

Abstract
Parthenolide is a naturally occurring terpene with promising anticancer properties, in particular in the context of acute myeloid leukemia (AML). Optimization of this natural product has been challenged by limited opportunities for the late-stage functionalization of this molecule without affecting the pharmacologically important α-methylene-γ-lactone moiety. Here, we report the further development and application of a chemoenzymatic strategy to afford a series of new analogs of parthenolide functionalized at the aliphatic positions C9 and C14. Several of these compounds were determined to be able to kill leukemia cells and patient-derived primary AML specimens with improved activity compared to parthenolide, exhibiting LC50 values in the low micromolar range. These studies demonstrate that different O−H functionalization chemistries can be applied to elaborate the parthenolide scaffold and that modifications at the C9 or C14 position can effectively enhance the antileukemic properties of this natural product. The C9-functionalized analogs 22a and 25b were identified as the most interesting compounds in terms of antileukemic potency and selectivity toward AML versus healthy blood cells.
Keywords: Parthenolide, Chemoenzymatic synthesis, Enzymatic hydroxylation, Anticancer activity, Leukemia, Late-stage C-H functionalization
Graphical abstract
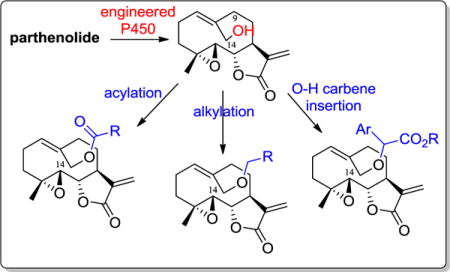
1. Introduction
Sesquiterpene lactones constitute a family of carbocyclic natural products isolated from various plant sources.1 Members of this family sharing an α-methylene-γ-lactone structural motif have recently attracted significant attention because of their promising anti-inflammatory and anticancer properties.2–4 Parthenolide (PTL; 1, Figure 1), in particular, was previously shown to be capable of inducing apoptosis in acute myeloid leukemia (AML) cells and reducing the viability of leukemia stem cells (LCSs).5–7 Because of the hierarchical organization of AML,8, 9 the ability to eradicate LCS is deemed to be a critical feature for the development of next-generation antileukemic drugs.5, 10–13 PTL has a complex mechanism of action, which likely involves targeting of multiple cellular components and pathways. The anticancer activity of this molecule has been associated with its ability to inhibit the NF-κB transcription factor,5, 14, 15 resulting in the down-regulation of anti-apoptotic genes under NF-κB-control. In addition, PTL was found to activate the p53 pathway,16 interfere with epigenetic regulation,16–18 and cause a depletion of glutathione levels in CD34+ AML cells, the latter leading to increased intracellular production of reactive oxygen species and oxidative stress.5, 19 After initial reports on the anticancer properties of PTL,5, 20 this and other sesquiterpene lactones were reported to possess antiproliferative activity against various types of cancer cells.21–28
Figure 1.
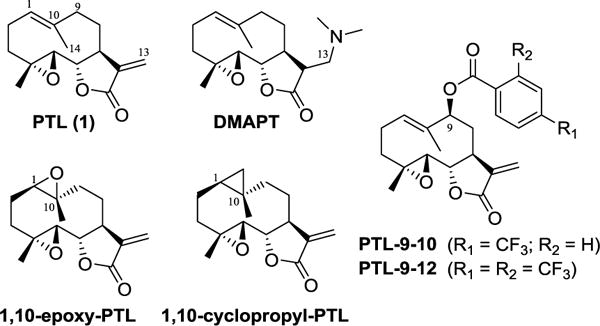
Chemical structure of parthenolide and previously reported parthenolide analogs.
Structural modification of the PTL scaffold has recently been pursued in an attempt to improve the poor water-solubility and pharmacokinetic properties of the molecule and/or to identify analogs with enhanced potency and selectivity. Studies in this area have mainly focused on modification of the C13 position due to its reactivity toward Michael addition (e.g., using amine-based nucleophiles),29–31 and its amenability to functionalization via Heck coupling.32 Since the α-methylene-γ-lactone moiety is critical for the biological activity of the molecule, however, C13 functionalizations have often resulted in a reduction or loss of anticancer potency.31, 32 As an exception, a C13-dimethylamino adduct of parthenolide (DMAPT, Figure 1) was found to possess similar antileukemic properties as PTL, while exhibiting significantly improved water solubility and pharmacokinetic profile upon oral administration in animal models of leukemia.29 Other modifications of this natural product have involved the regio- and stereoselective epoxidation33 and cyclopropanation27 of the C1-C10 double bond, resulting in PTL analogs (i.e., 1,10-epoxy-PTL and 1,10-cyclopropyl-PTL, respectively; Figure 1) with comparable anticancer properties as the parent compound.
As part of our ongoing studies on the late-stage C(sp3)−H functionalization of complex molecules via P450-mediated chemoenzymatic synthesis,34–36 we recently reported the development of two engineered cytochrome P450 catalysts for enabling the selective hydroxylation of position C9 and C14 positions in PTL.33 While the products of these enzymatic reactions, namely 9(S)-hydroxy-PTL and 14-hydroxy PTL, have low antileukemic activity (LC50 > 90 μM against primary AML samples), they represented valuable intermediates for the generation of C9- and C14-functionalized PTL analogs via chemical acylation. Among them, PTL derivatives carrying trifluoromethylated benzoyl groups appended to either the C9 or C14 hydroxyl group (e.g., PTL-9-10 and PTL-9-12; Figure 1) were found to possess improved antileukemic activity compared to PTL, as well as increased selectivity against AML cells versus normal cells (bone marrow isolates).33
In this report, we describe the synthesis and characterization of the antileukemic activity of novel analogs of PTL modified at the aliphatic position C9 and C14 by chemoenzymatic means. We report the development of an improved and scalable protocol for the enzymatic synthesis of the 9(S)-hydroxy- and 14-hydroxy-parthenolide intermediates and the successful application of different O−H functionalization chemistries for the late-stage diversification of the PTL scaffold. The antileukemic activity and cytotoxicity of the resulting analogs was investigated in cell-based assays involving a leukemia cell line, patient-derived AML samples, and healthy umbilical cord blood cells. These studies led to the discovery of new PTL-derived chemical agents with improved antileukemic properties as compared to parthenolide.
2. Results and discussion
2.1. Synthesis of 9(S)-hydroxy-PTL and 14-hydroxy-PTL via P450-catalyzed hydroxylation
CYP102A1 is a self-sufficient cytochrome P450 monooxygenases isolated from Bacillus megaterium.37 While the wild type enzyme recognizes long-chain (C12–C20) fatty acids as preferred substrates, engineered variants of this P450 have proven useful for the hydroxylation of a variety of non-native substrates,38, 39 including terpene natural products.34–36, 40–42 In previous work, we reported the development of a panel of CYP102A1-based catalysts useful for the late-stage oxyfunctionalization of PTL to give 1,10-epoxy-PTL (Figure 1), 9(S)-hydroxy-PTL (2), and 14-hydroxy-parthenolide (3) (Scheme 1).33 In particular, two different variants of this enzyme, called II-C5 and XII-D8, were engineered to afford the hydroxylation of either C9 or C14 site in PTL, respectively, with high regio- and stereoselectivity (C9: 68% regiosel., >99.9% de; C14: 95% regiosel.). More recently, we identified other CYP102A1 variants capable of hydroxylating PTL at the C14 position with good selectivity, including a variant called FL#46 (62% regiosel.; see Experimental Section for further details). In the context of the present study, FL#46 was preferred over XII-D8 due to its better expression levels in E. coli and higher catalytic turnovers in PTL oxidation (1,000 vs. 60 TON).
Scheme 1.
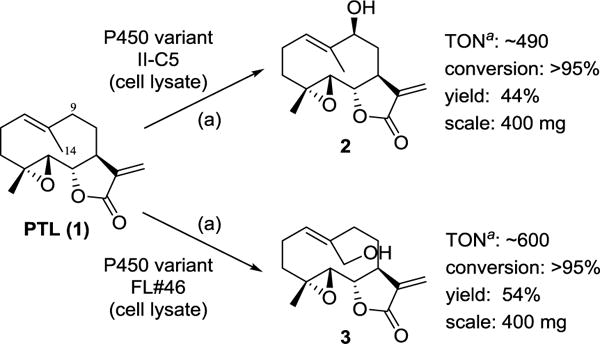
Synthesis of 9(S)-hydroxy-parthenolide (2) and 14-hydroxy-parthenolide (3) via P450-catalyzed transformation of PTL. Reaction conditions: 1 mM PTL, ~ 0.1 mol% P450, 2 μM PTDH, 150 μM NADP+, 50 mM sodium phosphite in 50 mM potassium phosphate buffer (pH 8.0), room temperature, 14 hours. a The representative values of catalytic turnovers (TON) correspond to the formation of the indicated hydroxylation product only. In both reactions, 1,10-epoxy-parthenolide is also produced.
Previously, PTL hydroxylation reactions were carried out using these P450s in purified form in the presence of a NADPH cofactor regeneration system consisting of a thermostable phosphite dehydrogenase (PTDH)43 and sodium phosphite as an inexpensive, sacrificial reductant.33 To streamline the synthesis of the hydroxylated PTL intermediates, we sought to develop an alternative protocol that could bypass the need of protein purification. Since CYP102A1-catalyzed hydroxylations have been previously achieved using E. coli whole-cell systems (i.e., intact P450-expressing cells),40, 44–46 we initially tested the viability of this approach in the context of PTL hydroxylation. Poor conversion of PTL to 2 and 3 (<5%) was obtained in whole-cell PTL biotransformations using II-C5- and FL#46-expressing E. coli cells, respectively. Because the recovery of unreacted PTL was also low (<25%), we attributed these results to side-reactions of the PTL molecule with components of the bacterial cell. More promisingly, we established that PTL transformation with these enzymes can be carried out directly in lysates of II-C5- or FL#46-expressing cells, supplemented with the PTDH-based cofactor regeneration system. Using this approach, preparative-scale (400 mg) PTL hydroxylation reactions could be successfully carried out, resulting in the isolation of 2 and 3 in 44% and 54% yield, respectively (Scheme 1). Notably, these isolated yields correspond to about 89% and 97%, respectively, of the theoretical maximal yields based on the enzyme selectivity at room temperature, thus indicating an excellent recovery of the hydroxylated products. A second oxidation product, corresponding to 1,10-epoxy-PTL, was also isolated during the preparation of 2 and 3 in 51% and 42% yield, respectively. The PTL biotransformation protocol could be further simplified by using lysates of E. coli cells expressing both the P450 and the PTDH enzyme via a two-plasmid system, thereby completing eliminating the need for protein purification. The co-expression of the PTDH protein, however, was also found to decrease the P450 levels in the cells, resulting in somewhat lower isolated yields (33% and 44% for 2 and 3, respectively). Overall, these optimized protocols furnished efficient and scalable routes for the production of the key, hydroxylated intermediates useful for synthesis of the C9- and C14-functionalized analogs.
2.2. Synthesis of PTL analogs via acylation
In our previous study, we found that acylated PTL analogs carrying aromatic substituents (i.e., benzoyl, p-(trifluoromethyl)-benzoyl, and m,m- or o,p-di(trifluoromethyl)-benzoyl groups) at either the C9 or C14 position possess improved antileukemic activity compared to PTL, while exhibiting comparably low cytotoxicity against healthy bone marrow cells.33 To further explore the influence of aromatic substituents on the antileukemic properties of PTL, additional analogs carrying different heteroaryl and biaryl groups at position C14 and C9 were synthesized (4–13, Scheme 2). These compounds were prepared via acylation of the hydroxylated PTL intermediate (2 or 3) using the appropriate acid chloride in the presence of triethylamine and catalytic amounts of 4-dimethylamino-pyridine (DMAP). Upon purification by flash chromatography, the target compounds were isolated in reasonable to good yields (20–55%).
Scheme 2.
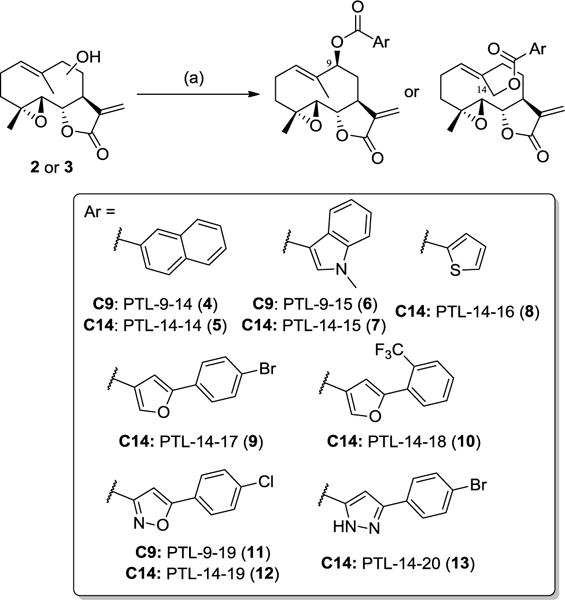
Synthesis of C9- and C14-functionalized PTL analogs via O−H acylation. (a) ArCOCl, Et3N, DMAP, room temperature, 2–4 hours.
