

Abstract

A remarkable aspect of enzyme evolution is the portability of catalytic mechanisms for fundamentally different chemical reactions. For example, aspartyl proteases, which contain two active site carboxylic acid groups, catalyze the hydrolysis of amide bonds, while glycosyltransferases (and glycosyl hydrolases), which often also contain two active site carboxylates, have evolved to form (or break) glycosidic bonds. However, neither catalyst exhibits cross-reactivity in the intracellular environment. The large, macromolecular architectures of these biocatalysts tailor their active sites to their precise, divergent functions. The analogous portability of a small-molecule catalyst for truly orthogonal chemical reactivity is rare. Herein, we report aspartic acid containing peptides that can be directed to different sectors of a substrate for which the danger of cross-reactivity looms large. A transiently formed aspartyl peracid catalyst can participate either as an electrophilic oxidant to catalyze alkene epoxidation or as a nucleophilic oxidant to mediate the Baeyer–Villiger oxidation (BVO) of ketones. We show in this study that an appended peptide sequence can dictate the mode of reactivity for this conserved catalytic functional group within a substrate that has the potential to undergo both alkene epoxidation and BVO; in both cases the additional aspects of chemical selectivity (regio- and stereoselectivity) are high. This sequence-dependent tuning of a common catalytic moiety for functional group selective reactions constitutes a biomimetic strategy that may impact late-stage diversification of complex polyfunctional molecules.
Short abstract
The sequence of an aspartyl peptide catalyst can dictate functional group selectivity within a complex substrate, wherein a transient peracid acts as either an electrophile or a nucleophile, catalyzing orthogonal chemical reactions.
Introduction
The management of competing chemical reactivity is a fundamental marvel of biochemistry.1−8 For synthetic chemists, however, it is a major challenge in complex molecular environments. Most chemical reactions target a unique mechanistic paradigm such that the pairing of one reactant’s potential complements the reactivity of another.9,10 Enzymes generally abide by these rules, but in many cases they have evolved to exhibit a higher level of selectivity, such that specific reactions can occur in the presence of many competing reaction pathways involving functional groups of similar reactivity. We posited that small-molecule catalysts might also exhibit orthogonal reactivity as a result of rational modification of the scaffolding in the vicinity of a catalytic moiety that would dominate the reactive, bond-forming function of the catalyst.11
We explored scenarios with the catalytic potential presented by the transient conversion of a terminal aspartyl residue to the corresponding aspartyl peracid by reaction of an activated acid with hydrogen peroxide (Figure 1a).12 The ambiphilic aspartyl peracid can deliver an O atom by one of two reaction pathways. In an epoxidation pathway, the peracid moiety is electrophilic (Figure 1a, path A); in the BVO pathway, the peracid is nucleophilic (Figure 1a, path B). Thus, the peracid moiety provides an opportunity to probe how the molecular environment surrounding a transiently generated reactive intermediate might fundamentally dictate a divergent chemical outcome. Recent studies from our laboratory have demonstrated the feasibility of each independent pathway (epoxidation and BVO). For example, utilizing combinatorial techniques, we discovered that peptide 1, Boc-Asp-Pro-Asn(Trt)-dPhe-Pro-Asn(Trt)-OMe, could catalyze the asymmetric epoxidation of many allylic alcohols, with enantioselectivities as high as 99:1 er (Figure 1a, path A).13,14 In a separate study, peptide 2, Boc-Asp-Pro-dLys(Boc)-dPro-Tyr(tBu)-OMe, was discovered to be an effective catalyst for certain BVO reactions.15 Perhaps most notably, catalyst 2 not only proved effective for the ring expansion of cyclic ketones but also favored the product of reversed migratory aptitude in a number of cases. For example, with phenylacetamide 3 the conventional, stoichiometric oxidant m-chloroperoxybenzoic acid (mCPBA) produces “normal” lactone 4 as the major product (2.5:1), while catalyst 2 delivers lactone 5 (1:28 in the stereochemically matched series; Figure 1a, path B). These BVO reactions also exhibit high levels of enantioselectivity and the hallmarks of kinetic resolution when racemic starting materials are employed (e.g., lactone 5 observed with 97:3 er).16
Figure 1.

