and P-gp inhibitors (ketoconazole [Study AI444-005; data on file] and cyclosporine [Study AI444-065]) [12]. Similarly, the extent of DDIs where DCV would be the precipitant of an interaction were investigated using sensitive probe substrates for CYP3A4 (midazolam [Study AI444-008]) [13] and the transporters P-gp (digoxin [Study AI444-027; data on file]), OATP1B1/B3, and BCRP (rosuvastatin [Study AI444-054]) [14].
Mechanistic Assessments of DCV DDIs (Object)
Reduced DCV Exposure via Induction of CYP3A4
Strong induction of CYP3A4 by steady-state rifampin resulted in significantly reduced DCV exposure (C max, 56% reduction; AUC, 79% reduction) to a degree likely to attenuate antiviral efficacy (Study AI444-012; data on file) and led to the contraindication of DCV co-administration with strong inducers of CYP3A4. Lesser reductions in DCV exposure during co-administration with moderate inducers, e.g., the antiretroviral efavirenz (described below), can be effectively and safely managed with dose modification (DCV 90 mg once daily [QD]).
Increased DCV Exposure via Inhibition of CYP3A4/P-gp
Co-administration of DCV with steady-state ketoconazole—a strong inhibitor of CYP3A4 and P-gp—resulted in a three-fold increase in DCV exposure (AUC; Study AI444-005; data on file). Results from this study support the dose modification of DCV to 30 mg QD during co-administration with strong inhibitors of CYP3A4. In a another DDI study with single-dose cyclosporine (Study AI444-065)—a strong P-gp inhibitor but weak CYP3A4 inhibitor—DCV AUC increased by only 40% during co-administration relative to administration alone, and suggests that co-administration of DCV with P-gp inhibitors without meaningful inhibitory effects on CYP3A4 is unlikely to have clinically relevant effects on DCV exposure [12]. Based on these data, no dose adjustment of DCV is recommended for co-administration with cyclosporine.
Mechanistic Assessments of DCV DDIs (Precipitant)
Specific DDIs with potential concomitant medications are described below including those studied, as well as predicted or expected interactions. Individually listed examples and associated dose modifications are based upon the European Medicines Agency and the US Food and Drug Administration product information. The requirement for dose modifications with CYP3A4 strong inhibitors (30 mg QD), moderate inducers (90 mg QD), or strong inducers (contraindicated) is consistent across all prescribing information documents globally.
Induction/Inhibition of CYP3A4 by DCV
A DDI study (AI444-008) investigating the interaction between steady-state DCV and single-dose midazolam (a sensitive CYP3A4 probe) demonstrated that DCV is unlikely to impact the exposure of concomitant medications through CYP3A4-related mechanisms; midazolam exposure was minimally decreased (13%) during co-administration, relative to administration alone [13]. This observation was further supported by the results of other direct DDI studies with CYP3A4 substrates (described below), including ethinyl estradiol, escitalopram, cyclosporine, tacrolimus, ASV, telaprevir, simeprevir, and buprenorphine.
Inhibition of P-gp by DCV
The co-administration of DCV with digoxin, both agents at steady-state, resulted in a 27% increase in digoxin AUC during co-administration, relative to administration alone (Study AI444-027; data on file). Similar increases in digoxin exposure were also noted when it was co-administered with DCV and ASV (also a P-gp inhibitor; Study AI447-040), suggesting that there is no additive risk of a DDI during co-administration of P-gp substrates with DCV when combined with ASV [15]. The observations from Studies AI444-027 (data on file) and AI447-040 suggest that caution should be used when DCV is co-administered with P-gp substrates with a narrow therapeutic index.
Inhibition of OATP1B1/B3 and/or BCRP by DCV
During co-administration with DCV at steady-state, single-dose rosuvastatin exposure was increased (C max, two-fold; AUC, 58% increase; Study AI444-054) by weak to moderate inhibition of OATP1B1/B3 and/or BCRP [14]. Based upon data during co-administration of DCV with ASV (a sensitive OATP substrate whose exposure is not meaningfully affected by DCV) [16], it is likely that the effect of DCV (a BCRP inhibitor) on the exposure of rosuvastatin (both an OATP and a BCRP substrate), is mediated primarily through BCRP. Caution is recommended during the co-administration of DCV and substrates of OATP1B1/B3 and/or BCRP with a narrow therapeutic index.
DCV DDIs and Dosing Guidance with Other Antiviral Agents
The DDI profile of DCV has been assessed with other HCV regimen partners and antiretroviral agents. GMRs of C max and AUC and associated 90% confidence intervals (CIs) from individual DDI studies are presented in Table 1.
Table 1.
