

Abstract
Background
Cupriavidus necator has attracted much attention as a platform for the production of polyhydroxyalkanoate (PHA) and other useful materials. Therefore, an appropriate modulation of gene expression is needed for producing the desired materials effectively. However, there is insufficient information on the genetic engineering techniques required for this in C. necator.
Results
We found that the disruption of a potential ribosome binding site (RBS) in the phaC1 gene in C. necator caused a small decrease in the PhaC1 expression level. We applied this result to finely regulate the expression of other genes. Several gene expression cassettes were constructed by combining three Escherichia coli derived promoters (PlacUV5, Ptrc and Ptrp) to the potential RBS of phaC1 or its disruptant, respectively. Their expression levels were then determined via a lacZ reporter assay in C. necator strains. The promoter strengths were both ranked similarly for the cells that were cultured with fructose or palm kernel oil as a sole carbon source (Ptrc ≥ PlacUV5 > Ptrp), both of which were much stronger than the phaC1 promoter. The disruption of RBS had minute attenuation effect on the expression level of these expression cassettes with E. coli promoters. Furthermore, they were used to finely regulate the (R)-3-hydroxyhexanoate (3HHx) monomer ratio in the production of poly[(R)-3-hydroxybutyrate-co-3-hydroxyhexanoate] (PHBHHx) via R-specific enoyl-CoA hydratases (PhaJs). The 3HHx composition in PHBHHx is crucial because it defines the thermal and mechanical properties of the resulting plastic material. The C. necator mutant strains, whose PhaJ expression was controlled under the gene expression cassettes, could be used to produce PHBHHx with various 3HHx compositions in the same culture conditions.
Conclusions
We constructed and evaluated several gene expression cassettes consisting of promoters and RBSs that finely regulate transcription and translation. These were then applied to finely modulate the monomer composition in the production of PHBHHx by recombinant C. necator.
Electronic supplementary material
The online version of this article (doi:10.1186/s12934-016-0583-7) contains supplementary material, which is available to authorized users.
Keywords: Polyhydroxyalkanoates, PHBHHx, Cupriavidus necator, Promoter, Ribosome binding site
Background
Cupriavidus necator (formerly Ralstonia eutropha) is a non-pathogenic gram-negative soil bacterium of the β-proteobacteria class. It is a well-known model organism for studying polyhydroxyalkanoate (PHA) synthesis and accumulation [1]. PHA has attracted industrial attention as an environmentally friendly material because it exhibits complete biodegradability and is a possible alternative to petroleum-based plastics [2]. Poly[(R)-3-hydroxybutyrate] (PHB) is the most widely investigated member of the PHA family. Cupriavidus necator can greatly accumulate PHB, which can constitute up to 80% of its dry cell weight, from renewable carbon sources [3]. Furthermore, the recombinant strains harboring the PHA synthase gene derived from Aeromonas caviae FA440 can produce poly[(R)-3-hydroxybutyrate-co-3-hydroxyhexanoate] (PHBHHx) with improved physical properties [4]. PHBHHx is more flexible than PHB, and its flexibility can be regulated by its monomer composition [5]. Increasing incorporation of (R)-3-hydroxyhexanoate (3HHx) as a second monomer results in increased flexibility of the copolymer. This characteristic is expected to be important for the practical application in many fields.
There has been a recent focus on C. necator as a host strain for the production of proteins and other metabolites. Srinivasan et al. reported that high yields of organophosphohydrolase protein could be obtained without the formation of inclusion bodies due to high levels of gene expression in high-cell density fermentation [6, 7]. In addition, some studies demonstrated that other useful materials such as cyanophycin, branched-chain alcohols, methyl ketones, or medium-chain-length fatty acids could be produced by genetically engineering C. necator strains to express various exogenous genes [8–11].