Aspartyl oxidation catalysts with different functional group reaction pathways: (a) catalytic cycles possible with aspartyl peptides under similar reaction conditions; (b) aspartyl peptide catalysts used in this study; (c) our proposed study featuring both a site of directed epoxidation and Baeyer–Villiger oxidation; (d) computations demonstrate the possibility of also examining the topological features of the proposed substrates 6 and 7; DIC = N,N′-diisopropylcarbodiimide, H2O2 = hydrogen peroxide, Bn = benzyl, Boc = tert-butoxycarbonyl, Trt = trityl, mCPBA = meta-chloroperoxybenzoic acid.
These results, while providing a certain set of chemical validations for these as yet unknown roles of aspartic acid in biochemistry, encouraged us to investigate whether the specific function of the aspartyl catalysts (epoxidation versus BVO) could be preserved, and in fact dictated by the peptide sequence in the situation of an intramolecular competition between multiple reactive sites in a single substrate.17 Therefore, we designed substrates (6-cis and 7-trans; Figure 1c) to test the ideas.18 These compounds were appealing since each (1) possesses attributes for both independent oxidation reactions; (2) could be compared as individual diastereomers; (3) could be obtained in its corresponding enantiopure form, in anticipation of the high degree of enantiospecificity exhibited in the reactions catalyzed by 1 and 2.19,20 Keto-olefins 6-cis and 7-trans are also intriguing in that each presents different topological features in their oxidation reactions, in addition to different conformational equilibria. Computations provided a quantitative description of these geometrical features (Figure 1d), showing that 6-cis exhibits an overwhelming preference for the mutually bis-equatorial conformation (99:1). On the contrary, 7-trans was found to display a 43:57 equilibrium distribution of conformers, suggesting that both conformations are amply populated under the reaction conditions, which adds an additional challenge for catalyst selectivity.21 The intrinsic reactivity of these compounds and their behavior in catalytic reactions are described below.
Results and Discussion
Our initial studies focused on benchmarking the intrinsic reactivity of compounds like 6 and 7 toward the generic oxidation reagent, m-chloroperoxybenzoic acid. To probe the fundamental BVO tendencies, acetate analogues 8-cis and 9-trans were examined (Scheme 1), expecting that acetylation of the hydroxyl group of 8/9 would effectively dampen the epoxidation pathway, on the basis of well-known electronic effects in olefin epoxidation.22 Under standard mCPBA conditions,15 substrate 8-cis proceeds to partial conversion furnishing lactones 10-normal and 11-reversed, albeit in a modest 4.3:1 ratio (Scheme 1, entry 1), along with some degree of unselective epoxidation. Under the same conditions, compound ent-9-trans was found to undergo a more efficient BVO with 90% conversion, with ent-12-normal and ent-13-reversed formed in an attenuated ratio of 2.3:1 (Scheme 1, entry 3).23,24 In this case, epoxide formation is not observed to an appreciable extent. Importantly, these findings suggest that the bis(equatorial)-substituted ketone (8-cis) is substantially less reactive than the axial/equatorial-substituted ketone (9-trans), likely due to intrinsic ground state destabilization in 9-trans. When the reaction is catalyzed by previously discovered BVO catalyst 2, minimal reactivity is observed with ketone 8-cis (Scheme 1, entry 5). However, ketone ent-9-trans undergoes highly efficient BVO with enhanced regioselectivity for ent-13-reversed over ent-12-normal, at a ratio of >99:1 (Scheme 1, entry 6). This excellent chemo- and regioselectivity persists when the catalyst loading is reduced (Scheme 1, entry 7). These results reveal a high level of reactivity between catalyst 2 and the ketone ent-9-trans, echoing the previously observed high level of stereochemical matching between 2 and ketone 3, the topology of which is stereochemically complementary. With these observations in hand, we thus proceeded with the trans series (e.g., 7-trans-like compounds) for the critical assessment of functional group selectivity under the influence of peptides 1 and 2.
