

Abstract
AIM
To investigate the changes in microbiota in feces of patients with ulcerative colitis (UC) and pouchitis using genomic technology.
METHODS
Fecal samples were obtained from UC patients with or without an ileal pouch-anal anastomosis (IPAA) procedure, as well as healthy controls. The touchdown polymerase chain reaction technique was used to amplify the whole V3 region of the 16S rRNA gene, which was transcribed from DNA extracted from fecal samples. Denaturing gradient gel electrophoresis was used to separate the amplicons. The band profiles and similarity indices were analyzed digitally. The predominant microbiota in different groups was confirmed by sequencing the 16S rRNA gene.
RESULTS
Microbial biodiversity in the healthy controls was significantly higher compared with the UC groups (P < 0.001) and IPAA groups (P < 0.001). Compared with healthy controls, the UC patients in remission and those in the mildly active stage, the predominant species in patients with moderately and severely active UC changed obviously. In addition, the proportion of the dominant microbiota, which was negatively correlated with the disease activity of UC (r = -6.591, P < 0.01), was decreased in pouchitis patients. The numbers of two types of bacteria, Faecalibacterium prausnitzii and Eubacterium rectale, were reduced in UC. Patients with pouchitis had an altered microbiota composition compared with UC patients. The microbiota from pouchitis patients was less diverse than that from severely active UC patients. Sequencing results showed that similar microbiota, such as Clostridium perfringens, were shared in both UC and pouchitis.
CONCLUSION
Less diverse fecal microbiota was present in patients with UC and pouchitis. Increased C. perfringens in feces suggest its role in the exacerbation of UC and pouchitis.
Keywords: Pouchitis, Intestinal flora, Ulcerative colitis, Disease activity index, Ileal pouch-anal anastomosis
Core tip: Dysbiosis in pouchitis might be similar to that observed in ulcerative colitis (UC). This study aimed to determine the altered microflora in patients with UC and pouchitis, and to investigate the relationship between them. We demonstrated the reduced biodiversity of the fecal microbiota in UC and pouchitis patients. The altered composition of the intestinal microbiota in UC and pouchitis included decreased numbers of two bacteria commonly observed in UC, and higher levels of Clostridium perfringens in both UC and pouchitis. The increase of this bacterium in feces suggested that it plays a role in exacerbating UC and pouchitis.
INTRODUCTION
One of the most common complications in ulcerative colitis (UC) patients who undergo ileal pouch-anal anastomosis (IPAA) surgery is pouchitis[1]. Interestingly, it is rarely seen in postoperative patients suffering from familial adenomatous polyposis. The gut microbiome plays a vital role in UC[2]. Antibiotics and probiotics are used to treat and prevent pouchitis[3]. The gut microbiome might play a vital role in the pathogenesis of UC[4].
However, direct evidence of the role of microflora in pathogenesis of pouchitis is lacking. Studies have shown variation in the microbiota in pouchitis and healthy controls; however, based on different culture methods and molecular biology techniques, no consensus was available[3]. Johnson et al[5] and Lim et al[6] showed no differences between pouchitis and no pouchitis (NP) groups. Some studies have suggested a reduction in bacterial diversity in pouchitis but not dysbiosis[7]. Other studies revealed an increase in bacterial diversity in pouchitis[8], such as increased numbers of Clostridium and Eubacterium[9], while others showed less Enterococcaceae in pouchitis[10]. The findings of the most recent study revealed that disorders caused by protective and harmful bacteria are associated with pouch inflammation[11]. The emergence of Ruminococcus gnavus (R. gnavus), Bacteriodes vulgatus and Clostridium perfringens (C. perfringens) and deficiency of Blautia and Roseburia in patients with UC before IPAA is closely related to pouchitis[12].
Denaturing gradient gel electrophoresis (DGGE) was reported to be useful to analyze changes in the composition of the intestinal microbiota[2]. We hypothesized that dysbiosis occurring in the pouch might be similar to that observed in UC. Thus, we determined the altered microflora in pouchitis and UC patients, and investigated the relationship between them.
MATERIALS AND METHODS
Patients and fecal samples
Patients who underwent IPAA for UC were recruited. Pouchitis was diagnosed based on symptoms, endoscopy and histology of the pouch. Patients underwent pouch endoscopy and biopsy. Physicians recorded clinical data, the pouch appearance and pathological manifestations based on the pouchitis disease activity index (PDAI)[13]. Antibiotic or other drug therapy was stopped to prevent variations in the microbiome 4 wk before collecting the fecal sample. A limited number of patients with pouches were excluded from the study because of antibiotic or probiotic usage for pouchitis or severe concomitant disease.
According to the PDAI, patients with IPAA were divided into two groups: NP, PDAI < 7 points (n = 11) and pouchitis, PDAI ≥ 7 points (n = 8). Matched fecal samples were obtained from healthy controls (n = 16) and from 41 UC patients who did not undergo IPAA. All the UC patients without a pouch underwent endoscopy. The Mayo scoring system for assessment of ulcerative colitis activity was employed to divide patients with UC into the remission group (n = 10), mild activity group (n = 11), moderate activity group (n = 10) and severe activity group (n = 10).
All fecal samples were collected at the hospital and preserved at 4 °C. Upon arrival at the laboratory, the samples were frozen at -80 °C within 12 h. This study was reviewed and approved by the Tianjin Medical University General Hospital Ethical Committee (China). Patient data are summarized in Table 1.
Table 1.