To improve the genetic construction of biosynthetic pathways, various expression systems in C. necator based on promoters and plasmid vectors have been evaluated. For instance, some popular and native promoters such as Plac, PlacUV5, Ptac, T7, PBAD, PphaC1, PphaP, PacoE, PacoD, PacoX, and PpdhE, were investigated. Furthermore, Ptac, which is known to be a constitutive strong promoter in Escherichia coli, functions well also in C. necator [12–16]. Additionally, highly stable vectors even under no antibiotic pressure have also been developed since various plasmid vectors are very unstable in C. necator H16. Sato et al. showed that artificial plasmids containing the partition locus and oriV28 region derived from the Cupriavidus metallidurans CH34 megaplasmid are highly stable in C. necator [17]. Moreover, Gruber et al. reported that the RP4 partitioning system confers stability on plasmids vectors [18]. They developed new plasmid vectors with varying combinations of replication origins and promoters, and demonstrated that the minireplicon derived from RSF1010 and the bacteriophage T5 promoter are superior in terms of stability and the expression level. It is also worthy to note that the T5 promoter is much stronger than Ptac.
In evaluating expression systems, greater significance is generally placed on the systems yielding higher intensity expression levels. However, if the expression is too strong, cell growth or the solubility of the resulting protein may be adversely affected. In addition, appropriately modulating gene expression is also required for fermentative production of materials that can be effectively produced through fine control of flux of their metabolic intermediates. Therefore, in the present study, we developed a gene expression cassette by combining common promoters derived from E. coli, PlacUV5, Ptrc, and Ptrp, in conjunction with a potential ribosome binding site (RBS) derived from the region upstream of phaC1 Cn or its disruptant. The expression levels of each cassette were examined via a reporter gene assay in C. necator recombinant strains and compared to PphaC1 combined with its native potential RBS or disruptant. Furthermore, these promoter-RBS gene expression cassettes were used to regulate the monomer ratio of PHBHHx produced in the recombinant C. necator.
Results
Evaluation of promoter-RBS gene expression cassettes in C. necator
The sequence AGAGAGA located 11 bases upstream of the C. necator PHA synthase (phaC1) gene initiation codon has been shown as a potential RBS [19]. To ascertain the function of this sequence, an expression plasmid with PhaC1 under the control of its native promoter in combination with its potential RBS (PphaC1RBS) or RBS disruptant (PphaC1dRBS) were constructed (Fig. 1) and introduced into the recombinant C. necator strain, H16/ds. Table 1 summarizes the bacterial strains and plasmids used in this study. The H16/ds strain is a phaC1 disruptant of the C. necator H16 strain, which was previously shown to be unable to produce PHAs [17].
Fig. 1.
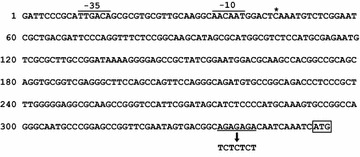
Nucleotide sequence of the 5′ upstream region of the phaC1 gene (PphaC1RBS). The promoter location, transcriptional start site, and translation initiation codon have been experimentally identified previously [19]. The putative −35 and −10 promoter region are overlined and labeled. An asterisk indicates the transcriptional start site. The translation initiation codon is boxed. The potential ribosome binding site (RBS) is underlined, and the arrow indicates the disrupted RBS sequence in PphaC1dRBS
Table 1.