2.3. Synthesis of PTL analogs via alkylation
Although the acylated PTL analogs described above were determined to be stable toward hydrolysis in cell-based activity assays, the ester linkage could represent a potential liability in the context of the future application of these compounds in animal models of the disease. These considerations concern in particular the C14-acylated analogs in reason of their potentially higher susceptibility to hydrolysis as a result of the less sterically hindered ester group as compared to the C9-acylated counterparts. Since 3 was found to possess drastically reduced antileukemic activity compared to PTL (LC50 = 100 μM against primary AML specimens)33, hydrolysis of the ester bond in the C14-acylated analogs would result in a dramatic loss of their biological activity. On the basis of these considerations, two additional C14-functionalized analogs we prepared in which a benzyl and a 4-(trifluoromethyl)-benzyl substituent are appended to the C14 site via a non-hydrolizable ether linkage (14–15, Scheme 3). These substituents are structurally related to the benzoyl substituents of the most active PTL analogs identified previously (e.g., PTL-9-10; Figure 1), but they lack the H-bond acceptor group provided by the carbonyl group in the latter compounds. The alkylation reactions could be realized in the presence of the corresponding alkyl bromides and silver oxide as additive, resulting in the isolation of 14 and 15 in 29–37% yields.
Scheme 3.
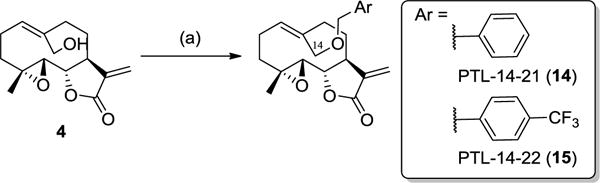
Synthesis of C14-functionalized PTL analogs via O−H alkylation. (a) ArCH2Br, Ag2O, THF, room temperature, 24 hours.
2.4. Synthesis of PTL analogs via O–H carbene insertion
The rhodium-catalyzed insertion of carbenoids into O−H bonds represents a viable route to the functionalization of alcohols under mild and neutral conditions.47 This strategy was deemed particularly attractive in the context of the late-stage derivatization of PTL in reasons of the relatively instability of this natural product at elevated temperature and/or under strongly acidic or alkaline conditions.48 Furthermore, as applied to the synthesis of PTL analogs (Scheme 4), other attractive features of this transformation included: (a) the functionalization of the −OH group in C9/C14 of PTL via a non-hydrolysable ether linkage; (b) the introduction a second ‘arm’ for functional elaboration of the appended substituent (i.e., via the carboxylic group), and (c) the creation of a new stereocenter upon the O−H carbene insertion step, thereby leading to the formation of two epimers with potentially different biological activity.
Scheme 4.
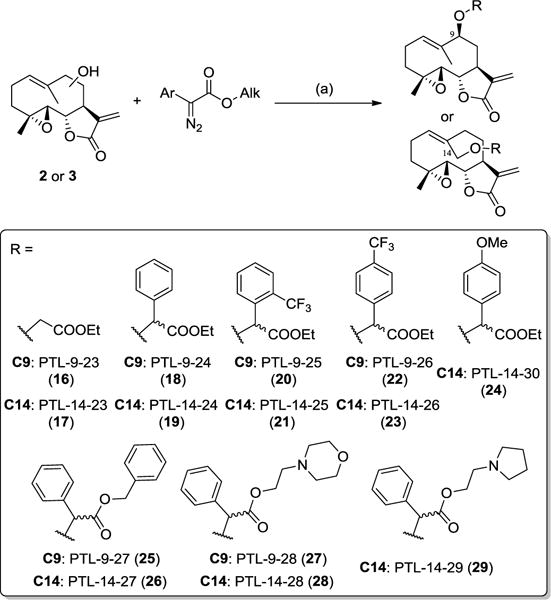
Synthesis of C14-functionalized PTL analogs via O−H carbene insertion. (a) 5mol% Rh2(OAc)4 in dichloromethane, room temperature, 2 hours.
The α-aryl-α-diazo-esters reagents for these reactions were prepared starting from the corresponding arylester derivatives upon reaction with p-acetamidobenzenesulfonyl azide (p-ABSA) and 1,8-diazabicycloundec-7-ene (DBU) in acetonitrile. Optimal conditions for the O−H carbene insertion reaction involved the use of 5 mol% of Rh2(OAc)4 catalyst in dichloromethane with slow addition of the α-diazo-ester in (over)stoichiometric amounts compared to the alcohol. As observed for the acylation reactions (2.2), derivatization of 3 using this chemistry was generally associated with faster kinetics and higher conversions as compared to 2. This trend is consistent with the expectedly higher reactivity of the hydroxyl group in 14-hydroxy-parthenolide (3) vs. that in 9(S)-hydroxy-parthenolide (2) as a result of steric effects. These reactivity differences notwithstanding, all but one of the α-diazo ester reagents tested, including sterically encumbered ones (e.g., benzyl α-phenyl-α-diazo ester), could be coupled to both of the hydroxylated PTL intermediates, resulting in the corresponding series of C9- and C14-functionalized analogs reported in Scheme 4 in moderate to good yields (25–46%).
Except for compounds 16 and 17, all of the O−H insertion products feature a newly introduced stereogenic center at the level of the α-carbon of the ester group. These compounds were purified by flash chromatography and tested as a mixture of the two epimers. For five of the most active analogs within these series, namely 18, 22, 23, 25, and 26, the two epimers were resolved by reverse-phase C18 high-pressure liquid chromatography (HPLC) and tested in pure stereoisomeric form. The isolated, enantiopure compounds failed to readily crystallize and the configuration of the newly generated stereocenter was therefore not determined at this stage.
2.5. Antileukemic activity of PTL analogs
The antileukemic activity of the PTL analogs was initially evaluated using the leukemia cell line M9-ENL1. M9-ENL1 cells are derived from lineage depleted (Lin−) human cord blood cells trasduced with a leukemogenic mixed-lineage leukemia (MLL)-eleven-nineteen leukemia (ENL) fusion gene.49 Injected in NOD/SCID mice, M9-ENL1 cells develop a rapid and fatal pro-B cell acute lymphoblastic leukemia (ALL) disease.11, 49 These cells show an enrichment of embryonic and B-cell progenitor gene sets and exhibit the hallmarks and gene expression patters typical of leukemia stem cells.11 To measure the compound activity on M9-ENL1, cells were treated for 24 hours in the presence or in the absence of the PTL analog, followed by measurement of cell viability using Annexin-V and 7-aminoactinomycin (7-AAD) stains. Half-maximal lethal concentration (LC50) values were obtained from the resulting dose-dependent cell viability curves after normalization to untreated controls (Table 1). In this assay, two of the most active C9-modified PTL analogs identified in the course of our previous work, namely PTL-9-10 and PTL-9-12 (Figure 1), were determined to have an LC50 value of 5 and 2 μM, respectively. These values fall in the same range of those measured for the same compounds in the presence of patient-derived primary AML samples (LC50 = 2.3 and 3.5 μM, respectively).33 In comparison, a LC50 of 5.5 μM was measured for the parent compound, PTL, against M9-ENL1 cells, a value that is slightly lower than that measured against primary AML specimens (LC50 = 6–10 μM)
Table 1.
Antileukemic activity of PTL and its C9- and C14-functionalized analogs as measured based on dose-dependent reduction of cell viability against M9-ENL1 cells. LC50 values are mean ± SD (n = 3).
| Compound | Name | Funct. site | Chemistry | LC50 (μM) |
|---|---|---|---|---|
| PTL | parthenolide | 5.4 ± 0.1 | ||
| 4 | PTL-09-14 | C9 | acylation | 4.5 ± 0.4 |
| 6 | PTL-09-15 | C9 | acylation | 3.8 ± 0.3 |
| 11 | PTL-09-19 | C9 | acylation | 2.6 ± 0.1 |
| 5 | PTL-14-14 | C14 | acylation | 13.8 ± 2.2 |
| 7 | PTL-14-15 | C14 | acylation | 8.2 ± 0.8 |
| 8 | PTL-14-16 | C14 | acylation | 5.9 ± 0.2 |
| 9 | PTL-14-17 | C14 | acylation | 3.8 ± 0.1 |
| 10 | PTL-14-18 | C14 | acylation | 3.7 ± 0.1 |
| 12 | PTL-14-19 | C14 | acylation | 13.7 ± 0.5 |
| 13 | PTL-14-20 | C14 | acylation | 2.9 ± 0.1 |
| 14 | PTL-14-21 | C14 | alkylation | 13.0 ± 1.6 |
| 15 | PTL-14-22 | C14 | alkylation | 4.3 ± 0.1 |
| 16 | PTL-09-23 | C9 | O−H inser. | 41.2 ± 1.2 |
| 18a | PTL-09-24-epi1 | C9 | O−H inser. | 2.3 ± 0.1 |
| 18b | PTL-09-24-epi2 | C9 | O−H inser. | 6.2 ± 1.8 |
| 20 | PTL-09-25 | C9 | O−H inser. | 3.9 ± 0.1 |
| 22a | PTL-09-26-epi1 | C9 | O−H inser. | 4.1 ± 1.1 |
| 22b | PTL-09-26-epi2 | C9 | O−H inser. | 3.3 ± 0.3 |
| 25a | PTL-09-27-epi1 | C9 | O−H inser. | 2.6 ± 0.3 |
| 25b | PTL-09-27-epi2 | C9 | O−H inser. | 3.3 ± 0.6 |
| 28 | PTL-09-28 | C9 | O−H inser. | 26.3 ± 7.9 |
| 17 | PTL-14-23 | C14 | O−H inser. | 23.5 ± 0.9 |
| 19 | PTL-14-24 | C14 | O−H inser. | 8.0 ± 0.6 |
| 21 | PTL-14-25 | C14 | O−H inser. | 3.4 ± 0.1 |
| 23a | PTL-14-26-epi1 | C14 | O−H inser. | 3.1 ± 0.2 |
| 23b | PTL-14-26-epi2 | C14 | O−H inser. | 4.1 ± 0.2 |
| 26a | PTL-14-27-epi1 | C14 | O−H inser. | 4.0 ± 0.5 |
| 26b | PTL-14-27-epi2 | C14 | O−H inser. | 8.0 ± 0.3 |
| 27 | PTL-14-28 | C14 | O−H inser. | 23.5 ± 1.5 |
| 29 | PTL-14-29 | C14 | O−H inser. | 14.8 ± 0.5 |
| 24 | PTL-14-30 | C14 | O−H inser. | 6.4 ± 1.7 |
As shown by the data reported in Table 1, several of the new acylated PTL analogs, and in particular those functionalized at the C9 position, retained potent antileukemic activity, exhibiting LC50 values below 5 μM. These results support the beneficial effect of aromatic substituents at either of these sites, which is in general agreement with the preliminary SAR data gathered in the context of the previously investigated PTL analogs.33 Furthermore, the low micromolar activity of analogs carrying larger aromatic substituents compared to the benzoyl-based groups in PTL-9-10 and PTL-9-12 (e.g., 6, 9, 11, and 13), suggest that there is significant tolerance in terms of steric accessibility in correspondence to both the C9 and C14 site of PTL. At the same time, larger aromatic substituents are clearly better tolerated at the C9 site vs. the C14 site, as indicated by the 2- to 3-fold lower LC50 values for the C9 analogs carrying a naphtyl (4), N-methyl-indolyl (6), and (4-chlorophenyl)isoxazole (11) group as compared to their C14-functionalized counterparts (i.e., 5, 7, and 12, respectively). Interestingly, the most active analogs (LC50 = 2.6–2.9 μM) emerging from this series of compounds, namely 11 and 13, share a heterocycle-containing biaryl group. These structural motifs represent ‘privileged’ scaffolds which are well known for their ability to establish favorable interactions with both hydrophobic/aromatic residues and polar groups in proteins.50, 51
As noted earlier, we were also interested in exploring the antileukemic activity of PTL analogs functionalized via a hydrolytically stable ether linkage. Interestingly, the C14-benzyl ether derivative 14 showed a 2-fold lower cytotoxicity against M9-ENL1 cells compared to its C14-benzoylated counterpart (LC50 = 13 vs. 8 μM). However, potent activity (LC50 = 4.3 μM) could be restored upon installation of a trifluoromethyl group in para position of the phenyl ring in the structurally related analog, 15. More extensive SAR information could be gathered from the activity tests with the series of C9- and C14-functionalized analogs obtained via O–H carbene insertion (16–29; Scheme 4). Both C9 and C14 analogs carrying an appended trifluoromethyl-substituted phenyl group (i.e., 20, 22, 21, 23) showed generally 2- to 3-fold lower LC50 than the phenyl-containing counterparts (18, 19). The beneficial effect of trifluoromethyl substituents is consistent with the trend observed for 15 vs. 14, and it could arise from an improved interaction of these molecules with the cellular targets as a result of their increased hydrophobicity. Furthermore, the greatly reduced in vitro potency of 16 and 17, which contain the side-chain carboxy ester group but lack the aromatic moiety, clearly support the notion that the aromatic substituent is primarily responsible for the improved antileukemic activity of these analogs as compared to PTL.