Demographic and clinical characteristics of the subjects
| Healthy controls |
UC (n = 41) |
Pouch (n = 19) |
|||||
| Remission | Mild | Moderate | Severe | Pouchitis | NP | ||
| Number of patients | 16 | 10 | 11 | 10 | 10 | 8 | 11 |
| Sex, n, M/F | 9/7 | 5/5 | 7/4 | 5/5 | 6/4 | 5/3 | 5/6 |
| Age (yr) | 46.2 ± 10.5 | 41.8 ± 9.2 | 43.9 ± 10.5 | 46 ± 8.2 | 40.8 ± 11.3 | 44.9 ± 15.6 | 47.8 ± 13.2 |
| UC duration (yr) | NA | 5.0 ± 1.6 | 7.0 ± 0.9 | 5.2 ± 1.7 | 4.0 ± 2.7 | NA | NA |
| Pouch duration (yr) | NA | NA | NA | NA | NA | 2.8 ± 1.5 | 3.9 ± 2.2 |
| BMI (kg/m2) | 24.5 ± 2.3 | 25.2 ± 1.6 | 24.8 ± 3.5 | 23.9 ± 2.7 | 24.4 ± 3.1 | 24.9 ± 2.2 | 24.3 ± 3.5 |
| Mayo score | NA | ≤ 2 | 4.2 ± 0.7 | 8.5 ± 1.2 | 11.5 ± 0.3 | ≥ 7 | < 7 |
| Age at colectomy | NA | NA | NA | NA | NA | 40.6 ± 12.9 | 42.4 ± 9.22 |
| Standard medication (%) | NA | 0 (0) | 5 (46.0) | 8 (80.0) | 10 (100) | 8 (100) | 3 (27.0) |
| Smoking (% at recruitment) | 10 (62.5) | 4 (40.0) | 3 (27.3) | 2 (20.0) | 3 (30.0) | 4 (50.0) | 5 (45.5) |
| Previous number of episodes of pouchitis (%) | NA | NA | NA | NA | NA | 4 (50.0) | 3 (27.0) |
| Number of patients with chronic pouchitis (%) | NA | NA | NA | NA | NA | 3 (37.5) | 2 (18.0) |
| Secondary causes of pouchitis (%) | NA | NA | NA | NA | NA | 2 (25.0) | 2 (19.0) |
UC: Ulcerative colitis; NA: Not available; Pouch: Ileal pouch established during ileal pouch-anal anastomosis surgery; pouchitis: Inflammation of ileal pouch.
Fecal DNA extraction
A Fecal DNA kit (Aidlab Biotechnologies Co, Ltd, Beijing, China) was used to isolate DNA from frozen feces individually, following the manufacturer’s guidelines and as previously described[14]. Following 1% agarose gel electrophoresis, the eluted DNA was quantified on a NanoDrop 2000 Spectrophotometer.
PCR amplification
The genomic DNA and universal primers including forward and reverse primers (AuGCT DNA technologies, Beijing, China) were employed to amplify the whole fragment V3 region of bacterial 16S rRNA gene.
After 15 cycles of thermocycling on a PCR system (Bio-Rad, Hercules, CA, United States) the amplified product was verified by 2% agarose electrophoresis. The amplified DNA was quantified on a NanoDrop 2000 Spectrophotometer, and recorded by a DH2000 gel imaging analysis.
DGGE for amplified 16S rRNA gene
DGGE was chosen to separate PCR amplicons according to the rules of Muyzer et al[15], with some modifications. A 10% polyacrylamide combined with Tris-acetate-EDTA (TAE) buffer was used for polyacrylamide gels with a denaturing gradient ranging from 30% to 70%. A stacking gel was added before polymerization of the denaturing gel, followed by appropriate comb insertion. Electrophoresis was performed at 200 V for 5 min and 85 V for 16 h in 0.5 × TAE buffer subsequently at a constant 60 °C. After staining with AgNO3[16], the gels were desiccated overnight at 60 °C.
Digital processing of DGGE profiles
Following the manufacturer’s instructions, DGGE profiles were analyzed digitally using Quantity One-4.6.5 in the UNIVERSAL HOODII Gel Imaging System (Beijing, China). After normalizing the gels according to the results for the healthy controls, the band in each sample was marked by the software, and manual corrections were conducted. The number of DGGE bands was shown as the mean ± SD. Based on the gray value, Dice similarity and UPGMA tree analyses were conducted using Quantity One software. Canoco software was used to conduct principal component analysis (PCA).
DGGE band extraction and sequencing
Based on the digital results, the bands distinguishing the groups were excised from the gel, purified and sequenced. The gel slice, in 15 μL of TE buffer, was heated at 65 °C for 10 min to elute the DNA from the gel. The DNA solution was amplified using the universal V3 primers F357+ a GC clamp and R518. DGGE gel expansion facilitated the purification of the bands. DGGE with an adjusted gradient of 32 was used to check the amplicons, which were excised at least three times until a single band was obtained. The DGGE was repeated to purify the PCR product before sequencing. In the final round, the amplicons were analyzed with the original sample profiles from which they were excised and analyzed visually for purification of the correct bands.
When the purified bands matched with the targeted bands, the amplicons sequenced using an ABI Prism system and primers R518 and F357 (without the clamp). BioNumerics software was used to analyze the sequences. BLAST homology searches were performed against the GenBank DNA database. According to BLAST results, the sequences of phylogenetic neighbor species, whose similarities were up to 90%, were included for reference in the cluster analysis using multiple sequence alignments. The purified band sequences were allocated to the most probable species according to the average linking method.
Statistical analysis
Quantity One software was used for Dice similarity analysis and UPGMA tree analysis. DGGE strips data are presented as mean ± SD. The Shapiro-Wilk test was used to test the normality of the band number and Dice analysis data. The content of each sample was similar. The homogeneity of variance was robust and highly efficient. The band number of DGGE strips showed a normal distribution, unlike the Dice analysis results. Therefore, the three groups were tested using a Student-Newman-Keuls test. The Bonferroni test was chosen to compare DGGE strips and band numbers among the five groups. An extension t test after non-parametric Kruskal-Wallis H test was employed to compare the Dice analysis results among multiple groups. The correlation between disease activity and bacterial count was assessed using the Spearman correlation coefficient. SPSS 19.0 software was employed to analyze all the data. Two-tailed tests were used in all analysis and P values of ≤ 0.05 were considered statistically significant.
RESULTS
Bacteria in fecal samples from UC patients
The demographic details of the study patients are shown in Table 1. DNA extracts from the fecal samples from different individuals presented variable number of bands after PCR-DGGE analysis. A band representing identical or similar sequences of the V3 regions of the 16S rRNA gene was observed, reflecting the dominant bacterial communities in the fecal samples.
Examination of digital DGGE profiles from healthy controls showed relative stability among different individuals (Figure 1). Profiles from UC patients shown in Figure 2 suggested significant variation in the position and number of bands compared with the healthy controls. The number of bands, which reflected the diverse microbiota, was 17 ± 3 in the 16 healthy controls and 13 ± 3 in the 41 UC patients (P = 0.001). Differences were also seen among the subgroups of UC patients (Figure 3). These results reveal that the number of predominant microbiota was negatively correlated with the Mayo classification (r = -6.591, P < 0.01). The Kruskal-Wallis H test showed greater similarity between groups than within the groups, which revealed variation in the predominant microbiota with clinical status (Table 2). UPGMA tree analysis showed similar results (Figure 4). PCA analysis of healthy and UC groups revealed large differences in the predominant species among the control group, remission and mild group, and the moderate and severe group (Figure 5).