An overview of DCV DDIs with HCV regimen partners and antiretroviral agents
| Drug/dose | Concomitant medication | DCV | |||||
|---|---|---|---|---|---|---|---|
| C max GMR (90% CI) | AUC GMR (90% CI) | AUC | C max GMR (90% CI) | AUC GMR (90% CI) | AUC | Dose adjustment | |
| HCV DAAs | |||||||
| Sofosbuvir (400 mg QD)a | 0.8 (0.77, 0.90) | 1.0 (0.95, 1.08) | ↔ | 0.88 (0.78, 0.99) | 0.95 (0.82, 1.10) | ↔ | None |
| Asunaprevir (200 mg BID during co-administration)b | 0.58 (0.45, 0.76) | 0.87 (0.73, 1.04) | ↔ | 1.07 (0.97, 1.18) | 1.20 (1.11, 1.30) | ↔ | None |
| Simeprevir (150 mg QD) | 1.39 (1.27, 1.52) | 1.44 (1.32, 1.56) | ↑ | 1.50 (1.39, 1.62) | 1.96 (1.84, 2.10) | ↑ | None |
| HIV integrase inhibitors | |||||||
| Dolutegravir (50 mg QD) | 1.29 (1.07, 1.57) | 1.33 (1.11, 1.59) | ↑ | 1.03 (0.84, 1.25) | 0.98 (0.83, 1.15) | ↔ | None |
| Raltegravir (400 mg BID)c | 1.04 (0.87, 1.23) | 0.94 (0.78, 1.14) | ↔ | Comparable to historic controls | ↔ | None | |
| Elvitegravir (unboosted) | NS | NS | ↔ | NS | NS | ↔ | None |
| HIV fusion inhibitors/CCR5 antagonists | |||||||
| Enfuvirtide/maraviroc | NS | NS | ↔ | NS | NS | ↔ | None |
| HIV NRTIs | |||||||
| Tenofovir (300 mg QD) | 0.95 (0.89, 1.02) | 1.10 (1.05, 1.15) | ↔ | 1.06 (0.98, 1.15) | 1.10 (1.01, 1.21) | ↔ | None |
| Lamivudine, zidovudine, emtricitabine, abacavir, didanosine, stavudine | NS | NS | ↔ | NS | NS | ↔ | None |
| HIV NNRTIs | |||||||
| Efavirenz (600 mg QD)d | NS | NS | NS | 0.83 (0.76, 0.92) | 0.68 (0.60, 0.78) | ↓ | 90 mg |
| Etravirine/neviripine | NS | NS | NS | NS | NS | ↓ | NR:90 mge |
| Rilpivirine | NS | NS | ↔ | NS | NS | ↔ | None |
| HIV protease inhibitors | |||||||
| Atazanavir/r (300/100 mg QD)e | NS | NS | NS | 1.35 (1.24, 1.47) | 2.10 (1.95, 2.26) | ↑ | 30 mg |
| Darunavir/rf | 0.97 (0.80, 1.17) | 0.90 (0.73, 1.11) | ↔ | 0.77 (0.70, 0.85) | 1.41 (1.32, 1.50) | ↔ | None |
| Lopinavir/rf | 1.22 (1.06, 1.41) | 1.15 (0.77, 1.72) | ↔ | 0.67 (0.61, 0.74) | 1.15 (1.07, 1.24) | ↔ | None |
| HIV PK-boosted fixed-dose combinations | |||||||
| Stribild | NS | NS | ↔ | NS | NS | ↑ | 30 mg |
All interaction studies used daclatasvir 60 mg QD, unless otherwise stated (daclatasvir AUC GMR [90% CI] dose normalized to 60 mg QD)
AUC area under curve, BID twice daily, CI confidence interval, C max maximal concentration, DAAs direct-acting antivirals, DCV daclatasvir, DDIs drug–drug interactions, GMR geometric mean ratio, HCV hepatitis C virus, HIV human immunodeficiency virus, NNRTIs non-nucleoside reverse transcriptase inhibitors, NRTIs nucleoside/nucleotide reverse transcriptase inhibitors, NR not recommended, NS not studied (effects on C max, AUC, and associated dose modifications are predicted), PK pharmacokinetic, QD once daily, ↓ exposure decreased, ↑ exposure increased, ↔ exposure unchanged (refers to clinically significant change)
aPlasma levels of GS-331007 (major circulating metabolite) were assessed
bDaclatasvir/asunaprevir 60 mg QD/600 mg BID for 7 days alone then 30 mg QD/200 mg BID during co-administration
cDCV 60 mg + ASV 100 mg BID (18/20 patients received rilpivirine/tenofovir/emtricitabine)
dDaclatasvir 60 or 120 mg QD
eCombination is not recommended in the EU label (lack of data), but dose modification (90 mg/day) is recommended in the US prescribing information
fDaclatasvir 20 mg QD
gDaclatasvir 30 mg QD
HCV DAA Regimen Partners
There are no clinically significant DDIs between DCV and ASV (Studies AI447-009 and AI447-011) [16, 17], ASV and BCV in combination (Study AI443-014) [18], or SOF (Study AI444-040) [19], and dose modifications during co-administration are not required. No dose adjustments are required during the co-administration of DCV with simeprevir (Study HCP1005) [20]. Steady-state co-administration with the strong CYP3A4 inhibitor, telaprevir, resulted in a 2.3-fold increase in DCV exposure (AUC) in Study AI444-067 (data on file); the interaction with boceprevir, also a strong inhibitor of CYP3A4, is predicted to be similar.
Antiretroviral Agents
The estimated global prevalence of HCV co-infection with human immunodeficiency virus (HIV) varies widely by geography and demography; among intravenous drug users, the co-infection rate may approach 100% [21]. The complex nature of many HIV regimens in terms of their DDI profile and the preference not to switch well-tolerated regimens in virally suppressed patients implies that the ideal HCV partner regimen for the treatment of co-infection has a low probability of DDIs. The probability of a DDI between DCV and antiretroviral agents often reflects the degree of impact the antiretroviral regimen has on CYP3A4 [22].
DCV in combination with SOF has been shown to be effective and generally well tolerated in phase 3 evaluations in an HIV/HCV genotype 1–4 co-infected patient population receiving all major HIV treatment regimens [7].
Protease Inhibitors
Differential DCV dosing guidance, based on the degree of CYP3A4 inhibition, exists during co-administration of DCV with ritonavir-boosted protease inhibitors. Co-administration of DCV with atazanavir/r (Study AI444-032) resulted in a 2.1-fold increase in DCV AUC during co-administration [23]; DCV dose modification to 30 mg QD is required during co-administration with ritonavir-boosted atazanavir. However, DCV dose modifications are not required during co-administration with unboosted atazanavir. Dose modification of DCV is not required during co-administration with darunavir/r or lopinavir/r, as lesser increases in dose-normalized DCV AUC were observed during co-administration with both darunavir/r (41% increase) and lopinavir/r (15% increase) in healthy volunteers in Study AI444-093 [24]. No clinically relevant changes in darunavir or lopinavir exposures were observed during co-administration with DCV (+pegIFN/RBV) in HIV/HCV co-infected patients receiving stable combination antiretroviral therapy (Study AI444-043 sub-study) [24]. Dosing recommendations with cobicistat-boosted regimens are aligned with those of ritonavir-boosted regimens [11].
