

Abstract
AIM
To evaluate the inhibitory effects of deferasirox (DFX) against hepatocellular carcinoma (HCC) through basic and clinical studies.
METHODS
In the basic study, the effect of DFX was investigated in three hepatoma cell lines (HepG2, Hep3B, and Huh7), as well as in an N-nitrosodiethylamine-induced murine HCC model. In the clinical study, six advanced HCC patients refractory to chemotherapy were enrolled. The initial dose of DFX was 10 mg/kg per day and was increased by 10 mg/kg per day every week, until the maximum dose of 30 mg/kg per day. The duration of a single course of DFX therapy was 28 consecutive days. In the event of dose-limiting toxicity (according to the Common Terminology Criteria for Adverse Events v.4.0), DFX dose was reduced.
RESULTS
Administration of DFX inhibited the proliferation of hepatoma cell lines and induced the activation of caspase-3 in a dose-dependent manner in vitro. In the murine model, DFX treatment significantly suppressed the development of liver tumors (P < 0.01), and significantly upregulated the mRNA expression levels of hepcidin (P < 0.05), transferrin receptor 1 (P < 0.05), and hypoxia inducible factor-1α (P < 0.05) in both tumor and non-tumor tissues, compared with control mice. In the clinical study, anorexia and elevated serum creatinine were observed in four and all six patients, respectively. However, reduction in DFX dose led to decrease in serum creatinine levels in all patients. After the first course of DFX, one patient discontinued the therapy. We assessed the tumor response in the remaining five patients; one patient exhibited stable disease, while four patients exhibited progressive disease. The one-year survival rate of the six patients was 17%.
CONCLUSION
We demonstrated that DFX inhibited HCC in the basic study, but not in the clinical study due to dose-limiting toxicities.
Keywords: Liver tumor, Hepatocellular carcinoma, Advanced stage, Iron-chelator, Deferasirox
Core tip: There are currently no established second-line chemotherapies for advanced hepatocellular carcinoma (HCC) patients. Iron chelators exert antiproliferative effects in several cancers. We demonstrated the inhibition of HCC by deferasirox (DFX) in the basic study. However, the efficacy of DFX in our clinical study could not be verified due to dose-limiting toxicities. Although iron chelators have promising therapeutic potential, further examinations are necessary to establish their clinical applications.
INTRODUCTION
Hepatocellular carcinoma (HCC) is the fifth most common cancer and the second leading cause of cancer-related deaths worldwide[1]. Although recent advances in treatment techniques have improved the prognosis of this malignancy, the prognosis of advanced HCC patients, especially in the case of vascular invasion and/or extrahepatic spread, remains poor[2,3]. For such patients, the multikinase inhibitor sorafenib is recommended as the current standard therapy worldwide[4,5]. On the other hand, hepatic arterial infusion chemotherapy (HAIC) is one of the recommended treatments in Japan[5]. Sorafenib is generally used to treat patients with Child-Pugh A score, while HAIC is indicated for those with Child-Pugh A or B scores. Therefore, both HAIC and sorafenib are first-line chemotherapy options for those with Child-Pugh A score. On the other hand, the options available for those with Child-Pugh B score are either HAIC or systemic chemotherapy with the exception of sorafenib[6]. However, there are currently no established second-line chemotherapies for advanced HCC patients.
Iron is essential for a number of cellular metabolic processes, including DNA synthesis[7]. It is also required for the proliferation of cancer cells before initiation of DNA synthesis[8]. Iron chelators are commonly used for the treatment of iron-overload disease. Although iron chelators are not classified as anticancer drugs, they exert antiproliferative effects in several cancers, including HCC[9-12]. We previously reported that deferoxamine (DFO) can prevent both liver fibrosis and development of preneoplastic lesions in rats[13,14]. We also performed a pilot study of DFO for HAIC in advanced HCC patients for the first time, and demonstrated the efficacy of this chelator[15]. Hence, DFO therapy may be used as second-line chemotherapy owing to its therapeutic potential in patients with deteriorated liver function. However, DFO cannot be administered orally, thus limiting its clinical application. Recently, deferasirox (DFX), a newly developed oral iron chelator, was shown to exert a powerful antiproliferative effect in human hepatoma cell culture[12], and on hepatocarcinogenesis in vivo[16]. We have also reported that DFX, like DFO, is able to prevent liver fibrosis and hepatocarcinogenesis in rats. In addition, we have shown that DFX can prevent the adverse effects of sorafenib[17]. Thus, DFX may represent a next-generation option for chemoprevention of HCC. However, there have been no in vivo or clinical studies of DFX against HCC. Therefore, the aim of this study is to evaluate the inhibitory effects of DFX against HCC, through both basic and clinical research.
MATERIALS AND METHODS
Basic research
Cell proliferation assay: Hepatoma cell lines (HepG2, Hep3B, and Huh7) were seeded at a density of 1.0 × 104 cells/well in a 96-well plate. Cell proliferation was measured using MTT [3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide] assay with CellTiter 96 AQueous One Solution Reagent (Promega, Madison, WI, United States). Cells were treated with DFX (0, 10, 20, 50, and 100 μmol/L) after 12 h, and were incubated for 24 h. The absorbance at 490 nm was measured to evaluate cell viability using a Bio-Rad plate reader (Hercules, CA, United States)[18].
Caspase-3 activity: Hepatoma cell lines were seeded at a density of 1.0 × 104 cells/well in a 96-well plate, and incubated in serum-free medium for 24 h. Then, the cells were treated with DFX (0, 10, 20, 50, and 100 μM) and incubated for another 24 h in serum-free medium. The activity of caspase-3 was determined using a caspase-3 colorimetric assay kit (MBL, Nagoya, Japan). This assay measured the cleavage of a specific colorimetric caspase substrate, DEVD-pNA, which releases p-nitroaniline (pNA). Free pNA produces a yellow color that was detected by a spectrophotometer at 405 nm[18].
