

Abstract
This study investigated several types of eye movements that rely on the function of brainstem-cerebellar pathways specifically (vestibular-ocular reflexes) or on widely distributed pathways of the brain (horizontal pursuit and saccade eye movements). Although eye movements that rely on higher brain regions have been studies fairly extensively in autism, eye movements dependent on brainstem and cerebellum have not. This study involved 79 individuals with autism and 62 typical controls aged 5 to 52 years with IQ scores above 70. No differences between the autism and control groups were present on the measures of vestibular ocular reflexes, or on saccade velocity or accuracy. The autism group was significantly slower to initiate saccades, which was most prominent in the 8-18 year old age range. These findings provide the most substantial evidence to date of the functional integrity of brainstem and cerebellar pathways in autism, suggesting that the histopathological abnormalities described in these structures may not be associated with intrinsic dysfunction but rather reflect developmental alterations related to forebrain cortical systems formation. The increase in saccade latency adds to the substantial evidence of altered function and maturation of cortical systems in autism.
Objective
This study assessed the functionality of vestibular, pursuit and saccade circuitry in autism across a wide age range.
Methods
Subjects were 79 individuals with autism (AUT) and 62 controls (CON) aged 5 to 52 years with IQ scores > 70. For vestibular testing, earth-vertical axis rotation was performed in darkness and in a lighted visual surround with a fixation target. Ocular motor testing included assessment of horizontal saccades and horizontal smooth pursuit.
Results
No between-group differences were found in vestibular reflexes or in mean saccade velocity or accuracy. Saccade latency was increased in the AUT group with significant age-related effects in the 8-18 year old subgroups. There was a trend toward decreased pursuit gain without age effects.
Conclusions
Normal vestibular-induced eye movements and normal saccade accuracy and velocity provide the most substantial evidence to date of the functional integrity of brainstem and cerebellar pathways in autism, suggesting that the histopathological abnormalities described in these structures may not be associated with intrinsic dysfunction but rather reflect developmental alterations related to forebrain cortical systems formation. Increased saccade latency with age effects adds to the extensive existing evidence of altered function and maturation of cortical systems in autism.
Keywords: Autism, Ocular motility, Brainstem, Clinical neurophysiology, Eye movements
INTRODUCTION
Over the past decade, imaging, genetic, and cognitive research has generated compelling evidence of disturbances in the development and function of cortical systems as a central pathophysiological mechanism for autism. Longitudinal magnetic resonance imaging (MRI) studies of children under the age of 3 years later confirmed to have autism documented increased cerebral grey and white matter volumes with the largest volume increases involving frontal, temporal and cingulate cortex [Schumann et al., 2010]. Longitudinal MRI studies of infants at high genetic risk for developing autism identified an accelerated rate of brain growth beginning around 6 to 9 months of age and plateauing around 2 years of age [Hazlett et al., 2005]. Longitudinal diffusion tensor imaging of these infants reported aberrant cerebral white matter tract development in 12 of 15 fiber tracts at 7 months of age; tract development continued in a complex deviant pattern until the last images at 24 months of age [Wolff et al., 2012]. Functional MRI (fMRI) studies of adolescents and adults with autism have reported reduced and altered cortical systems connectivity across a broad range of cognitive and motor tasks linking structural alterations in connectivity to functional alterations and behavior [Mostofsky et al., 2009; Schipul et al., 2011]. Genetic studies have identified a rapidly growing number of risk genes in autism that implicate a number of neurobiologic processes involved in the formation and function of cortical systems [McFadden & Minshew, 2013; Parikshak et al., 2013; Willsey et al., 2013]. The demonstration of a common pattern of alterations in higher order abilities across domains in the presence of intact basic abilities in children and adults with autism [Minshew et al., 1997; Williams et al., 2006] and the simultaneous emergence of all manifestations of autism in the first two years of life [Rogers, 2009] consistent with a disorder of neuronal organization [Volpe, 2008] provided a connection between the genetic and imaging findings on the one hand and the cognitive and behavioral manifestations of autism on the other.
Despite the coalescence of recent evidence around aberrant development and function of cortical systems in autism, there is a long and persisting history of theories proposing a central role for brainstem-cerebellar circuits in the pathophysiology of ASD with two such theories dominating the field for decades. The earliest such theory proposed that autism resulted from dysfunction in the brainstem-cerebellar vestibular system [Ornitz & Ritvo, 1968a,b], which was superseded by a theory proposing dysfunction in the cerebellar vermis resulting in shifting attention abnormalities as the basis for the manifestations of autism [Courchesne et al., 1994]. The basic tenets of these theories were not supported by subsequent research [Minshew et al., 1999; Scott et al., 2009; Williams et al., 2013]. Interest in the role of the cerebellum in autism was renewed by postmortem brain studies that cumulatively reported near universal reduction in Purkinje cell number in the posterolateral neocerebellar cortex and adjacent areas of archicerebellar cortex without retrograde cell loss in the inferior olivary nuclei, indicating prenatal loss of the Purkinje cells [Kemper & Bauman, 1998; Whitney et al., 2008]. Documentation of a normal number of basket cells and stellate interneurons in the molecular layer of the cerebellum provided evidence that the Purkinje cells were generated, migrated to their correct destination, and then died [Whitney et al., 2009]. The deep cerebellar nuclei exhibited abnormally large and more numerous neurons in child brains and small pale neurons in decreased numbers in adult brains, suggesting abnormalities in the postnatal development of these cells [Kemper & Bauman, 1998]. In apparent contrast to the cellular findings, neuroimaging studies were nearly consistent in reporting increased volume of the cerebellar hemispheres that was proportionate to the increase in cerebral volume [Scott et al., 2009]. In addition to this structural evidence, several reports emerged attributing eye movement abnormalities in autism to intrinsic cerebellar dysfunction [Johnson et al., 2012; Mosconi et al., 2013; Stanley-Cary et al., 2011; Takarae et al., 2004]. These findings led to a focus on the contribution of the cerebellum to non-motor cognitive functions [Fatemi et al., 2012; Strick et al., 2009]. The identification of autism risk genes, such as En2 (Engrailed homeobox 2) [James et al., 2013] and Met (Met proto-oncogene) [Abrahams & Geschwind, 2008; Rossman et al., 2014; Schaaf & Zoghbi, 2011] known to be involved in the development of the cerebellum and hindbrain suggested that the emphasis should shift from theories espousing localized dysfunction of the cerebellum to the investigation of mechanisms of hind brain and forebrain development and of the widely distributed circuitry interconnecting them [Fatemi et al., 2012; Rogers et al., 2013]. These mechanisms are not yet well delineated.