Figure 1.
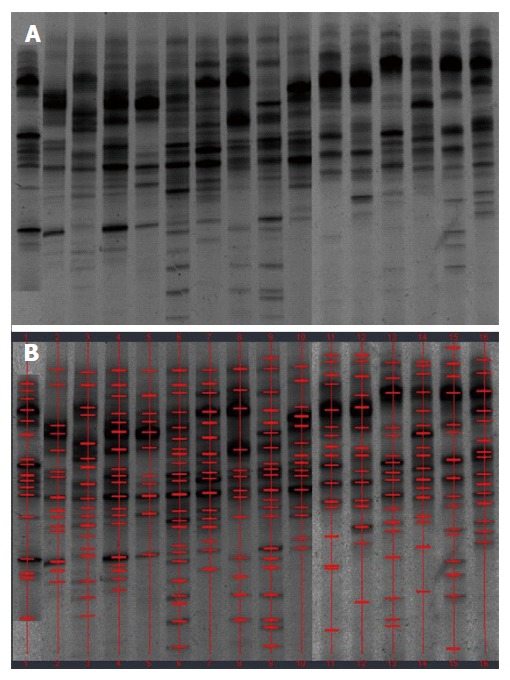
Denaturing gradient gel electrophoresis profiles of fecal samples from healthy controls. A: Denaturing gradient gel electrophoresis (DGGE) profiles; B: Marked DGGE profiles. DGGE bands showed relative stability among different individuals.
Figure 2.
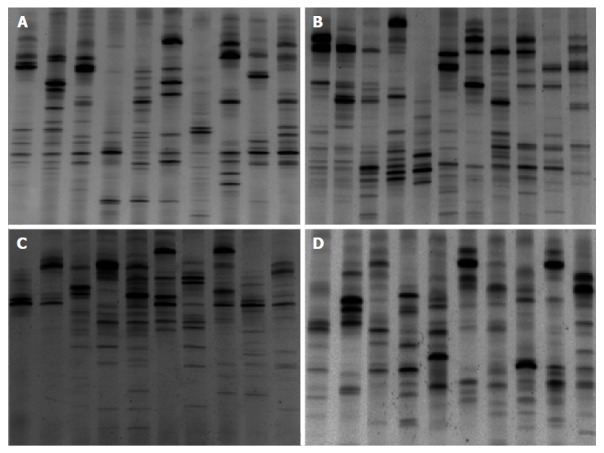
Denaturing gradient gel electrophoresis profiles showed microbial biodiversity in different ulcerative colitis groups. A: Ulcerative colitis (UC) patients in remission; B: UC patients in the mildly active stage; C: UC patients in the moderately active stage; D: UC patients in the severely active stage.
Figure 3.
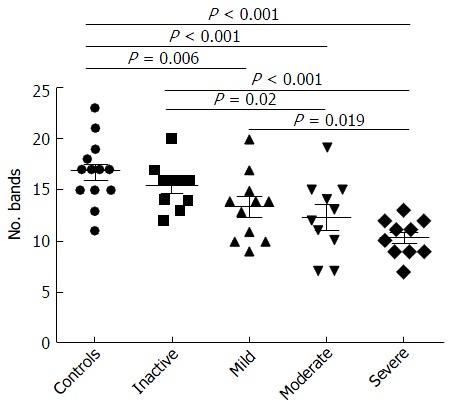
Number of bands in denaturing gradient gel electrophoresis profiles of samples obtained from 41 ulcerative colitis patients. The number of bands decreased significantly from healthy controls to severe ulcerative colitis.
Table 2.
Dice analysis of healthy control and ulcerative colitis subgroups
| Group | Control | Remission | Mild | Moderate | Severe |
| Control | (49.79 ± 11.24)%a | (35.32 ± 14.86)% | (30.13 ± 11.23)% | (31.98 ± 16.48)% | (28.18 ± 14.99)% |
| Remission | (42.89 ± 18.29)%a | (32.79 ± 13.68)% | (30.22 ± 15.28)% | (26.28 ± 13.94)% | |
| Mild | (41.83 ± 16.38)%a | (29.89 ± 13.10)% | (28.31 ± 18.39)% | ||
| Moderate | (43.45 ± 21.32)%a | (28.88 ± 13.69)% | |||
| Severe | (37.12 ± 19.98)%a |
P < 0.05; result from similarity in the same group vs similarity in different groups.
Figure 4.
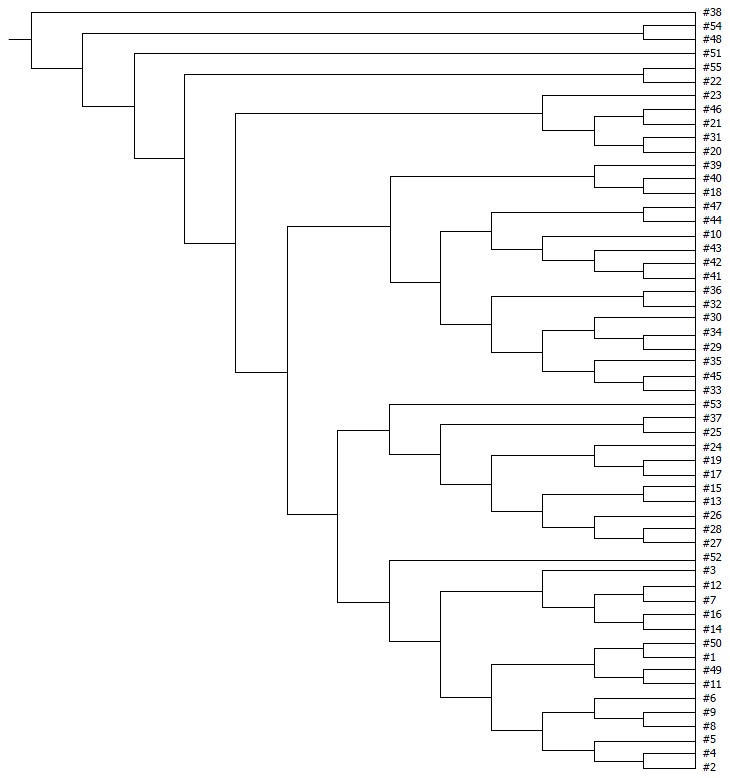
UPGMA tree analysis of healthy controls and ulcerative colitis patients at different stages. 1-16: Healthy controls; 17-26: Ulcerative colitis (UC) patients in remission; 27-37: UC patients in the mildly active stage; 38-47: UC patients in the moderately active stage; 48-57: UC patients in the severely active stage. UPGMA tree analysis showed a significant difference among groups of healthy controls and UC patients at different stages.