Animals and experimental protocol: Animal care was performed in accordance with the animal ethics requirements of Yamaguchi University School of Medicine; the approval ID of the experimental protocol was 21-025. Seven-week-old female C57 BL/6 mice (20-30 g) were purchased from Nippon SLC (Shizuoka, Japan) and housed in a room under controlled temperature (25 °C) and lighting (12-h light, 12-h dark) at the Animal Experiment Facility of Yamaguchi University School of Medicine.
Male mice received a single intraperitoneal injection of 10 μg/g body weight of N-nitrosodiethylamine (DEN) (Sigma-Aldrich Japan, Tokyo, Japan) at 14 d of age. Incidence of liver tumors was histologically evaluated five months after DEN injection[19]. The mice were divided into two groups (n = 10 per group): normal diet only (control group) and normal diet with DFX (DFX group) (Figure 1). At 20 wk after DEN injection, DFX (20 mg/kg per day) was administered orally for a period of 3 mo until week 32. Food intake of mice in each group was measured. To equalize the total food intake in all groups, additional food was not supplied until all food had been consumed.
Figure 1.
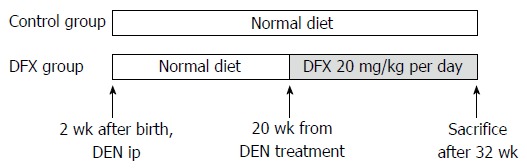
Protocol for the murine model of hepatocarcinogenesis. The model was induced by injection of 10 μg/g of DEN at 14 d of age. In the deferasirox (DFX) group, 20 mg/kg of DFX was administered orally for 3 mo and fed with normal diet. In the control group, the same amount of normal diet was administered. After 3 mo (at week 32), the mice were sacrificed and underwent autopsy examination.
Histology and immunohistochemical examination: Sections (3 μm thick) of the mouse liver were fixed in 4% paraformaldehyde (Muto; Tokyo, Japan) for 24 h and embedded in paraffin. The sections were processed for hematoxylin and eosin (H&E) staining. Tumor area of the liver in H&E stain was quantified using a Keyence BIOREVO BZ9000 microscope (Osaka, Japan) and was expressed as percentage of the total specimen area.
Real-time quantitative polymerase chain reaction: Expression of hepcidin, transferrin receptor 1, and hypoxia inducible factor-1α (HIF-1α) mRNA between tumor and non-tumor tissues was evaluated by real-time polymerase chain reaction (PCR) as described previously[17]. Briefly, RNA extraction was performed using an RNeasy Mini kit (Qiagen GmbH, Hilden, Germany) according to the manufacturer’s protocol. The primers used were as follows:
Mouse hepcidin: sense (5′-AGAGCTGCAGCCTTTGCAC-3′),
antisense (5′-GAAGATGCAGATGGGGAAGT-3′);
Mouse transferrin receptor 1: sense (5′-GGTGATCCATACACACCTGGCTT-3′),
antisense (5′-TGATGACTGAGATGGCGGAA-3′);
Mouse HIF-1α: sense (5′-GCGTGCATGTCTAATCTGTTCC-3′),
antisense (5′-GATTCTGACATGCCACATAGCTC-3′);
Mouse β-actin: sense (5′-TGACAGGATGCAGAAGGAGA-3′),
antisense (5′-GCTGGAAGGTGGACAGTGAG-3′).
PCR amplification was performed in triplicate using the following cycle conditions: 40 cycles of 90 °C for 30 s, 55-60 °C for 45 s, and 72 °C for 1 min. Gene expression levels were analyzed using β-actin as the reference gene.
Mice survival and serum components: To analyze mice survival in the control (n = 10) and DFX (n = 9) groups, mice were fed normal diet and administered the same dose of DFX (20 mg/kg per day) for approximately one year. We used the Kaplan-Meier estimator of survival, and verified the survival function against lifetime data.
Serum samples were obtained by eye puncture method at 32 weeks (n = 8 for control group, n = 5 for DFX group). In all experiments, serum total protein, total bilirubin, albumin, alanine aminotransferase (ALT), aspartate aminotransferase (AST), alkaline phosphatase (ALP), blood urea nitrogen (BUN), and creatinine levels were measured using an analyzer for clinical chemistry (SPOTCHEM EZ SP-4430; Arkray, Kyoto, Japan).
In addition, we measured body weight of the mice at 32 wk.
Clinical research
Patients: The eligibility criteria for inclusion in this study were as follows: age > 20 years; Child-Pugh score A or B; leukocyte count > 2500/mm3; platelet count > 5000/mm3; hemoglobin level > 9 g/dL; prothrombin activity > 50%; total bilirubin < 3 mg/dL; serum creatinine < 2 mg/dL; unresectable HCC due to extensive, locally advanced disease, bilobar disease, extrahepatic metastasis, or vascular tumor thrombosis; refractory to chemotherapy; and Eastern Cooperative Oncology Group performance status of 0 or 1[20]. The exclusion criteria were as follows: severe complicating disease; concomitant malignancy; a history of allergy; pregnancy/lactation; a history of interstitial pneumonia; chronic respiratory failure; and severe general condition.
Six patients who were admitted to our hospital enrolled in this study between April 2014 and July 2015. The diagnosis of HCC was performed based on imaging results and elevated serum levels of alpha-fetoprotein and/or des-gamma-carboxy prothrombin.
This study (H25-148) was approved by the Institutional Review Board of Yamaguchi University Hospital, and written informed consent was obtained from all patients. The study protocol was conducted according to the principles of the 1975 Declaration of Helsinki. The trial was registered online (http://www.umin.ac.jp/) (UMIN 000013451).