Importantly, the results corresponding to the 25 epimers, which are the most active analogs within this series, indicated that further functionalization of the ‘carboxylate arm’ is compatible with high antileukemic activity (LC50 = 2.6–3.3 μM). These results prompted us to explore a sub-series of PTL derivatives, in which a polar group (i.e., morpholine, pyrrolidine) is appended to the molecule via the carboxylate moiety. In previous studies, the limited water solubility of PTL was significantly improved by preparing a C13-dimethylamino adduct of PTL (DMAPT; Figure 1) via Michael addition of the dimethylamine to the α-methylene-γ-lactone moiety.29 Since this moiety is also critical for the anticancer activity of the natural product,27, 31 we envisioned that incorporating a tertiary amine functionality into the C9/C14-linked substituent could provide an alternative strategy to improve the water solubility of the PTL analogs without altering the critical, electrophilic C13 site. Accordingly, compounds 27, 28, and 29 were synthesized, which showed improved solubility in buffered solutions at neutral pH. Notably, these analogs maintained the ability to kill M9-ENL1 cells in the low micromolar range (LC50 = 15–25 μM), although their activity was reduced by about 3 to 5 folds compared to PTL.
As noted earlier, the O–H insertion reaction creates a new stereogenic center resulting in the formation of two epimeric products. Since preliminary tests identified 18, 22, 23, 25, and 26 as the most promising analogs in this series, the two epimeric forms of these compounds were resolved, isolated, and tested individually. As shown by the data in Table 2 the two epimers exhibited comparable antileukemic activity in all cases except for 18 and 26, for which one of the epimers showed about three and two fold lower LC50, respectively, than the other. These results thus indicate that the configuration of the α-carbon atom in the C9- or C14-linked substituents has in most cases only moderate influence on the biological activity of these molecules.
Table 2.
Cytotoxicity (LC50) of PTL and selected PTL analogs against patient-derived primary Acute Myeloid Leukemia samples (AML01) and human umbilical cord blood cells (hUBC) as determined based on cell viability assays. IC50 values refer to the compound inhibitory activity on hUBC as measured via a plate-based colony formation assay. LC50 and IC50 values are mean ± SD (n = 3). n.d. = not determined.
| Compound | Name | AML01 LC50 (μM) |
hUBC LC50 (μM) |
hUBC IC50 (μM) |
|---|---|---|---|---|
| PTL | parthenolide | 6.1 ± 0.3 | >20 (42)a | 12.5 ± 0.6 |
| 13 | PTL-14-20 | 2.2 ± 0.1 | n.d. | n.d. |
| 23a | PTL-14-26-epi1 | 2.5 ± 0.1 | 5.4 ± 0.5 | 7.5 ± 0.7 |
| 23b | PTL-14-26-epi2 | 3.1 ± 0.3 | >5 (8.8)a | 4.6 ± 0.5 |
| 26a | PTL-14-27-epi1 | 3.2 ± 0.1 | 3.7 ± 0.4 | 6.2 ± 0.6 |
| 26b | PTL-14-27-epi2 | 5.1 ± 0.5 | 5.5 ± 0.6 | 5.4 ± 0.5 |
| 28 | PTL-14-28 | 22.4 ± 5.6 | n.d. | n.d. |
| 29 | PTL-14-29 | 23.6 ± 9.3 | n.d. | n.d. |
| 24 | PTL-14-30 | 8.5 ± 1.0 | n.d. | n.d. |
| 18a | PTL-9-24-epi1 | 2.6 ± 0.2 | n.d. | n.d. |
| 18b | PTL-9-24-epi2 | 10.0 ± 2.1 | n.d. | n.d. |
| 22a | PTL-9-26-epi1 | 3.4 ± 0.3 | >5 (7.3)a | 13.2 ± 1.0 |
| 22b | PTL-9-26-epi2 | 2.6 ± 0.2 | 2.7 ± 0.3 | 2.4 ± 0.2 |
| 25a | PTL-9-27-epi1 | 5.1 ± 1.0 | >5 (12.2)a | 31.6 ± 5 |
| 25b | PTL-9-27-epi2 | 3.1 ± 0.1 | >5 (10.9)a | 5.0 ± 0.4 |
| 27 | PTL-9-28 | 40 ± 20 | n.d. | n.d. |
Extrapolated values beyond concentration range tested.
Next, the most promising PTL analogs from both the C9 and C14 series were tested for their ability to kill patient-derived AML primary samples. As summarized by the data reported in Table 2, these compounds exhibited potent cytotoxicity also in the context of these cells, with LC50 values falling in the low micromolar range. Similar experiments with the less active analogs 27 and 29 further evidenced a very good correlation between the cytotoxic activities observed with primary AML specimens and those measured with M9-ENL1 cells.
2.6. Toxicity against human umbilical cord blood cells
The PTL analogs with the most promising antileukemic properties (i.e., 22, 23, 25, 26) were then further evaluated with respect to their toxicity against healthy blood cells by means of a cell viability assay with human umbilical cord blood cell samples from consent donors. In addition, these compounds were tested in a colony formation inhibition assay, which provides a means to assess the stem cell potential of normal hematopoietic cells. In these assays, the parent molecule PTL was determined to exhibit an estimated LC50 of 42 μM and IC50 of 12.5 μM, respectively (Table 2). Compared to PTL, the analogs were generally found to possess enhanced toxicity against human umbilical cord blood cells, with LC50 values ranging between 3 and 12 μM. In contrast, the ability of these compounds to inhibit colony formation was comparable or only slightly increased as compared to that of PTL (Table 2). On the basis of both the antileukemic activity and toxicity studies, the C9-functionalized analogs 22a and 25b emerged as the most interesting compounds from these studies. 25-epi2, in particular, not only possesses improved antileukemic properties compared to PTL but its LC50 against human umbilical cord blood remains 3.5-fold lower than that against primary AML samples, thus indicating a good degree of selectivity toward leukemia cells over healthy cells. Importantly, the branched structure of the C9 substituent in 25b combined with the accessibility of structurally diverse aryl substituents at this site as derived from the SAR information gained with the other analogs suggest that there exist significant opportunities for further optimization of this compound via structural modification of both the aryl and carboxylate ‘arm’.
3. Conclusions
A series of novel C9- and C14-functionalized PTL analogs was synthesized and evaluated for anti-proliferative activity against leukemia cells, primary AML samples, and toxicity against healthy blood cells and hematopoietic cells. Several members of these libraries were determined to be capable of killing leukemia cells with improved activity compared to the parent compound parthenolide, exhibiting LC50 values in the low micromolar range (< 3–4 μM). Expanding upon our previous findings, these studies demonstrate not only that different O−H functionalization chemistries can be applied to diversify this scaffold, but also that late-stage derivatization at the C9 or C14 position can effectively enhance the antileukemic properties of this natural product. The C9-functionalized analogs 22a and 25b emerged as the most interesting compounds from the present study in terms of antileukemic potency and selectivity toward AML versus normal cells. Finally, the discovery of improved PTL analogs where the C9 or C14 substituent is introduced via a non-hydrolyzable ether linkage bears particular relevance toward the future development and application of these parthenolide-based agents in the context of in vivo studies.
4. Materials and Methods
4.1. Reagents and substrates
All solvents and reagents were purchased from commercial suppliers (Sigma-Aldrich, TCI, Fluka, and Tocris Bioscience) and used without any further purification, unless stated otherwise. All dry reactions were carried out under argon or nitrogen in oven-dried glassware with magnetic stirring using standard gas-light syringes, cannulae and septa. Silica gel chromatography purifications were carried out using AMD Silica Gel 60 230–400 mesh. 1H and 13C NMR spectra were measured on a Bruker DPX-400 (operating at 400 MHz for 1H and 100 MHz for 13C) or a Bruker DPX-500 (operating at 500 MHz for 1H and 125 MHz for 13C). Mass spectrometry data were collected via direct infusion on a Thermo Scientific LTQ Velos ESI/ion-trap mass spectrometer. HPLC purifications were carried out using a Shimadzu LC-2010A HT instrument equipped with a Grace, Vision HT-C18 HL, 5μ column and using Solvent A (H2O with 1% trifluoroacetic acid) and Solvent B (CH3CN with 1% trifluoroacetic acid) at a flow rate of 1 mL/min. Gradient method: 10% solvent B for 2 min, from 10% to 25% solvent B over 0.5 min, from 25% to 80% solvent B over 26 min, from 80% to 90% solvent B over 2 min, then 90% solvent B for 3 min. Gas chromatography analyses were carried out on a Shimadzu GC2010 using a Restek RTX-5 column (15 m × 0.25 mm × 0.25 μm film) and a FID detector. The following separation method was used: 1 μL injection, 200 °C inlet, 300 °C detector, 130 °C oven, 12 °C/min gradient to 290 °C and 290 °C for 2 min.
4.2. Expression of P450 variants
P450s were expressed from pCWori-based vectors as described previously.34 The CYP102A1 variant FL#46 contains the following mutations: R47C, V78A, F87I, K94I, P142S, T175I, A184V, F205C, S226R, H236Q, E252G, R255S, A290V, and L353V. Briefly, the cultures were grown in Terrific Broth (TB) medium (ampicillin, 100 mg L−1) at 37°C and 200 rpm until OD600 reached 0.6 and then induced with 0.5 mM β-D-1-thiogalactopyranoside (IPTG) and 0.3 mM δ-aminolevulinic acid (ALA). After induction, the cultures were shaken at 150 rpm and 27°C and harvested by centrifugation (4,000 rpm) after 20 hours. The cells were resuspended in 50 mM potassium phosphate buffer (pH 8.0) and stored at −80°C for up to 3 months. Cell lysates were prepared by sonication followed by clarification via centrifugation (14,000 rpm) and used immediately for the enzymatic hydroxylation reactions. PTDH was expressed from pET15b-based vectors in BL21(DE3) cells and purified using Ni-affinity chromatography according to the published procedure.43 E. coli cells expressing both P450 and PTDH were prepared by co-transformation of BL21(DE3) cells with the pCWori vector encoding for the P450 enzyme and the pET21b vector encoding for PTDH. Cell cultures were prepared as described above with the exception that induction was carried out using 0.5 mM IPTG, 0.3 mM ALA and 0.06% L-arabinose.
4.3. Enzymatic hydroxylation reactions
4.3.1. General procedure for enzymatic hydroxylation using P450 lysate
To prepare 2, a 1.6 L scale reaction was set up by the slow addition of 400 mg parthenolide (1) dissolved in 32 ml dimethylsulfoxide (DMSO) (final concentration 1 mM, 2% DMSO) to a 50 mM phosphate buffer (pH 8.0) solution. The cell lysate (~ 100 mL from 4 liters of cell culture) containing the P450 variant II-C5 (final P450 concentration ~ 1 μM) was added to the mixture followed by the addition of PTDH (2 μM), NADP+ (150 μM), and sodium phosphite (50 mM). The reaction mixture was divided into four 2-L Erlenmeyer flasks and shaken for 14 hours at 25°C in an incubator shaker (100 rpm). The crude product was extracted with dichloromethane (3 × 400 mL), dried with sodium sulfate and concentrated under reduced pressure. The crude solid was purified by flash chromatography (hexanes/ethyl acetate: 1/2) to afford 2 (187 mg, 44% (89% of theoretical maximum)) and 1,10-epoxy-parthenolide (217 mg, 51%). To prepare 3, an identical procedure was followed with the exception that cell lysate containing the P450 variant FL#46 (final concentration ~1 μM) was used. Purification of the crude solid by flash chromatography (hexanes/ethyl acetate: 1/2) yielded 3 (230 mg, 54% (97% of theoretical maximum)) and 1,10-epoxy-parthenolide (179 mg, 51%).
4.3.2. General procedure for enzymatic hydroxylation using E. coli cells expressing both the P450 and PTDH enzyme
A similar procedure as described in Section 4.3.1 was used for preparation of 2 and 3 using cell lysates containing the P450 biocatalyst (either II-C5 or FL#46, respectively) and PTDH. Briefly, a 0.2 L scale reaction was set up by the slow addition of 50 mg parthenolide (1) in 4 ml DMSO (final concentration 1 mM, 2% DMSO) to a 50 mM phosphate buffer (pH 8.0) solution. The cell lysate was added to the parthenolide solution followed by addition of NADP+ (150 μM) and sodium phosphite (50 mM). The reaction mixture was shaken for 14 hours at 25°C in an incubator shaker (100 rpm). The crude product was extracted with dichloromethane (3 × 50 mL), dried with sodium sulfate and concentrated under reduced pressure. The crude solid was purified by flash chromatography (hexanes/ethyl acetate: 1/2) to afford 2 (35 mg, 33%) or 3 (47 mg, 44%).
4.3.3. General procedure for whole-cell parthenolide hydroxylation
Cell expressing P450 variant IIC5 or FL#46 were prepared as described previously (Section 4.2) and resuspended in nitrogen-free M9 minimal media. To prepare the hydroxylated products 2 or 3, the P450-expressing cells were diluted in 100 ml nitrogen-free M9 minimal media to a final OD600 of 5. Parthenolide (25 mg dissolved in 2 ml DMSO) was added to the cell suspension, which was shaken at 100 rpm for 16 hours at 25°C. After centrifugation (4,000 rpm), the supernatant was extracted with dichloromethane and the extract was analyzed by gas chromatography.