Figure 5.
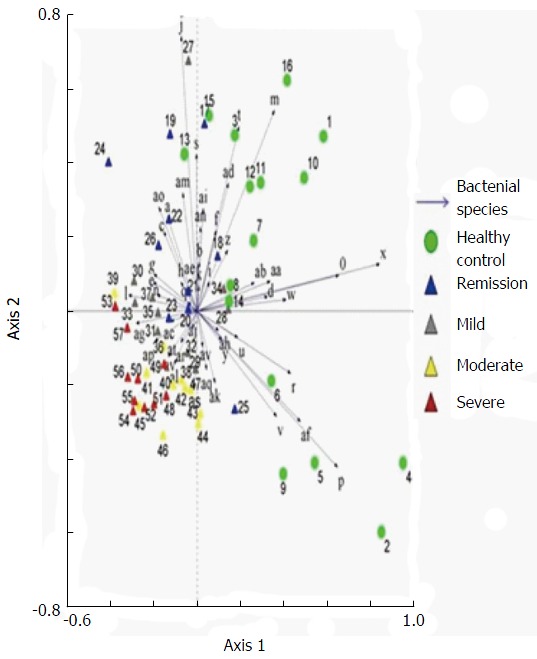
Principal component analysis of denaturing gradient gel electrophoresis microbial profiles in fecal samples of healthy controls and ulcerative colitis patients at different stages. Clustering of similar microbial profiles showed systematic differences among different groups.
Sequencing results after purification (based on digital DGGE profiles) showed the presence of a greater number of C. perfringens and fewer Faecalibacterium prausnitzii (F. prausnitzii) and Eubacterium rectale (E. rectal) in UC group compared with the control group. C. perfringens was present predominantly in severe UC.
Bacteria in fecal samples of pouchitis patients
Significant changes occurred in the position and number of bands from patients with pouchitis when compared with NP and healthy controls (Figure 6). Differences in the number of bands in the controls (17 ± 3 bands), NP (11 ± 3 bands) and pouchitis (8 ± 2 bands) are shown in Figure 7 (ANOVA test). A Bonferroni test showed greater similarity between groups than within groups, suggesting differences in the predominant species in the healthy controls, NP and pouchitis groups (Table 3). These results suggested that patients with pouchitis had an altered microbiota composition compared with healthy individuals. UPGMA tree analysis showed similar results (Figure 8). PCA analysis of healthy control and pouchitis groups revealed great variation in the predominant species in pouchitis compared with non-pouchitis and healthy controls, which also differed from each other (Figure 9).
Figure 6.
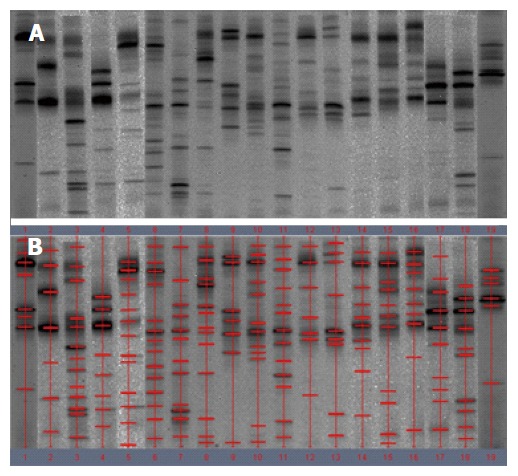
Denaturing gradient gel electrophoresis profiles of fecal samples from patients with pouchitis. A: Denaturing gradient gel electrophoresis (DGGE) profiles; B: Marked DGGE profiles. DGGE bands revealed the relative stability of the microbiota in pouchitis group.
Figure 7.
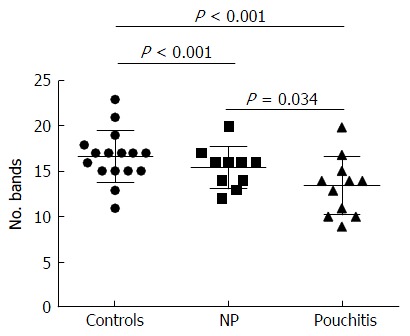
Number of bands in denaturing gradient gel electrophoresis profiles of samples obtained from patients receiving surgery. The number of bands was reduced significantly in pouchitis compared with the control group and the no pouchitis (NP) group.
Table 3.
Dice analysis of healthy control and pouchitis subgroups
| Group | Control | Pouchitis | NP |
| Control | (49.79 ± 11.24)%a | (25.33 ± 11.13)% | (28.86 ± 14.23)% |
| Pouchitis | (35.43 ± 13.30)%a | (20.87 ± 12.31)% | |
| NP | (35.39 ± 10.80)%a |
P < 0.05; result from similarity in the same group vs similarity in different groups. NP: No pouchitis.
Figure 8.