Study design and treatment protocol: This study was designed as a dose-escalation trial in which patients received continuous oral administration of DFX at doses ranging from 5 to 30 mg/kg per day. The initial dose of DFX was 10 mg/kg per day, based on the minimal dose used for the treatment of iron-overload disease. If patients did not experience dose-limiting toxicity (DLT) that was beyond the grade 3 adverse event (AE) within one week, the dose of DFX was increased by 10 mg/kg per day every week, until it reached 30 mg/kg per day. On the other hand, if a serious clinical toxicity was observed, the dose of DFX was either decreased (grade 3 AE) or the treatment discontinued (grade 4 AE). Nevertheless, because renal dysfunction developed in the initial three cases, we reduced the DFX dose if the estimated glomerular filtration rate (eGFR) decreased to less than 50% of the baseline level.
A single course of DFX treatment consisted of DFX administration for 28 continuous days. After the treatment was suspended for one or two weeks and its safety was confirmed, DFX was administered at the maintenance dose (5-15 mg/kg per day) in the outpatient department. Patients continued treatment until disease progression, withdrawal of consent, or in cases of intolerability. Toxicity was graded using the Common Terminology Criteria for Adverse Events v.4.0 (CTCAE v.4.0)[21]. Tumor measurements were performed at baseline and at the end of every course using dynamic computed tomography or magnetic resonance imaging, and the evaluation of the response to the treatment was classified according to the mRECIST guideline[22]. When repeated DFX treatment was performed, the best response was considered for response evaluation. Survival time was defined as the interval between DFX administration and the last follow-up or death. The follow-up period ended on December 31, 2015. The primary endpoint was safety, and the secondary endpoint was tumor response and overall survival.
Statistical analysis
Statistical significance was assessed using student’s t-test for biochemical and histological results, and the log-rank test for survival analysis. Human results are expressed as mean ± SD, differences with P < 0.05 were considered significant. All analyses were performed using the JMP ver. 10.0 software package (SAS Institute, Cary, NC, United States).
RESULTS
Basic research
Effect of DFX on hepatoma cell lines: Administration of DFX inhibited the proliferation of all hepatoma cell lines in a dose-dependent manner (Figure 2A). In addition, DFX also induced the activity of caspase-3 in a similar manner (Figure 2B).
Figure 2.
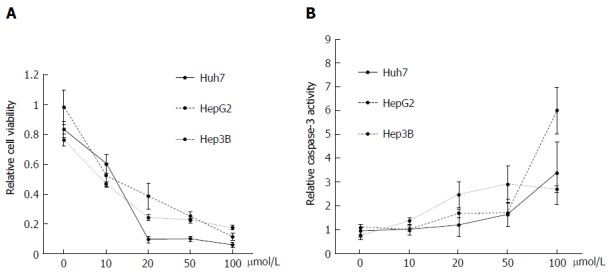
Antitumor effects of deferasirox in hepatoma cell lines. A: Deferasirox (DFX) exhibited antiproliferative effects against each cell line in a dose-dependent manner as revealed by MTT assay. Bars represent SD; B: Colorimetric assay of caspase-3 activity showing activation of caspase-3 by DFX in a dose-dependent manner. Bars represent SD.
Effect of DFX on tumor in murine model: Figure 3A (control group) and B (DFX group) show macroscopic images of tumor formation in mice liver. The total tumor area was significantly reduced (P < 0.01) in the DFX group compared with the control group (Figure 3C).
Figure 3.
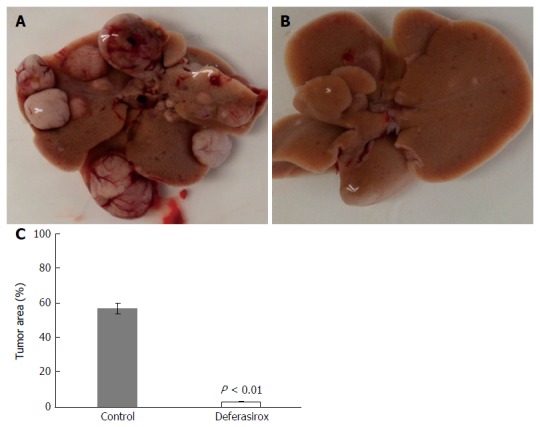
Inhibition of liver tumor formation by deferasirox. Macroscopic images of liver tumors in the (A) control and (B) deferasirox (DFX) groups. Evaluation of (C) tumor area percentage of the total specimen area in control and DFX-treated mice. Bars represent SD.
Effect of DFX on the expression of iron-related genes: Hepcidin, transferrin receptor 1, and HIF-1α mRNA expression levels in both tumor and non-tumor areas were significantly higher (P < 0.05) in the DFX-treated group than the control group (Figure 4A and C).
Figure 4.
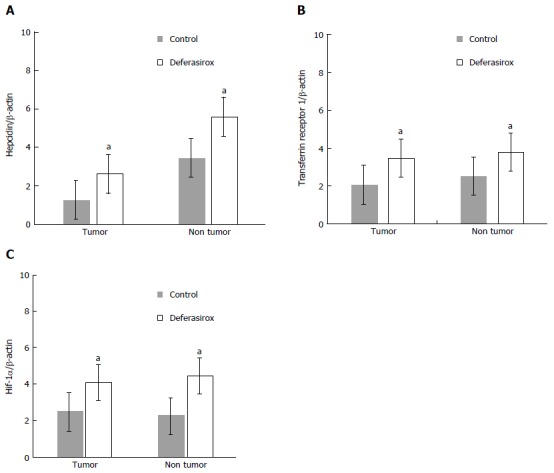
Regulation of iron-related gene expressions by deferasirox in a murine model. Real-time RT-PCR data of the expressions of (A) hepcidin, (B) transferrin receptor 1, and (C) HIF-1α in tumor and non-tumor tissues. Bars indicate SD. aP < 0.05.
Effect of DFX on mice survival and serum components: Table 1 shows the levels of different serum components in DFX and control groups. Mice in the DFX group had significantly lower level of serum ALT than those in the control group (P < 0.05). However, there were no significant differences in the levels of total protein, albumin, total bilirubin, AST, ALP, BUN, creatinine, and body weight between both groups. Liver dysfunction and renal dysfunction were not observed in the DFX group in this murine model.