The purpose of the present study was to provide a better perspective on the possible contributions of localized dysfunction of brainstem-cerebellar circuitry in autism to the pathophysiology of autism through a more extensive evaluation of its functional status than is currently available and a limited assessment of the status of cortical systems in the same subjects using eye movement procedures. Age effects were evaluated by studying both children and adults.
METHODS
Subjects
The subjects for this study consisted of 79 verbal individuals (IQ scores>70) with autism (AUT) and 62 typical control individuals (CON) between the ages of 5 and 52 years. Subjects with autism were recruited from an outpatient population and controls were recruited from the community to have the same socioeconomic level as subjects with autism. Subjects with autism met all cut offs and overall scores for autism on the Autism Diagnostic Interview (ADI) [Lord et al., 1994] and the Autism Diagnostic Observation Schedule (ADOS) [Lord et al., 1989] and all had historical evidence of delayed and disordered language development. The diagnosis of autism was confirmed by expert clinical opinion.
Potential subjects with autism were excluded if found to have an associated neurologic, genetic, infectious or other disorder causative of autism, or if taking medications, such as benzodiazipines, lithium or phenothiazines, known to affect the measurements under study. Potential control subjects were excluded if they had evidence of congenital or developmental abnormalities, acquired brain injury, learning or language disability, current or past history of psychiatric or neurologic disorder or an immediate family history of developmental disability, anxiety disorder or of autism in first-, second-, or third-degree relatives. All subjects in both groups had Full Scale Intelligence Quotient (FSIQ), Verbal Intelligence Quotient (VIQ) and Performance Intelligence Quotient (PIQ) scores above 70.
This study was approved by the University of Pittsburgh Institutional Review Board. All subjects and/or their legal guardians completed informed consent and assent procedures.
Procedures
Ocular-motor screening was performed to assess horizontal saccades and horizontal smooth pursuit. For vestibular testing, earth vertical axis rotation was performed in darkness to assess the vestibular-ocular reflex (VOR) and in lighted visual surround with a fixation target to assess visual-vestibular interaction.
Recording Techniques
Eye movements were recorded using bitemporal electrooculography and were performed separately for each eye. Eye movement signals were amplified by a DC-coupled amplifier with a cut-off frequency of 40 Hz and recorded by computer at a sampling rate of 100 Hz. Calibration was performed before each test by having subjects fixate a target placed ±15° horizontally.
Vestibular testing
Earth vertical axis rotation in the dark was performed using both sinusoidal rotation and constant velocity rotation [Baloh, Sills, & Honrubia, 1979]. Sinusoidal rotations were performed using several frequencies, i.e. 0.05 Hz, 0.1 Hz, 0.5 Hz, and 1.0 Hz, each at 50 deg/sec peak velocity and several amplitudes using 0.1 Hz rotation with a peak velocity of 25, 50, 100, and 150 deg/sec. At 0.05 Hz and 0.1 Hz, five cycles of rotation were delivered. At 0.5 Hz and 1.0 Hz, ten cycles of rotation were delivered. Data analysis for sinusoidal testing yielded VOR gain and phase at each frequency [Furman & Kamerer, 1989]. Gain is defined as the ratio of eye velocity to head velocity. Phase is a measure of the temporal synchrony between head velocity and eye velocity. The gain and phase data from all frequencies were combined using a first-order low-pass system model and this yielded an estimate of sinusoidal VOR sensitivity and sinusoidal VOR time constant [Baloh et al., 1984]. VOR sensitivity is a measure of the magnitude of VOR response overall. VOR time constant was estimated from the sinusoidal data and represents the theoretical time that would be required for slow-phase eye velocity to decline to 37% of its initial value after the onset of a velocity step stimulus.
Constant velocity rotations were delivered using 90 deg/sec rotations in both clockwise (CW) and counterclockwise (CCW) directions with an initial acceleration of 10 deg/sec/sec. Rotation was continued for 60 sec after which the subjects were decelerated at 100 deg/sec/sec to a stop. Eye movements were analyzed to yield the VOR time constant using an exponential fit [Baloh & Honrubia, 1990]. To assess semicircular canal-otolith interaction, constant velocity earth-vertical axis rotations were performed while subjects wore darkened goggles. Using the same stimuli noted above for constant velocity rotation, immediately following cessation of rotation, subjects were pitched forward by approximately 30 degrees to change their orientation with respect to gravity and induce so-called “pitch dumping” [Hain, Zee, & Maria, 1988]. Data analysis again yielded the VOR time constant using an exponential fit. A head tilt ratio was calculated using the following formula ([CW time constant with tilt/ CW time constant without tilt]+[CCW time constant with tilt/CCW time constant without tilt])/2.
Visual-vestibular interaction was assessed using sinusoidal earth-vertical axis rotation with a visual stimulus [Baloh et al., 1982]. Rotation was performed at 0.05 Hz with a peak velocity of 50 deg/sec. Visual-vestibulo-ocular reflex (VVOR) responses, which measure responses to congruent visual and vestibular stimuli, were performed by rotating subjects while they viewed a lighted visual surround. The visual surround consisted of vertical stripes completely surrounding the rotational chair. The stripes were earth-stationary during rotation. To assess VOR fixation, subjects were rotated sinusoidally at 0.05 Hz with a peak velocity of 50 deg/sec while they looked at a small laser target placed directly in front of them that rotated with the chair. Eye movements recorded during visual-vestibular interaction testing were used to compute the sinusoidal VVOR gain and sinusoidal VOR fixation gain.
Saccades and smooth pursuit
Ocular motor screening was performed by asking subjects to look at a small laser target moving horizontally. For saccades, the target moved randomly in time and direction with an excursion that was within ±15 degrees horizontally [Baloh & Honrubia, 1976]. Eye movements were analyzed by estimating saccade latency, accuracy and velocity using standard techniques [Baloh & Honrubia, 1976]. Note that the sampling rate of 100 Hz was considered adequate for estimating saccade accuracy and velocity [Wierts et al., 2008] whereas saccade latency was accurate only to 10 msec. Ocular pursuits were assessed by having subjects follow a small laser target moving at 0.2 Hz sinusoidally with a peak velocity of 22.6 deg/sec. Eye movements were analyzed to yield smooth pursuit gain.