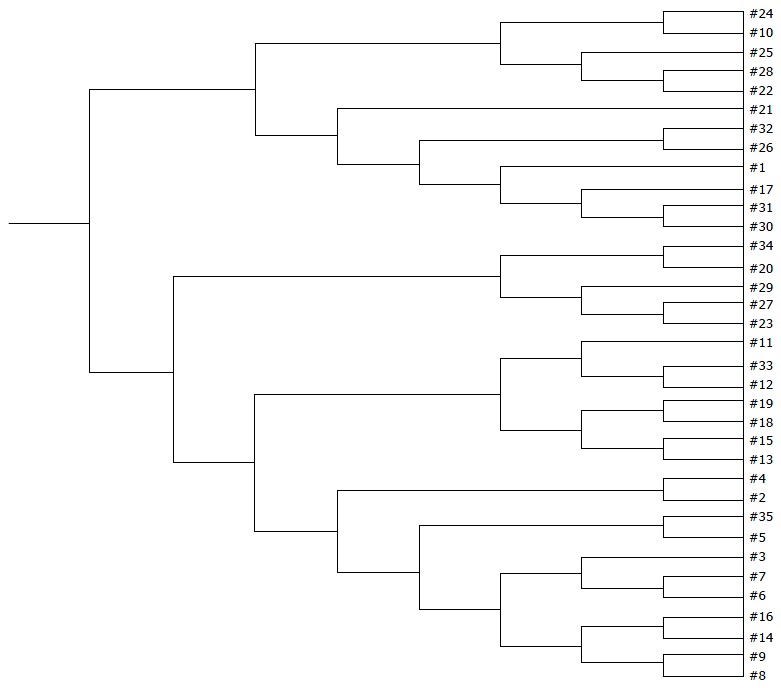
UPGMA tree analysis of healthy controls and postoperative patients. 1-16: Healthy controls; 17-27: UC patients without pouchitis after IPAA; 28-35: Patients with pouchitis. UPGMA tree analysis showed a significant difference among the three groups.
Figure 9.
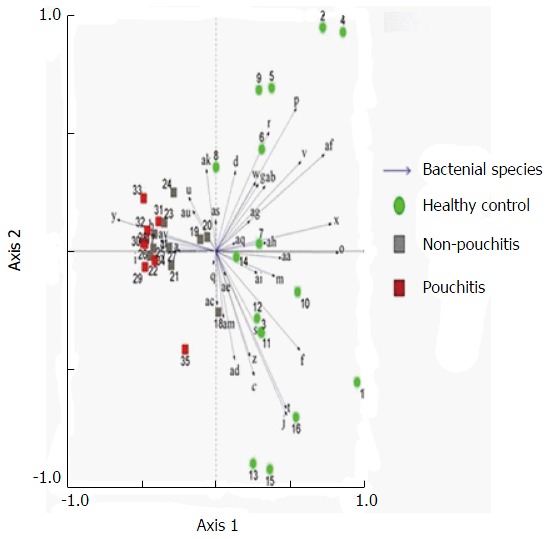
Principal component analysis of denaturing gradient gel electrophoresis microbial profiles in fecal samples of healthy controls and patients with pouch (with or without pouchitis). Clustering of similar microbial profiles showed significant differences among the three groups.
Sequencing results after purification (based on digital DGGE profiles) showed fewer E. rectale and more C. perfringens in the pouchitis group compared with the NP and control groups.
As shown in Table 4, the DGGE profiles in pouchitis patients varied significantly from UC in remission to the severe state, while the NP group of patients differed from UC in remission. The results showed that patients with a pouch have an altered microbiota diversity compared with UC patients (Figure 10). The diversity of the microbiota from pouchitis patients was lower than that in severe UC patients. A normal pouch can be can be present along with mild, moderate and severe UC. The sequencing results for the UC and pouchitis groups showed that they shared a similar microbiota, such as C. perfringens.
Table 4.
Bacterial diversity comparison
| Healthy controls |
UC |
Pouch |
|||||
| Remission | Mild | Moderate | Severe | NP | Pouchitis | ||
| Healthy controls | - | 0.298 | 0.006a | 0.001a | 0.000a | 0.000a | 0.000a |
| Remission UC | 0.298 | - | 0.113 | 0.020a | 0.000a | 0.014a | 0.003a |
| Mild UC | 0.006a | 0.113 | - | 0.404 | 0.019a | 0.007a | 0.125 |
| Moderate UC | 0.001a | 0.020a | 0.404 | - | 0.128 | 0.009a | 0.448 |
| Severe UC | 0.000a | 0.000a | 0.019a | 0.128 | - | 0.019a | 0.496 |
| NP: PDAI < 7 | 0.000a | 0.014a | 0.007a | 0.009a | 0.019a | - | 0.034a |
| Pouchitis: PDAI ≥ 7 | 0.000a | 0.003a | 0.005a | 0.448 | 0.496 | 0.034a | - |
P < 0.05. NP: No pouchitis; UC: Ulcerative colitis; Pouch: Ileal pouch established during ileal pouch-anal anastomosis surgery; pouchitis: Inflammation of ileal pouch; PDAI: Pouchitis Disease Activity Index.
Figure 10.
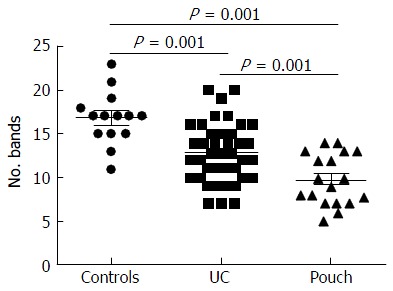
Number of bands in denaturing gradient gel electrophoresis profiles of samples obtained from all subjects. The number of bands was reduced significantly in pouch group (UC patients who underwent IPAA, with or without pouchitis) compared with the control group and the UC group. UC: Ulcerative colitis.