Table 1.
Comparison of body weight and serum data between deferasirox and control groups
| Body weight | TP | Albumin | T-bil | AST | ALTa | ALP | BUN | CRE | |
| (g) | (g/dL) | (g/dL) | (mg/dL) | (IU/L) | (IU/L) | (IU/L) | (mg/dL) | (mg/dL) | |
| Control | 40.7 ± 4.1 | 5.6 ± 0.4 | 2.8 ± 0.2 | 0.5 ± 0.1 | 95.8 ± 33.9 | 92.3 ± 43.1 | 175.1 ± 23.2 | 25.5 ± 2.9 | 0.4 ± 0.1 |
| DFX | 42.8 ± 4.7 | 5.2 ± 0.5 | 2.7 ± 0.1 | 0.6 ± 0.3 | 87.6 ± 20.1 | 29.5 ± 13.8 | 162.2 ± 35.9 | 30.2 ± 1.6 | 0.6 ± 0.1 |
Mean ± SD.
P < 0.05. DFX: Deferasirox; AST: Aspartate aminotransferase; ALT: Alanine aminotransferase; ALP: Alkaline phosphatase; BUN: Blood urea nitrogen; TP: Total protein; CRE: Creatinine.
Survival analysis of mice in the two groups over approximately one year showed that DFX-treated mice survived significantly longer (P < 0.01) than control mice (Figure 5).
Figure 5.
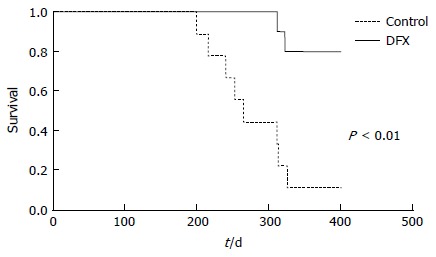
Cumulative survival rates of control and deferasirox-treated mice in a murine hepatocellular carcinoma model. Deferasirox (DFX)-treated mice showed significantly higher survival rate than control mice (P < 0.01).
Clinical research
Patient characteristics: The patients’ characteristics are shown in Table 2. There were four male and two female patients with an average age of 70 years (range, 60 to 80). Five patients had hepatitis C virus infection, while one had hepatitis B virus infection. According to the Child-Pugh classification, half of the patients were classified as class A, and the other half as class B. The tumor stages were classified as III (n = 2) and IVA (n = 4), according to the Liver Cancer Study Group of Japan[23].
Table 2.
Patients’ characteristics
| Case | Sex | Age | Etiology | Child-Pugh score (Points) | Stage1 | BCLC2 | Maintenance dose (mg/kg per day) | Administration period (d) | Response |
AFP (ng/mL) |
DCP (mAU/mL) |
||
| Baseline | After 1 course | Baseline | After 1 course | ||||||||||
| 1 | M | 80 | HCV | B (7) | IVA | C | 10 | 17 | PD | 1918 | - | 57 | - |
| 2 | M | 73 | HBV | B (7) | III | C | 10 | 35 | PD | 127402 | 260627 | 73 | 61 |
| 3 | M | 72 | HBV | B (8) | III | B | 5 | 248 | SD | 189 | 394 | 85 | 25 |
| 4 | M | 65 | HBV | A (5) | IVA | C | 10 | 28 | PD | 603 | 814 | 9995 | 16793 |
| 5 | F | 70 | HCV | A (6) | IVA | C | 15 | 29 | PD | 5580 | 8434 | 19958 | 15321 |
| 6 | F | 60 | HCV | A (6) | IVA | C | 15 | 28 | PD | 6172 | 7671 | 4606 | 2283 |
According to Liver Cancer Study Group of Japan;
According to Barcelona Clinic Liver Cancer. HCV: Hepatitis C virus; HBV: Hepatitis B virus; AFP: Alfha-fetoprotein; DCP: Des-gamma-carboxy prothrombin. PD: Progressive disease; SD: Stable disease.
Safety and tolerability of DFX: Table 3 summarizes the adverse events observed during the trial. Although all patients showed renal dysfunction, their conditions improved with reduction in DFX dose (Figure 6A and B). Elevated creatinine levels observed in patients were classified into different AE grades; grade 3 in one patient, grade 2 in four patients, and grade 1 in one patient. Anorexia was observed in four patients (grade 2 in two patients, grade 1 in two patients). However, there were no treatment-related deaths.
Table 3.
Summary of adverse effects (mean ± SD)
| Case | Creatinine at baseline (mg/dL) | eGFR at baseline (mL/min/1.73 m2) | Adverse effects (grade) | Onset time of grade 2 adverse effects |
| 1 | 1.19 | 45.6 | Elevated creatinine (3) | Week 2 |
| Anorexia (2) | Week 2 | |||
| 2 | 0.70 | 83.7 | Elevated creatinine (2) | Week 2 |
| Anorexia (2) | Week 3 | |||
| 3 | 1.00 | 56.9 | Elevated creatinine (2) | Week 3 |
| 4 | 0.75 | 80.2 | Elevated creatinine (2) | Week 3 |
| 5 | 0.63 | 70.2 | Elevated creatinine (2) | Week 4 |
| Anorexia (1) | ||||
| 6 | 0.43 | 111.5 | Anorexia (1) | |
| Elevated creatinine (1) |
Figure 6.
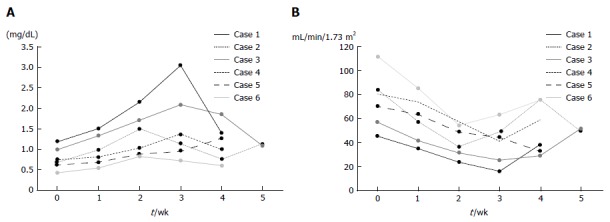
Changes in serum creatinine levels and eGFR of patients administered with deferasirox. A: All patients showed gradual increase in creatinine levels after initiation of DFX treatment, until reduction in DFX dose or discontinuation of treatment. Creatinine levels in all patients were improved by dose reduction. B: All patients showed gradual decrease in eGFR after initiation of DFX treatment, until reduction in DFX dose or discontinuation of treatment.