Statistical Analysis
Summary statistics included frequencies and percentages for categorical variables, and means, standard deviations, medians, and ranges for continuous variables for the study and control groups. Saccade and pursuit data were adjusted for age and Full Scale IQ. To test the differences in the distributions between the two groups, a chi-squared test was used for categorical variables and a Wilcoxon rank-sum test was used for continuous variables, due to the skewness of the distributions. We assessed the group difference by a multivariate regression adjusting for age and Full Scale IQ. When outliers existed, we fitted a robust regression; otherwise we used a linear regression, with outcome measures transformed by Box-Cox transformation if the measure was non-normal. Locally weighted scatterplot smoothing curves were drawn for the measurement with significant difference between groups. Analysis of age effects was accomplished using a series of age moderator models followed by more focused analyses within a priori determined age groups. Age bins were chosen to correspond to age analyses in other studies, in particular the study that subdivided subjects between 8 and 33 years of age into three age bins (Luna et al., 2004), and other major developmental milestones: 5-7 (n=33), 8-12 (n=24), 13-17 (n=44), 18-33 (n=26), 34-53 (n=14) years. Levene's test was used to examine potential significant differences in variability in smooth pursuit gain. Levene's test for equality of variances was used to compare sample variance estimates for the ASD and control groups. This commonly used test provides a direct comparison of variability in eye movement data between individuals with ASD and control participants by calculating within-group variance statistics and testing the magnitude of their difference between groups using an F distribution with k - 1 and N - k numerator and denominator degrees of freedom, respectively, where k = the number of groups examined. The result is an analytic approach that will test the hypothesis that group variances are equal, and reject this hypothesis in the case of a significant critical F-value.
RESULTS
Demographic data are provided in Table 1. There were no statistical differences between the groups with regard to age, gender, Verbal IQ score and SES. Mean Full Scale IQ was significantly lower (p=0.05) in the AUT group, but the 5 point difference between group means was within the confidence intervals for the instruments. Likewise, the 5 point difference in group means for Performance IQ (p=0.06) was also within the confidence intervals for the instrument. Thus, neither IQ score difference was clinically meaningful.
Table 1.
Demographic data
| Variables | AUT n=79 | Control n=62 | p-value |
|---|---|---|---|
| Age: | n=79 | n=62 | |
| mean±sd | 17.0±10.4 | 16.5±10.4 | 0.81 |
| median(range) | 15.2(46.5) | 14.4(46.4) | |
| Gender: | n=79 | n=62 | |
| Male | 71(89.9%) | 56(90.3%) | 0.93 |
| Female | 8(10.1%) | 6(9.7%) | |
| Full IQ: | n=79 | n=62 | |
| mean±sd | 94.8±15.0 | 99.8±14.4 | 0.05 |
| median(range) | 95.0(66.0) | 98.0(64.0) | |
| Verbal IQ: | n=79 | n=60 | |
| mean±sd | 96.0±17.2 | 100.1±14.7 | 0.10 |
| median(range) | 95.0(78.0) | 101.0(65.0) | |
| Performance IQ: | n=79 | n=60 | |
| mean±sd | 94.2±13.5 | 98.57±13.3 | 0.06 |
| median(range) | 93.0(56.0) | 100.0(63.0) | |
| SES | n=79 | n=60 | |
| mean±sd | 3.5±1.6 | 3.6±1.6 | 0.86 |
| median(range) | 3.0 (6.0) | 4.0 (6.0) |
Vestibular Testing
The results of vestibular testing are provided in Table 2. Assessment of the vestibulo-ocular reflex, using constant velocity and earth-vertical axis rotation in darkness, indicated no differences in gain, sensitivity, and time constant between the two groups. Semicircular canal-otolith interaction assessed using post-rotatory head tilt also showed no differences between the two groups. There were also no differences in visual-vestibular interaction assessed using congruent and conflicting visual stimulation.
Table 2.
Vestibulo-ocular reflex data
| Variables | AUT n=79 | Control n=62 | Adjusted p-valuea |
|---|---|---|---|
| Sinusoidal VOR Sensitivity | n=67 | n=56 | |
| mean±sd | 0.69±0.17 | 0.65±0.18 | 0.43 |
| median(range) | 0.66(0.97) | 0.65(0.73) | |
| Sinusoidal VOR Time Constant | n=66 | n=56 | |
| mean±sd (sec) | 37.0±45.3 | 56.2±133.5 | 0.59 |
| median(range) (sec) | 21.2(287.5) | 22.1(939.7) | |
| Constant Velocity VOR Gain | n=69 | n=53 | |
| mean±sd | 0.67±0.19 | 0.68±0.19 | 0.81b |
| median(range) | 0.62(0.81) | 0.66(0.87) | |
| Constant Velocity Time Constant | n=65 | n=56 | |
| mean±sd (sec) | 15.8±4.2 | 16.0±4.4 | 0.80b |
| median(range) (sec) | 15.7(19.3) | 15.4(20.9) | |
| Head Tilt Ratio | n=65 | n=56 | |
| mean±sd | 0.64±0.34 | 0.56±0.21 | 0.82 |
| median(range) | 0.56(1.83) | 0.54(0.99) | |
| Sinusoidal VOR Fixation Ratio | n=67 | n=53 | |
| mean±sd | 0.14±0.07 | 0.16±0.08 | 0.40 |
| median(range) | 0.13(0.55) | 0.14(0.32) | |
| Sinusoidal VVOR Gain | n=69 | n=53 | |
| mean±sd | 0.87±0.23 | 0.89±0.16 | 0.12 |
| median(range) | 0.83(1.31) | 0.87(0.68) |
Age and Full Scale IQ adjusted.
p-values are from linear regression. If not specified, p-values are from robust regression.
Saccades, Pursuit, Variance, and Age Effects
The results of saccade and pursuit testing are presented in Table 3. No between group differences were found for saccade velocity or accuracy. Saccade latency was significantly increased in the AUT group (p=0.01). Pursuit gain was not significantly different between groups, but there was a trend (p 0.06) toward decreased gain in the AUT group. There was significantly greater variability in pursuit gain in the AUT group compared to the CON group using Levine's test. Analysis of age effects with age moderator models did not reveal significant group differences for either saccade latency or pursuit gain. Focused analyses between a priori chosen age groups revealed significant increases in saccade latency for the 8-12 year old and 13-17 year old subgroups (Table 4). Figure 1 provides scatter plots for saccade latency as a function of age.
Table 3.