DISCUSSION
In this study, we focused on UC patients after IPAA surgery, and specifically compared patients developing pouch inflammation with those without surgery. Our digital analysis of stool samples showed that the predominant microbiota in UC patients was reduced compared with the healthy group. Sequence analysis showed more C. perfringens and less F. prausnitzii and E. rectale in the UC group. Levels of E. rectale (a butyrate-producing bacteria) were significantly reduced on UC mucosa[17], and had high age dependence. High clinical activity indices, as well as sigmoidoscopy scores, were associated with E. rectale[18]. Vermeiren demonstrated fewer E. rectale shown in UC patients via a dynamic gut model of the mucin environment[17]. C. perfringens, a Gram-positive, anaerobic, spore-forming bacillus, is found in the intestinal contents of both animals and humans[19]. C. perfringens is an intestinal commensal organism as well as a pathogen, for example, via production of toxins that damage the host tissues[20]. C. perfringens exerts proteolytic and mucinase activity, both of which could mediate the pathogenesis of inflammatory bowel disease (IBD)[21]. C. perfringens found in IBD patients, which is thought to be an important factor during the immunopathogenesis of IBD, could result from dysbiosis[22]. Falk showed that there were more C. perfringens in pouchitis patients[24]. Another study found that 21% of the total bacteria in colonic specimens collected from patients with UC belonged to clostridia of clusters I, II and XI, which were not found in the control groups[23]. We should keep in mind that ileum tissues of UC patients were the origin of the present pouches. F. prausnitzii is the most host species-specific microbe in the study of IBD. Sokol et al[25] studied a small group of 17 UC patients and reported a reduction in F. prausnitzii in active UC patients. A strong anti-inflammatory effect of F. prausnitzii has been demonstrated both in vitro and in vivo[26]. Machiels et al[27] observed a significant inverse correlation between disease activity and numbers of F. prausnitzii, indicating that a deficiency of this species provokes or enhance inflammation. F. prausnitzii produces high concentrations of butyrate, a vital energy source for colonocytes, which also prevents mucosal atrophy. Consequently, butyrate improves the mucosal barrier function of the colon. Furthermore, butyrate exhibits immunomodulatory and anti-inflammatory effects by downregulating pro-inflammatory cytokines[28]. Our data showed that bacterial biodiversity in feces decreased distinctly with the severity of Mayo classification compared with healthy controls. Studies have demonstrated that the mucosal biopsies from patients with active Crohn’s disease (CD) or active UC showed reduced bacterial diversity after analysis of 16S rRNA genes[29]. Furthermore, Manichanh et al[30] reported a reduction in the phylum Firmicutes in CD in remission using an extensive metagenomic analysis. Consistent with previous studies, our results confirmed that bacterial diversity was reduced in fecal samples from UC patients at different grades, and demonstrated changes in the microbial composition among subgroups in UC. The decreased biodiversity in UC might disrupt the stability of gut ecosystem. The results revealed that changes in the predominant bacteria were consistent with the Mayo classification. Therefore, we suggest that the fecal microflora in UC patients is reduced in aggravated intestinal lesions. A previous study by Wills et al[31] reported patient-specific shifts in microbial composition in UC patients showing altered pathological activity over time. The changes were more pronounced in CD cases than in UC patients, suggesting their role in the inflammatory process of UC.
By contrast, the number of bands on the DGGE profiles from pouchitis patients varied between UC and healthy controls. We showed a decrease in bacterial diversity and reduced abundance of predominant bacteria in UC pouches. R. gnavus infection, especially occurring as the predominant microbiota before colectomy, was shown to increase the risk of pouchitis 1 year after IPAA[12]. R. gnavus produces the bacteriocin ruminococcin A, which inhibits the growth of phylogenetically-related species and various bifidobacterial and clostridial species[32]. Ruminococcin A also degrades intestinal mucin[33] and induced α-galactosidase and β-glucuronidase activity in vitro[34]. β-glucuronidase activity generates toxic metabolites in the colon, which provoke local inflammation. Png et al[35] observed an increase in mucolytic bacteria, including R. gnavus, in biopsies of patients with UC and CD. Our data are supported by reports from several groups that analyzed fecal or biopsy samples using different DNA-based methods[7], further confirming the association between changes in microbiota and pouchitis. By contrast, the variability in endogenous factors, including secretion of mucins, defensins, cytokines and immunoglobulins, might also affect the composition of predominant bacterial species in UC and pouchitis. However, data about the effects of these secretions effects on the variability of UC is limited. Studies involving UC have revealed that a high percentage of fecal bacteria (about 30%-40% of dominant species) belong to unusual genera in healthy populations[36]. Hypothetically, the decreased number of stable commensals in individuals that are genetically susceptible to pouchitis would break this first line of natural defense against potentially invasive bacteria, resulting in inflammation.
In conclusion, our research demonstrated reduced biodiversity of fecal microbiota in UC and pouchitis patients. There were fewer F. prausnitzii and E. rectale in UC, more R. gnavus in pouchitis and more C. perfringens in both UC and pouchitis.
COMMENTS
Background
Pouchitis is a commonly-seen complication in patients with ulcerative colitis (UC) following ileal pouch-anal anastomosis (IPAA) procedure. The gut microbiome is considered to play a vital role in the occurrence and development of UC. In addition, antibiotics and probiotics have already been used to treat and prevent pouchitis. However, direct evidence of dysbacteriosis in pouchitis is lacking.
Research frontiers
Gut microbiota is hot topic in field of intestinal inflammation, especially in inflammatory bowel disease (IBD), where data are often conflicting.
Innovations and breakthroughs
The authors demonstrated the role of the reduced diversity and the changed composition of intestinal microbiota in the pathogenesis of pouchitis. Reduced levels of Faecalibacterium prausnitzii and Eubacterium rectal were confirmed in UC patients, and Clostridium perfringens was significantly increased in UC and pouchitis.
Applications
These findings provide new clues to better understand the pathogenesis of pouchitis in UC patients subjected to IPAA, although the findings will not be used immediately in the treatment of pouchitis.
Terminology
Pouchitis: An inflammation in the intestinal pouch, which is established following the proctocolectomy IPAA procedure for patients with UC to prevent permanent abdominal fistula. Pouchitis could have serious consequences, such as bloody diarrhea and even ileal pouch failure.
Peer-review
The manuscript definitely deals with a hot topic in the IBD and intestinal inflammation field, where data are often conflicting. The deep insight and detailed description made by the authors of the diversity of microbiota in healthy people and patients will provide new clues to better understand the pathogenesis of pouchitis following IPAA for UC.
Footnotes
Manuscript source: Invited manuscript
Specialty type: Gastroenterology and hepatology
Country of origin: China
Peer-review report classification
Grade A (Excellent): 0
Grade B (Very good): 0
Grade C (Good): C, C
Grade D (Fair): 0
Grade E (Poor): 0
Institutional review board statement: The study was reviewed and approved by Tianjin Medical University General Hospital Institutional Review Board, Tianjin 300052, China.
Conflict-of-interest statement: This work was presented as a poster at the 10th International Congress on Autoimmunity in Leipzig, Germany, April 6-10, 2016. We have no financial relationships to disclose.
Data sharing statement: No additional data are available.