Bacterial strains and gene expression plasmids
| Strain or plasmid | Descriptiona | Source or reference |
|---|---|---|
| C. necator | ||
| H16 | Wild type | ATCC17699 |
| H16/ds | H16 derivative; ΔphaC1 | 17 |
| 005dZ126 | H16 derivative; ΔphaC1::phaC AcNSDG, ΔphaZ1, ΔphaZ2, ΔphaZ6 | 27 |
| CnC1A | 005dZ126, PphaC1RBS-phaJ4a | This study |
| CnTRPDA | 005dZ126, PtrpdRBS-phaJ4a | This study |
| CnTRPA | 005dZ126, PtrpRBS-phaJ4a | This study |
| CnUV5DA | 005dZ126, PlacUV5dRBS-phaJ4a | This study |
| CnUV5A | 005dZ126, PlacUV5RBS-phaJ4a | This study |
| CnTRCDA | 005dZ126, PtrcdRBS-phaJ4a | This study |
| CnC1DB | 005dZ126, PphaC1dRBS-phaJ4b | This study |
| CnC1B | 005dZ126, PphaC1RBS-phaJ4b | This study |
| CnTRPB | 005dZ126, PtrpRBS-phaJ4b | This study |
| CnUV5B | 005dZ126, PlacUV5RBS-phaJ4b | This study |
| CnTRCB | 005dZ126, PtrcRBS-phaJ4b | This study |
| E. coli | ||
| JM109 | recA1 endA1 gyrA96 thi hsdR17 supE44 relA1 Δ(lac-proAB)/F’ [traD36 proAB + lacI q lacZΔM15] | Takara |
| S17-1 | recA pro hsdR RP4-2-Tc::Mu-Km::Tn7 | ATCC47055 |
| Plasmids | ||
| pCUP3 | Stable plasmid vector in C. necator, Kmr | 17 |
| pCUP3-PphaC1RBS-phaC1 | PphaC1RBS-phaC1 expression cassette cloned into pCUP3 | This study |
| pCUP3-PphaC1dRBS-phaC1 | PphaC1dRBS-phaC1 expression cassette cloned into pCUP3 | This study |
| pCUP3-RBS-lacZ | RBS-lacZ cloned into pCUP3 | This study |
| pCUP3-PphaC1RBS-lacZ | PphaC1RBS-lacZ expression cassette cloned into pCUP3 | This study |
| pCUP3-PlacUV5RBS-lacZ | PlacUV5RBS-lacZ expression cassette cloned into pCUP3 | This study |
| pCUP3-PtrpRBS-lacZ | PtrpRBS-lacZ expression cassette cloned into pCUP3 | This study |
| pCUP3-PtrcRBS-lacZ | PtrcRBS-lacZ expression cassette cloned into pCUP3 | This study |
| pCUP3-dRBS-lacZ | dRBS-lacZ cloned into pCUP3 | This study |
| pCUP3-PphaC1dRBS-lacZ | PphaC1dRBS-lacZ expression cassette cloned into pCUP3 | This study |
| pCUP3-PlacUV5dRBS-lacZ | PlacUV5dRBS-lacZ expression cassette cloned into pCUP3 | This study |
| pCUP3-PtrpdRBS-lacZ | PtrpdRBS-lacZ expression cassette cloned into pCUP3 | This study |
| pCUP3-PtrcdRBS-lacZ | PtrcdRBS-lacZ expression cassette cloned into pCUP3 | This study |
aRBS, potential ribosome binding site of phaC1; dRBS, RBS disruptant
The strains harboring the PhaC1 expression plasmids were cultured with palm kernel oil as a carbon source, and their PHA synthase activities were measured (Fig. 2). Although PphaC1dRBS showed a reduction in PHA synthase activity, conveniently, it was still considered highly active since it was more than 50% of the PphaC1RBS activity. Therefore, we tried to apply the disruption of this potential RBS for the modulation of the gene expression.
Fig. 2.
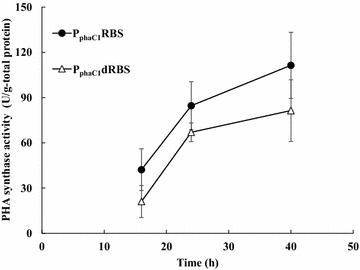
Polyhydroxyalkanoate (PHA) synthase activity. Cells were cultured in mineral salt medium with 0.129 w/v% (NH4)2SO4 and 1.5 w/v% palm kernel oil. The values shown are averages of triplicate experiments. Error bars indicate standard deviations
For the construction of the promoter-RBS gene expression cassettes, a 19-bp sequence (GCAGAGAGACAATCAAATC) containing the potential RBS derived from the region upstream of the phaC1 gene, or its RBS disruptant, were attached to the E. coli promoters (Fig. 3). Then, the plasmids with LacZ under the control of the resulting cassettes were constructed and introduced into the 005dZ126 strain, which is the C. necator H16 derivative native PHA synthase gene substituted with PHBHHx synthase gene.
Fig. 3.