Integrase Inhibitors
The lack of clinically meaningful steady-state DDIs between DCV and unboosted integrase inhibitors has been demonstrated with raltegravir in HCV–HIV co-infected patients (ANRS HC30 QUADRIH study) [25] and with dolutegravir in healthy subjects (Study NCT02082808) [26]. In both studies, no clinically meaningful changes to the exposure of the integrase inhibitor were observed (raltegravir exposure was reduced by 6% and dolutegravir exposure increased by 33%), and DCV exposures were either comparable with historic controls (ANRS HC30 QUADRIH study) [25] or unaffected (Study NCT02082808) [26]. The slight increase in dolutegravir exposure was thought to arise via P-gp/BCRP inhibition by DCV. No dose modifications are required during co-administration with unboosted integrase inhibitors.
Nucleoside/Nucleotide Reverse Transcriptase Inhibitors
The observation that steady-state exposures of both DCV and tenofovir (administered as tenofovir disoproxil fumarate) were unaffected during co-administration in healthy subjects relative to dosing alone (Study AI444-033) [23], coupled with the mechanistic assumption that other nucleoside/nucleotide reverse transcriptase inhibitors (NRTIs) (e.g., emtricitabine) are not metabolized to a large extent by CYP enzymes, nor are they know to be common precipitants of DDIs, supports the recommendation that DCV dose modifications are not required during co-administration with NRTIs. Although direct study data are not available, it is predicted that co-administration of tenofovir alafenamide fumarate (TAF) would not affect DCV exposure (TAF is not an inhibitor or inducer of CYP3A4 or P-gp) or vice versa (strong inhibitors of P-gp/BCRP may affect TAF absorption).
Non-Nucleoside Reverse Transcriptase Inhibitors
Differential dosing guidance, based on the degree of CYP3A4 induction, exists for DCV when co-administered with non-nucleoside reverse transcriptase inhibitors. Rilpivirine is not an inducer of CYP3A4 and no meaningful DDI with DCV is predicted (no dosing adjustments during co-administration are required). Steady-state efavirenz, a CYP3A4 inducer, reduced DCV exposure (AUC) by 32% when co-administered in healthy subjects (Study AI444-034) [23] and, as such, DCV dose modification to 90 mg QD during co-administration is required to maintain DCV exposures similar to those achieved at the therapeutic dose of 60 mg QD. DDIs between DCV and etravirine and nevipirine have not been studied, but reduced DCV exposure resulting from CYP3A4 induction is predicted; co-administration is currently not recommended in the EU prescribing information, whereas dose modification to DCV 90 mg QD is recommended in the US label.
Fusion Inhibitors and CCR5 Antagonists
DDIs between DCV and fusion inhibitors (e.g., enfuvirtide) and CCR5 antagonists (e.g., maraviroc) have not been studied directly. A review of the metabolic profiles of enfuvirtide (a peptide that undergoes catalytic breakdown to constituent amino acids) and maraviroc (primarily metabolized by CYP3A4) suggests that no meaningful DDIs during co-administration with DCV are likely to occur.
DCV DDIs and Dosing Guidance with Commonly Prescribed Concomitant Medications
The DDI profile of DCV has been assessed with other commonly prescribed and over-the-counter medications, e.g., immunosuppressants, narcotic analgesics, sedatives, antidepressants, cardiovascular medications, and gastric acid-reducing agents. GMRs of C max and AUC and associated 90% CIs from individual DDI studies are presented in Table 2.
Table 2.
An overview of DCV DDIs with other common concomitant medications
| Drug/dose | Concomitant medication | Daclatasvir | |||||
|---|---|---|---|---|---|---|---|
| C max GMR (90% CI) | AUC GMR (90% CI) | AUC | C max GMR (90% CI) | AUC GMR (90% CI) | AUC | Dose adjustment | |
| Immunosuppressants | |||||||
| Cyclosporine (400 mg single dose) | 0.96 (0.91, 1.02) | 1.03 (0.97, 1.09) | ↔ | 1.04 (0.94, 1.15) | 1.40 (1.29, 1.53) | ↔ | None |
| Tacrolimus (5 mg single dose) | 1.05 (0.90, 1.23) | 1.00 (0.87, 1.15) | ↔ | 1.07 (1.02, 1.12) | 1.05 (1.03, 1.07) | ↔ | |
| Sirolimus/mycophenolate mofetil | NS | NS | ↔ | NS | NS | ↔ | |
| Narcotic analgesics | |||||||
| Buprenorphine/naloxone (8/2 to 24/6 mg QD)a | 1.30 (1.03, 1.64)/1.65 (1.38, 1.99) | 1.31 (1.15, 1.48)/1.62 (1.33, 1.96) | ↔/↔ | Comparable to historic data | ↔ | None | |
| NS | NS | ↔ | |||||
| Methadone (40–120 mg QD)b | 1.07 (0.97, 1.18) | 1.08 (0.94, 1.24) | ↔ | ||||
| Sedatives | |||||||
| Midazolam (5 mg single dose) | 0.95 (0.88, 1.04) | 0.87 (0.83, 0.92) | ↔ | NS | NS | ↔ | None |
| Triazolam/alprazolam | NS | NS | ↔ | NS | NS | ↔ | |
| Antidepressants | |||||||
| Escitalopram (10 mg QD) | 1.00 (0.92, 1.08) | 1.05 (1.02, 1.08) | ↔ | 1.14 (0.98, 1.32) | 1.12 (1.01, 1.26) | ↔ | None |
| Antibacterials | |||||||
| Azithromycin/ciprofloxacin | NS | NS | ↔ | NS | NS | ↔ |
None None/caution 90 mg |
| Erythromycin | NS | NS | ↔ | NS | NS | ↑ | |
| Clarithromycin/telithromycin | NS | NS | ↔ | NS | NS | ↑ | |
| Antifungals | |||||||
| Fluconazole | NS | NS | ↔ | NS | NS | ↑ |
None 30 mg 30 mg |