Scheme 1. Examination of Substituent Orientation and Baeyer–Villiger Oxidation Tendencies.

Standard conditions with mCPBA: substrate (1.0 equiv), mCPBA (1.0 equiv), chloroform, magnetic stirring. Standard conditions with peptide 2: substrate (1 equiv), 2 (10 mol %), 4-dimethylaminopyridine (DMAP; 10 mol %), aqueous H2O2 (30 wt %; 2.0 equiv), N,N′-diisopropylcarbodiimide (DIC; 3.0 equiv), chloroform, 4 °C, 24 h, magnetic stirring. bAverage from two trials; determined by uncalibrated HPLC integrations; value in parentheses refers to isolated yield (see Supporting Information for details). cOther oxidation products were detected.
To benchmark the intrinsic reactivity of keto-olefins with the requisite allylic alcohol 14-trans (with no artificially introduced attenuation of epoxidation reactivity), we once again examined reactions with mCPBA as a preamble to reactions with peptides 1 and 2 (Scheme 2, entry 1).25 When a stoichiometric amount of mCPBA is used, the reaction proceeds unselectively with respect to both functional group selectivity and regioselectivity, producing a myriad of products (15–22). Notably, with octanoic acid as a catalyst under conditions analogous to those employed for catalysts 1 and 2, only trace reactivity is observed. Furthermore, the use of protected aspartic acid as a catalyst (Boc-Asp(OH)-OMe) also resulted in low catalytic activity, favoring primarily epoxide products 15 and 16 (Scheme 2, entry 3). Collectively, these results demonstrate the ineffectiveness of the above catalysts in controlling the complex reaction coordinate that substrate 14-trans presents.
Scheme 2. Chemoselectivity Evaluation and Optimization.

Standard conditions with mCPBA: substrate (1.0 equiv), mCPBA (1.0 equiv), chloroform (0.05 M), 4 °C, magnetic stirring. Standard conditions with peptide 1: substrate (1 equiv), 1 (10 mol %), 4-dimethylaminopyridine (10 mol %), 1-hydroxybenzotriazole (10 mol %), aqueous H2O2 (30 wt %; 2.0 equiv), N,N′-diisopropylcarbodiimide (3.0 equiv), chloroform (0.05 M), 4 °C, magnetic stirring. Standard conditions with peptide 2: substrate (1 equiv), 2 (10 mol %), 4-dimethylaminopyridine (10 mol %), H2O2 (30 wt %; 2.0 equiv), N,N′-diisopropylcarbodiimide (3.0 equiv), chloroform (0.1 M), 4 °C, magnetic stirring. bAverage from two trials; determined by uncalibrated HPLC integrations of the crude reaction media (see Supporting Information for details). cMixing @ 30 rpm. d15 + 16 only.
However, when peptide 1 is used as a catalyst in the reaction, a strikingly different outcome is obtained. Under these conditions, substrate 14-trans is consumed quantitatively, and the major product is monoepoxide 15, formed with >99:1 diastereoselectivity over 16 (Scheme 2, entry 4). Thus, peptide 1, as a function of its secondary structure, has exerted near total control of the complex reaction coordinate potential presented by multifunctional substrate 14, showcasing highly unique functional group selectivity. Examination of substrate 14-trans with the enantiomer of this peptide, ent-1, reveals a configuration mismatch of the olefinic side arm, resulting in lower levels of conversion and erosion of the diastereoselectivity being observed (1:4.8; 15:16; Scheme 2, entry 5); interestingly, as with catalyst 1, products derived from the BVO reaction are not detected.