Saccade and pursuit data
| Variables | AUT n=79 | Control n=62 | Adjusted p-valuea |
|---|---|---|---|
| Saccade Latency (msec) | n=78 | n=61 | |
| mean±sd | 207±47 | 190±36 | 0.01 |
| median(range) | 199(263) | 184(197) | |
| Saccade Accuracy | n=78 | n=61 | |
| mean±sd | 73±14 | 75±12 | 0.18b |
| median(range) | 73(82) | 74(64) | |
| Saccade Velocity (deg/sec) | n=77 | n=60 | |
| mean±sd | 477±105 | 462±75 | 0.38 |
| median(range) | 481(532) | 454(356) | |
| Smooth Pursuit Gain | n=79 | n=61 | |
| mean±sd | 0.77±0.24 | 0.83±0.15 | 0.06b |
| median(range) | 0.78(1.35) | 0.84(0.60) |
Age and Full Scale IQ adjusted.
p-values are from linear regression. If not specified, p-values are from robust regression.
Table 4.
Age effects on saccade latency and pursuit gain
| Age bin (years) | AUT n=79 | Control n=62 | Adjusted p-value |
|---|---|---|---|
| 5 – 7 | n=18 | n=15 | |
| Saccade latency | 0.305 | ||
| mean±sd | 246.39±51.30 | 224.42±35.08 | |
| Smooth pursuit gain | 0.122 | ||
| mean±sd | 0.68±0.23 | 0.77±0.12 | |
| 8 – 12 | n=15 | n=9 | |
| Saccade latency | .028 | ||
| mean±sd | 217.53±46.12 | 190.00±37.85 | |
| Smooth pursuit gain | .799 | ||
| mean±sd | 0.77±0.28 | 0.78±0.15 | |
| 13 – 17 | n=23 | n=21 | |
| Saccade latency | .017 | ||
| mean±sd | 195.06±37.43 | 172.00±21.48 | |
| Smooth pursuit gain | .378 | ||
| mean±sd | 0.79±0.26 | 0.86±0.15 | |
| 18 – 33 | n=15 | n=11 | |
| Saccade latency | .962 | ||
| mean±sd | 182.2±29.09 | 177.11±18.40 | |
| Smooth pursuit gain | .445 | ||
| mean±sd | 0.82±0.21 | 0.89±0.11 | |
| 34 – 52 | n=8 | n=6 | |
| Saccade latency | .899 | ||
| mean±sd | 180.06±28.69 | 185.79±48.75 | |
| Smooth pursuit gain | .449 | ||
| mean±sd | 0.76±0.24 | 0.88±0.21 |
Fig.1.
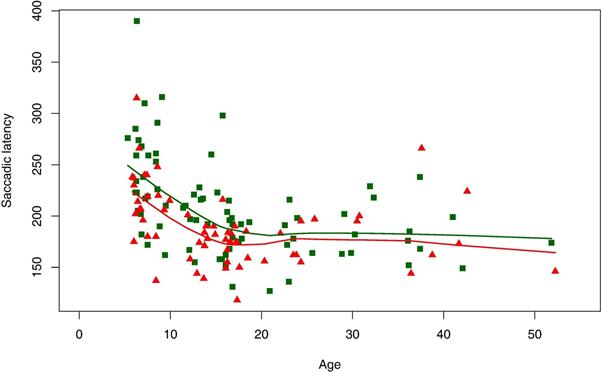
Saccade latency measures for AUT group (green squares) and control group (red triangles), and locally weighted scatterplot smoothing curves for AUT group (green line) and control group (red line).
DISCUSSION
The present study is unique in evaluating multiple aspects of eye movements within a single group of individuals with autism. It also constitutes the most extensive functional assessment to date of posterior fossa circuitry in autism using eye movement procedures. The main findings of this study are the absence of significant differences in the vestibulo-ocular reflex, visual-vestibular interaction, and semicircular canal-otolith interaction and in saccade accuracy and velocity, and the presence of a significant increase in saccade latency with evidence of delayed maturation.
Vestibular Eye Movements
To our knowledge, this is the first study evaluating visual-vestibular interaction in autism. We found no between-group differences in VOR gain and phase, suggesting normal peripheral vestibular function. There were no differences in visual-vestibular interaction or in semicircular canal-otolith interaction. The normal tilt suppression is consistent with a prior report of 13 children with autism (9-17 years; FS IQ 83-146) compared to 10 typical controls (7-15 years; FS IQ 86-137) [Goldberg et al., 2000]. These findings support the functional integrity of vestibular cerebellar and brainstem structures and pathways in children and adults with autism, including the cerebellar flocculus, nodulus and uvula [Baloh & Halmagyi, 1996; Leigh & Zee, 2006]. These findings suggest that the histopathological alterations commonly reported in autism in the cerebellum and sometimes in the brainstem [Bauman & Kemper, 2005; Schumann et al, 2010] may not be associated with intrinsic dysfunction of these structures and pathways, but rather secondary reflections of developmental alterations in cortical systems development and function. Histopathologic alterations in limbic structures in the presence of normal hippocampal memory function have similarly been interpreted as secondary to under-development of cortical connectivity rather than intrinsic dysfunction of these structures [Minshew & Goldstein, 1993].
Pursuit Eye Movements
Smooth pursuit gain was not statistically different between those with and without autism in this study, and there were no between group differences in age effects. The increased variability in pursuit gain may reflect etiologic heterogeneity at the genetic level, which has been suggested as an explanation for the heterogeneity in brain size and growth in autism [Lainhart et al., 2006; O'Roak et al., 2012].
Only two other studies using contemporary diagnostic and ocular motor procedures have explored smooth pursuit eye movements in autism. One study of 16 children with autism or pervasive developmental disorder not otherwise specified (age 10.9±2.0; FSIQ 93+17) compared to 18 typical children (age 10.0±1.5; FSIQ 96±10) found no differences in pursuit eye movements [Kemner et al., 2004]. A study of 60 adolescents and adults with autism (20±11 years; FSIQ102±16) and 94 matched controls (19±11 years; FSIQ 108±13) reported a significant decrease in pursuit gain in the autism group [Takarae et al., 2004]. This study had 19 fewer subjects in their autism group, 32 more controls, and a narrower age range than our study, all of which may have contributed to their detection of significant differences in pursuit gain compared to the trend level finding of our study. This finding was further characterized with ramp and oscillating targets and three tasks, which revealed two types of dysfunction underlying the decrease in smooth pursuit gain. The first finding consisted of poorer closed loop smooth pursuit gain related to dysfunction in perceptual feedback about target tracking error and dynamic predictions about target location; these functions are dependent upon on effective communication among multiple cortical and subcortical regions including frontal cortex, basal ganglia and cerebellum. The second finding entailed a hemi-field open loop deficit indicating dysfunction in left extra-striate areas or pathways carrying visual motion information forward to sensorimotor areas. A recent fMRI study of two pursuit tasks investigating the neural basis of these deficits in visual motion processing and visual sensorimotor control in autism confirmed local alterations in V5 circuitry and reduced top down modulation of sensory processing in V5 [Takarae et al., 2014].