| Ketoconazole (400 mg QD)c | NS | NS | ↔ | 1.57 (1.31, 1.88) | 3.00 (2.62, 3.44) | ↑ | |
| Itraconazole/posaconazole/voriconazole | NS | NS | ↔ | NS | NS | ↑ | |
| Antimycobacterials | |||||||
| Rifampicin (600 mg QD) | NS | NS | ↔ | 0.44 (0.40, 0.48) | 0.21 (0.19, 0.23) | ↓ | Contraindicated |
| Hormonal contraceptives | |||||||
| Ethinylestradiol (35 μg QD × 21 days) + norgestimate (0.18/0.215/0.25 mg QD for 7/7/7 days)d | 1.11 (1.02, 1.20) | 1.01 (0.95, 1.07) | ↔ | NS | NS | ↔ | None |
| 1.06 (0.99, 1.14)/1.07 (0.99, 1.16) | 1.12 (1.06, 1.17)/1.12 (1.02, 1.23) | ↔/↔ | NS | NS | ↔ | ||
| Cardiac glycosides | |||||||
| Digoxin (0.125 mg QD) | 1.65 (1.52, 1.80) | 1.27 (1.20, 1.34) | ↑ | NS | NS | ↔ |
None Titrate digoxin |
| HMG-CoA reductase inhibitors | |||||||
| Rosuvastatin (10 mg QD) | 2.04 (1.83, 2.26) | 1.58 (1.44, 1.74) | ↑ | NS | NS | ↔ |
None Caution |
| Anticoagulants | |||||||
| Warfarin | NS | NS | ↔ | NS | NS | ↔ |
None None/monitor safety |
| Dabigatran etexilate | NS | NS | ↑ | NS | NS | ↔ | |
All interaction studies used daclatasvir 60 mg QD, unless otherwise stated (daclatasvir AUC GMR [90% CI] dose normalized to 60 mg QD)
AUC area under curve, CI confidence interval, C max maximal concentration, DCV daclatasvir, DDIs drug–drug interactions, GMR geometric mean ratio, HMG-CoA reductase 3-hydroxy-3-methyl-glutaryl-coenzyme A reductase, NS not studied (effects on C max, AUC, and associated dose modifications are predicted), QD once daily, ↓ exposure decreased, ↑ exposure increased, ↔ exposure unchanged (refers to clinically significant change)
aBuprenorphine/naloxone was assessed as buprenorphine/norbuprenorphine
bAssessed as R-methadone
cDaclatasvir 10 mg single dose
dNorgestimate is presented as norelgestromin/norgestrel
Immunosuppressants
As HCV is a chronic disease of the liver that ultimately results in hepatic failure, it is likely that DCV will be co-administered with immunosuppressants in patients who have received organ transplantation.
As described above, steady-state DCV exposure was not affected to a meaningful degree when administered to healthy subjects in combination with single doses of cyclosporine (DCV AUC increased by 40% during co-administration) or tacrolimus (DCV AUC increased by 5% during co-administration) sufficient to mimic steady-state concentrations (Study AI444-065); cyclosporine and tacrolimus exposures were unaffected (≤5% changes in C max and AUC during co-administration) [12]. No meaningful DDIs are predicted with sirolimus (primarily metabolized by CYP3A4 and also a substrate of P-gp) and mycophenolate mofetil (metabolized by both UDP-glucuronosyltransferases [UGTs] and CYP3A4).
The lack of requirement for DCV dose adjustments during co-administration with immunosuppressants is also further supported by the observation that DCV (in combination with SOF ± RBV) is highly effective and generally well tolerated in a phase 3 evaluation in post-transplant recipients with recurrent HCV infection (N = 53); furthermore, only one patient required minor dosage adjustment of immunosuppressive medications (tacrolimus dose reduced from 3.5 to 3.0 mg), and there were no events of graft rejection [6].
Narcotic Analgesics
Approximately, 70% of the global population of intravenous drug users are infected with HCV [27]. The effect of steady-state DCV on the exposure of oral opioids (methadone and buprenorphine/naloxone) administered at stable doses was assessed in non-HCV-infected, opioid-dependent adults in Study AI444-064 [28]. During co-administration, the exposures (AUCs) of DCV and R-methadone (active methadone enantiomer) were unaffected, and clinically insignificant increases in the exposures of buprenorphine (31%) and norbuprenorphine (62%) were observed; the 90% GMRs of opioid AUCs were contained entirely within the literature-derived ranges of no effect (R-methadone, 0.7–1.23; buprenorphine and norbuprenorphine, 0.5–2.0) and no dose adjustments during co-administration were required. Pharmacodynamic measures of opioid activity were also unaffected [28].
Sedatives
The lack of a clinically relevant interaction between DCV and the benzodiazepine, midazolam, has been described previously (Study AI444-008) [13], and co-administration does not require dose modification. Similarly, no meaningful DDIs between DCV and either triazolam or alprazolam—both of which are primarily metabolized by CYP3A4—are predicted, and thus no dose adjustments during co-administration are recommended.
Antidepressants
Selective serotonin reuptake inhibitors (SSRIs) are effective in the management of the depressive episodes that are common in patients with HCV infection [29, 30]. During co-administration in healthy subjects (Study AI444-084), steady-state DCV and representative SSRI (escitalopram) exposures were unaffected to a meaningful degree (DCV and SSRI AUCs were increased by 12% and 5%, respectively, during co-administration; data on file), and thus dose modifications during co-administration are not required.
The herbal supplement, St. John’s wort (Hypericum perforatum), is a known strong inducer of CYP3A4 and thus contraindicated for concomitant use with DCV.