We then turned our attention to the question of whether or not the previously discovered BVO catalyst 2 might exhibit orthogonal reactivity. The first experiment provided a cautionary tale. When substrate 14-trans is exposed to peptide 2 under the original catalytic conditions, the reaction proceeds to 38% conversion; however, instead of the anticipated preference for BVO products, we observe 6:1 preference for epoxides 15 and 16 over lactones 17 and 18. The diastereoselectivity for the epoxidation is also poor (1:1), and the regioisomeric preference among the lactones is discouraging, at a level of 6:1 favoring the normal over the reversed isomers (Scheme 2, entry 6). Based on these results, we speculated that the originally studied enantiomer of peptide 2 was stereochemically mismatched with bifunctional substrate 14-trans.18 Therefore, we prepared ent-2 and re-examined its performance under the same conditions, and striking differences were observed (Scheme 2, entries 7–10).
Under these previously established reaction conditions,15 we now observed an improvement in BVO selectivity such that near parity between the epoxidation versus BVO pathways was observed (Scheme 2, entry 7), with a 1.0:0.8 preference for epoxides 15 and 16 over lactones 17 and 18. Of particular note, the preference for the lactone derived from reversed migratory aptitude was restored (1:26). Optimization of this result was guided by analysis of 1H-1H NMR derived structures of peptides 1 and 2 that rationalized the observations in our initial studies of enantioselective epoxidation and regio- and enantioselective BVO in our initial studies (Figure 2). In the case of 1-catalyzed epoxidation, hydroxyl-directed epoxidation through hydrogen bonding is consistent with previously reported enantioselectivity in epoxidation reactions (Figure 2a).14 In the case of 2-catalyzed BVO, a favorable interaction of the amido group with catalyst 2 is consistent with our recent observations (Figure 2b). This hypothesis was stimulated by analysis of an experimentally derived (ROESY) solution structure of 2,26 along with our study of catalyst analogues.15 A critical role for the lysine side chain, along with the schematic secondary structure shown, contributes to a view of competing hydrogen bonding networks as catalyst ent-2 operates on substrate 14-trans (Figure 2c). One network promotes hydroxyl-directed epoxidation (the blue arrow); the second network promotes amide-directed BVO (the red arrow). Critically, the blue arrow implies an electrophilic peracid; the red arrow implies a nucleophilic peracid. Our analysis culminated in the hypothesis that disruption of the hydroxyl-directed epoxidation pathway in blue might amplify selectivity for the BVO pathway with catalyst ent-2, if the red network could be preserved.
Figure 2.
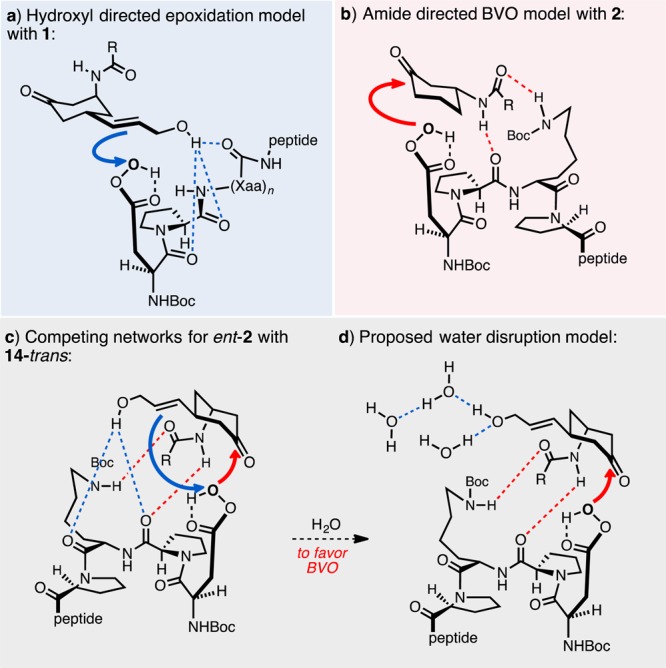
Representative models for the competing reaction pathways: (a) hypothetical model for hydroxyl-directed epoxidation; (b) hypothetical model for amide-directed Baeyer–Villiger oxidation; (c) presentation of a competing H-bonding network; (d) proposed water disruption of the H-bonding network.