DLT of DFX was observed in patients treated with high doses of the drug (≥ 20 mg/kg per day). Two patients experienced DLT at a dose of 30 mg/kg per day, and another four patients at 20 mg/kg per day. After the first course of DFX, all patients required dose reductions and one patient discontinued treatment due to intolerance associated with anorexia.
Tumor response and survival: We assessed tumor response to DFX in all patients except one who discontinued the treatment (Case 1). One patient exhibited stable disease (SD), and four patients exhibited progressive disease (PD) (Table 2). One patient who had multinodular HCCs without portal vein tumor thrombus (Case 3) maintained SD for eight months (Figure 7).
Figure 7.
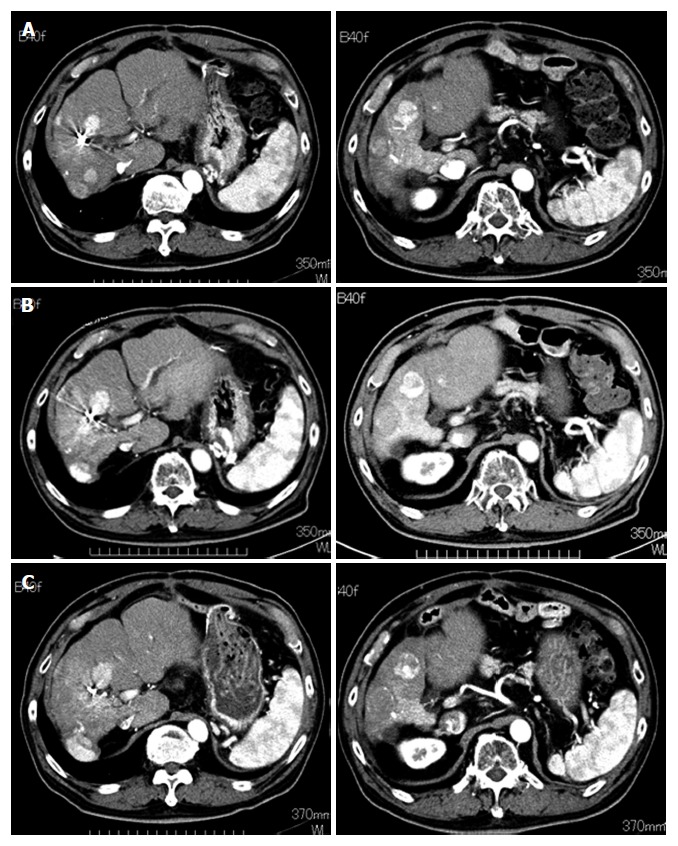
Progress of hepatocellular carcinoma in one patient (case 3) who maintained stable disease for eight months. The tumor size did not increase during the DFX administration period. A: Before deferasirox (DFX) administration; B: After one course of DFX therapy; C: After four courses of DFX therapy.
The one-year cumulative survival rate of the six patients was 17%, and the median survival time was 271 d. Five patients died of cancer-related disease, and one remained alive.
DISCUSSION
Sorafenib has been used worldwide as first-line chemotherapy for advanced HCC patients. However, there are currently no established second-line chemotherapies for such patients. We have been researching the efficacy of iron chelators such as DFO and DFX in basic and clinical studies[13-15,17]. We demonstrated that DFO therapy can be considered as second-line chemotherapy for advanced HCC patients refractory to chemotherapy, showing that the response to DFO therapy and the one-year survival rate were both 20%[15]. DFX is a newly developed oral iron chelator for the treatment of iron-overload disease. We demonstrated that DFX can prevent liver fibrosis and hepatocarcinogenesis in a choline-deficient L-amino acid (CDAA) diet-induced rat model of liver injury[17]. However, we have not examined the efficacy of DFX in a HCC model. Therefore, we investigate in the present study, the effect of DFX against HCC through basic and clinical research.
In the basic study, we demonstrated the inhibitory effects of DFX against HCC both in vitro and in vivo. Administration of DFX inhibited the proliferation of three hepatoma cell lines (HepG2, Hep3B, and Huh7) in a dose-dependent manner, and induced apoptosis through an increase in caspase-3 activity (Figure 2). Previous reports have shown that DFX inhibited the cell cycle at the G0/G1 and S phases, and induced apoptosis in the Huh7 human hepatoma cell line[12,24]. We showed that DFX notably inhibited the development of liver tumors and significantly upregulated the mRNA expression levels of hepcidin, transferrin receptor 1, and HIF-1α in both tumor and non-tumor areas, compared with control (Figures 3 and 4). Iron chelators can induce hypoxia, which in turn upregulates HIF-1. Hypoxia inducible factor-1 is composed of two subunits, an α subunit that is regulated by the hypoxic state and a constitutively expressed β subunit. It also activates downstream effectors such as transferrin receptor 1, N-myc downstream regulated gene-1, and p53[9]. Subsequently, hepcidin is transcriptionally activated by p53[25]. It has been reported that hepcidin mRNA levels were significantly lower in liver tumor tissues than in the surrounding and control tissues in the DEN-induced murine hepatocarcinogenic model used in this study[26]. Furthermore, it was shown that the hepatic hepcidin expression decreased in tissues with cirrhosis and in HCC patients[27]. In our study, hepcidin mRNA levels were also reduced in tumor tissues before DFX treatment. As hepcidin is an iron homeostasis regulator, DFX can potentially improve iron homeostasis in liver tumors. Therefore, our findings suggested that DFX might inhibit tumor growth via hypoxia-associated factors and by regulating iron homeostasis through the upregulation of hepcidin.