Anti-Infectives
Though not studied, dosing recommendations during co-administration of DCV with antibacterials are based on the predicted impact of the antibacterial on CYP3A4. DCV dose modification to 30 mg QD is required during co-administration with anti-infectives that are strong CYP3A4 inhibitors (e.g., clarithromycin and telithromycin), whereas no DCV dose adjustments are required during co-administration with anti-infectives that are moderate or weak CYP3A4 inhibitors. It should be noted that inhibitory effects of such anti-infectives may be dose dependent and therefore each case should be reviewed on a case-by-case basis.
Antifungals and Antimycobacterials
As previously discussed, DCV dose modification is required during co-administration with the antifungal ketoconazole (strong CYP3A4 inhibitor; DCV 30 mg QD) and co-administration with the antimycobacterial, rifampicin (strong CYP3A4 inducer), is contraindicated. DCV dosing recommendations or contraindications when co-administered with further antifungals that are strong CYP3A4 inhibitors (e.g., itraconazole, posaconazole, voriconazole [DCV 30 mg QD]), moderate CYP3A4 inhibitors (e.g., fluconazole [no dose adjustments]), or other antimycobacterials that are strong CYP3A4 inducers (e.g., rifabutin and rifapentine [contraindicated]), are based on mechanistic assumptions and direct studies have not been conducted.
Hormonal Contraceptives
DCV has embryotoxic and teratogenic effects in animal species. As such, prevention of pregnancy during DCV treatment and prevention in female partners of male patients on therapy is essential. Teratogenic effects have also been observed with RBV [31], and extreme care should be taken to avoid pregnancy in female patients receiving DCV-based regimens supplemented with RBV.
Co-administration of DCV at steady state with an ethinyl estradiol- and norgestimate-containing oral contraceptive (Ortho Tri-Cyclen®, Ortho-McNeil-Janssen Pharmaceuticals, Inc, Titusvile, USA) did not result in clinically relevant differences in ethinyl estradiol, norelgestromin, or norgestrel exposures (Study AI444-020; 90% CIs were contained within the boundary of bioequivalence) [32]. An oral contraceptive containing ethinyl estradiol 35 μg and norgestimate 0.180/0.215/0.250 mg is recommended with DCV. Other oral contraceptives have not been studied.
Cardiovascular Agents
The majority of patients infected with HCV were born between the years of 1945–1965 and, therefore, given the mean age of the HCV population, the concomitant administration of DCV with cardiovascular agents is likely [33]. There are no predicted DDIs or cautionary requirements when DCV is co-administered with angiotensin-converting enzyme inhibitors, angiotensin II receptor blockers, or phosphodiesterase-5 inhibitors.
Antiarrhythmics
The requirement for caution during co-administration of DCV with digoxin due to DCV-based inhibition of P-gp resulting in increased digoxin exposure (AUC, 27% increase) has been described above (Study AI444027; data on file).
Cases of severe bradycardia and heart block have been observed when DCV is co-administered with SOF and concomitant amiodarone (CYP3A4 substrate/inhibitor, P-gp inhibitor) with or without other drugs that lower heart rate; similar observations have also been made with other SOF-based regimens [34]. Cases are potentially life threatening; therefore, amiodarone should only be used in patients on DCV and SOF when other alternative antiarrhythmic treatments are not tolerated or are contraindicated. Close monitoring is required when the concomitant use of amiodarone is considered to be unavoidable; patients who are identified as being at high risk of bradyarrhythmia should be continuously monitored for 48 h in an appropriate clinical setting. Furthermore, due to the long half-life of amiodarone, appropriate monitoring should also be carried out for patients who have discontinued amiodarone within the past few months and are to be initiated on DCV in combination with SOF [11]. The exact mechanism of this toxicity is not established and a direct study has not been conducted.
Calcium Channel Blockers
The interaction between DCV and calcium channel blockers has not been studied directly, and the recommendation for caution during co-administration due to predicted increases in DCV exposure is based upon inhibition of CYP3A4 (diltiazem, nifedipine, and amlodipine) or CYP3A4 and P-gp (verapamil).
Anticoagulants
There is no predicted DDI between DCV and warfarin (metabolized primarily by CYP3A4 [R-isomer] and CYP2C9 [S-isomer]); however, the exposure of dabigatran etexilate is predicted to be increased by P-gp inhibition by DCV during co-administration; thus, safety monitoring due to the narrow therapeutic index of dabigatran is recommended.
HMG-CoA Reductase Inhibitors
The recommendation of caution during co-administration of DCV with HMG-CoA reductase inhibitors, in general, is based on the observed increase in rosuvastatin exposure in Study AI444-054 (OATP/BCRP inhibition by DCV) as described above [14], and similar effects with the OATP and/or BCRP substrates, atorvastatin, fluvastatin, simvastatin, pitavastatin, and pravastatin are predicted.
Systemic Corticosteroids
Systemic dexamethasone is a strong inducer of CYP3A4 and therefore co-administration with DCV is contraindicated.
Anticonvulsants
Concomitant administration of anticonvulsants that are strong inducers of CYP3A4 (e.g., carbamazepine, oxcarbazepine, phenobarbital, and phenytoin) is contraindicated for co-administration with DCV.
Gastric Acid-Reducing Agents
DCV, among other NS5a inhibitors, demonstrates pH-dependent solubility and therefore assessments of the impact of proton pump inhibitor (PPI), omeprazole, and the H2 receptor antagonist (H2RAs), famotidine, on DCV exposure during co-administration were conducted (Studies AI444-024 and AI444-009, respectively).
Concomitant steady state omeprazole (40 mg QD) or single-dose famotidine (40 mg QD) did not affect DCV exposure to a meaningful extent during co-administration with DCV (DCV AUC reduced by 16% and 18% during co-administration with omeprazole [35] and famotidine [data on file], respectively), and dose modifications during co-administration are not required.