Saccades
The present study found a significant increase in saccade latency in the AUT group, and is the first study in autism to demonstrate this. Though saccade latency is known to vary with level of attention, the intact functioning of our subjects on the other ocular motor tests does not support reduced attention or poor comprehension of test requirements as the basis for this finding. Our study also found a significant age effect between 8 and 17 years of age, suggesting a lag in maturation of these pathways that is consistent with the delay in maturation of postural control previously reported in these subjects. The abnormalities in postural control resulted from dysfunction within the widely distributed network for multi-modality sensory integration [Minshew et al., 2004].
Other studies (discussed below) have reported normal saccade latency but have used targets to a few fixed locations. Our study used targets that moved randomly in time and position rather than to only a few fixed locations repeatedly. For our task, latency was increased in the subjects with autism, suggesting that visual processing was delayed when target location was unknown, thereby placing greater demands on rapid on-line processing of visual information. The only other study that used a protocol similar to ours investigated saccades to a visual target moving randomly in time and position in a range of ±20 degrees in 14 children with autism with IQ scores above 60 and 20 controls with typical cognitive functioning, all between 5 and 12 years of age [Pensiero et al., 2009]. This study found no changes in saccade velocity or accuracy, but reported the same 18 msec increase in mean saccade latency in their autism group as we found in our study. This increase was not statistically significant in their sample, which may reflect a sample size that was one-third the size of the sample in our study. Our sampling rate of 100 Hz limited the precision of estimating saccade latency to 10 msec. This limitation applied to both controls and subjects in the AUT group and, thus, would not be considered likely to influence the comparisons between the two groups. Also, the difference in saccade latency between the two groups in our study agrees with the same measure in an earlier study [Pensiero et al., 2009] in which they used a sampling rate of 500 Hz with a resolution greater than 0.2 deg to measure saccadic eye movements.
Frontal and parietal cortex, cingulate gyrus, and superior colliculus are all thought to be involved in saccade latency [Leigh & Zee, 2006] and all except the superior colliculus have been implicated in autism by neuroimaging and neuropathological studies [Schumann et al., 2010; Schumann et al., 2011]. An fMRI study has reported reduced activation bilaterally in frontal and supplementary eye fields, posterior parietal cortex, visual cortex and the cerebellar hemispheres in subjects with autism relative to controls during a visually guided saccade task, thus supporting a distributed network level localization for disturbances in this ocular motor function [Takarae et al., 2007]. In support of a distributed network level localization in autism is a study by Fan et al. [Fan et al., 2009] wherein the pupillary light reflex (PLR) was measured in a group of 24 participants with ASD and compared with the PLR in a control group. Similar to our finding regarding saccadic latency, the participants with ASD in the Fan et al. study showed prolonged PLR latency.
Our study did not find evidence of significant abnormalities in saccade accuracy or velocity. Abnormalities in saccade accuracy are classically seen in disorders affecting the cerebellar dorsal vermis, i.e. lobules VI and VII, and the fastigial nucleus [Ciuffreda & Tannen, 1995]. The normal saccade accuracy in our population suggests no major involvement of these brain areas. Abnormalities in saccade velocity can be caused by pontine lesions affecting the paramedian pontine reticular formation or by lesions in the upper midbrain. Normal saccade velocity in this study suggests no major localized dysfunction in these areas.
Studies of saccades to fixed locations have reported either no abnormalities in latency, velocity and accuracy, or abnormalities in saccade accuracy which were initially attributed to dysfunction in cerebellar circuitry. A number of these studies reported normal visually guided saccades in the presence of deficits in volitional saccades. A study of 26 verbal adolescent and adult subjects with autism (20.2±8.5 years; IQ 105±13) and 26 controls (20.0±8.7 years; IQ 101±18) with IQ scores above 80 reported normal latency, accuracy, peak velocity, and duration of visually guided saccades at a fixed amplitude of ±10, ±20, and ±30 degrees and deficits on two volitional saccade tasks, indicating dysfunction in prefrontal cortex and its connections to parietal cortex [Minshew et al., 1999]. A subsequent study of 61 subjects with autism and 61 control subjects aged 8 to 33 years with IQ scores above 80 replicated these findings; subdivision of these subjects into three age groups (8-12 year olds, 13-17 year olds, and 18-33 year olds) found no between-group differences in saccade latency, velocity or accuracy except for a decrease in saccade accuracy only in the youngest AUT group (8-12 years) [Luna et al., 2007]. Two small studies of children with autism with IQ scores above 60 compared to typical children reported deficits in saccade accuracy, e.g., hypometric saccades in one study [Rosenhall et al., 1988] and normal saccade metrics in the other [Kemner et al., 2004].
A few studies have subdivided subjects according to early or current language abnormalities reflecting reports of gene contributions specifically associated with disordered language in autism. Results were inconsistent with regard to relationship between language and saccade accuracy and with some reporting hypometric saccades [Takarae et al., 2004; Johnson et al., 2012] and others normal saccade accuracy [Kelly et al., 2013]. Saccade accuracy was investigated in further detail in a study using square wave jerks during visual fixation of central and eccentric targets and of foveopetal ocular drift during fixation of eccentric targets in 52 individuals with autism (17+9 years; FSIQ 106+13) and 52 typical controls (18+9 years; FSIQ 109+12) [Nowinski et al., 2005]. No abnormalities were found in visual fixation providing evidence of the functional integrity of brainstem-cerebellar circuitry involved in ocular motor control even though histopathologic abnormalities have been reported in these structures in autism. Abnormal metrics of intrusive saccades were observed in the autism group and were interpreted as possible evidence of altered functional connectivity in cortico-cerebellar circuitry.
The basis for the different results related to saccade latency in our study compared with previous studies likely relates to differences in methodology with our study using targets that moved unpredictably in time and space, thus increasing the processing demands on cortical systems that provide input to the brainstem-cerebellar oculomotor centers about target location and motion. Several studies found abnormalities in saccade accuracy though we did not; these abnormalities were ultimately traced with more specialized paradigms to cortical systems dysfunction rather than to intrinsic dysfunction in cerebellar brainstem pathways as were abnormalities reported in pursuit gain. Hence, although intrinsic dysfunction in cerebellum and brainstem have been repeatedly hypothesized to play a major role in the pathophysiology of the autism syndrome, recent research emphasizes dysfunction and dysmaturation at the network level in widely distributed networks of which these structures are part [Mostofsky et al., 2009].