Based on the results of the basic study, we proposed a clinical study of DFX therapy in advanced HCC patients refractory to chemotherapy. To our knowledge, this is the first report on DFX therapy for HCC in a clinical trial setting. To determine the recommended therapeutic dose for DFX therapy, we started at 10 mg/kg per day based on the minimal dose used in the treatment of iron-overload disease, and gradually increased the dose to a maximum of 30 mg/kg per day, if proven safe. Unfortunately, we could not continuously administer a high dose of DFX (20-30 mg/kg per day) due to AEs. Thus, the administered maintenance dose of DFX was 5-15 mg/kg per day.
Renal dysfunction, as indicated by elevated creatinine levels was observed in all six patients, while anorexia which was well tolerated, was observed in four patients (Table 3). Porter et al[28] reported that increased serum creatinine related to DFX was observed in 39.7% of patients with myelodysplastic syndrome and other transfusion-dependent anemias, most frequently at doses between 20 and 30 mg/kg per day; however, this increase was not progressive. Although there were no reports on the Japanese population exclusively, Kohgo et al[29] reported that the incidence of increased serum creatinine was 23.5% in patients from five countries, including Japan (Japanese patients, n = 53; non-Japanese patients, n = 49)[29]. In our clinical study, all six patients exhibited elevated creatinine levels (AE grade, 1-3) upon DFX administration at a dose of 20 mg/kg per day (n = 4) and 30 mg/kg per day (n = 2). However, the creatinine levels decreased after the dose was reduced (Figure 6A). In addition, DFX was associated with a higher risk of acute renal failure than DFO in a large Asian population[30]. In fact, our data showed that the incidence of increased serum creatinine was 10% (1 in 10 patients) in DFO therapy[15] and 100% in DFX therapy; however, these studies only involved small populations. A possible reason for the difference in the risk of renal dysfunction between DFX and DFO therapies is the difference in their half-lives; DFX has a relatively long half-life compared with DFO.
We demonstrated that a DFX dose of 20 mg/kg per day inhibited the development of liver tumors in a murine model. A previous report also confirmed the efficacy of DFX using the same dose in an esophageal cancer xenograft mice model[31]. However, there were no responders for DFX therapy in our clinical study; one patient exhibited SD, and four patients exhibited PD. On the other hand, it has been reported that the iron chelation activity of DFX increased in a dose-dependent manner; a decrease in serum ferritin was observed at a dose of 20 and 30 mg/kg per day, but not at 5 and 10 mg/kg per day[28]. Supplementary Table 1 shows changes in serum ferritin before and after a single course of DFX therapy. Although three patients had normal range of ferritin at baseline, there were no significant changes in serum ferritin level between baseline and one course after treatment (P = 0.12). Thus, DFX dosage of 5-15 mg/kg per day may not be adequate for not only tumor inhibition, but also for iron chelation in patients with HCC and liver cirrhosis. However, it is problematic to increase the effective dose of DFX to 20-30 mg/kg per day because of elevated creatinine levels in patients, in accordance to the treatment protocol of this clinical study; thus, a normal level of eGFR should be included as an eligibility criterion in future clinical studies. Alternatively, we previously reported that the combination therapy of DFX and sorafenib markedly inhibited liver fibrosis and hepatocarcinogenesis with a significant reduction of the AEs associated with sorafenib in a CDAA-induced rat model[17]. The efficacy of this combination therapy was also demonstrated based on in vitro and in vivo studies by other researchers[32]. If the effectiveness of DFX and sorafenib combination therapy can be verified in clinical trials, a low dose of DFX may be used as a novel HCC therapy.
In conclusion, we demonstrated the inhibitory effects of DFX against HCC in a basic study designed to investigate its antiproliferative potential. However, the efficacy of DFX in the clinical study could not be demonstrated because of inadequate doses due to DLT. Although iron chelators have promising therapeutic potential, further examinations are necessary to establish their clinical applications.
ACKNOWLEDGMENTS
We thank Mrs. Mariko Yamada, Mrs. Ihoko Fujimoto, and Mrs. Hiromi Kurose for their technical assistance and support.
COMMENTS
Background
Sorafenib is recommended as the current standard therapy for advanced hepatocellular carcinoma (HCC) patients and has been generally administered as first-line chemotherapy for those with Child-Pugh A score. However, no established second-line chemotherapies are available for patients with Child-Pugh B score or those refractory to sorafenib. Iron is essential for a number of cellular metabolic processes including DNA synthesis. It is also required for the proliferation of cancer cells before initiation of DNA synthesis. Although iron chelators are not classified as anticancer drugs, they exert antiproliferative effects in several cancers, including HCC.
Research frontiers
The authors previously reported that deferoxamine (DFO) can prevent both liver fibrosis and development of preneoplastic lesions in rats. We also performed a pilot study of DFO in advanced HCC patients for the first time, in which we demonstrated the efficacy of this chelator. However, DFO cannot be administered orally, thus limiting its clinical application. Recently, deferasirox (DFX), a newly developed oral iron chelator, was shown to exert a potent antiproliferative effect against human hepatoma cell culture and hepatocarcinogenesis in vivo. We have also reported that DFX, like DFO, was able to prevent liver fibrosis and hepatocarcinogenesis in rats.
Innovations and breakthroughs
There have been no in vivo or clinical studies of DFX against HCC. The authors investigated for the first time, the inhibitory effects of DFX against HCC through both basic and clinical research.
Applications
The authors demonstrated the inhibitory effects of DFX against HCC in a basic study. However, the efficacy of DFX in the clinical study could not be verified owing to dose-limiting toxicity in patients. Although iron chelators have promising therapeutic potential, further examinations are necessary to establish their clinical applications.
Terminology
DFO and DFX are iron chelators that are commonly used for the treatment of iron-overload disease. DFO is ordinarily administered by intravenously, whereas DFX is a newly developed oral iron chelator.
Peer-review
The manuscript is a combination of experimental and clinical results, it is difficult to compare the experimental cell lines derived from cells of HCC. There are no substantial criticisms to the present work.