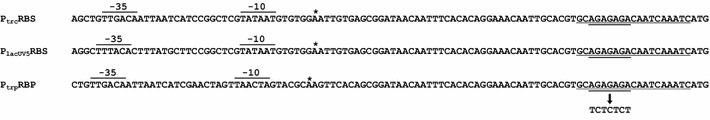
Structure of gene expression cassettes with the E. coli promoters, PtrcRBS, PlacUV5RBS, and PtrpRBS. The −35 and −10 promoter regions are overlined and labeled. Asterisks indicate the transcriptional start sites in E. coli. The 19-bp sequences derived from the region upstream of phaC1 are underlined, and the potential ribosome binding sites (RBS) are double-underlined. In their RBS disruptants, PtrcdRBS, PlacUV5dRBS, and PtrpdRBS, the potential RBS sequence (AGAGAGA) was changed to TCTCTCT
First, the recombinant strains harboring the lacZ gene cloned downstream of the expression cassette with the potential RBS (AGAGAGA) were cultured with fructose or palm kernel oil as a sole carbon source, and the promoter strengths were compared with that of PphaC1 by measuring the resulting β-galactosidase activity (Fig. 4). Although there was about a twofold higher activity with fructose than with palm kernel oil, the rank order of the promoter strengths in C. necator was the same (Ptrc ≥ PlacUV5 > Ptrp > PphaC1) in both conditions. The strongest promoter, Ptrc, exhibited expression that was at least 20-fold higher than that of PphaC1 with palm kernel oil. Finally, as expected, promoter-less constructs had minimal activity.
Fig. 4.
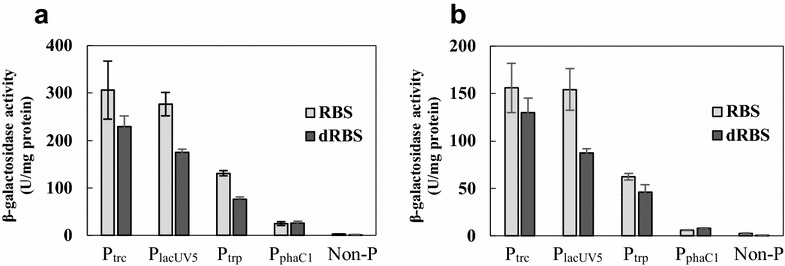
Activity of gene expression cassettes determined by lacZ reporter assay. Cells were grown with fructose (a) or palm kernel oil (b) as a sole carbon source. Triplicate samples from each of the two independent experiments were measured by optical absorbance (at 570 nm) of the chromogenic product, chlorophenol red. Data are presented as mean with error bars indicating the standard deviation. Non-P, non-promoter construct; RBS, with potential ribosome binding site of phaC1; dRBS, RBS disruptant
Secondly, the effects of RBS disruption on gene expression were also examined (Fig. 4). The RBS-disrupted expression cassettes with the E. coli promoters, and even non-promoter construct, showed a decline in the activity. The diminution rate was not significant as with the PHA synthase activity in PphaC1dRBS (Fig. 2), and the RBS disruptants with the E. coli promoters still maintained the activity of more than 55%, respectively. On the other hand, by disrupting the potential RBS with PphaC1, the β-galactosidase activity was not changed or slightly increased in both culture conditions unexpectedly. Since attenuation was not observed, this suggests that the potential RBS sequence did not function as a RBS in this construct.
Controlling 3HHx composition of PHBHHx using the expression cassettes
Previously, the characteristics and role of R-specific enoyl-CoA hydratases (PhaJs) in the synthesis pathway of PHA containing medium-chain length monomer units have been reported [20–22]. The PHBHHx synthesis pathway from plant oil via PhaJ is shown in Fig. 5. Cupriavidus necator H16 has two phaJ genes, which encode enzymes capable of efficiently catalyzing the conversion from β-oxidation intermediate (2-hexenoyl-CoA) to a precursor of the 3HHx monomer unit [(R)-3-hydroxyhexanoyl-CoA], named phaJ4a and phaJ4b, respectively [23]. Thus, we hypothesize that it would be possible to control the 3HHx composition of PHBHHx via the regulation of PhaJ expression when plant oil is used as a carbon source. Therefore, we constructed chromosomal mutant strains, in which the gene expression cassettes described above were placed immediately up-stream of the phaJ4a or phaJ4b genes in 005dZ126, and then attempted to produce PHBHHx with fine modulated 3HHx composition from palm kernel oil in flask experiments (Table 2).