4.4. Synthetic methods
4.4.1. General procedure for acylation reactions
To a solution of 2 or 3 in anhydrous dichloromethane (5 mL) under argon atmosphere was added 4-dimethylaminopyridine (0.5 equiv.), triethylamine (10 equiv.), and the corresponding acid chloride (5 equiv.). The reaction mixture was stirred at room temperature until complete disappearance of the starting material. At this point, the reaction mixture was added with saturated sodium bicarbonate solution (5 mL) and extracted with dichloromethane (2 × 10 mL). The organic layer was dried over sodium sulfate, concentrated under reduced pressure, and the acylated product was isolated by flash chromatography using a ethyl acetate:hexane mixture (1:1).
4.4.2. General procedure for alkylation reactions
A mixture of 2 or 3 (1 equiv.) and Ag2O (2 equiv.) in THF (5 mL) was stirred at room temperature under an argon atmosphere. To this solution was added the benzyl alcohol (1.5 equiv.) and the mixture was stirred for 24 hours at room temperature. After completion of the reaction, the solvent was removed in vacuo, and the crude product was purified by flash chromatography using a ethyl acetate:hexane mixture (1:1).
4.4.3. General procedure for preparation of 2-diazoacetate reagents
4.4.3.1. Synthesis of 2-aryl-acetate esters
The corresponding alcohol (1.5 equiv.) and acid (1 equiv.) were dissolved in dry dichloromethane (15 mL). The solution was cooled to 0 °C and 1-ethyl-3-(3-dimethylaminopropyl) carbodiimide (EDC) (1.5 equiv.) and catalytic amounts of DMAP were added. The reaction mixture was stirred at room temperature overnight. The reaction was then diluted with water and extracted with dichloromethane (2 × 20 mL). The combined organic extract was dried with Na2SO4 and concentrated under reduced pressure. The desired 2-aryl-acetate ester product was obtained through purification by flash column chromatography using a ethyl acetate:hexane mixture (1:4).
4.4.3.2. Synthesis of 2-aryl-2-diazo-acetate esters
All the diazo reagents except for the commercially available ethyl 2-diazo-acetate (EDA) used for preparation of 16 and 17 were prepared according to the following method. The appropriate 2-aryl-acetate ester from 4.4.3.1. (1 equiv.) and para-acetamidobenzene-sulfonyl azide (1.2 equiv.) were dissolved in acetonitrile (10 mL) and cooled to 0°C under nitrogen atmosphere. Then, DBU (1.5 equiv.) was added dropwise under stirring. The mixture was stirred overnight at room temperature. The solvent was evaporated under reduced pressure and the residue was extracted with ethyl acetate (2 × 20 mL). The organic layer was further washed with brine (10 mL), dried with Na2SO4, concentrated under reduced pressure and crude was purified via flash chromatography using a ethyl acetate:hexane mixture (1:9).
4.4.4. General procedure for O–H carbene insertion reactions
A mixture of 2 or 3 (1 equiv.) and Rh2(OAc)4 (5 mol%) in dry dichloromethane (3 mL) was stirred at room temperature under argon atmosphere. To this solution was added the appropriate 2-aryl-2-diazo-acetate ester from 4.4.3.2. (2 equiv.) in 2 mL of dichloromethane dropwise over 15–20 minutes. The reaction mixture was then stirred for additional 2 hours at room temperature. After completion of the reaction as determined by TLC, the solvent was removed under reduced pressure and the crude product was purified via flash chromatography using a ethyl acetate:hexane mixture (1:1).
4.5. Compound characterization data
4.5.1. 9(S)-hydroxy-parthenolide (2)
1H NMR (500 MHz, CDCl3): δ 1.34 (s, 3 H), 1.76 (s, 3 H), 1.97−2.06 (m, 1 H), 2.15−2.27 (m, 4 H), 2.50 (dq, 1 H, J = 5.2, 12.4 Hz), 2.70 (d, 1 H, J = 8.7 Hz), 2.83−2.90 (m, 1 H), 3.86 (t, 1 H, J = 8.5 Hz), 4.27 (dd, 1 H, J = 2.2, 10.5 Hz), 5.42 (d, 1 H, J = 11.3 Hz), 5.69 (d, 1 H, J = 3.2 Hz), 6.36 (d, 1 H, J = 3.6 Hz); 13C NMR (125 MHz, CDCl3): δ 10.9, 17.4, 23.9, 36.1, 38.0, 44.5, 61.4, 66.2, 79.7, 81.5, 121.6, 126.5, 136.6, 138.3, 168.8; HRMS (ESI) calcd for C15H20O4 [M+H]+ m/z: 265.14; found: 265.14.
4.5.2. 14-hydroxy-parthenolide (3)
1H NMR (500 MHz, CDCl3): δ 1.31 (s, 3 H), 1.32−1.38 (m, 1 H), 1.82−1.1.92 (m, 1 H), 2.09–2.16 (m, 1 H), 2.20− 2.32 (m, 3 H), 2.50 (dq, 1 H, J = 5.0, 13.4 Hz), 2.82–2.90 (m, 3 H), 3.92 (t, 1 H, J = 8.7 Hz), 4.16 (d, 1 H, J = 11.3 Hz), 4.46 (d, 1 H, J = 11.8 Hz), 5.43 (dd, 1 H, J = 4.1, 12.4 Hz), 5.68 (d, 1 H, J = 3.2 Hz), 6.39 (d, 1 H, J = 3.8 Hz); 13C NMR (125 MHz, CDCl3): δ =16.9, 23.7, 31.4, 36.3, 47.3, 59.8, 61.1, 66.2, 82.4, 121.4, 129.0, 137.8, 139.2, 169.3. HRMS (ESI) calcd for C15H20O4 [M+H]+ m/z: 265.14; found: 265.14.
4.5.3. (3aS,5S,9aR,10aS,10bS,E)-6,9a-dimethyl-3-methylene-2oxo2,3,3a,4,5,8,9,9a,10a,10bdecahydrooxireno[2′,3′:9,10]cycl odeca [1,2-b]furan-5-yl 2-naphthoate (4)
Standard procedure was applied using 9(S)-hydroxy-parthenolide (2) (10 mg, 0.030 mmol), 4-dimethylaminopyridine (0.015 mmol), triethylamine (0.30 mmol), and substituted acid chloride (0.15 mmol). Isolated PTL-09-14: 6 mg, 37 % yield. 1H NMR (400 MHz, CDCl3): δ = 8.66 (s, 1H), 8.11−8.09 (m, 1H), 8.03 (d, J = 6.4 Hz, 1H), 7.96 (d, J = 6.8 Hz, 2H), 7.68 (t, J = 5.6 Hz, 1H), 7.63 (t, J = 6.4 Hz, 1H), 6.46 (d, J = 2.8 Hz, 1H), 5.82 (d, J = 2.4 Hz, 1H), 5.70 (d, J = 1.6 Hz, 1H), 5.59−5.57 (m, 1H), 4.00 (t, J = 7.2 Hz, 1H), 3.09 (brs, 1H), 2.84 (d, J = 7.2 Hz, 1H), 2.30−2.25 (m, 5H), 1.95 (s, 3H), 1.43 (s, 3H), 1.36−1.32 (m, 1H) ppm, MS (ESI) calcd for C26H26O5 [M+Na]+ m/z: 441.17; found: 441.1.
4.5.4. ((3aR,9aR,10aS,10bS,Z)-9a-methyl-3-methylene-2-oxo 2,3,3a,4,5,8,9,9a,10a,10bdecahydrooxireno[2′,3′:9,10]cyclodec a[1,2-b]furan-6-yl)methyl 2-naphthoate (5)
Standard procedure was applied using 14-hydroxy-parthenolide (3) (8 mg, 0.030 mmol), 4-dimethylaminopyridine (0.015 mmol), triethylamine (0.30 mmol), and substituted acid chloride (0.15 mmol). Isolated PTL-14-14: 5 mg, 39 % yield. 1H NMR (400 MHz, CDCl3): δ = 8.57 (s, 1H), 8.02 (d, J = 8.8 Hz, 1H), 7.94−7.87 (m, 3H), 7.62−7.55 (m, 2H), 6.36 (d, J = 3.2 Hz, 1H), 5.63 (d, J = 3.2 Hz, 1H), 5.61 (dd, J = 4.0, 8.4 Hz, 1H), 5.15 (d, J = 12 Hz, 1H), 4.90 (d, J = 12.0 Hz, 1H), 3.95 (t, J = 8.4 Hz, 1H), 2.86−2.77 (m, 3H), 2.72−2.61 (m, 1H), 2.38−2.35(m, 1H), 2.27−2.18(m, 3H), 1.95−1.87 (m, 1H), 1.39−1.31 (m, 4H) ppm, 13C NMR (125 MHz, CDCl3): δ = 169.0, 166.6, 138.9, 135.6, 133.5, 131.9, 131.1, 129.3, 128.5, 128.4, 127.8, 126.9, 126.8, 124.9, 121.5, 82.4, 66.2, 61.6, 61.0, 47.3, 36.6, 36.2, 31.2, 24.0, 17.0 ppm, MS (ESI) calcd for C26H26O5 [M+Na]+ m/z: 441.17; found: 441.2.
4.5.5. (3aS,5S,9aR,10aS,10bS,E)-6,9a-dimethyl-3-methylene-2-oxo-2,3,3a,4,5,8,9,9a,10a,10b decahydrooxireno [2′,3′:9,10] cyclodeca[1,2-b]furan-5-yl 1-methyl-1H-indole-3-carboxylate (6)
Standard procedure was applied using 9(S)-hydroxy-parthenolide (2) (10 mg, 0.030 mmol), 4-dimethylaminopyridine (0.015 mmol), triethylamine (0.30 mmol), and substituted acid chloride (0.15 mmol). Isolated PTL-09-15: 3 mg, 18 % yield. 1H NMR (400 MHz, CDCl3): δ = 8.20 (d, J = 4.4 Hz, 1H), 7.87 (d, J = 3.2 Hz, 1H), 7.42−7.35 (m, 3H), 6.44 (s, 1H), 5.80 (s, 1H), 5.67 (d, J = 8.8 Hz, 1H), 5.77 (d, J = 8.0 Hz. 1H), 3.98−3.90 (m, 4H), 3.08 (brs, 1H), 2.87−2.81 (m, 1H), 2.58−2.10(m, 5H), 1.93 (s, 3H), 1.42 (s, 3H), 1.41−1.32 (s, 1H) ppm, MS (ESI) calcd for C25H27NO5 [M+Na]+ m/z: 444.2; found: 444.1.
4.5.6. ((3aR,9aR,10aS,10bS,Z)-9a-methyl-3-methylene-2-oxo-2,3,3a,4,5,8,9,9a,10a,10b-decahydrooxireno[2′,3′:9,10]cyclodeca [1,2-b]furan-6-yl)methyl 1-methyl-1H-indole-3-carboxylate (7)
Standard procedure was applied using 14-hydroxy-parthenolide (3) (7 mg, 0.026 mmol), 4-dimethylaminopyridine (0.013 mmol), triethylamine (0.26 mmol), and substituted acid chloride (0.13 mmol). Isolated PTL-14-15: 4 mg, 36 % yield. 1H NMR (400 MHz, CDCl3): δ = 8.13 (d, J = 7.6 Hz, 1H), 7.74 (s, 1H), 7.37−7.27 (m, 3H), 6.34 (d, J = 3.6 Hz, 1H), 5.61 (d, J = 2.8 Hz, 1H), 5.55 (dd, J = 4.0, 9.6 Hz, 1H), 5.10 (d, J = 12 Hz, 1H), 4.85 (d, J = 12 Hz, 1H), 3.93 (t, J = 8.8 Hz, 1H), 3.84 (s, 3H), 2.82−2.78 (m, 3H), 2.64−2.59 (m, 1H), 2.36−2.13 (m, 4H), 1.88−1.84 (m, 1H), 1.33 (s, 3H), 1.30−1.08 (m,1H) ppm, 13C NMR (125 MHz, CDCl3): δ = 178.2, 169.1, 151.1, 139.0, 137.2, 135.1, 134.2, 131.2,123.0, 122.1, 121.5, 116.1, 109.9, 82.4, 66.2, 61.1, 59.9, 47.3, 36.5, 36.2, 33.5, 31.1, 23.9, 17.0 ppm, MS (ESI) calcd for C25H27NO5 [M+Na]+ m/z: 444.2; found: 444.3.