Peer-review started: June 24, 2016
First decision: August 8, 2016
Article in press: August 30, 2016
P- Reviewer: Caviglia R, Pastorelli L S- Editor: Yu J L- Editor: Stewart G E- Editor: Zhang FF
References
- 1.Morgan XC, Kabakchiev B, Waldron L, Tyler AD, Tickle TL, Milgrom R, Stempak JM, Gevers D, Xavier RJ, Silverberg MS, et al. Associations between host gene expression, the mucosal microbiome, and clinical outcome in the pelvic pouch of patients with inflammatory bowel disease. Genome Biol. 2015;16:67. doi: 10.1186/s13059-015-0637-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Martinez C, Antolin M, Santos J, Torrejon A, Casellas F, Borruel N, Guarner F, Malagelada JR. Unstable composition of the fecal microbiota in ulcerative colitis during clinical remission. Am J Gastroenterol. 2008;103:643–648. doi: 10.1111/j.1572-0241.2007.01592.x. [DOI] [PubMed] [Google Scholar]
- 3.McLaughlin SD, Clark SK, Tekkis PP, Nicholls RJ, Ciclitira PJ. The bacterial pathogenesis and treatment of pouchitis. Therap Adv Gastroenterol. 2010;3:335–348. doi: 10.1177/1756283X10370611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Wu H, Shen B. Pouchitis: lessons for inflammatory bowel disease. Curr Opin Gastroenterol. 2009;25:314–322. doi: 10.1097/MOG.0b013e32832b36eb. [DOI] [PubMed] [Google Scholar]
- 5.Johnson MW, Rogers GB, Bruce KD, Lilley AK, von Herbay A, Forbes A, Ciclitira PJ, Nicholls RJ. Bacterial community diversity in cultures derived from healthy and inflamed ileal pouches after restorative proctocolectomy. Inflamm Bowel Dis. 2009;15:1803–1811. doi: 10.1002/ibd.21022. [DOI] [PubMed] [Google Scholar]
- 6.Lim M, Adams JD, Wilcox M, Finan P, Sagar P, Burke D. An assessment of bacterial dysbiosis in pouchitis using terminal restriction fragment length polymorphisms of 16S ribosomal DNA from pouch effluent microbiota. Dis Colon Rectum. 2009;52:1492–1500. doi: 10.1007/DCR.0b013e3181a7b77a. [DOI] [PubMed] [Google Scholar]
- 7.McLaughlin SD, Walker AW, Churcher C, Clark SK, Tekkis PP, Johnson MW, Parkhill J, Ciclitira PJ, Dougan G, Nicholls RJ, et al. The bacteriology of pouchitis: a molecular phylogenetic analysis using 16S rRNA gene cloning and sequencing. Ann Surg. 2010;252:90–98. doi: 10.1097/SLA.0b013e3181e3dc8b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Komanduri S, Gillevet PM, Sikaroodi M, Mutlu E, Keshavarzian A. Dysbiosis in pouchitis: evidence of unique microfloral patterns in pouch inflammation. Clin Gastroenterol Hepatol. 2007;5:352–360. doi: 10.1016/j.cgh.2007.01.001. [DOI] [PubMed] [Google Scholar]
- 9.Zella GC, Hait EJ, Glavan T, Gevers D, Ward DV, Kitts CL, Korzenik JR. Distinct microbiome in pouchitis compared to healthy pouches in ulcerative colitis and familial adenomatous polyposis. Inflamm Bowel Dis. 2011;17:1092–1100. doi: 10.1002/ibd.21460. [DOI] [PubMed] [Google Scholar]
- 10.Scarpa M, Grillo A, Faggian D, Ruffolo C, Bonello E, D’Incà R, Scarpa M, Castagliuolo I, Angriman I. Relationship between mucosa-associated microbiota and inflammatory parameters in the ileal pouch after restorative proctocolectomy for ulcerative colitis. Surgery. 2011;150:56–67. doi: 10.1016/j.surg.2011.02.009. [DOI] [PubMed] [Google Scholar]
- 11.Reshef L, Kovacs A, Ofer A, Yahav L, Maharshak N, Keren N, Konikoff FM, Tulchinsky H, Gophna U, Dotan I. Pouch Inflammation Is Associated With a Decrease in Specific Bacterial Taxa. Gastroenterology. 2015;149:718–727. doi: 10.1053/j.gastro.2015.05.041. [DOI] [PubMed] [Google Scholar]
- 12.Machiels K, Sabino J, Vandermosten L, Joossens M, Arijs I, de Bruyn M, Eeckhaut V, Van Assche G, Ferrante M, Verhaegen J, et al. Specific members of the predominant gut microbiota predict pouchitis following colectomy and IPAA in UC. Gut. 2015 doi: 10.1136/gutjnl-2015-309398. Epub ahead of print. [DOI] [PubMed] [Google Scholar]
- 13.Sandborn WJ, Tremaine WJ, Batts KP, Pemberton JH, Phillips SF. Pouchitis after ileal pouch-anal anastomosis: a Pouchitis Disease Activity Index. Mayo Clin Proc. 1994;69:409–415. doi: 10.1016/s0025-6196(12)61634-6. [DOI] [PubMed] [Google Scholar]
- 14.Vanhoutte T, Huys G, Brandt E, Swings J. Temporal stability analysis of the microbiota in human feces by denaturing gradient gel electrophoresis using universal and group-specific 16S rRNA gene primers. FEMS Microbiol Ecol. 2004;48:437–446. doi: 10.1016/j.femsec.2004.03.001. [DOI] [PubMed] [Google Scholar]
- 15.Muyzer G, de Waal EC, Uitterlinden AG. Profiling of complex microbial populations by denaturing gradient gel electrophoresis analysis of polymerase chain reaction-amplified genes coding for 16S rRNA. Appl Environ Microbiol. 1993;59:695–700. doi: 10.1128/aem.59.3.695-700.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Sanguinetti CJ, Dias Neto E, Simpson AJ. Rapid silver staining and recovery of PCR products separated on polyacrylamide gels. Biotechniques. 1994;17:914–921. [PubMed] [Google Scholar]
- 17.Vermeiren J, Van den Abbeele P, Laukens D, Vigsnaes LK, De Vos M, Boon N, Van de Wiele T. Decreased colonization of fecal Clostridium coccoides/Eubacterium rectale species from ulcerative colitis patients in an in vitro dynamic gut model with mucin environment. FEMS Microbiol Ecol. 2012;79:685–696. doi: 10.1111/j.1574-6941.2011.01252.x. [DOI] [PubMed] [Google Scholar]