The impact of gastric acid-reducing agent co-administration on the exposure of steady-state DCV has also been assessed in a population pharmacokinetic (PK) analysis (data on file) of patient data from 14 clinical studies, including nine phase 2 and five phase 3 studies; approximately, 19% (n = 514/2768) of the analysis population were exposed to gastric acid-reducing agents (PPIs or acid modifiers). Gastric acid-reducing agents were identified as a statistically significant covariate on DCV absorption in the full model (at P < 0.05; data on file), indicating a slower absorption in the presence of gastric acid-reducing agents. However, this effect was not retained in the final model, as the impact on the absorption rate was considered marginal (<22%) and the predicted clinical impact on DCV AUC was expected to be small and not clinically meaningful. Furthermore, univariate screening suggests no effect of gastric acid-reducing agents on overall DCV exposure.
Based on findings from direct DDI studies and the population PK analysis discussed above, dose modifications during co-administration of DCV with either PPIs or H2RAs are not warranted, and no meaningful DDI between DCV and antacids is predicted. Proton pump inhibitor use at any dose was permitted in the phase 3 studies of DCV + SOF in combination.
Herbal Remedies and Food Supplements
Several herbal remedies and food supplements are known to modify CYP3A4 activity and may therefore alter DCV exposure during co-administration. Popular botanical supplements that contain substantial quantities of moieties which act as mechanism-based inhibitors of CYP3A4 include, but are not limited to, Golden-seal (Hydrastis canadensis; commonly used in the USA as a preventative for common colds and upper respiratory tract infections) and Schisandra spp. (traditionally used hepatoprotectant supplements in China, Japan, and Russia) [36]. Other foodstuffs that are known inhibitors of CYP3A4, and may increase exposure of DCV, include grapefruit and Seville oranges [37].
In addition to herbal and dietary supplements which may increase DCV exposure via CYP3A4 inhibition, others may induce or modulate the induction of CYP3A4 and thus potentially reduce the antiviral activity of DCV. Such extracts include St. John’s wort (Hypericum perforatum; a strong inducer of CYP3A4 and thus contraindicated for concomitant use with DCV) [36] and taurine (commonly found in body-building supplements and energy drinks) [38].
It is suggested that prior to the initiation of DCV-based therapy, health-care professionals discuss with the patient the use of herbal preparations and the consumption of food and drink known to impact CYP3A4 activity.
Real-World Experiences
Real-world DDI frequency data for DCV are limited; however, DDI frequencies with NS5A-containing regimens were discussed in the report of a German outpatient clinic cohort study in HCV mono-infected patients (N = 261) [39]. Consistent with the DDI profile for DCV, fewer DDIs requiring dose adjustment and/or close monitoring were predicted when real-life concomitant medications were reviewed in recipients of DCV/SOF (95 patients [36.4%]) than with other NS5A inhibitor-based DAA regimens; a single patient (0.4%) was receiving concomitant medication that would be contraindicated with DCV/SOF. The most frequently predicted DDIs with a regimen of DCV/SOF were with thyroid hormones (n = 43, 16.5%), dihydropyridine derivatives (n = 28, 10.7%), and alpha and beta blockers (n = 14, 5.4%). Proton pump inhibitors were the most commonly predicted precipitants of a DDI with other NS5A inhibitor-based DAA regimens [39].
Conclusions
The DDI studies conducted during the development program of DCV demonstrate that it has few or no clinically relevant DDIs with HCV DAA regimen partners [16–20], first-line antiretrovirals [23–26], immunosuppressants [12], narcotic analgesics [28], gastric acid-reducing agents [35], or other common concomitant medications (e.g., hormonal contraceptives [32], antidepressants [SSRIs; data on file], and benzodiazepine sedatives) [13]. Furthermore, real-world data, although limited, suggest that the regimen of DCV/SOF has a favorable DDI profile, compared with other NS5A inhibitor-based regimens [39].
When unavoidable DDIs are present, the predictability of CYP3A4-based DDIs combined with the flexibility to dose adjust when co-administering DCV with strong CYP3A4 inhibitors (DCV 30 mg QD) or moderate CYP3A4 inducers (DCV 90 mg QD) helps clinicians to effectively manage these DDIs. Co-administration of DCV with strong CYP3A4 and P-gp inducers, such as anticonvulsants (e.g., carbamazepine), systemic corticosteroids, antimycobacterials (e.g., rifampin), and the herbal supplement, St John’s wort, is contraindicated.
In conclusion, the robust clinical pharmacology program of DCV has demonstrated that DCV has few or no clinically relevant DDIs with medications with which it is likely to be co-administered, and the majority of DDIs that do occur can be predicted and easily managed.
Electronic supplementary material
Below is the link to the electronic supplementary material.
{kind=link}
Acknowledgments
Sponsorship, article processing charges, and the open access charge for this study were funded by Bristol-Myers Squibb. Editorial assistance with the drafting of this manuscript was provided by A. Stead of Articulate Science, Manchester, UK, and funded by Bristol-Myers Squibb. The authors acknowledge and thank all the physicians, associated health-care professionals, and patients who took part in the studies listed within this manuscript. All named authors meet the International Committee of Medical Journal Editors (ICMJE) criteria for authorship for this manuscript, take responsibility for the integrity of the work as a whole, and have given final approval to the version to be published. All authors had full access to all of the data in this study and take complete responsibility for the integrity of the data and accuracy of the data analysis.
Disclosures
Tushar Garimella was an employee of Bristol-Myers Squibb at the time the studies were conducted and may be a shareholder. Xiaoli You was an employee of Bristol-Myers Squibb at the time the studies were conducted and may be a shareholder. Reena Wang was an employee of Bristol-Myers Squibb at the time the studies were conducted and may be a shareholder. Shu-Pang Huang was an employee of Bristol-Myers Squibb at the time the studies were conducted and may be a shareholder. Hamza Kandoussi was an employee of Bristol-Myers Squibb at the time the studies were conducted and may be a shareholder. Marc Bifano was an employee of Bristol-Myers Squibb at the time the studies were conducted and may be a shareholder. Richard Bertz was an employee of Bristol-Myers Squibb at the time the studies were conducted and may be a shareholder. Timothy Eley was an employee of Bristol-Myers Squibb at the time the studies were conducted and may be a shareholder.