4.5.7. ((3aR,9aR,10aS,10bS,Z)-9a-methyl-3-methylene-2-oxo-2,3,3a,4,5,8,9,9a,10a,10b-decahydrooxireno[2′,3′:9,10]cyclodeca [1,2-b]furan-6-yl)methyl thiophene-2-carboxylate (8)
Standard procedure was applied using 14-hydroxy-parthenolide (3) (8 mg, 0.030 mmol), 4-dimethylaminopyridine (0.015 mmol), triethylamine (0.30 mmol), and substituted acid chloride (0.15 mmol). Isolated PTL-14-16: 6 mg, 53 % yield. 1H NMR (400 MHz, CDCl3): δ = 7.80 (d, J = 3.6 Hz, 1H), 7.57 (d, J = 4.8 Hz, 1H), 7.12 (t, J = 4.8 Hz, 1H), 6.35 (d, J = 3.2 Hz, 1H), 5.63 (d, J = 2.8 Hz, 1H), 5.56 (dd, J = 3.6, 8.8 Hz, 1H), 4.98 (d, J = 12 Hz, 1H), 4.85 (d, J = 12 Hz, 1H), 3.97 (t, J = 8.8 Hz, 1H), 2.84−2.80 (m, 2H), 2.69−2.78 (m,1H), 2.63−2.52 (m, 1H), 2.41−2.32 (m, 3H), 2.24−2.17 (m,1H), 2.01−1.82 (m, 1H), 1.33−1.29 (m, 4H) ppm, 13C NMR (125 MHz, CDCl3): δ = 169.1, 161.9, 139.0, 133.9, 133.1, 132.9, 132.7, 132.2, 128.0, 121.5, 82.5, 66.2, 62.3, 61.0, 47.2, 36.9, 36.2, 31.7, 24.1,16.9 ppm, MS (ESI) calcd for C20H22O5S [M+Na]+ m/z: 397.1; found: 397.2.
4.4.8. ((3aR,9aR,10aS,10bS,Z)-9a-methyl-3-methylene-2-oxo-2,3,3a,4,5,8,9,9a,10a,10b-decahydrooxireno[2′,3′:9,10]cyclodeca [1,2-b]furan-6-yl)methyl 5-(4-bromophenyl)furan-2-carboxylate (9)
Standard procedure was applied using 14-hydroxy-parthenolide (3) (10 mg, 0.037 mmol), 4-dimethylaminopyridine (0.018 mmol), triethylamine (0.37 mmol), and substituted acid chloride (0.18 mmol). Isolated PTL-14-17: 6 mg, 31 % yield. 1H NMR (400 MHz, CDCl3): δ = 7.57−7.52 (m, 4H), 7.24 (d, J = 3.2 Hz, 1H), 6.74 (d, J = 3.6 Hz, 1H), 6.37 (d, J = 3.2 Hz, 1H), 5.63 (d, J =2.4 Hz, 1H), 5.58 (dd, J = 3.6, 8.8 Hz, 1H), 4.98 (d, J =12.0 Hz, 1H), 4.90 (d, J= 12 Hz, 1H), 4.0 (t, J= 8.4 Hz, 1H), 2.85−2.81 (m, 2H), 2.71−2.68 (m, 1H), 2.55−2.47 (m, 1H), 2.35−2.32 (m, 1H), 2.25−2.20 (m, 3H), 2.07−1.98 (m, 1H), 1.37−1.29 (m, 4H) ppm, 13C NMR (125 MHz, CDCl3): δ = 169.0, 158.4, 156.8, 143.4, 139.0, 132.9, 132.5, 132.1, 128.1, 126.1, 123.4, 121.5, 120.5, 107.4, 82.2, 66.2, 62.0, 61.1, 47.3, 37.0, 36.2, 31.6, 24.1, 16.9 ppm, MS (ESI) calcd for C26H25BrO6 [M+Na]+ m/z: 535.1; found: 535.2.
4.4.9. ((3aR,9aR,10aS,10bS,Z)-9a-methyl-3-methylene-2-oxo-2,3,3a,4,5,8,9,9a,10a,10b-decahydrooxireno[2′,3′:9,10]cyclodeca [1,2-b]furan-6-yl)methyl 5-(2-(trifluoromethyl)phenyl)furan-2-carboxylate (10)
Standard procedure was applied using 14-hydroxy-parthenolide (3) (10 mg, 0.037 mmol), 4-dimethylaminopyridine (0.018 mmol), triethylamine (0.37 mmol), and substituted acid chloride (0.18 mmol). Isolated PTL-14-18: 7 mg, 37 % yield. 1H NMR (400 MHz, CDCl3): δ = 7.78 (d, J = 7.6 Hz, 1H), 7.71 (d, J = 7.6 Hz, 1H), 7.62 (t, J = 8.0 Hz, 1H), 7.53 (t, J = 6.8 Hz, 1H), 7.28−7.25 (m, 1H), 6.78 (s, 1H), 6.32 (d, J = 2.8 Hz, 1H), 5.59−5.54 (m, 2H), 4.99 (d, J =12 Hz, 1H), 4.89 (d, J = 12 Hz, 1H), 3.97 (t, J = 8.8 Hz, 1H), 2.82−2.79 (m, 1H), 2.71−2.68 (m, 1H), 2.59−2.45 (m, 1H), 2.34−2.32 (m, 1H), 2.24−2.18 (m, 3H), 2.03−1.97 (m, 2H), 1.38−1.28 (m, 4H) ppm, 13C NMR (125 MHz, CDCl3): δ = 169.1, 158.4, 154.3, 139.0, 133.0, 132.4, 132.0, 130.5, 129.3, 126.8, 121.4, 120.0, 114.7, 112.1, 82.2, 66.2, 62.2, 61.1, 47.3, 46.0, 37.1, 36.2, 31.5, 24.2, 16.9 ppm, MS (ESI) calcd for C27H25F3O6[M+Na]+ m/z: 525.1; found: 525.0.
4.4.10. (3aR,5S,9aR,10aS,10bS,E)-6,9a-dimethyl-3-methylene-2-oxo-2,3,3a,4,5,8,9,9a,10a,10b-decahydrooxireno[2′,3′:9,10] cyclodeca[1,2-b]furan-5-yl 5-(4-chlorophenyl)isoxazole-3-carboxylate (11)
Standard procedure was applied using 9(S)-hydroxy-parthenolide (2) (10 mg, 0.037 mmol), 4-dimethylaminopyridine (0.018 mmol), triethylamine (0.37 mmol), and substituted acid chloride (0.18 mmol). Isolated PTL-9-19: 9 mg, 47 % yield. 1H NMR (400 MHz, CDCl3): δ = 7.75 (d, J = 8.0 Hz, 2H), 7.48 (d, J = 8.4 Hz, 2H), 6.90 (s, 1H), 6.38 (s, 1H), 5.71 (s, 1H), 5.65 (d, J = 10.8 Hz, 1H), 5.52 (d, J = 10.8 Hz, 1H), 3.92 (t, J = 8.8 Hz, 1H), 2.99 (brs, 1H), 2.75 (d, J = 8.8 Hz, 1H), 2.57−2.18 (m, 5H), 1.84 (s, 3H), 1.38−1.27 (m, 4H) ppm, 13C NMR (125 MHz, CDCl3): δ = 170.8, 168.4, 158.8, 156.7, 137.7, 137.1, 132.3, 129.5, 129.0, 127.2, 124.9, 122.1, 100.2, 82.5, 81.3, 65.9, 61.3, 44.0, 35.9, 35.7, 23.8, 17.3, 11.8 ppm, MS (ESI) calcd for C25H24ClNO6 [M+Na]+ m/z: 492.1; found: 492.1.
4.4.11. ((3aR,9aR,10aS,10bS,Z)-9a-methyl-3-methylene-2-oxo-2,3,3a,4,5,8,9,9a,10a,10b-decahydrooxireno[2′,3′:9,10]cyclodeca [1,2-b]furan-6-yl)methyl 5-(4-chlorophenyl)isoxazole-3-carboxylate (12)
Standard procedure was applied using 14-hydroxy-parthenolide (3) (10 mg, 0.037 mmol), 4-dimethylaminopyridine (0.018 mmol), triethylamine (0.37 mmol), and substituted acid chloride (0.18 mmol). Isolated PTL-14-19: 9 mg, 47 % yield. 1H NMR (400 MHz, CDCl3): δ = 7.73 (d, J = 8.4 Hz, 2H), 7.48 (d, J = 8.8 Hz, 2H), 6.90 (s, 1H), 6.35 (s, 1H), 5.62 (s, 1H), 5.58 (brs, 1H), 5.08 (d, J =12 Hz, 1H), 4.93 (d, J = 12 Hz, 1H), 4.02 (t, J = 8.4 Hz, 1H), 2.84−2.79 (m, 2H), 2.73−2.70 (m, 1H), 2.61−2.52 (m,1H), 2.36−2.34 (m, 1H), 2.25−2.19 (m, 3H), 2.07−1.95 (m,1H), 1.38−1.33 (m, 1H), 1.24 (s, 3H) ppm, 13C NMR (125 MHz, CDCl3): δ = 170.8, 169.1, 159.8, 156.5, 138.9, 137.1, 133.1, 132.3, 129.5, 127.1, 124.8, 121.5, 100.1, 82.1, 66.3, 62.9, 61.0, 47.3, 36.9, 36.1, 31.3, 24.2, 16.9 ppm, MS (ESI) calcd for C25H24ClNO6 [M+Na]+ m/z: 492.12; found: 492.3.
4.4.12. ((3aR,9aR,10aS,10bS,Z)-9a-methyl-3-methylene-2-oxo-2,3,3a,4,5,8,9,9a,10a,10b-decahydrooxireno[2′,3′:9,10]cyclodeca [1,2-b]furan-6-yl)methyl 3-(4-bromophenyl)-1H-pyrazole-5-carboxylate (13)
Standard procedure was applied using 14-hydroxy-parthenolide (3) (10 mg, 0.030 mmol), 4-dimethylaminopyridine (0.015 mmol), triethylamine (0.30 mmol), and substituted acid chloride (0.15 mmol). Isolated PTL-14-13: 8 mg, 41 % yield. 1H NMR (400 MHz, CDCl3): δ = 7.60−7.54 (m, 4H), 7.05 (s, 1H), 6.33 (brs, 1H), 5.61−5.56 (m, 2H), 5.06 (d, J = 12 Hz, 1H), 4.84 (d, J = 12 Hz, 1H), 4.03−3.99 (m, 1H), 2.83−2.81 (m, 2H), 2.76−2.67 (m, 1H), 2.61−2.53 (m, 1H), 2.51−2.50 (m, 1H), 2.35−2.16 (m, 3H), 1.89−1.84 (m, 1H), 1.59−1.46 (m, 1H), 1.39−1.25 (m, 4H) ppm, 13C NMR (125 MHz, CDCl3): δ = 169.4, 160.5, 138.9, 132.8, 132.7, 132.1, 129.3, 127.4, 127.2, 122.8, 121.7, 105.7, 82.2, 66.2, 61.8, 61.2, 47.2, 36.6, 36.1, 31.2, 24.1, 17.0, MS (ESI) calcd for C25H25BrN2O5[M+Na]+ m/z: 535.1; found: 535.0.
4.4.13. (3aR,9aR,10aS,10bS,Z)-6-((benzyloxy)methyl)-9a-methyl-3-methylene-3a,4,5,8,9,9a,10a,10b-octahydrooxireno[2′,3′:9,10] cyclodeca[1,2-b]furan-2(3H)-one (14)
Standard procedure was applied using 14-hydroxy-parthenolide (3) (10 mg, 0.037 mmol), Ag2O (18 mg, 0.074 mmol) and benzyl alcohol (6 mg, .056 mmol). Isolated PTL derivative PTL-14-21: 4 mg 29 % yield. 1H NMR (400 MHz, CDCl3): δ = 7.34−7.25 (m, 5H), 6.33 (d, J = 3.2 Hz, 1H), 5.61 (d, J = 2.8 Hz, 1H), 5.43−5.40 (m, 1H), 4.53 (d, J = 11.6 Hz, 1H), 4.43 (d, J = 11.6 Hz, 1H), 4.09−4.02 (m, 2H), 3.87−3.84 (m, 1H), 2.78−2.72 (m, 3H), 2.44−2.39 (m, 1H), 2.22−2.04 (m, 4H), 1.87−1.83 (m, 1H), 1.24−1.21 (m, 4H) ppm, 13C NMR (125 MHz, CDCl3): δ = 169.2, 139.2, 137.8, 135.6, 129.9, 128.4, 127.8, 127.7, 121.3, 82.3, 72.4, 67.1, 66.2, 61.1, 47.3, 37.0, 36.3, 31.5, 23.8, 16.9 ppm, MS (ESI) calcd for C22H26O4 [M+Na]+ m/z: 377.2; found: 377.4.
4.4.14. (3aR,9aR,10aS,10bS,Z)-9a-methyl-3-methylene-6-(((4-(trifluoromethyl)benzyl)oxy)methyl)-3a,4,5,8,9,9a,10a,10b-octahydrooxireno[2′,3′:9,10]cyclodeca[1,2-b]furan-2(3H)-one (15)
Standard procedure was applied using 14-hydroxy-parthenolide (3) (10 mg, 0.037 mmol), Ag2O (18 mg, .074) and substituted benzyl alcohol (9 mg, 0.056 mmol). Isolated PTL derivative PTL-14-22: 6 mg 37 % yield. 1H NMR (400 MHz, CDCl3): δ = 7.61 (d, J = 8.0 Hz, 2H), 7.42 (d, J = 8.0 Hz, 2H), 6.34 (d, J = 3.2 Hz, 1H), 5.62 (d, J = 2.4 Hz, 1H), 5.47 (d, J = 8.8 Hz, 1H), 4.58 (d, J = 12.4 Hz, 1H), 4.49 (d, J = 12.4 Hz, 1H), 4.13−4.06 (m, 2H), 3.87 (t, J = 8.8 Hz, 1H), 2.79 (m, 3H), 2.44−2.36 (m, 1H), 2.25−2.03 (m, 4H), 1.88−1.79 (m, 1H), 1.37−1.22 (m, 4H) ppm, 13C NMR (125 MHz, CDCl3): δ = 169.1, 142.0, 139.1, 135.2, 130.3, 127., 125.4, 121.4, 82.3, 71.5, 67.5, 66.2, 61.0, 47.3, 36.8, 36.3, 31.5, 23.8, 16.9 ppm, MS (ESI) calcd for C23H25F3O4 [M+Na]+ m/z: 445.2; found: 445.3.