Fig. 5.
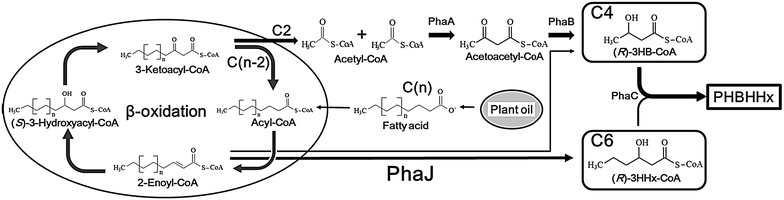
PHBHHx synthesis pathway with R-specific enoyl-CoA hydratase. PhaA, 3-ketoacyl-CoA thiolase; PhaB, NADPH-dependent acetoacetyl-CoA reductase; PhaC, PHA synthase; PhaJ, R-specific enoyl-CoA hydratase; (R)-3HB-CoA, (R)-3-hydroxybutyryl-CoA; (R)-3HHx-CoA, (R)-3-hydroxyhexanoyl-CoA
Table 2.
PHBHHx production by phaJ gene expression-controlled strains
| Strain | Dry cell weight (mg/mL) | PHA content (% dry cell weight) | PHA (mg/mL) | Real cell massa (mg/mL) | 3HHx composition (mol%) |
|---|---|---|---|---|---|
| 005dZ126 | 19.1 ± 0.1 | 84.2 ± 0.2 | 16.1 ± 0.1 | 3.0 ± 0.1 | 2.8 ± 0.1 |
| CnC1A | 19.3 ± 0.2 | 84.7 ± 2.0 | 16.4 ± 0.2 | 2.9 ± 0.4 | 5.3 ± 0.1 |
| CnTRPDA | 19.1 ± 0.6 | 84.2 ± 2.7 | 16.1 ± 0.0 | 3.0 ± 0.6 | 7.8 ± 0.0 |
| CnTRPA | 19.1 ± 0.3 | 82.3 ± 1.5 | 15.8 ± 0.0 | 3.4 ± 0.3 | 8.6 ± 0.1 |
| CnUV5DA | 19.2 ± 0.5 | 83.4 ± 0.9 | 16.0 ± 0.3 | 3.2 ± 0.3 | 9.1 ± 0.0 |
| CnUV5A | 18.7 ± 0.8 | 84.0 ± 1.1 | 15.7 ± 0.4 | 3.0 ± 0.3 | 9.9 ± 0.2 |
| CnTRCDA | 19.1 ± 0.5 | 82.1 ± 0.2 | 15.7 ± 0.3 | 3.4 ± 0.1 | 9.6 ± 0.0 |
| CnC1DB | 18.8 ± 0.2 | 85.0 ± 0.6 | 16.0 ± 0.1 | 2.8 ± 0.2 | 5.8 ± 0.2 |
| CnC1B | 19.3 ± 0.5 | 84.2 ± 0.7 | 16.3 ± 0.3 | 3.0 ± 0.2 | 6.7 ± 0.6 |
| CnTRPB | 18.8 ± 0.1 | 84.7 ± 1.3 | 15.9 ± 0.3 | 2.9 ± 0.2 | 10.0 ± 0.1 |
| CnUV5B | 19.0 ± 0.0 | 84.1 ± 1.9 | 16.0 ± 0.3 | 3.0 ± 0.4 | 10.6 ± 0.0 |
| CnTRCB | 18.2 ± 0.4 | 84.0 ± 3.3 | 15.3 ± 1.0 | 2.9 ± 0.5 | 10.7 ± 0.1 |
The cells were cultured in mineral salt medium with 0.129 w/v% (NH4)2SO4 and 1.5 w/v% palm kernel oil for 72 h. Data represent mean ± SD from three experiments performed in triplicate
aReal cell mass = dry cell minus polyhydroxyalkanoates (PHAs)
Within the same gene expression cassette, PhaJ4b brought about a higher 3HHx composition than PhaJ4a. This would result from the differences not only in their specific activities but also in the total expression level of PhaJs in the cells since phaJ4a is expressed also under its native promoter, but phaJ4b does not [23, 24].