Footnotes
Manuscript source: Invited manuscript
Specialty type: Gastroenterology and hepatology
Country of origin: Japan
Peer-review report classification
Grade A (Excellent): A
Grade B (Very good): 0
Grade C (Good): C, C
Grade D (Fair): 0
Grade E (Poor): 0
Institutional review board statement: This study (H25-148) was approved by the Institutional Review Board of Yamaguchi University Hospital. The study protocol was designed according to the principles of the 1975 Declaration of Helsinki.
Clinical trial registration statement: This study is single arm, non-randomized, open-labeled study. The trial was registered online (http://www.umin.ac.jp/) (UMIN 000013451).
Informed consent statement: Written informed consent was obtained from all patients prior to enrollment.
Conflict-of-interest statement: No potential conflicts of interest relevant to this article were reported.
Data sharing statement: No additional data are available.
Peer-review started: June 29, 2016
First decision: August 19, 2016
Article in press: September 28, 2016
P- Reviewer: Peng SY, Srichairatanakool S, Zielinski J S- Editor: Qi Y L- Editor: A E- Editor: Zhang FF
References
- 1.GLOBOCAN 2012: Estimated cancer incidence, Mortality and prevalence worldwide in 2012. Available from: http://globocan.iarc.fr/Default.aspx.
- 2.El-Serag HB, Marrero JA, Rudolph L, Reddy KR. Diagnosis and treatment of hepatocellular carcinoma. Gastroenterology. 2008;134:1752–1763. doi: 10.1053/j.gastro.2008.02.090. [DOI] [PubMed] [Google Scholar]
- 3.Mauer K, O’Kelley R, Podda N, Flanagan S, Gadani S. New treatment modalities for hepatocellular cancer. Curr Gastroenterol Rep. 2015;17:442. doi: 10.1007/s11894-015-0442-4. [DOI] [PubMed] [Google Scholar]
- 4.Bruix J, Sherman M. Management of hepatocellular carcinoma. Hepatology. 2005;42:1208–1236. doi: 10.1002/hep.20933. [DOI] [PubMed] [Google Scholar]
- 5.Arii S, Sata M, Sakamoto M, Shimada M, Kumada T, Shiina S, Yamashita T, Kokudo N, Tanaka M, Takayama T, et al. Management of hepatocellular carcinoma: Report of Consensus Meeting in the 45th Annual Meeting of the Japan Society of Hepatology (2009) Hepatol Res. 2010;40:667–685. doi: 10.1111/j.1872-034X.2010.00673.x. [DOI] [PubMed] [Google Scholar]
- 6.Yamasaki T, Saeki I, Sakaida I. Efficacy of iron chelator deferoxamine for hepatic arterial infusion chemotherapy in advanced hepatocellular carcinoma patients refractory to current treatments. Hepatol Int. 2014;8 Suppl 2:492–498. doi: 10.1007/s12072-013-9515-3. [DOI] [PubMed] [Google Scholar]
- 7.Andrews NC. Disorders of iron metabolism. N Engl J Med. 1999;341:1986–1995. doi: 10.1056/NEJM199912233412607. [DOI] [PubMed] [Google Scholar]
- 8.Brodie C, Siriwardana G, Lucas J, Schleicher R, Terada N, Szepesi A, Gelfand E, Seligman P. Neuroblastoma sensitivity to growth inhibition by deferrioxamine: evidence for a block in G1 phase of the cell cycle. Cancer Res. 1993;53:3968–3975. [PubMed] [Google Scholar]
- 9.Yu Y, Gutierrez E, Kovacevic Z, Saletta F, Obeidy P, Suryo Rahmanto Y, Richardson DR. Iron chelators for the treatment of cancer. Curr Med Chem. 2012;19:2689–2702. doi: 10.2174/092986712800609706. [DOI] [PubMed] [Google Scholar]
- 10.Torti SV, Torti FM. Iron and cancer: more ore to be mined. Nat Rev Cancer. 2013;13:342–355. doi: 10.1038/nrc3495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kicic A, Chua AC, Baker E. Effect of iron chelators on proliferation and iron uptake in hepatoma cells. Cancer. 2001;92:3093–3110. doi: 10.1002/1097-0142(20011215)92:12<3093::aid-cncr10107>3.0.co;2-b. [DOI] [PubMed] [Google Scholar]
- 12.Chantrel-Groussard K, Gaboriau F, Pasdeloup N, Havouis R, Nick H, Pierre JL, Brissot P, Lescoat G. The new orally active iron chelator ICL670A exhibits a higher antiproliferative effect in human hepatocyte cultures than O-trensox. Eur J Pharmacol. 2006;541:129–137. doi: 10.1016/j.ejphar.2006.05.001. [DOI] [PubMed] [Google Scholar]
- 13.Sakaida I, Hironaka K, Uchida K, Okita K. Iron chelator deferoxamine reduces preneoplastic lesions in liver induced by choline-deficient L-amino acid-defined diet in rats. Dig Dis Sci. 1999;44:560–569. doi: 10.1023/a:1026661508553. [DOI] [PubMed] [Google Scholar]
- 14.Jin H, Terai S, Sakaida I. The iron chelator deferoxamine causes activated hepatic stellate cells to become quiescent and to undergo apoptosis. J Gastroenterol. 2007;42:475–484. doi: 10.1007/s00535-007-2020-5. [DOI] [PubMed] [Google Scholar]
- 15.Yamasaki T, Terai S, Sakaida I. Deferoxamine for advanced hepatocellular carcinoma. N Engl J Med. 2011;365:576–578. doi: 10.1056/NEJMc1105726. [DOI] [PubMed] [Google Scholar]
- 16.Kaji K, Yoshiji H, Kitade M, Ikenaka Y, Noguchi R, Shirai Y, Aihara Y, Namisaki T, Yoshii J, Yanase K, et al. Combination treatment of angiotensin II type I receptor blocker and new oral iron chelator attenuates progression of nonalcoholic steatohepatitis in rats. Am J Physiol Gastrointest Liver Physiol. 2011;300:G1094–G1104. doi: 10.1152/ajpgi.00365.2010. [DOI] [PubMed] [Google Scholar]