4.4.15. ethyl2-(((3aR,5S,9aR,10aS,10bS,E)-6,9a-dimethyl-3-methylene-2-oxo-2,3,3a,4,5,8,9,9a,10a,10b-decahydrooxireno [2′,3′:9,10]cyclodeca[1,2-b]furan-5-yl)oxy)acetate (16)
Standard procedure was applied using 9(S)-hydroxy-parthenolide (2) (5 mg, 0.018 mmol), Rh2(OAc)4 (5 mol%) and ethyldiazoacetate (036 mmol). Isolated PTL derivative PTL-09-23: 2 mg 35 % yield. 1H NMR (400 MHz, CDCl3): δ = 6.36 (d, J = 3.2 Hz, 1H), 5.73 (d, J = 3.2 Hz, 1H), 5.45 (d, J = 10 Hz, 1H), 4.23 (q, J = 6.8 Hz, 2H), 4.02−3.82 (m, 3H), 2.85−2.80 (m, 1H), 2.68 (d, J = 8.8 Hz, 1H), 2.41−2.37 (m, 1H), 2.27−2.16 (m, 3H), 2.05−1.98 (m, 2H), 1.76 (s, 3H), 1.41−1.27 (m, 7H) ppm, 13C NMR (125 MHz, CDCl3): δ = 170.2, 168.7, 138.0, 129.9, 121.8, 86.8, 81.2, 66.1, 64.7, 61.3, 60.9, 44.8, 36.1, 36.5, 24.1, 22.7, 17.4, 14.2, 11.1 ppm, MS (ESI) calcd for C19H26O6 [M+Na]+ m/z: 373.2; found: 373.3.
4.4.16. ethyl2-(((3aR,9aR,10aS,10bS,Z)-9a-methyl-3-methylene-2-oxo-2,3,3a,4,5,8,9,9a,10a,10b-decahydrooxireno[2′,3′:9,10] cyclodeca[1,2-b]furan-6-yl)methoxy)acetate (17)
Standard procedure was applied using 14-hydroxy-parthenolide (5 mg, 0.018 mmol), Rh2(OAc)4 (5 mol%) and ethyldiazoacetate (0.036 mmol), Isolated PTL derivative PTL-14-23: 3 mg, 45 % yield, 1H NMR (400 MHz, CDCl3): δ = 6.33 (d, J = 4.0 Hz, 1H), 5.62 (brs, 1H), 5.48 (d, J = 12 Hz, 1H), 4.24−3.98 (m, 6H), 3.91 (t, J = 8.0 Hz, 1H), 2.80−2.78 (m, 3H), 2.50−2.44 (m, 1H), 2.26−2.05 (m, 4H), 2.00−1.89 (m,1H), 1.30−1.09 (m, 7H) ppm, 13C NMR (125 MHz, CDCl3): δ = 170.2, 169.2, 139.2, 134.9, 130.9, 121.3, 82.2, 68.3, 67.3, 66.2, 61.1, 60.9, 47.4, 37.0, 36.3, 31.5, 23.8, 16.9, 14.2 ppm, MS (ESI) calcd for C19H26O6 [M+Na]+ m/z: 373.2; found: 373.4.
4.4.17. ethyl2-(((3aR,5S,9aR,10aS,10bS,E)-6,9a-dimethyl-3-methylene-2-oxo-2,3,3a,4,5,8,9,9a,10a,10b-decahydrooxireno [2′,3′:9,10]cyclodeca[1,2-b]furan-5-yl)oxy)-2-phenylacetate (18)
Standard procedure was applied using 9(S)-hydroxy-parthenolide (2) (10 mg, 0.037 mmol), Rh2(OAc)4 (5 mol %) and phenylethyldiazoacetate (0.074 mmol), Isolated PTL-09-24 (mixture of diasteroisomers): 5 mg, 32% yield. epimer 1 (HPLC tR 21.4 min.) =1H NMR (400 MHz, CDCl3): δ = 7.41−7.37 (m, 5H), 6.32 (d, J =3.2 Hz, 1H), 5.62 (d, J = 3.2 Hz, 1H), 5.30−5.26 (m, 1H), 4.72 (s, 1H), 4.19−4.08 (m, 2H), 3.85 (t, J = 8.4 Hz, 1H), 3.66 (d, J = 10.8 Hz, 1H), 2.70−2.66 (m, 1H), 2.58−2.49 (m, 1H), 2.32−2.05 (m, 4H), 1.91−1.83 (m, 1H), 1.61 (s, 3H), 1.28 (s, 3H), 1.24−1.10 (m, 4H) ppm, epimer 2 (HPLC tR 22.3 min.) = 1H NMR (400 MHz, CDCl3): δ = 7.53−7.35 (m, 5H), 6.38 (s, 1H), 5.79 (s, 1H), 5.63−5.57 (m, 1H), 5.17 (s, 1H), 4.18 (br s, 2H), 4.02−4.00 (m, 1H), 3.86−3.84 (m, 1H), 2.77−2.70 (m, 2H), 2.52−2.49 (m, 2H), 2.40−1.93 (m, 3H), 1.60 (s, 3H), 1.41 (s, 3H), 1.33−1.07 (m, 4H) ppm, MS (ESI) calcd for C25H30O6 [M+Na]+ m/z: 449.2; found: 449.3.
4.4.18. ethyl 2-(((3aR,9aR,10aS,10bS,Z)-9a-methyl-3-methylene-2-oxo-2,3,3a,4,5,8,9,9a,10a,10b-decahydrooxireno[2′,3′:9,10] cyclodeca[1,2-b]furan-6-yl)methoxy)-2-phenylacetate (19)
Standard procedure was applied using 14-hydroxy-parthenolide (10 mg, 0.037 mmol), Rh2(OAc)4 (0.8 mg, 5 mol %) and phenylethyldiazoacetate (14 mg, 0.074 mmol), Isolated PTL-14-24 (mixture of diasteroisomers): 6 mg 38 % yield. 1H NMR (400 MHz, CDCl3): δ = 7.38−7.35 (m, 10H), 6.33−6.32 (m, 2H), 5.62−5.61 (m, 2H), 5.46−5.43 (m, 2H), 4.84 (s, 1H), 4.80 (s, 1H), 4.22−4.02 (m, 8H), 3.92 (t, J = 8.4 Hz, 1H), 3.83 (t, J = 8.8 Hz, 1H), 2.81−2.71 (m, 5H), 2.27−2.23 (m, 1H), 2.18−2.03 (m, 10H), 1.97−1.88 (m, 2H), 1.29−1.17 (m, 11H), 1.10 (s, 3H) ppm, MS (ESI) calcd for C25H30O6 [M+Na]+ m/z: 449.2; found: 449.3.
4.4.19. ethyl2-(((3aR,5S,9aR,10aS,10bS,E)-6,9a-dimethyl-3-methylene-2-oxo-2,3,3a,4,5,8,9,9a,10a,10b-decahydrooxireno [2′,3′:9,10]cyclodeca[1,2-b]furan-5-yl)oxy)-2-(2-(trifluoromethyl)phenyl)acetate (20)
Standard procedure was applied using 9(S)-hydroxy-parthenolide (2) (5 mg, 0.018 mmol), Rh2(OAc)4 (5 mol %) and substituted diazoacetate (0.037 mmol), Isolated PTL-09-25 (mixture of diasteroisomers): 2.7 mg 28 % yield. 1H NMR (400 MHz, CDCl3): δ = 7.82−7.47 (m, 8H), 6.39−6.31 (m, 2H), 5.79 (brs, 1H), 5.56−5.31 (m, 3H), 5.16 (s, 2H), 4.13 (brs, 6H), 3.85−3.84 (m, 1H), 3.65 (brs, 1H), 2.97−2.88 (m, 4H), 2.68−2.48 (m, 4H), 2.26−2.03 (m, 6H), 1.63 (s, 6H), 1.34−1.15 (m, 14H) ppm, MS (ESI) calcd for C26H29F3O6 [M+Na]+ m/z: 517.2; found: 517.4.
4.4.20 ethyl2-(((3aR,9aR,10aS,10bS,Z)-9a-methyl-3-methylene-2-oxo-2,3,3a,4,5,8,9,9a,10a,10b-decahydrooxireno[2′,3′:9,10] cyclodeca[1,2-b]furan-6-yl)methoxy)-2-(2-(trifluoromethyl) phenyl)acetate (21)
Standard procedure was applied using 14-hydroxy-parthenolide (10 mg, 0.037 mmol), Rh2(OAc)4 (5 mol %) and substituted diazoacetate (0.074 mmol), Isolated PTL-14-25 (mixture of diasteroisomers): 6 mg 31 % yield. 1H NMR (400 MHz, CDCl3): δ = 7.68−7.45 (m, 8H), 6.32 (s, 2H), 5.60 (d, J = 11.6 Hz, 2H), 5.46 (d, J = 12.8 Hz, 2H), 5.23 (d, J = 9.2 Hz, 2H), 4.29−4.03 (m, 7H), 3.87−3.74 (m, 3H), 2.78−2.76 (m, 4H), 2.45−1.88 (m, 14H), 1.30−1.17 (m, 14H) ppm, MS (ESI) calcd for C26H29F3O6[M+Na]+ m/z: 517.2; found: 517.3.
4.4.21. ethyl 2-(((3aR,5S,9aR,10aS,10bS,E)-6,9a-dimethyl-3-methylene-2-oxo-2,3,3a,4,5,8,9,9a,10a,10b-decahydrooxireno [2′,3′:9,10]cyclodeca[1,2-b]furan-5-yl)oxy)-2-(4-(trifluoromethyl) phenyl)acetate (22)
Standard procedure was applied using 9(S)-hydroxy-parthenolide (2) (10 mg, 0.037 mmol), Rh2(OAc)4 (5 mol %) and substituted diazoacetate (0.074 mmol), Isolated PTL-09-26 (mixture of diasteroisomers): 8 mg, 42 % yield. epimer 1 (HPLC tR 24.2 min.) = 1H NMR (400 MHz, CDCl3): δ = 7.67 (d, J = 8.0 Hz, 2H), 7.57 (d, J = 8.0 Hz, 2H), 6.35 (d, J = 3.5 Hz, 1H), 5.64 (d, J = 3.5 Hz, 1H), 5.32−5.30 (m, 1H), 4.80 (s, 1H), 4.22−4.09 (m, 2H), 3.86 (t, J = 8.5 Hz, 1H), 3.66 (d, J = 10.5 Hz, 1H), 2.73−2.69 (m, 1H), 2.60−2.52 (m, 2H), 2.34−2.07 (m, 4H), 1.74 (s, 3H), 1.35 (s, 3H), 1.25−1.16 (m, 4H) ppm, epimer 2 (HPLC tR 25.6 min.) = 1H NMR (400 MHz, CDCl3): δ 7.63−7.57 (m, 4H), 6.40 (d, J = 3.5 Hz, 1H), 5.80 (d, J = 3.5 Hz, 1H), 5.51−5.49 (m, 1H), 4.80 (s, 1H), 4.22−4.14 (m, 2H), 4.05 (d, 1H), 3.88 (t, J = 8.5 Hz, 1H), 2.90−2.85 (m, 1H), 2.70 (d, J = 9.0 Hz, 1H), 2.58−2.51 (m, 2H), 2.31−2.28 (m, 1H), 2.21−2.16 (m, 1H), 2.08−2.01 (m, 1H), 1.58 (s, 3H), 1.34 (s, 3H), 1.25−1.21 (m, 4H) ppm, MS (ESI) calcd for C26H29F3O6 [M+Na]+ m/z: 517.2; found: 517.4.