The increased 3HHx compositions of PHBHHx in these recombinant strains, except for CnC1DB, were correlated with the activity determined by a LacZ assay of the expression cassettes inserted upstream of phaJ4a and phaJ4b. Although CnC1DB produced PHBHHx with a lower 3HHx composition than CnC1B, this was inconsistent with the results from the LacZ reporter assay, but was in agreement with that of the PhaC1 assay (Fig. 2). Therefore, future studies are required to resolve this discrepancy.
In addition, the three PhaJ4a-regulated strains (CnTRPA, CnTRPDA, and CnC1A), four PhaJ4b-regulated strains (CnTRCB, CnTRPB, CnC1B and CnC1DB), and the 005dZ126 strain were used for PHBHHx production by high cell density fermentation. These strains were cultured with feeding palm kernel oil in a jar fermenter for 68 h. The dry cell weight and PHA production under this experimental condition were almost the same as in any strain, and reached approximately 215 and 175 g/L, respectively (Fig. 6). Also in such a high-level production, the rank order of 3HHx compositions of PHBHHx at every sampling point both in the PhaJ4a- and in the PhaJ4b-regulated strains was the same as in the flask experiments, although their values were higher.
Fig. 6.
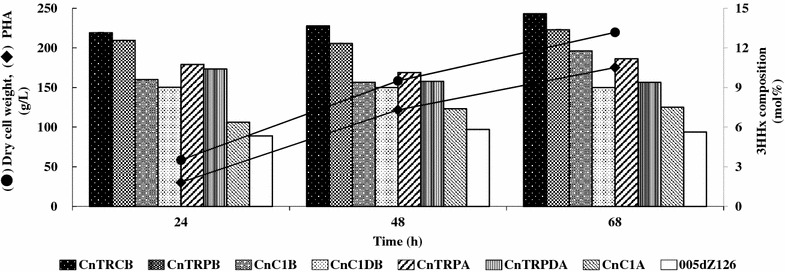
PHBHHx production by high-cell-density fermentation. Dry cell weight (filled circle) and PHA (filled diamond) in 005dZ126 are shown as representatives, and the values in all the other strains were within ±6 g/L of each of them. The bars indicate 3HHx composition of PHBHHx for each strain
In conclusion, we successfully produced, in high yield under the same culture conditions, PHBHHx with various compositions of 3HHx via regulation of PhaJs expression using various expression cassettes.
Thermal properties of PHBHHx with various 3HHx compositions
Differential scanning calorimetry was used to analyze various PHBHHx samples, each containing different 3HHx compositions produced by the PhaJ expression-controlled strains, as well as the PHBHHx produced by 005dZ126 with PHB as a standard of comparison (Table 3). Melting temperature in the first heating process (T m) decreased as the 3HHx composition increased. In addition, the crystallinity (X c) calculated from the melting enthalpy (ΔH m) of the PHBHHx was much smaller than that of PHB and gradually decreased as 3HHx composition increased. These results indicate that the crystallization of PHB was inhibited by incorporation of 3HHx co-monomer.
Table 3.