To disrupt putative hydrogen bonds to the substrate hydroxyl group, we hypothesized that an increased concentration of H2O might effectively compete with hydroxyl-directed epoxidation to a greater extent than it would perturb the amide-directed BVO (Figure 2d). This speculation is in keeping with theories about hydrogen bond strength that point to the importance of complementarity of hydrogen bond donors and acceptors.27 A systematic study with additional H2O content supports these notions (Figure 3). Our findings reveal a linear relationship between the overall water content of the reaction medium and BVO/epoxide selectivity when using the BVO peptide catalyst ent-2: the more H2O relative to H2O2, the higher the lactone/epoxide ratio. Unambiguously, H2O plays an important role in these reactions, and the outcome is consistent with our hypothesis. With modest increases in H2O content (15–100 extra equivalents of H2O, relative to standard 30 wt % H2O2), overall conversion of substrate 14-trans remains high (70–75%) with a slight increase (2–3-fold, relative to Scheme 2, entry 7) in selectivity increase toward BVO. Strikingly, further increasing of the equivalents of H2O leads to even greater selectivity for BVO (4–5-fold), albeit at some expense of overall conversion (50–58%).28 The erosion of conversion may be due to consumption of the carbodiimide reagent, DIC, and its adducts leading to a buildup of urea byproduct, 1,3-diisopropylurea, a competent H-bond donor/acceptor group.29
Figure 3.
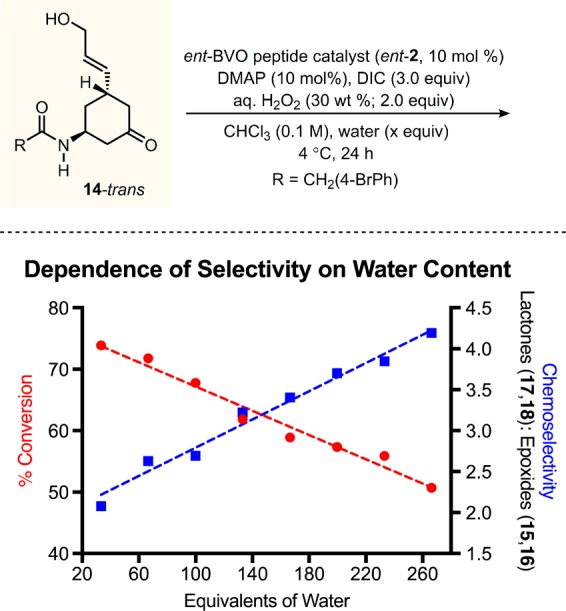
Systematic study with additional H2O added to the reaction media. Standard conditions with peptide 2: 14 (1 equiv), 2 (10 mol %), 4-dimethylaminopyridine (10 mol %), H2O2 (30 wt %; 2.0 equiv), N,N′-diisopropylcarbodiimide (3.0 equiv total; 1 equiv @ 0 h, 4 h, 8 h), chloroform (0.1 M wrt 14), water (n + 15 equiv starting at 30 equiv), 4 °C, mixing @ 30 rpm, 24 h. Conversion and selectivity were determined by uncalibrated HPLC integrations of the crude reaction media (see Supporting Information for details).
These observations enabled further optimization for the BVO pathway. Thus, a transition from aqueous solutions of 30 wt % H2O2 to 3 wt % H2O2 in H2O, ensuring optimum mixing of the biphasic reaction mixture, lead to rather dramatic results, such that substrate 14-trans is converted to the corresponding oxidation products with total control over migratory aptitude (1:>50; Scheme 2, entry 8). Further dilution of the reaction mixture further increases the functional group selectivity leading to enhanced results, such that 53% conversion is observed, with a 1:12.5 ratio of epoxide to BVO products, again with total reversal of migratory aptitude (Scheme 2, entry 9). Finally, execution of these conditions with an increase in catalyst loading provides a high converting reaction with the desired functional group selectivity (Scheme 2, entry 10; see Supporting Information for complete screening of conditions). Under these conditions, the BVO products 17 and 18 are also observed to participate in overoxidation to a somewhat greater extent (19–22), modifying the ratios observed at lower conversion.30 Overall, the BVO selectivity reversal observed using catalyst ent-2 for the oxidation of 14-trans, under high-H2O conditions, appears to be due to a combination of both the catalyst’s unique secondary structure and medium effects where the epoxidation and BVO pathways are differentially modulated by the demands of H2O in this situation of competing hydrogen bond networks.