Conclusions
The findings of this and prior studies reviewed above provide substantial support for the functional integrity of localized brainstem-cerebellar circuitry in autism, suggesting that histopathological abnormalities may not be associated with intrinsic dysfunction of these structures. Rather, the evidence suggests that these structural alterations more likely reflect altered development and maturation of cortical systems. Elucidation of the mechanisms underlying hindbrain and forebrain development and of the widely distributed circuitry interconnecting them will likely provide new insights into these findings in autism.
Acknowledgments
Supported by NIH (HD055748 from the Eunice Kennedy Shriver National Institute of Child Health & Human Development). Statistical analysis supported by Grant Number 2UL1 RR024153-06 from the National Center for Research Resources (NCRR), a component of the National Institutes of Health (NIH) and NIH Roadmap for Medical Research. We express our appreciation to the individuals and families who gave generously of their time and courage to participate in these imaging studies. This research was supported by the Autism Center of Excellence Grant HD055748 from the National Institute of Child Health and Human Development and the Clinical and Translational Science Institute supported by the National Institutes of Health through Grant Numbers UL1RR024153 and UL1TR000005.
Footnotes
Authors and contributions:
Joseph M. Furman, MD PhD: Design/conceptualization of study, analysis/interpretation of data, drafting/revising manuscript.
Maria Joana Osorio, MD: Drafting/revising manuscript and analysis/interpretation of data.
Nancy J. Minshew, MD: Design/conceptualization of study, analysis/interpretation of data, manuscript drafting/revising.
Disclosures of financial author's relationships:
Joseph M. Furman, MD PhD: Nothing to disclose.
Maria Joana Osorio, MD: Nothing to disclose.
Nancy J. Minshew, MD: Nothing to disclose.
Contributor Information
Joseph M. Furman, Departments of Otolaryngology and Neurology, University of Pittsburgh School of Medicine.
Maria Joana Osorio, Division of Child Neurology, Children's Hospital of Pittsburgh, University of Pittsburgh School of Medicine.
Nancy J. Minshew, Departments of Psychiatry & Neurology, University of Pittsburgh School of Medicine.
References
- Abrahams BS, Geschwind DH. Advances in autism genetics: on the threshold of a new neurobiology. Nature Reviews. Genetics. 2008;9(5):341–355. doi: 10.1038/nrg2346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baloh RW, Honrubia V, Konrad HR. Periodic alternating nystagmus. Brain. 1976;99(1):11–26. doi: 10.1093/brain/99.1.11. [DOI] [PubMed] [Google Scholar]
- Baloh RW, Honrubia V. Reaction time and accuracy of the saccadic eye movements of normal subjects in a moving target task. Aviation Space and Environmental Medicine. 1976;47:1165–1167. [Google Scholar]
- Baloh RW, Jenkins HA, Honrubia V, Yee RD, Lau CG. Visual-vestibular interaction and cerebellar atrophy. Neurology. 1979;29(1):116–119. doi: 10.1212/wnl.29.1.116. [DOI] [PubMed] [Google Scholar]
- Baloh RW, Sills AW, Honrubia V. Impulsive and sinusoidal rotatory testing: a comparison with results of caloric testing. Laryngoscope. 1979;89:646–654. doi: 10.1288/00005537-197904000-00013. [DOI] [PubMed] [Google Scholar]
- Baloh RW, Henn V, Jager J. Habituation of the human vestibulo-ocular reflex with low-frequency harmonic acceleration. American Journal of Otolaryngology. 1982;3(4):235–241. doi: 10.1016/s0196-0709(82)80061-6. [DOI] [PubMed] [Google Scholar]
- Baloh RW, Yee RD, Jenkins H, Honrubia V. Nystagmus and vertigo: clinical approaches to the patient with dizziness. Academic Press; Orlando, Fl: 1982. Quantitative assessment of visual-vestibular interaction using sinusoidal rotatory stimuli. pp. 231–239. [Google Scholar]
- Baloh RW, Sakala SM, Yee RD, Langhofer L, Honrubia V. Quantitative vestibular testing. Otolaryngology—Head and Neck Surgery. 1984;92(2):145–150. doi: 10.1177/019459988409200205. [DOI] [PubMed] [Google Scholar]
- Baloh RW, Honrubia V, Yee RD, Hess K. Changes in human vestibulo ocular reflex after loss of peripheral sensitivity. Annals of Neurology. 1984;16:222–228. doi: 10.1002/ana.410160209. [DOI] [PubMed] [Google Scholar]
- Baloh RW, Honrubia V. Clinical neurophysiology of the vestibular system (2nd edition) F.A. Davis Co.; Philadelphia: 1990. [PubMed] [Google Scholar]
- Baloh RW, Halmagyi GM. Disorders of the vestibular system, Neuropsychology. Oxford University Press; New York: 1996. [Google Scholar]
- Bauman ML, Kemper TL. Structural brain anatomy in autism: What is the evidence? In: Bauman ML, Kemper TL, editors. The neurobiology of autism, 2e. The Johns Hopkins University Press; Baltimore: 2005. pp. 121–135. [Google Scholar]
- Ciuffreda KJ, Tannen B. Eye movement basics for the clinician. Mosby; St. Louis: 1995. [Google Scholar]
- Courchesne E, Townsend J, Akshoomoff NA, Saitoh O, Yeung-Courchesne R, Lincoln AJ, et al. Impairment in shifting attention in autistic and cerebellar patients. Behavioral Neuroscience. 1994;108(5):848–865. doi: 10.1037//0735-7044.108.5.848. [DOI] [PubMed] [Google Scholar]
- Fan X, Miles JH, Takahashi N, Yao G. Abnormal transient pupillary light reflex in individuals with autism spectrum disorders. Journal of Autism and Developmental Disorders. 2009;39(11):1499–1508. doi: 10.1007/s10803-009-0767-7. [DOI] [PubMed] [Google Scholar]
- Fatemi SH, Aldinger KA, Ashwood P, Bauman ML, Blaha CD, Blatt GJ, et al. Consensus paper: pathological role of the cerebellum in autism. Cerebellum. 2012;11(3):777–807. doi: 10.1007/s12311-012-0355-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Furman JM, Kamerer DB. Rotational responses in patients with bilateral caloric reduction. Acta Otolaryngologica (Stockh) 1989;108:355–361. doi: 10.3109/00016488909125539. [DOI] [PubMed] [Google Scholar]