Compliance with Ethics Guidelines
All procedures followed were in accordance with the ethical standards of the responsible committee on human experimentation (institutional and national) and with the Helsinki Declaration of 1964, as revised in 2013. Informed consent was obtained from all patients for being included in the studies.
Open Access
This article is distributed under the terms of the Creative Commons Attribution-NonCommercial 4.0 International License (http://creativecommons.org/licenses/by-nc/4.0/), which permits any noncommercial use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
Footnotes
Enhanced content
To view enhanced content for this article go to http://www.medengine.com/Redeem/FED6F0607B3FA4B5.
References
- 1.World Health Organization. Guidelines for the screening, care and treatment of persons with hepatitis C infection. Geneva, Switzerland. http://www.who.int/hiv/pub/hepatitis/hepatitis-c-guidelines/en/. Accessed April 15, 2015. [PubMed]
- 2.Kiser JJ, Burton JR, Jr, Everson GT. Drug–drug interactions during antiviral therapy for chronic hepatitis C. Nat Rev Gastroenterol Hepatol. 2013;10:596–606. doi: 10.1038/nrgastro.2013.106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.McPhee F, Suzuki Y, Toyota J, et al. High sustained virologic response to daclatasvir plus asunaprevir in elderly and cirrhotic patients with hepatitis C virus genotype 1b without baseline NS5A polymorphisms. Adv Ther. 2015;32:637–649. doi: 10.1007/s12325-015-0221-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Poordad F, Sievert W, Mollison L, et al. Fixed-dose combination therapy with daclatasvir, asunaprevir, and beclabuvir for noncirrhotic patients with HCV genotype 1 infection. JAMA. 2015;313:1728–1735. doi: 10.1001/jama.2015.3860. [DOI] [PubMed] [Google Scholar]
- 5.Muir AJ, Poordad F, Lalezari J, et al. Daclatasvir in combination with asunaprevir and beclabuvir for hepatitis C virus genotype 1 infection with compensated cirrhosis. JAMA. 2015;313:1736–1744. doi: 10.1001/jama.2015.3868. [DOI] [PubMed] [Google Scholar]
- 6.Poordad F, Schiff ER, Vierling JM, et al. Daclatasvir with sofosbuvir and ribavirin for hepatitis C virus infection with advanced cirrhosis or post-liver transplant recurrence. Hepatology. 2016;63:1493–1505. doi: 10.1002/hep.28446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wyles DL, Ruane PJ, Sulkowski MS, et al. Daclatasvir plus sofosbuvir for HCV in patients coinfected with HIV-1. N Engl J Med. 2015;373:714–725. doi: 10.1056/NEJMoa1503153. [DOI] [PubMed] [Google Scholar]
- 8.Leroy V, Angus P, Bronowicki JP, et al. Daclatasvir, sofosbuvir, and ribavirin for hepatitis C virus genotype 3 and advanced liver disease: a randomized phase III study (ALLY-3 +) Hepatology. 2016;63:1430–1441. doi: 10.1002/hep.28473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kumada H, Suzuki Y, Ikeda K, et al. Daclatasvir plus asunaprevir for chronic HCV genotype 1b infection. Hepatology. 2014;59:2083–2091. doi: 10.1002/hep.27113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Manns M, Pol S, Jacobson I, et al. All-oral dual therapy with daclatasvir and asunaprevir in patients with HCV genotype 1b infection: phase 3 study results. J Hepatol. 2014;60(Suppl 1):S524–S525. doi: 10.1016/S0168-8278(14)61461-6. [DOI] [Google Scholar]
- 11.Bristol-Myers Squibb. Daklinza (daclatasvir) prescribing information 2016.
- 12.Bifano M, Adamczyk R, Hwang C, Kandoussi H, Marion A, Bertz RJ. An open-label investigation into drug–drug interactions between multiple doses of daclatasvir and single-dose cyclosporine or tacrolimus in healthy subjects. Clin Drug Investig. 2015;35:281–289. doi: 10.1007/s40261-015-0279-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bifano M, Sevinsky H, Stonier M, Jiang H, Bertz RJ. Daclatasvir, an HCV NS5A replication complex inhibitor, has minimal effect on pharmacokinetics of midazolam, a sensitive probe for cytochrome P450 3A4. Rev Antivir Ther Infect Dis. 2013;6:17. [Google Scholar]
- 14.Bertz R. Bristol-Myers Squibb HCV full development portfolio overview. Plenary presentation presented at the 14th International Workshop on Clinical Pharmacology, April 22–24, 2014, Amsterdam, The Netherlands.
- 15.Garimella T, Adamczyk R, Stonier M, et al. Effect of steady-state daclatasvir plus asunaprevir on the single-dose pharmacokinetics of the P-glycoprotein substrate digoxin in healthy adult subjects. Presented at ID Week 2014, October 8–12, 2014; Philadelphia, USA.
- 16.Eley T, Sevinsky H, Huang SP, et al. The pharmacokinetics of daclatasvir and asunaprevir administered in combination in studies in healthy subjects and patients infected with hepatitis C virus. Clin Drug Investig. 2014;34:661–671. doi: 10.1007/s40261-014-0219-9. [DOI] [PubMed] [Google Scholar]
- 17.Eley T, Garimella T, Li W, Bertz RJ. Asunaprevir: a review of preclinical and clinical pharmacokinetics and drug–drug interactions. Clin Pharmacokinet. 2015;54:1205–1222. doi: 10.1007/s40262-015-0299-6. [DOI] [PubMed] [Google Scholar]
- 18.Wang X, Li W, Huang S, et al. Evaluation of pharmacokinetic drug–drug interaction (DDI) between BMS-791325, an NS5B non-nucleoside polymerase inhibitor, daclatasvir and asunaprevir in triple combination in HCV genotype 1-infected patients [Abstract] J Hepatol. 2013;58(Suppl 1):S184. doi: 10.1016/S0168-8278(13)60453-5. [DOI] [Google Scholar]
- 19.Eley T, You X, Huang S, et al. Evaluation of drug interaction potential between daclatasvir and sofosbuvir. Rev Antivir Ther Infect Dis. 2013;6:16. [Google Scholar]
- 20.Janssen Therapeutics. OLYSIO™ (simeprevir) Summary of product characteristics. 2014. http://www.ema.europa.eu/docs/en_GB/document_library/EPAR_-_Product_Information/human/002777/WC500167867.pdf. Accessed June 2016.