The critical role of the secondary structure, in light of the optimized high-H2O reaction conditions, prompted us to evaluate several truncated peptide-based analogues of catalyst ent-2 under the optimized reaction conditions. The sequential deletion of amino acids from the C-terminal end of the catalyst ent-2 delivers incrementally inferior chemo- and regioselectivity along with reduced reaction efficiency. As shown in Scheme 3, catalyst 24 wherein the C-terminal dTyr(tBu)-OMe of ent-2 is replaced with a methyl ester (Scheme 3, entry 2) results in both a significant loss of functional group selectivity (1.3:1.0, epoxide:lactone) and a decrease in control of the migratory aptitude of the BVO products (1.0:14.1; 17:18) in addition to lower levels of conversion. Reminiscent of an observation that emerged in our earlier work that the dPro-Xaa-Pro motif is a crucial sequence element for BVO, truncated peptide 25, which lacks the i + 3 Pro, leads to a further erosion in conversion and chemoselectivity with epoxides 15 and 16 being favored over lactones 17 and 18 (3.3:1.0) and a total loss of control in migratory aptitude with a slight preference toward the “normal” lactone 17 (1.1:1.0; 17:18; Scheme 3, entry 3). Moreover, deletion of the i + 2 Lys(Boc) residue, where Boc = tert-butyloxycarbonyl, results in further reduced reaction efficiency (Scheme 3, entry 4) on par with the results of the monomer Boc-Asp(OH)-OMe (Scheme 2, entry 3). Collectively, these results demonstrate that the selectivity for the BVO reaction and attendant regio- and stereoselectivity are the unambiguous results of interaction of 14 with the peptide catalyst and its complete secondary structure.
Scheme 3. Selectivities Observed with Truncated Peptides.

Standard conditions with peptide 2: substrate (1 equiv), 2 (10 mol %), 4-dimethylaminopyridine (10 mol %), aqueous H2O2 (3 wt %; 2.0 equiv), N,N′-diisopropylcarbodiimide (3.0 equiv), chloroform (0.033 M), 4 °C, mixing @ 30 rpm, 24 h. bAverage from two trials; determined by uncalibrated HPLC integrations of the crude reaction media (see Supporting Information for details).
Conclusion
It is thus established that aspartyl peptides can catalyze either epoxidation reactions as electrophilic oxidants or BV oxidations as nucleophilic oxidants, with accompanying diastereo- and regioselectivity, when presented with an intramolecular choice. The molecular context in which the aspartyl residue is placed—the peptide sequence that defines the catalyst secondary structure—provides the capacity to control orthogonal functional group selectivity in an intramolecular competition experiment. The ability to dial in functional group selectivity, while controlling attendant stereoselectivity issues, is a dimension that may create new possibilities for orchestrating the assembly of complex molecules, and for complex scaffold diversification. Perhaps more fundamentally, the peptide-regulated selectivity for specific functional groups within a multifunctional substrate signals control over the mechanistic behavior of common catalytic machinery, which at present seems to be known primarily only in the context of enzymes.31
Acknowledgments
We are grateful to the National Institute of General Medical Sciences of the NIH (GM-096403) for support. All computational work was supported by the facilities and staff of the Yale University Faculty of Arts and Sciences High Performance Computing Center, and by the National Science Foundation under Grant No. CNS 08-21132 that partially funded acquisition of the facilities. C.R.S. gratefully acknowledges the National Science Foundation Graduate Research Fellowship program for funding. We also thank Dr. Brandon Q. Mercado for X-ray crystallographic analyses. We also thank Anthony J. Metrano and Dr. Julian Tirado-Rives for assistance with the computational data.