DSC analysis of PHBHHx with various 3HHx composition
| 3HHx composition (mol%) | First heating process | Cooling process | Second heating process | |||||
|---|---|---|---|---|---|---|---|---|
| T m (°C) | ΔH m (J/g) | Xc (%) | T g (°C) | T c (°C) | ΔH c (J/g) | T cc (°C) | ΔH cc (J/g) | |
| 0 (PHB)a | 175.1 | 103.0 | 70.5 | 0.4 | 77.4 | 80.2 | – | – |
| 2.7 | 150.8 | 72.9 | 49.9 | −2.3 | 50.2 | 25.0 | 50.3 | 21.7 |
| 5.2 | 142.0 | 70.8 | 48.5 | −2.9 | 41.0 | 3.6 | 58.8 | 47.1 |
| 5.9 | 138.9 | 69.5 | 47.6 | −2.4 | – | – | 68.2 | 43.3 |
| 7.1 | 133.5 | 66.3 | 45.4 | −2.1 | – | – | 74.5 | 27.2 |
| 7.9 | 131.1 | 67.5 | 46.2 | −2.2 | – | – | 76.7 | 20.2 |
| 9.1 | 116.4 | 65.9 | 45.1 | −3.4 | – | – | 77.0 | 12.2 |
| 10.0 | 114.7 | 60.4 | 41.4 | −3.6 | – | – | 78.9 | 1.9 |
| 10.8 | 113.5 | 55.8 | 38.2 | −4.7 | – | – | 79.9 | 1.2 |
T m, melting temperature; ΔH m, melting enthalpy; Xc, crystallinity; T g, glass transition temperature; T c, crystallization temperature; ΔH c, crystallization enthalpy; T cc, cold-crystallization temperature; ΔH cc, cold-crystallization enthalpy
aPurchased from Sigma-Aldrich
Furthermore, the crystallization temperature (T c) and enthalpy (ΔH c) in the cooling process, as well as the cold-crystallization temperature (T cc) and enthalpy (ΔH cc) in second heating process, were also investigated since these parameters reflect the crystallization rate. A large T c peak in PHB was observed at 77.4 °C with a ΔH c value of 80.2 J/g, which was 78% of ΔH m, whereas a T cc peak was not observed. This suggests that almost all the possible crystallization of PHB under our analysis conditions occurred in the cooling process; that is, PHB was able to crystallize well. On the other hand, no T c peak was observed for PHBHHx with ≥5.9 mol% 3HHx, and even ΔH cc decreased as the 3HHx composition increased. Therefore, these results suggest that PHBHHx with a high 3HHx composition hardly crystallized under our experimental conditions after once it was melted.
Discussion
In this study, we attempted to establish a fine gene expression regulation system using combinations of several promoters with either a potential RBS or a disrupted RBS in C. necator. The 5′ upstream region of the PHB synthase gene (phaC1) has been previously analyzed [19]. The C. necator gene phaC1 is organized in an operon together with the other PHB-biosynthetic genes phaA (3-ketoacyl-CoA thiolase) and phaB (NADPH-dependent acetoacetyl-CoA reductase), with a relatively long 5′ untranslated region (5′ UTR) of 307 bases in the resulting transcript. The location of the promoter, transcription start site, and the translational initiation codon of phaC1 have been confirmed experimentally, but not the function of the potential RBS (AGAGAGA located 11 bases upstream of the initiation codon). Therefore, we tested the effect of this potential RBS in regulating protein expression levels, and tried to use it for the construction of gene expression cassettes.
The results of the disruption experiments of the potential RBS by measuring PHA synthase (PhaC1) activity suggest that this RBS sequence has comparatively little ability to interact with the ribosome because the disruption caused insignificant decreases in the resulting activity (Fig. 2). A gene may have multiple RBSs contributing to its expression; for instance, the pyrC gene in E. coli K-12 has two potential RBS sequences [25]. The attenuation effect of one of them on the expression is significant, but the other is not. Thereby, the phaC1 gene in C. necator may also has other RBS which assumes the leading role.
The gene expression cassettes containing various E. coli promoters and the potential RBS of phaC1 (AGAGAGA) were successfully used in C. necator, where their expressions were 5 to 25-fold higher than that of the PphaC1RBS (Fig. 4). Moreover, the RBS disruptants were constructed to vary the expression activities. The RBS disruption caused a reduction in expression levels (Fig. 4) and resulted in a lower 3HHx composition, correlating with the activities of the gene expression cassettes inserted immediately upstream of phaJ (Table 2; Fig. 6).
In the LacZ reporter assay with PphaC1RBS, the disruption of RBS did not cause a decrease in the expression level (Fig. 4). The reason why the attenuating effect was not observed in this experiment is unclear, but the PhaC1 expression and PhaJ expression expected from 3HHx composition with PphaC1RBS were reduced by the RBS disruption as described above. Therefore, it may be specific to the combination of PphaC1RBS and the lacZ, and may be due to influence of some sort of interaction between the long 5′ UTR of PphaC1RBS and the lacZ gene on the translation or the stability of the mRNA based on the overall structure containing the 5′ UTR (including the RBS region) and the ORF sequence.