- 18.Fite A, Macfarlane S, Furrie E, Bahrami B, Cummings JH, Steinke DT, Macfarlane GT. Longitudinal analyses of gut mucosal microbiotas in ulcerative colitis in relation to patient age and disease severity and duration. J Clin Microbiol. 2013;51:849–856. doi: 10.1128/JCM.02574-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Varga J, Stirewalt VL, Melville SB. The CcpA protein is necessary for efficient sporulation and enterotoxin gene (cpe) regulation in Clostridium perfringens. J Bacteriol. 2004;186:5221–5229. doi: 10.1128/JB.186.16.5221-5229.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hatheway CL. Toxigenic clostridia. Clin Microbiol Rev. 1990;3:66–98. doi: 10.1128/cmr.3.1.66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Pruteanu M, Hyland NP, Clarke DJ, Kiely B, Shanahan F. Degradation of the extracellular matrix components by bacterial-derived metalloproteases: implications for inflammatory bowel diseases. Inflamm Bowel Dis. 2011;17:1189–1200. doi: 10.1002/ibd.21475. [DOI] [PubMed] [Google Scholar]
- 22.Hansen JJ. Immune Responses to Intestinal Microbes in Inflammatory Bowel Diseases. Curr Allergy Asthma Rep. 2015;15:61. doi: 10.1007/s11882-015-0562-9. [DOI] [PubMed] [Google Scholar]
- 23.Kleessen B, Kroesen AJ, Buhr HJ, Blaut M. Mucosal and invading bacteria in patients with inflammatory bowel disease compared with controls. Scand J Gastroenterol. 2002;37:1034–1041. doi: 10.1080/003655202320378220. [DOI] [PubMed] [Google Scholar]
- 24.Falk A, Olsson C, Ahrné S, Molin G, Adawi D, Jeppsson B. Ileal pelvic pouch microbiota from two former ulcerative colitis patients, analysed by DNA-based methods, were unstable over time and showed the presence of Clostridium perfringens. Scand J Gastroenterol. 2007;42:973–985. doi: 10.1080/00365520701204238. [DOI] [PubMed] [Google Scholar]
- 25.Sokol H, Seksik P, Furet JP, Firmesse O, Nion-Larmurier I, Beaugerie L, Cosnes J, Corthier G, Marteau P, Doré J. Low counts of Faecalibacterium prausnitzii in colitis microbiota. Inflamm Bowel Dis. 2009;15:1183–1189. doi: 10.1002/ibd.20903. [DOI] [PubMed] [Google Scholar]
- 26.Sokol H, Pigneur B, Watterlot L, Lakhdari O, Bermúdez-Humarán LG, Gratadoux JJ, Blugeon S, Bridonneau C, Furet JP, Corthier G, et al. Faecalibacterium prausnitzii is an anti-inflammatory commensal bacterium identified by gut microbiota analysis of Crohn disease patients. Proc Natl Acad Sci USA. 2008;105:16731–16736. doi: 10.1073/pnas.0804812105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Machiels K, Joossens M, Sabino J, De Preter V, Arijs I, Eeckhaut V, Ballet V, Claes K, Van Immerseel F, Verbeke K, et al. A decrease of the butyrate-producing species Roseburia hominis and Faecalibacterium prausnitzii defines dysbiosis in patients with ulcerative colitis. Gut. 2014;63:1275–1283. doi: 10.1136/gutjnl-2013-304833. [DOI] [PubMed] [Google Scholar]
- 28.Segain JP, Raingeard de la Blétière D, Bourreille A, Leray V, Gervois N, Rosales C, Ferrier L, Bonnet C, Blottière HM, Galmiche JP. Butyrate inhibits inflammatory responses through NFkappaB inhibition: implications for Crohn’s disease. Gut. 2000;47:397–403. doi: 10.1136/gut.47.3.397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ott SJ, Musfeldt M, Wenderoth DF, Hampe J, Brant O, Fölsch UR, Timmis KN, Schreiber S. Reduction in diversity of the colonic mucosa associated bacterial microflora in patients with active inflammatory bowel disease. Gut. 2004;53:685–693. doi: 10.1136/gut.2003.025403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Manichanh C, Rigottier-Gois L, Bonnaud E, Gloux K, Pelletier E, Frangeul L, Nalin R, Jarrin C, Chardon P, Marteau P, et al. Reduced diversity of faecal microbiota in Crohn’s disease revealed by a metagenomic approach. Gut. 2006;55:205–211. doi: 10.1136/gut.2005.073817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wills ES, Jonkers DM, Savelkoul PH, Masclee AA, Pierik MJ, Penders J. Fecal microbial composition of ulcerative colitis and Crohn’s disease patients in remission and subsequent exacerbation. PLoS One. 2014;9:e90981. doi: 10.1371/journal.pone.0090981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Dabard J, Bridonneau C, Phillipe C, Anglade P, Molle D, Nardi M, Ladiré M, Girardin H, Marcille F, Gomez A, et al. Ruminococcin A, a new lantibiotic produced by a Ruminococcus gnavus strain isolated from human feces. Appl Environ Microbiol. 2001;67:4111–4118. doi: 10.1128/AEM.67.9.4111-4118.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Dethlefsen L, Eckburg PB, Bik EM, Relman DA. Assembly of the human intestinal microbiota. Trends Ecol Evol. 2006;21:517–523. doi: 10.1016/j.tree.2006.06.013. [DOI] [PubMed] [Google Scholar]
- 34.Cervera-Tison M, Tailford LE, Fuell C, Bruel L, Sulzenbacher G, Henrissat B, Berrin JG, Fons M, Giardina T, Juge N. Functional analysis of family GH36 α-galactosidases from Ruminococcus gnavus E1: insights into the metabolism of a plant oligosaccharide by a human gut symbiont. Appl Environ Microbiol. 2012;78:7720–7732. doi: 10.1128/AEM.01350-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Png CW, Lindén SK, Gilshenan KS, Zoetendal EG, McSweeney CS, Sly LI, McGuckin MA, Florin TH. Mucolytic bacteria with increased prevalence in IBD mucosa augment in vitro utilization of mucin by other bacteria. Am J Gastroenterol. 2010;105:2420–2428. doi: 10.1038/ajg.2010.281. [DOI] [PubMed] [Google Scholar]
- 36.Seksik P, Rigottier-Gois L, Gramet G, Sutren M, Pochart P, Marteau P, Jian R, Doré J. Alterations of the dominant faecal bacterial groups in patients with Crohn’s disease of the colon. Gut. 2003;52:237–242. doi: 10.1136/gut.52.2.237. [DOI] [PMC free article] [PubMed] [Google Scholar]