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acscentsci.6b00237.
Synthetic procedures, descriptions of compounds, analytical methods, computational methods, and crystallographic data (PDF)
Crystallographic data are deposited with the Cambridge Crystallographic Data Centre under the accession numbers CCDC 1480519 and 1480521.
The authors declare no competing financial interest.
Supplementary Material
References
- Anantharaman V.; Aravind L.; Koonin E. V. Emergence of diverse biochemical activities in evolutionarily conserved structural scaffolds of proteins. Curr. Opin. Chem. Biol. 2003, 7, 12–20. 10.1016/S1367-5931(02)00018-2. [DOI] [PubMed] [Google Scholar]
- Tang J.; Wong R. N. S. Evolution in the Structure and Function of Aspartic Proteases. J. Cell. Biochem. 1987, 33, 53–63. 10.1002/jcb.240330106. [DOI] [PubMed] [Google Scholar]
- Lairson L. L.; Henrissat B.; Davies G. J.; Withers S. G. Glycosyltransferases: structures, functions, and mechanisms. Annu. Rev. Biochem. 2008, 77, 521–555. 10.1146/annurev.biochem.76.061005.092322. [DOI] [PubMed] [Google Scholar]
- Yoon T. P.; Jacobsen E. N. Privileged Chiral Catalysts. Science 2003, 299, 1691–1693. 10.1126/science.1083622. [DOI] [PubMed] [Google Scholar]
- Peris G.; Jakobsche C. E.; Miller S. J. Aspartate-Catalyzed Asymmetric Epoxidation Reactions. J. Am. Chem. Soc. 2007, 129, 8710–8711. 10.1021/ja073055a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peris G.; Miller S. J. A Nonenzymatic Acid/Peracid Catalytic Cycle for the Baeyer-Villiger Oxidation. Org. Lett. 2008, 10, 3049–3052. 10.1021/ol8010248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williamson J. R. Cooperativity in macromolecular assembly. Nat. Chem. Biol. 2008, 4, 458–465. 10.1038/nchembio.102. [DOI] [PubMed] [Google Scholar]
- Mahadevi A. S.; Sastry G. N. Cooperativity in Noncovalent Interactions. Chem. Rev. 2016, 116, 2775–2825. 10.1021/cr500344e. [DOI] [PubMed] [Google Scholar]
- Anslyn E. V.; Dougherty D. A.. Part II: Reactivity, Kinetics, and Mechanisms. In Modern Physical Organic Chemistry; University Science Books: Sausalito, CA, 2005; pp 355–753. [Google Scholar]
- Harper K. C.; Sigman M. S. Predicting and optimizing asymmetric catalyst performance using the principles of experimental design and steric parameters. Proc. Natl. Acad. Sci. U. S. A. 2011, 108, 2179–2183. 10.1073/pnas.1013331108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Que L. Jr.; Tolman W. B. Biologically inspired oxidation catalysis. Nature 2008, 455, 333–340. 10.1038/nature07371. [DOI] [PubMed] [Google Scholar]
- Jakobsche C. E.; Peris G.; Miller S. J. Functional Analysis of an Aspartate-Based Epoxidation Catalyst with Amide-to-Alkene Peptidomimetic Catalyst Analogues. Angew. Chem., Int. Ed. 2008, 47, 6707–6711. 10.1002/anie.200802223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lichtor P. A.; Miller S. J. Combinatorial evolution of site- and enantioselective catalyst for polyene epoxidation. Nat. Chem. 2012, 4, 990–995. 10.1038/nchem.1469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abascal N. C.; Lichtor P. A.; Giuliano M. W.; Miller S. J. Function-oriented investigations of a peptide-based catalyst that mediates enantioselective allylic alcohol epoxidation. Chem. Sci. 2014, 5, 4504–4511. 10.1039/C4SC01440E. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Romney D. K.; Colvin S. M.; Miller S. J. Catalyst Control over Regio- and Enantioselectivity in Baeyer-Villiger Oxidations of Functionalized Ketones. J. Am. Chem. Soc. 2014, 136, 14019–14022. 10.1021/ja508757g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kagan H. B.; Fiaud J. C.. Kinetic Resolution. In Topics in Stereochemistry; Eliel E. L., Wilen S. H., Eds.; John Wiley & Sons, Inc.: Hoboken, NJ, 1988: Vol. 18. [Google Scholar]
- Giuliano M. W.; Miller S. J. Site-Selective Reactions with Peptide-Based Catalysts. Top. Curr. Chem. 2015, 372, 157–201. 10.1007/128_2015_653. [DOI] [PubMed] [Google Scholar]
- The selection of phenylacetamide was based on our previous results for BVO chemistries. See references (14) and (15) for the scope of these transformations.