- Goldberg MC, Landa R, Lasker A, Cooper L, Zee DS. Evidence of normal cerebellar control of the Vestibulo-Ocular Reflex (VOR) in children with high-functioning autism. Journal of Autism Developmental Disorders. 2000;30:519–524. doi: 10.1023/a:1005631225367. [DOI] [PubMed] [Google Scholar]
- Hain TC, Zee DS, Maria BL. Tilt suppression of vestibulo-ocular reflex in patients with cerebellar lesions. Acta Otolaryngology. 1988;105(1-2):13–20. doi: 10.3109/00016488809119440. [DOI] [PubMed] [Google Scholar]
- Hazlett HC, Poe M, Gerig G, et al. Magnetic resonance imaging and head circumference study of brain size in autism: birth through age 2 years. Archives of General Psychiatry. 2005;62:1366–1376. doi: 10.1001/archpsyc.62.12.1366. [DOI] [PubMed] [Google Scholar]
- James SJ, Shpyleva S, Melnyk S, Pavliv O, Pogribny IP. Complex epigenetic regulation of engrailed-2 (EN-2) homeobox gene in the autism cerebellum. Translational Psychiatry. 2013;3:e232. doi: 10.1038/tp.2013.8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson BP, Rinehart NJ, Papadopoulos N, et al. A closer look at visually guided saccades in autism and Asperger's disorder. Frontiers in Integrative Neuroscience. 2012;6:99. doi: 10.3389/fnint.2012.00099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelly DJ, Walker R, Norbury CF. Deficits in volitional oculomotor control align with language status in autism spectrum disorders. Developmental Science. 2013;16:56–66. doi: 10.1111/j.1467-7687.2012.01188.x. [DOI] [PubMed] [Google Scholar]
- Kemner C, van der Geest JN, Verbaten MN, van Engeland H. In search of neurophysiological markers of pervasive developmental disorders: smooth pursuit eye movements? Journal of Neural Transmission. 2004;111:1617–1626. doi: 10.1007/s00702-004-0164-5. [DOI] [PubMed] [Google Scholar]
- Kemper TL, Bauman ML. Neuropathology of infantile autism. Journal of Neuropathology and Experimental Neurology. 1998;57:645–652. doi: 10.1097/00005072-199807000-00001. [DOI] [PubMed] [Google Scholar]
- Lainhart JE, Bigler ED, Bocian M, et al. Head circumference and height in autism: a study by the collaborative program of excellence in autism. American Journal of Medical Genetics. Part A. 2006;140:2257–2274. doi: 10.1002/ajmg.a.31465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leigh RJ, Zee DS, editors. The Neurology of Eye Movements. 4e. Oxford University Press; New York: 2006. [Google Scholar]
- Lord C, Rutter M, Goode S, et al. Autism diagnostic observation schedule: a standardized observation of communicative and social behavior. Journal of Autism Developmental Disorders. 1989;19:185–212. doi: 10.1007/BF02211841. [DOI] [PubMed] [Google Scholar]
- Lord C, Rutter M, Le Couteur A. Autism Diagnostic Interview-Revised: a revised version of a diagnostic interview for caregivers of individuals with possible pervasive developmental disorders. Journal of Autism Developmental Disorders. 1994;24:659–685. doi: 10.1007/BF02172145. [DOI] [PubMed] [Google Scholar]
- Luna B, Doll SK, Hegedus SJ, Minshew NJ, Sweeney JA. Maturation of executive function in autism. Biological Psychiatry. 2007;61:474–481. doi: 10.1016/j.biopsych.2006.02.030. [DOI] [PubMed] [Google Scholar]
- McFadden K, Minshew NJ. Evidence for dysregulation of axonal growth and guidance in the etiology of ASD. Frontiers in Human Neuroscience. 2013;7:671. doi: 10.3389/fnhum.2013.00671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Minshew NJ, Goldstein G. Is autism an amnesic disorder? Biological Psychiatry. 1993;33(Suppl 6A):105A. doi: 10.1016/0006-3223(93)90017-8. [DOI] [PubMed] [Google Scholar]
- Minshew NJ, Goldstein G, Siegel DJ. Neuropsychologic functioning in autism: profile of a complex information processing disorder. Journal of the International Neuropsychological Society. 1997;3:303–316. [PubMed] [Google Scholar]
- Minshew NJ, Luna B, Sweeney JA. Oculomotor evidence for neocortical systems but not cerebellar dysfunction in autism. Neurology. 1999;52:917–922. doi: 10.1212/wnl.52.5.917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Minshew NJ, Sung K, Jones BL, Furman JM. Underdevelopment of the postural control system in autism. Neurology. 2004;63:2056–2061. doi: 10.1212/01.wnl.0000145771.98657.62. [DOI] [PubMed] [Google Scholar]
- Mosconi MW, Luna B, Kay-Stacey M, et al. Saccade adaptation abnormalities implicate dysfunction of cerebellar-dependent learning mechanisms in Autism Spectrum Disorders (ASD). PLoS One. 2013;8:e63709. doi: 10.1371/journal.pone.0063709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mostofsky SH, Powell SK, Simmonds DJ, Goldberg MC, Caffo B, Pekar JJ. Decreased connectivity and cerebellar activity in autism during motor task performance. Brain. 2009;132:2413–2425. doi: 10.1093/brain/awp088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nowinski CV, Minshew NJ, Luna B, Takarae Y, Sweeney JA. Oculomotor studies of cerebellar function in autism. Psychiatry Research. 2005;5(137):11–19. doi: 10.1016/j.psychres.2005.07.005. [DOI] [PubMed] [Google Scholar]
- Ornitz EM, Ritvo ER. Neurophysiologic mechanism underlying perceptual inconstancy in autistic and schizophrenic children. Archives of General Psychiatry. 1986a;19:22–27. doi: 10.1001/archpsyc.1968.01740070024004. [DOI] [PubMed] [Google Scholar]
- Ornitz EM, Ritvo ER. Perceptual inconstancy in early infantile autism. Archives of General Psychiatry. 1986b;18:76–98. doi: 10.1001/archpsyc.1968.01740010078010. [DOI] [PubMed] [Google Scholar]
- O'Roak BJ, Vives L, Fu W, Egertson JD, Stanaway IB, Phelps IG, et al. Multiplex targeted sequencing identifies recurrently mutated genes in autism spectrum disorders. Science. 2012;338:1619–1622. doi: 10.1126/science.1227764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parikshak NN, Luo R, Zhang A, et al. Integrative functional genomic analyses implicate specific molecular pathways and circuits in autism. Cell. 2013;155:1008–1021. doi: 10.1016/j.cell.2013.10.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pensiero S, Fabbro F, Michieletto P, Accardo A, Brambilla P. Saccadic characteristics in autistic children. Functional Neurology. 2009;24:153–158. [PubMed] [Google Scholar]