- 21.Platt L, Easterbrook P, Gower E, et al. Prevalence and burden of HCV co-infection in people living with HIV: a global systematic review and meta-analysis. Lancet Infect Dis. 2016;16:797–808. doi: 10.1016/S1473-3099(15)00485-5. [DOI] [PubMed] [Google Scholar]
- 22.Tseng A, Foisy M. Important drug–drug interactions in HIV-infected persons on antiretroviral therapy: an update on new interactions between HIV and non-HIV drugs. Curr Infect Dis Rep. 2012;14:67–82. doi: 10.1007/s11908-011-0229-1. [DOI] [PubMed] [Google Scholar]
- 23.Bifano M, Hwang C, Oosterhuis B, et al. Assessment of pharmacokinetic interactions of the HCV NS5A replication complex inhibitor daclatasvir with antiretroviral agents: ritonavir-boosted atazanavir, efavirenz and tenofovir. Antivir Ther. 2013;18:931–940. doi: 10.3851/IMP2674. [DOI] [PubMed] [Google Scholar]
- 24.Gandhi Y, Adamczyk R, Wang R, et al. Assessment of drug–drug interactions between daclatasvir and darunavir/ritonavir or lopinavir/ritonavir. Presented at the 16th International Workshop on Clinical Pharmacology of HIV and Hepatitis Therapy 015, May 26–28, 2015, Washington DC, USA.
- 25.Barrail-Tran A, Vincent C, Furlan V, et al. Raltegravir pharmacokinetics in patients on asunaprevir-daclatasvir. Antimicrob Agents Chemother. 2015;59:7903–7905. doi: 10.1128/AAC.01603-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Song I, Jerva F, Zong J, et al. Evaluation of drug interactions between dolutegravir and daclatasvir in healthy subjects. Abstract 79 presented at the 16th International Workshop on Clinical Pharmacology of HIV and Hepatitis Therapy, May 26–28, 2015, Washington, DC.
- 27.Nelson PK, Mathers BM, Cowie B, et al. Global epidemiology of hepatitis B and hepatitis C in people who inject drugs: results of systematic reviews. Lancet. 2011;378:571–583. doi: 10.1016/S0140-6736(11)61097-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Garimella T, Wang R, Luo WL, et al. Assessment of drug–drug interactions between daclatasvir and methadone or buprenorphine-naloxone. Antimicrob Agents Chemother. 2015;59:5503–5510. doi: 10.1128/AAC.00478-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Zacks S, Beavers K, Theodore D, et al. Social stigmatization and hepatitis C virus infection. J Clin Gastroenterol. 2006;40:220–224. doi: 10.1097/00004836-200603000-00009. [DOI] [PubMed] [Google Scholar]
- 30.Gleason OC, Yates WR, Philipsen MA. Major depressive disorder in hepatitis C: an open-label trial of escitalopram. Prim Care Companion J Clin Psychiatry. 2005;7:225–230. doi: 10.4088/PCC.v07n0503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Genentech USA I. COPEGUS™ (ribavirin) package insert. 2013.
- 32.Bifano M, Sevinsky H, Hwang C, et al. Effect of the coadministration of daclatasvir on the pharmacokinetics of a combined oral contraceptive containing ethinyl estradiol and norgestimate. Antivir Ther. 2014;19:511–519. doi: 10.3851/IMP2718. [DOI] [PubMed] [Google Scholar]
- 33.American Association for the Study of Liver Diseases and the Infectious Diseases Society of America. Recommendations for testing, managing, and treating hepatitis C. 2016. http://www.hcvguidelines.org. Accessed Sept 5, 2016. [DOI] [PMC free article] [PubMed]
- 34.Gilead Sciences Inc. Sovaldi (sofosbuvir) US prescribing information. 2015.
- 35.Bifano M, Connolly S, Hwang C, et al. The effect of co-administration of the proton-pump inhibitor omeprazole on the pharmacokinetics of daclatasvir in healthy subjects. Presented at the 48th Annual Meeting of the European Association for the Study of the Liver, April 24–28, 2013, Amsterdam, The Netherlands.
- 36.Gurley BJ, Fifer EK, Gardner Z. Pharmacokinetic herb–drug interactions (part 2): drug interactions involving popular botanical dietary supplements and their clinical relevance. Planta Med. 2012;78:1490–1514. doi: 10.1055/s-0031-1298331. [DOI] [PubMed] [Google Scholar]
- 37.Malhotra S, Bailey DG, Paine MF, Watkins PB. Seville orange juice-felodipine interaction: comparison with dilute grapefruit juice and involvement of furocoumarins. Clin Pharmacol Ther. 2001;69:14–23. doi: 10.1067/mcp.2001.113185. [DOI] [PubMed] [Google Scholar]
- 38.Matsuda H, Kinoshita K, Sumida A, et al. Taurine modulates induction of cytochrome P450 3A4 mRNA by rifampicin in the HepG2 cell line. Biochim Biophys Acta. 2002;1593:93–98. doi: 10.1016/S0167-4889(02)00345-2. [DOI] [PubMed] [Google Scholar]
- 39.Höner Zu Siederdissen C, Maasoumy B, Marra F, et al. Drug–drug interactions with novel all oral interferon-free antiviral agents in a large real-world cohort. Clin Infect Dis. 2016;62:561–567. doi: 10.1093/cid/civ973. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.