4.4.22. ethyl 2-(((3aR,9aR,10aS,10bS,Z)-9a-methyl-3-methylene-2-oxo-2,3,3a,4,5,8,9,9a,10a,10b-decahydrooxireno[2′,3′:9,10] cyclodeca[1,2-b]furan-6-yl)methoxy)-2-(4-(trifluoromethyl) phenyl)acetate (23)
Standard procedure was applied using 14-hydroxy-parthenolide (3) (10 mg, 0.037 mmol), Rh2(OAc)4 (5 mol %) and substituted diazoacetate (0.074 mmol), Isolated PTL-14-26 (mixture of diasteroisomers): 8 mg 42 % yield. Epimer 1 (HPLC tR 24.2 min), 1H NMR (400 MHz, CDCl3): δ = 7.65 (d, J = 6.4 Hz, 2H), 7.54 (d, J = 6.4 Hz, 2H), 6.35 (d, J = 2.8 Hz, 1H), 5.63 (d, J = 2.4 Hz, 1H), 5.51−5.49 (m, 1H), 4.88 (s, 1H), 4.19−4.07 (m, 4H), 3.82−3.81 (m, 1H), 2.82−2.77 (m, 3H), 2.28−2.09 (m, 5H), 1.92−1.87 (m, 1H), 1.31−1.20 (m, 4H), 1.14 (s, 3H) ppm. Epimer 2 (HPLC tR 24.8 min.), 1H NMR (400 MHz, CDCl3): δ = 7.64 (d, J = 6.4 Hz, 2H), 7.55 (d, J = 6.4 Hz, 2H), 6.34 (d, J = 2.8 Hz, 1H), 5.62 (d, J = 2.4 Hz, 1H), 5.50−5.47 (m, 1H), 4.91 (s, 1H), 4.27−4.13 (m, 4H), 3.90−3.88 (m, 1H), 2.80−2.72 (m, 2H), 2.23−2.07 (m, 5H), 1.94−1.86 (m, 1H),1.30−1.21 (m, 8H) ppm, MS (ESI) calcd for C26H29F3O6 [M+Na]+ m/z: 517.2; found: 517.4.
4.4.23. Ethyl 2-(4-methoxyphenyl)-2-(((3aR,9aR,10aS,10bS,Z)-9a-methyl-3-methylene-2-oxo-2,3,3a,4,5,8,9,9a,10a,10b-decahydrooxireno[2′,3′:9,10]cyclodeca[1,2-b]furan-6-yl)methoxy)acetate (24)
Standard procedure was applied using 14-hydroxy-parthenolide (3) (10 mg, 0.037 mmol), Rh2(OAc)4 (5 mol %) and substituted diazoacetate (0.074 mmol), Isolated PTL-14-30 (mixture of diasteroisomers): 5 mg 29 % yield. 1H NMR (400 MHz, CDCl3): δ 7.38−7.28 (m, 4H), 6.96−6.88 (m, 4H), 6.30 (s, 2H), 5.60−5.58 (m, 2H), 5.43−5.40 (m, 2H), 5.25 (s, 1H), 5.20 (s, 1H), 4.19−4.05 (m, 8H), 3.88−3.81 (m, 10H), 2.78−2.74 (m, 3H), 2.52−2.41 (m, 2H), 2.31−2.26 (m, 1H), 2.16−1.90 (m, 10H), 1.38 (s, 3H), 1.35−1.08 (m, 8H), 0.85 (s, 3H) ppm, MS (ESI) calcd for C26H32O7 [M+Na]+ m/z: 479.2; found: 479.3.
4.4.24. benzyl 2-(((3aR,5S,9aR,10aS,10bS,E)-6,9a-dimethyl-3-methylene-2-oxo-2,3,3a,4,5,8,9,9a,10a,10b-decahydrooxireno [2′,3′:9,10]cyclodeca[1,2-b]furan-5-yl)oxy)-2-phenylacetate (25)
Standard procedure was applied using 9(S)-hydroxy-parthenolide (2) (10 mg, 0.037 mmol), Rh2(OAc)4 (5 mol %) and substituted diazoacetate (0.074 mmol), Isolated PTL-09-27: 6 mg 32 % yield. Epimer 1 (HPLC tR 24.6 min.), 1H NMR (500 MHz, CDCl3): δ = 7.44 (d, J = 5.4 Hz, 2H), 7.35−7.26 (m, 8H), 6.35 (d, J = 2.8 Hz, 1H), 5.69 (d, J = 2.8 Hz, 1H), 5.29 (d, J = 9.2 Hz, 1H), 5.20−5.13 (m, 2H), 4.79 (s, 1H), 3.91 (d, J = 8.4 Hz, 1H), 3.82 (t, J = 6.8 Hz, 1H), 2.72 (brs, 1H), 2.60 (d, J = 6.4 Hz, 1H), 2.51−2.41 (m, 2H), 2.23−2.15 (m, 2H), 2.01−1.96 (m, 1H), 1.58 (s, 3H), 1.31 (s, 3H), 1.25−1.14 (m, 1H) ppm. Epimer 2 (HPLC tR 25.3 min.), 1H NMR (500 MHz, CDCl3): δ = 7.42−7.36 (m, 5H), 7.29−7.26 (m, 3H), 7.17−7.16 (m, 2H), 6.32 (d, J = 2.8 Hz, 1H), 5.62 (d, J = 2.8 Hz, 1H), 5.29−5.27 (m, 1H), 5.16−5.07 (m, 2H), 4.80 (s, 1H), 3.84 (t, J = 6.8 Hz, 1H), 3.69−3.67 (m, 1H), 2.67 (brs, 1H), 2.57−2.51 (m, 2H), 2.33−2.04 (m, 4H), 1.71 (s, 3H), 1.33 (s, 3H), 1.20−1.14 (m, 1H) ppm, MS (ESI) calcd for C30H32O6[M+Na]+ m/z: 511.2; found: 511.2.
4.4.25. benzyl2-(((3aR,9aR,10aS,10bS,Z)-9a-methyl-3-methylene-2-oxo-2,3,3a,4,5,8,9,9a,10a,10b-decahydrooxireno[2′,3′:9,10] cyclodeca[1,2-b]furan-6-yl)methoxy)-2-phenylacetate(26)
Standard procedure was applied using 14-hydroxy-parthenolide (3) (10 mg, 0.037 mmol), Rh2(OAc)4 (0.8 mg, 5 mol %) and substituted diazoacetate (19 mg, 0.074 mmol), Isolated PTL-14-27: 6 mg 32 % yield. Epimer 1 (HPLC tR 24.5 min.), 1H NMR (500 MHz, CDCl3): δ = 7.36−7.20 (m, 10H), 6.33 (d, J = 3.5 Hz, 1H), 5.60 (d, J = 3.5 Hz, 1H), 5.42−5.40 (m, 1H), 5.17−5.13 (m, 3H), 4.88 (s, 1H), 4.05−4.04 (m, 1H), 3.80 (t, J = 8.5 Hz, 1H), 2.77−2.73 (m, 2H), 2.31−2.21 (m, 7H), 1.25−1.19 (m, 1H), 1.09 (s, 3H) ppm. Epimer 2 (HPLC tR 24.9 min.), 1H NMR (500 MHz, CDCl3): δ = 7.42−7.21 (m, 10H), 6.33 (d, J = 4.0 Hz, 1H), 5.60 (d, J = 4.0 Hz, 1H), 5.42−5.40 (m, 1H), 5.15−5.11 (m, 3H), 4.88 (s, 1H), 4.06 (s, 1H), 3.80 (t, 1H), 2.77−2.73 (m, 2H), 2.31−2.03 (m, 6H), 1.90−1.83 (m, 1H), 1.25−1.15 (m, 1H), 1.09 (s, 3H) ppm, MS (ESI) calcd for C30H32O6 [M+Na]+ m/z: 511.2; found: 511.3.
4.4.26 2-morpholinoethyl2-(((3aR,5S,9aR,10aS,10bS,E)-6,9a-dimethyl-3-methylene-2-oxo-2,3,3a,4,5,8,9,9a,10a,10b-decahy drooxireno[2′,3′:9,10]cyclodeca[1,2-b]furan-5-yl)oxy)-2-phenyl acetate (27)
Standard procedure was applied using 9(S)-hydroxy-parthenolide (2) (10 mg, 0.037 mmol), Rh2(OAc)4 (5 mol %) and substituted diazoacetate (0.074 mmol), Isolated PTL-09-28 (mixture of diasteroisomers): 5 mg 25 % yield. 1H NMR (400 MHz, CDCl3): 7.54−7.29 (m, 10H), 6.39−6.38 (m, 1H), 6.31−6.30 (m, 1H), 5.79−5.77 (m, 1H), 5.61 (m, 1H), 5.50−5.47 (m, 1H), 5.28−5.25 (m, 1H), 5.11 (s, 2H), 4.29−4.20 (m, 2H), 3.85 (brs, 2H), 3.47−3.71 (m, 8H), 3.58−3.57 (m, 2H), 2.96−2.91 (m, 4H), 2.69−1.98 (m, 24H), 1.72 (s, 3H), 1.59 (s, 3H), 1.33−1.12 (m, 8H) ppm, MS (ESI) calcd for C29H37NO7 [M+H]+ m/z: 512.3; found: 512.3.
4.4.27 2-morpholinoethyl 2-(((3aR,9aR,10aS,10bS,Z)-9a-methyl-3-methylene-2-oxo-2,3,3a,4,5,8,9,9a,10a,10b-decahydrooxireno [2′,3′:9,10]cyclodeca[1,2-b]furan-6-yl)methoxy)-2-phenylacetate (28)
Standard procedure was applied using 14-hydroxy-parthenolide (10 mg, 0.037 mmol), Rh2(OAc)4 (5 mol %) and substituted diazoacetate (0.074 mmol), Isolated PTL-14-28 (mixture of diasteroisomers): 9 mg 46 % yield. 1H NMR (400 MHz, CDCl3): δ = 7.43−7.30 (m, 10H), 6.32 (brs, 2H), 5.61−5.59 (m, 2H), 5.28 (brs, 2H), 4.85 (s, 1H), 4.82 (s, 1H), 4.36−4.05 (m, 9H), 3.92−3.80 (m, 2H), (brs, 8H), 2.79−2.74 (m, 6H), 2.53 (brs, 8H), 2.41−2.07 (m, 16H), 1.29−1.24 (m, 6H), 1.10 (s, 1H) ppm, MS (ESI) calcd for C29H37NO7 [M+H]+ m/z: 512.3; found: 512.3.
4.4.28. 2-(pyrrolidin-1-yl)ethyl 2-(((3aR,5S,9aR,10aS,10bS,E)-6,9a-dimethyl-3-methylene-2-oxo-2,3,3a,4,5,8,9,9a,10a,10b-decahydrooxireno[2′,3′:9,10]cyclodeca[1,2-b]furan-5-yl)oxy)-2-phenylacetate (29)
Standard procedure was applied using 14-hydroxy-parthenolide (3) (10 mg, 0.037 mmol), Rh2(OAc)4 (5 mol %) and substituted diazoacetate (0.074 mmol), Isolated PTL-14-29 (mixture of diasteroisomers): 5 mg 27 % yield. 1H NMR (400 MHz, CDCl3): δ 7.39−7.34 (m, 10H), 6.32 (s, 2H), 5.59 (s, 2H), 5.46−5.43 (m, 2H), 4.83−4.85 (m, 2H), 4.10 (t, J = 10.4 Hz, 4H), 3.89−3.82 (m, 2H), 3.78−3.75 (m, 2H), 2.79−2.66 (m, 12H), 2.23−1.49 (m, 12H), 1.29−1.10 (m, 24H) ppm, MS (ESI) calcd for C29H37NO6 [M+H]+ m/z: 496.2; found: 496.3.
4.6. Cell culture and isolation
For all cell culture studies, cells were kept in a 37 °C humidified incubator with 5% CO2. Human acute myeloid leukemia and umbilical cord samples were obtained after informed consent from volunteer donors. Live mononuclear cells were isolated by subjection to Ficoll-Paque (GE Healthcare Bio-Sciences) density gradient and either cryopreserved (leukemia samples) or cultured without cryopreservation (umbilical cord samples). The viability of cryopreserved leukemia samples after thawing was 40 – 70%.
4.7. Cytotoxicity studies with M9-ENL-1 cells
The stem-cell-like cell line M9-ENL1 was utilized for initial drug screening. Cells were plated at a density of 106 cells/mL in alpha-MEM culture media (Invitrogen) supplemented with 5% human plasma, 20% FBS, and the cytokines SCF, IL-3, IL-7, and FLT3 ligand (Peprotech) and penicillin/streptomycin. Compounds were diluted into the culture media from a DMSO stock solution. At least three doses were included for each compound and parthenolide was included in all screens as a positive control. After twenty-four hours of drug exposure, cells were collected for flow cytometry. Cells were stained with Annexin V and 7-aminoactinomycin (7-AAD) to quantify the percentage of nonapoptotic (negative for both stains) cells. Analyses were conducted in triplicate.
4.8. Cytotoxicity studies with primary AML specimens
Thawed primary AML samples were cultured in serum-free media (SFM) prepared with Iscove’s MDM supplemented with 20% BIT 9500 serum substitute (StemCell Technologies), LDL, beta-mercaptoethanol, and penicillin/streptomycin for at least one hour after thawing before the addition of test compounds. Cytotoxicity studies were performed as described above.
4.9. Human umbilical cord blood cell cytotoxicity tests
Donated human umbilical cord blood samples were obtained within 48 hours of collection. Cell culture and drug treatment was performed in SFM for twenty-four hours as above. For analysis, cells were stained with fluorescent labels for CD45 and CD34 to identify the hematopoietic progenitor compartment as well as DAPI to exclude non-viable cells via flow cytometry. The percentage of CD34+CD45dim cells in the DAPI-negative compartment was compared between drug-treated and vehicle-treated samples to determine toxicity to normal hematopoietic progenitor cells.
4.10. Colony growth inhibition assay
After the 24-hour treatment, an aliquot of each treatment sample of human umbilical cord blood was washed with phosphate-buffered saline (PBS) and resuspended in methycellulose media at a density of 50,000 cells/mL. Each sample was plated in triplicate and stored in a humidified incubator for several weeks until colonies were visible. Colonies were counted under a dissecting microscope and the number of colonies was compared between drug-treated and vehicle-treated samples to determine the relative impairment of colony-forming ability.