- Masamune S.; Choy W.; Petersen J. S.; Sita L. R. Double Asymmetric Synthesis and a New Strategy for Stereochemical Control in Organic Synthesis. Angew. Chem., Int. Ed. Engl. 1985, 24, 1–30. 10.1002/anie.198500013. [DOI] [Google Scholar]
- Our choice of the specific enantiomeric series is predicated on our previously determined cases of stereochemical “matching” and mismatching. See ref (15). See also Supporting Information for substrate synthesis.
- See Supporting Information for computational data.
- Johnson R. A.; Sharpless K. B.. Addition Reactions with Formation of Carbon-Oxygen Bonds: (ii) Asymmetric Methods of Epoxidation. In Comprehensive Organic Synthesis: Oxidation; Trost B. M., Ley S. V., Fleming I., Eds.; Elsevier: Oxford, 1991; Vol. 7, pp 389–436. [Google Scholar]
- The synthetic route employed to access 8 and 9 utilizes a common, chiral synthetic intermediate where the stereochemistry of the olefin sidearm is dictated by the starting material. Therefore, for stereochemical consistency with respect to the amido group, ent-9-trans was examined which is derived from the enantiomer of the starting material, see Supporting Information for details.
- The term “ent” refers to the corresponding enantiomer.
- For practical reasons, we elected to proceed in the study with the enantiomer of 14 that was derived from commercially available (S)-3-cyclohexenecarboxylic acid.
- Abascal N. C.; Miller S. J. Solution structures and molecular associations of a peptide-based catalyst for the stereoselective Baeyer–Villiger oxidation. Org. Lett. 2016, 10.1021/acs.orglett.6b02282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rablen P. R.; Lockman J. W.; Jorgensen W. L. Ab Initio Study of Hydrogen-Bonded Complexes of Small Molecules with Water. J. Phys. Chem. A 1998, 102, 3782–3797. 10.1021/jp980708o. [DOI] [Google Scholar]
- As a substitute to water, we screened a variety of aliphatic alcohols that resulted in poor yields and selectivities, in addition to esterification of the “activated” aspartyl acid catalyst; see Supporting Information.
- Additional water can compete with H2O2 for the “activated” aspartyl acid catalyst thus reverting back to the parent aspartyl acid catalyst ent-2.
- Schreiber S. L.; Schreiber T. S.; Smith D. B. Reactions That Proceed with a Combination of Enantiotopic Group and Diastereotopic Face Selectivity Can Deliver Products with Very High Enantiomeric Excess: Experimental Support of a Mathematical Model. J. Am. Chem. Soc. 1987, 109, 1525–1529. 10.1021/ja00239a036. [DOI] [Google Scholar]
- Knowles R. R.; Jacobsen E. N. Attractive noncovalent interactions in asymmetric catalysis: Links between enzymes and small molecule catalysts. Proc. Natl. Acad. Sci. U. S. A. 2010, 107 (48), 20678–20685. 10.1073/pnas.1006402107. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.