- 17.Yamamoto N, Yamasaki T, Takami T, Uchida K, Fujisawa K, Matsumoto T, Saeki I, Terai S, Sakaida I. Deferasirox, an oral iron chelator, prevents hepatocarcinogenesis and adverse effects of sorafenib. J Clin Biochem Nutr. 2016;58:202–209. doi: 10.3164/jcbn.15-127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Saeki I, Terai S, Fujisawa K, Takami T, Yamamoto N, Matsumoto T, Hirose Y, Murata Y, Yamasaki T, Sakaida I. Bortezomib induces tumor-specific cell death and growth inhibition in hepatocellular carcinoma and improves liver fibrosis. J Gastroenterol. 2013;48:738–750. doi: 10.1007/s00535-012-0675-z. [DOI] [PubMed] [Google Scholar]
- 19.Takami T, Kaposi-Novak P, Uchida K, Gomez-Quiroz LE, Conner EA, Factor VM, Thorgeirsson SS. Loss of hepatocyte growth factor/c-Met signaling pathway accelerates early stages of N-nitrosodiethylamine induced hepatocarcinogenesis. Cancer Res. 2007;67:9844–9851. doi: 10.1158/0008-5472.CAN-07-1905. [DOI] [PubMed] [Google Scholar]
- 20.Oken MM, Creech RH, Tormey DC, Horton J, Davis TE, McFadden ET, Carbone PP. Toxicity and response criteria of the Eastern Cooperative Oncology Group. Am J Clin Oncol. 1982;5:649–655. [PubMed] [Google Scholar]
- 21.National Cancer Institute. Common Terminology Criteria for Adverse Events v.4.0. Bethesda 2009 [Google Scholar]
- 22.Lencioni R, Llovet JM. Modified RECIST (mRECIST) assessment for hepatocellular carcinoma. Semin Liver Dis. 2010;30:52–60. doi: 10.1055/s-0030-1247132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kudo M, Kitano M, Sakurai T, Nishida N. General Rules for the Clinical and Pathological Study of Primary Liver Cancer, Nationwide Follow-Up Survey and Clinical Practice Guidelines: The Outstanding Achievements of the Liver Cancer Study Group of Japan. Dig Dis. 2015;33:765–770. doi: 10.1159/000439101. [DOI] [PubMed] [Google Scholar]
- 24.Lescoat G, Chantrel-Groussard K, Pasdeloup N, Nick H, Brissot P, Gaboriau F. Antiproliferative and apoptotic effects in rat and human hepatoma cell cultures of the orally active iron chelator ICL670 compared to CP20: a possible relationship with polyamine metabolism. Cell Prolif. 2007;40:755–767. doi: 10.1111/j.1365-2184.2007.00468.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Weizer-Stern O, Adamsky K, Margalit O, Ashur-Fabian O, Givol D, Amariglio N, Rechavi G. Hepcidin, a key regulator of iron metabolism, is transcriptionally activated by p53. Br J Haematol. 2007;138:253–262. doi: 10.1111/j.1365-2141.2007.06638.x. [DOI] [PubMed] [Google Scholar]
- 26.Youn P, Kim S, Ahn JH, Kim Y, Park JD, Ryu DY. Regulation of iron metabolism-related genes in diethylnitrosamine-induced mouse liver tumors. Toxicol Lett. 2009;184:151–158. doi: 10.1016/j.toxlet.2008.11.002. [DOI] [PubMed] [Google Scholar]
- 27.Kessler SM, Barghash A, Laggai S, Helms V, Kiemer AK. Hepatic hepcidin expression is decreased in cirrhosis and HCC. J Hepatol. 2015;62:977–979. doi: 10.1016/j.jhep.2014.10.046. [DOI] [PubMed] [Google Scholar]
- 28.Porter J, Galanello R, Saglio G, Neufeld EJ, Vichinsky E, Cappellini MD, Olivieri N, Piga A, Cunningham MJ, Soulières D, et al. Relative response of patients with myelodysplastic syndromes and other transfusion-dependent anaemias to deferasirox (ICL670): a 1-yr prospective study. Eur J Haematol. 2008;80:168–176. doi: 10.1111/j.1600-0609.2007.00985.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kohgo Y, Urabe A, Kilinç Y, Agaoglu L, Warzocha K, Miyamura K, Lim LC, Glaser S, Wang C, Wiktor-Jedrzejczak W. Deferasirox Decreases Liver Iron Concentration in Iron-Overloaded Patients with Myelodysplastic Syndromes, Aplastic Anemia and Other Rare Anemias. Acta Haematol. 2015;134:233–242. doi: 10.1159/000381893. [DOI] [PubMed] [Google Scholar]
- 30.Huang WF, Chou HC, Tsai YW, Hsiao FY. Safety of deferasirox: a retrospective cohort study on the risks of gastrointestinal, liver and renal events. Pharmacoepidemiol Drug Saf. 2014;23:1176–1182. doi: 10.1002/pds.3657. [DOI] [PubMed] [Google Scholar]
- 31.Ford SJ, Obeidy P, Lovejoy DB, Bedford M, Nichols L, Chadwick C, Tucker O, Lui GY, Kalinowski DS, Jansson PJ, et al. Deferasirox (ICL670A) effectively inhibits oesophageal cancer growth in vitro and in vivo. Br J Pharmacol. 2013;168:1316–1328. doi: 10.1111/bph.12045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Urano S, Ohara T, Noma K, Katsube R, Ninomiya T, Tomono Y, Tazawa H, Kagawa S, Shirakawa Y, Kimura F, et al. Iron depletion enhances the effect of sorafenib in hepatocarcinoma. Cancer Biol Ther. 2016;17:648–656. doi: 10.1080/15384047.2016.1177677. [DOI] [PMC free article] [PubMed] [Google Scholar]