Acknowledgments
This work was supported by the Leukemia and Lymphoma Society Translational Research Grant LLS-6116-14 and in part by the U.S. National Institute of Health grant GM098628 awarded to R.F.. MS instrumentation was supported by the U.S. National Science Foundation grant CHE-0946653.
Abbreviations
- PTL
parthenolide
- AML
acute myeloid leukemia
- LCS
leukemia stem cells
- DMAPT
dimethylamino-parthenolide
- PTDH
phosphite dehydrogenase
- NADPH
nicotinamide adenine dinucleotide phosphate (reduced)
- IPTG
β-D-1-thiogalactopyranoside
- δ-ALA
δ-aminolevulinic acid
- TB
terrific broth
- DMSO
dimethylsulfoxide
- DMAP
4-dimethylamino-pyridine
- EDA
ethyl 2-diazo-acetate
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References and notes
- 1.Breitmaier E. Terpenes: Flavors, Fragrances, Pharmaca, Pheromones. Weinheim: Wiley-VCH Verlag GmbH; 2006. [Google Scholar]
- 2.Ghantous A, Gali-Muhtasib H, Vuorela H, Saliba NA, Darwiche N. Drug Discov Today. 2010;15:668. doi: 10.1016/j.drudis.2010.06.002. [DOI] [PubMed] [Google Scholar]
- 3.Merfort I. Curr Drug Targets. 2011;12:1560. doi: 10.2174/138945011798109437. [DOI] [PubMed] [Google Scholar]
- 4.Kreuger MR, Grootjans S, Biavatti MW, Vandenabeele P, D’Herde K. Anticanc Drugs. 2012;23:883. doi: 10.1097/CAD.0b013e328356cad9. [DOI] [PubMed] [Google Scholar]
- 5.Guzman ML, Rossi RM, Karnischky L, Li XJ, Peterson DR, Howard DS, Jordan CT. Blood. 2005;105:4163. doi: 10.1182/blood-2004-10-4135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Guzman ML, Jordan CT. Expert Opin Biol Th. 2005;5:1147. doi: 10.1517/14712598.5.9.1147. [DOI] [PubMed] [Google Scholar]
- 7.Ghantous A, Sinjab A, Herceg Z, Darwiche N. Drug Discov Today. 2013 doi: 10.1016/j.drudis.2013.05.005. [DOI] [PubMed] [Google Scholar]
- 8.Lapidot T, Sirard C, Vormoor J, Murdoch B, Hoang T, Cacerescortes J, Minden M, Paterson B, Caligiuri MA, Dick JE. Nature. 1994;367:645. doi: 10.1038/367645a0. [DOI] [PubMed] [Google Scholar]
- 9.Hope KJ, Jin L, Dick JE. Nat Immunol. 2004;5:738. doi: 10.1038/ni1080. [DOI] [PubMed] [Google Scholar]
- 10.Guzman ML, Li X, Corbett CA, Rossi RM, Bushnell T, Liesveld JL, Hebert J, Young F, Jordan CT. Blood. 2007;110:4436. doi: 10.1182/blood-2007-05-088815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.McDermott SP, Eppert K, Notta F, Isaac M, Datti A, Al-Awar R, Wrana J, Minden MD, Dick JE. Blood. 2012;119:1200. doi: 10.1182/blood-2011-01-330019. [DOI] [PubMed] [Google Scholar]
- 12.Lagadinou ED, Sach A, Callahan K, Rossi RM, Neering SJ, Minhajuddin M, Ashton JM, Pei S, Grose V, O’Dwyer KM, Liesveld JL, Brookes PS, Becker MW, Jordan CT. Cell Stem Cell. 2013;12:329. doi: 10.1016/j.stem.2012.12.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hartwell KA, Miller PG, Mukherjee S, Kahn AR, Stewart AL, Logan DJ, Negri JM, Duvet M, Jaras M, Puram R, Dancik V, Al-Shahrour F, Kindler T, Tothova Z, Chattopadhyay S, Hasaka T, Narayan R, Dai M, Huang C, Shterental S, Chu LP, Haydu JE, Shieh JH, Steensma DP, Munoz B, Bittker JA, Shamji AF, Clemons PA, Tolliday NJ, Carpenter AE, Gilliland DG, Stern AM, Moore MA, Scadden DT, Schreiber SL, Ebert BL, Golub TR. Nature Chem Biol. 2013;9:840. doi: 10.1038/nchembio.1367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Garcia-Pineres AJ, Castro V, Mora G, Schmidt TJ, Strunck E, Pahl HL, Merfort I. J Biol Chem. 2001;276:39713. doi: 10.1074/jbc.M101985200. [DOI] [PubMed] [Google Scholar]
- 15.Hehner SP, Heinrich M, Bork PM, Vogt M, Ratter F, Lehmann V, Schulze-Osthoff K, Droge W, Schmitz ML. J Biol Chem. 1998;273:1288. doi: 10.1074/jbc.273.3.1288. [DOI] [PubMed] [Google Scholar]
- 16.Gopal YN, Chanchorn E, Van Dyke MW. Mol Cancer Ther. 2009;8:552. doi: 10.1158/1535-7163.MCT-08-0661. [DOI] [PubMed] [Google Scholar]
- 17.Liu Z, Liu S, Xie Z, Pavlovicz RE, Wu J, Chen P, Aimiuwu J, Pang J, Bhasin D, Neviani P, Fuchs JR, Plass C, Li PK, Li C, Huang TH, Wu LC, Rush L, Wang H, Perrotti D, Marcucci G, Chan KK. J Pharmacol Exp Ther. 2009;329:505. doi: 10.1124/jpet.108.147934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Nakshatri H, Appaiah HN, Anjanappa M, Gilley D, Tanaka H, Badve S, Crooks PA, Mathews W, Sweeney C, Bhat-Nakshatri P. Cell Death Dis. 2015;6:e1608. doi: 10.1038/cddis.2014.569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Pei S, Minhajuddin M, Callahan KP, Balys M, Ashton JM, Neering SJ, Lagadinou ED, Corbett C, Ye H, Liesveld JL, O’Dwyer KM, Li Z, Shi L, Greninger P, Settleman J, Benes C, Hagen FK, Munger J, Crooks PA, Becker MW, Jordan CT. J Biol Chem. 2013;288:33542. doi: 10.1074/jbc.M113.511170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Nakshatri H, Rice SE, Bhat-Nakshatri P. Oncogene. 2004;23:7330. doi: 10.1038/sj.onc.1207995. [DOI] [PubMed] [Google Scholar]
- 21.Janecka A, Wyrebska A, Gach K, Fichna J, Janecki T. Drug Discov Today. 2012;17:561. doi: 10.1016/j.drudis.2012.01.013. [DOI] [PubMed] [Google Scholar]
- 22.Srivastava SK, Abraham A, Bhat B, Jaggi M, Singh AT, Sanna VK, Singh G, Agarwal SK, Mukherjee R, Burman AC. Bioorg Med Chem Lett. 2006;16:4195. doi: 10.1016/j.bmcl.2006.05.083. [DOI] [PubMed] [Google Scholar]
- 23.Nasim S, Pei S, Hagen FK, Jordan CT, Crooks PA. Bioorg Med Chem. 2011;19:1515. doi: 10.1016/j.bmc.2010.12.045. [DOI] [PubMed] [Google Scholar]
- 24.Zhang Q, Lu Y, Ding Y, Zhai J, Ji Q, Ma W, Yang M, Fan H, Long J, Tong Z, Shi Y, Jia Y, Han B, Zhang W, Qiu C, Ma X, Li Q, Shi Q, Zhang H, Li D, Zhang J, Lin J, Li LY, Gao Y, Chen Y. J Med Chem. 2012;55:8757. doi: 10.1021/jm301064b. [DOI] [PubMed] [Google Scholar]
- 25.An Y, Guo W, Li L, Xu C, Yang D, Wang S, Lu Y, Zhang Q, Zhai J, Fan H, Qiu C, Qi J, Chen Y, Yuan S. PLoS One. 2015;10:e0116202. doi: 10.1371/journal.pone.0116202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Janganati V, Ponder J, Jordan CT, Borrelli MJ, Penthala NR, Crooks PA. J Med Chem. 2015;58:8896. doi: 10.1021/acs.jmedchem.5b01187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kempema AM, Widen JC, Hexum JK, Andrews TE, Wang D, Rathe SK, Meece FA, Noble KE, Sachs Z, Largaespada DA, Harki DA. Bioorg Med Chem. 2015;23:4737. doi: 10.1016/j.bmc.2015.05.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hexum JK, Becker CM, Kempema AM, Ohlfest JR, Largaespada DA, Harki DA. Bioorg Med Chem Lett. 2015;25:2493. doi: 10.1016/j.bmcl.2015.04.058. [DOI] [PubMed] [Google Scholar]
- 29.Guzman ML, Rossi RM, Li XJ, Corbett C, Hassane DC, Bushnell T, Carroll M, Sullivan E, Neelakantan S, Crooks PA, Jordan CT. Blood. 2006;108:74a. [Google Scholar]
- 30.Nasim S, Crooks PA. Bioorg Med Chem Lett. 2008;18:3870. doi: 10.1016/j.bmcl.2008.06.050. [DOI] [PubMed] [Google Scholar]
- 31.Neelakantan S, Nasim S, Guzman ML, Jordan CT, Crooks PA. Bioorg Med Chem Lett. 2009;19:4346. doi: 10.1016/j.bmcl.2009.05.092. [DOI] [PubMed] [Google Scholar]
- 32.Han C, Barrios FJ, Riofski MV, Colby DA. J Org Chem. 2009;74:7176. doi: 10.1021/jo901533e. [DOI] [PubMed] [Google Scholar]
- 33.Kolev JN, O’Dwyer KM, Jordan CT, Fasan R. ACS Chem Biol. 2014;9:164. doi: 10.1021/cb400626w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Zhang K, El Damaty S, Fasan R. J Am Chem Soc. 2011;133:3242. doi: 10.1021/ja109590h. [DOI] [PubMed] [Google Scholar]
- 35.Zhang K, Shafer BM, Demars MD, 2nd, Stern HA, Fasan R. J Am Chem Soc. 2012;134:18695. doi: 10.1021/ja3073462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kolev JN, Zaengle JM, Ravikumar R, Fasan R. Chembiochem. 2014;15:1001. doi: 10.1002/cbic.201400060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Narhi LO, Fulco AJ. J Biol Chem. 1987;262:6683. [PubMed] [Google Scholar]
- 38.Fasan R. ACS Catal. 2012;2:647. [Google Scholar]
- 39.Whitehouse CJ, Bell SG, Wong LL. Chem Soc Rev. 2012;41:1218. doi: 10.1039/c1cs15192d. [DOI] [PubMed] [Google Scholar]
- 40.Sowden RJ, Yasmin S, Rees NH, Bell SG, Wong LL. Org Biomol Chem. 2005;3:57. doi: 10.1039/b413068e. [DOI] [PubMed] [Google Scholar]
- 41.Kille S, Zilly FE, Acevedo JP, Reetz MT. Nat Chem. 2011;3:738. doi: 10.1038/nchem.1113. [DOI] [PubMed] [Google Scholar]
- 42.Le-Huu P, Heidt T, Claasen B, Laschat S, Urlacher VB. ACS Catal. 2015;5:1772. [Google Scholar]
- 43.McLachlan MJ, Johannes TW, Zhao H. Biotechnol Bioeng. 2008;99:268. doi: 10.1002/bit.21546. [DOI] [PubMed] [Google Scholar]
- 44.Urlacher VB, Makhsumkhanov A, Schmid RD. Appl Microbiol Biot. 2006;70:53. doi: 10.1007/s00253-005-0028-4. [DOI] [PubMed] [Google Scholar]
- 45.Fasan R, Chen MM, Crook NC, Arnold FH. Angew Chem Int Ed Engl. 2007;46:8414. doi: 10.1002/anie.200702616. [DOI] [PubMed] [Google Scholar]
- 46.Fasan R, Crook NC, Peters MW, Meinhold P, Buelter T, Landwehr M, Cirino PC, Arnold FH. Biotechnol Bioeng. 2011;108:500. doi: 10.1002/bit.22984. [DOI] [PubMed] [Google Scholar]
- 47.Miller DJ, Moody CJ. Tetrahedron. 1995;51:10811. [Google Scholar]
- 48.Jin P, Madieh S, Augsburger LL. Aaps Pharmscitech. 2007;8:200. doi: 10.1208/pt0802043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Barabe F, Kennedy JA, Hope KJ, Dick JE. Science. 2007;316:600. doi: 10.1126/science.1139851. [DOI] [PubMed] [Google Scholar]
- 50.Horton DA, Bourne GT, Smythe ML. Chem Rev. 2003;103:893. doi: 10.1021/cr020033s. [DOI] [PubMed] [Google Scholar]
- 51.Hajduk PJ, Bures M, Praestgaard J, Fesik SW. J Med Chem. 2000;43:3443. doi: 10.1021/jm000164q. [DOI] [PubMed] [Google Scholar]