- Rogers SJ. What are infant siblings teaching us about autism in infancy? Autism Research. 2009;2:125–137. doi: 10.1002/aur.81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rogers TD, McKimm E, Dickson PE, Goldowitz D, Blaha CD, Mittleman G. Is autism a disease of the cerebellum? An integration of clinical and pre-clinical research. Frontiers in System Neuroscience. 2013;7:15. doi: 10.3389/fnsys.2013.00015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenhall U, Johansson E, Gillberg C. Oculomotor findings in autistic children. Journal of Laryngology and Otology. 1988;102:435–439. doi: 10.1017/s0022215100105286. [DOI] [PubMed] [Google Scholar]
- Rossman IT, Lulu L, Morgan KM, DiGiovine M, Van Buskirk EK, Kamdar S, et al. Molecular Autism. 2014;5:9. doi: 10.1186/2040-2392-5-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schaaf CP, Zoghbi HY. Solving the autism puzzle a few pieces at a time. Neuron. 2011;70(5):806–808. doi: 10.1016/j.neuron.2011.05.025. [DOI] [PubMed] [Google Scholar]
- Schipul SE, Keller TA, Just MA. Inter-regional brain communication and its disturbance in autism. Frontiers in System Neuroscience. 2011;5:10. doi: 10.3389/fnsys.2011.00010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schumann CM, Bloss CS, Barnes CC, et al. Longitudinal MRI study of cortical development through early childhood in autism. Journal of Neuroscience. 2010;30:4419–4427. doi: 10.1523/JNEUROSCI.5714-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schumann CM, Noctor SC, Amaral DG. Neuropathology of autism spectrum disorders: postmortem studies. In: Amaral DG, Dawson G, Geschwind DH, editors. Autism spectrum disorders. Oxford University Press; New York: 2011. pp. 539–565. [Google Scholar]
- Scott JA, Schumann CM, Goodlin-Jones BL, Amaral DG. A comprehensive volumetric analysis of the cerebellum in children and adolescents with autism spectrum disorder. Autism Research. 2009;2(5):246–257. doi: 10.1002/aur.97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stanley-Cary C, Rinehart N, Tonge B, White O, Fielding J. Greater disruption to control voluntary saccades in autistic disorder than Asperger's Disorder: evidence for greater cerebellar involvement in autism? Cerebellum. 2011;10:70–80. doi: 10.1007/s12311-010-0229-y. [DOI] [PubMed] [Google Scholar]
- Strick P, Dum R, Fiez J. Cerebellum and nonmotor function. Annual Review of Neuroscience. 2009;32(1):413–434. doi: 10.1146/annurev.neuro.31.060407.125606. [DOI] [PubMed] [Google Scholar]
- Takarae Y, Minshew NJ, Luna B, Krisky CM, Sweeney JA. Pursuit eye movement deficits in autism. Brain. 2004;127:2584–2594. doi: 10.1093/brain/awh307. [DOI] [PubMed] [Google Scholar]
- Takarae Y, Minshew NJ, Luna B, Sweeney JA. Oculomotor abnormalities parallel cerebellar histopathology in autism. Journal of Neurology, Neurosurgery, and Psychiatry. 2004;75:1359–1361. doi: 10.1136/jnnp.2003.022491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takarae Y, Minshew NJ, Luna B, Sweeney JA. Atypical involvement of frontostriatal systems during sensorimotor control in autism. Psychiatry Research. 2007;156:117–127. doi: 10.1016/j.pscychresns.2007.03.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takarae Y, Luna B, Minshew NJ, Sweeney JA. Visual motion processing and visual sensorimotor control in autism. Journal of the International Neuropsychological Society. 2014;20:113–122. doi: 10.1017/S1355617713001203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Volpe JJ. Neuronal proliferation, migration, organization, and myelination. In: Volpe JJ, editor. Neurology of the Newborn. Saunders Elsevier; Philadelphia: 2008. p. 51. [Google Scholar]
- Whitney ER, Kemper TL, Bauman ML, Rosene DL, Blatt GJ. Cerebellar Purkinje cells are reduced in a subpopulation of autistic brains: a stereological experiment using calbindin-D28k. Cerebellum. 2008;7(3):406–416. doi: 10.1007/s12311-008-0043-y. [DOI] [PubMed] [Google Scholar]
- Whitney ER, Kemper TL, Rosene DL, Bauman ML, Blatt GJ. Density of cerebellar basket and stellate cells in autism: Evidence for a late developmental loss of Purkinje cells. Journal of Neuroscience Research. 2009;87:2245–2254. doi: 10.1002/jnr.22056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wierts R, Janssen MJ, Kingma H. Measuring saccade peak velocity using a low-frequency sampling rate of 50 Hz. IEEE Transactions on Biomedical Engineering. 2008;55(12):2840–2842. doi: 10.1109/TBME.2008.925290. [DOI] [PubMed] [Google Scholar]
- Williams DL, Goldstein G, Minshew NJ. Neuropsychologic functioning in children with autism: further evidence for disordered complex information-processing. Child Neuropsychology. 2006;12:279–298. doi: 10.1080/09297040600681190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams DL, Vladimir LC, Mason RA, Keller TA, Minshew NJ, Just MA. Brain function differences in language processing in children and adults with autism. Autism Research. 2013;6(4):288–302. doi: 10.1002/aur.1291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Willsey AJ, Sanders SJ, Mingfeng L, et al. Coexpression networks implicate human midfetal deep cortical projection neurons in the pathogenesis of autism. Cell. 2013;155:997–1007. doi: 10.1016/j.cell.2013.10.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolff JJ, Gu H, Gerig G, et al. Differences in white matter fiber tract development present from 6 to 24 months in infants with autism. American Journal of Psychiatry. 2012;169:589–600. doi: 10.1176/appi.ajp.2011.11091447. [DOI] [PMC free article] [PubMed] [Google Scholar]