The present study demonstrated that the disruption of the potential phaC1 RBS downstream of the promoters resulted in an attenuation of expression levels. Therefore, this can be used to design other expression cassettes in combination with various promoters to be able to further finely regulate expression levels. In the future, we would like to investigate other functional RBSs in C. necator, and the further optimization of gene expression may become available also through the use of RBS libraries.
Furthermore, the thermal properties of PHA are important from a practical application perspective. The incorporation of 3HHx co-monomers in PHA results in a lower melting temperature and lower crystallinity (Table 3), resulting in an increase in elongation [5]. A decrease in the melting temperature is favorable for the improvement of the process window because the melting temperature of PHB is too close to its thermal degradation point [26], and its flexibility is essential for broad practical applications of PHA. Therefore, these characteristics of PHBHHx, with melting temperatures and flexibilities regulated by its 3HHx composition, are preferable as plastic material. On the other hand, consideration should also be given to its ability to crystallize. A lower crystallization rate influences the productivity in the melt processing. The priority of required properties in the plastic material varies by application and processing method. For example, when PHBHHx is used in films, high elongation may be more beneficial than crystallization rate, whereas for blow molding of PHBHHx bottles, a balance of both parameters would be important. Moreover, if other polymers are blended in as a component, the polymers used for blending can compensate for properties that are lacking in the selected PHBHHx. Hence, the control of properties by finely modulating the 3HHx composition is key to using PHBHHx successfully in a wide variety of applications.
Frequent changes in manufacturing conditions should be avoided in the industrial fermentation production for its stable operation. The recombinant C. necator strains, constructed in the present study, were able to produce PHBHHx with varying 3HHx composition under the same fermentation conditions in flask (Table 2) and even in high cell density fed-batch culture (Fig. 6). Therefore, we will be able to produce PHBHHx with any required 3HHx composition not only in the laboratory, but also on a commercial scale by using these strains properly, though there is a possibility that the 3HHx composition of PHBHHx will be influenced by the fermenter size, culture conditions such as aeration and agitation, culture medium materials and so on.
Conclusions
In this work, we showed that the disruption of the potential RBS of the gene phaC1 results in small decreases in the expression level. Therefore, we utilized this observation to construct the promoter-RBS gene expression cassettes. The expression cassettes comprised of any one of the E. coli promoters (PlacUV5, Ptrc and Ptrp) or the phaC1 promoter in conjunction with the potential RBS or its disruptant exhibited various expression activities in C. necator. We then applied these constructs to control the monomer ratio in PHBHHx by regulating PhaJs expression, and demonstrated that they allowed to produce PHBHHx with fine modulated 3HHx composition in recombinant C. necator. The observations from this study could raise the possibility of using C. necator as a platform for producing useful materials.
Methods
Bacterial strains, plasmids, and culture conditions
The strains and gene expression plasmids used in this study are shown in Table 1. The plasmids for chromosomal recombination are shown in Table S1 (Additional file 1). All E. coli strains were grown in Luria–Bertani (LB) medium. Escherichia coli strains JM109 and S17-1 were used for plasmid construction and as donors in intergeneric conjugation experiments, respectively. All C. necator strains were grown in modified basal mineral (MBM) medium as described previously [17].
For enzyme activity assays, the recombinant C. necator strains were cultured in 500-mL flask containing 50 mL of the mineral salts medium [27] with 0.129 w/v% (NH4)2SO4 and 1.5 w/v% fructose or palm kernel oil as a sole carbon source for 72 h. To produce PHBHHx, the recombinant C. necator strains were cultured using palm kernel oil as described above.
All E. coli and C. necator strains were cultured at 37 and 30 °C, respectively. When need, kanamycin was added to LB medium and MBM medium at 50 and 100 mg/L, respectively, to maintain the plasmids.
Construction of recombinant C. necator strains for evaluation of gene expression cassettes via enzyme activity assay
The oligonucleotides used for the constructions of the gene expression plasmids are listed in Table S2 (Additional file 1). The phaC1 expression plasmids pCUP3-PphaC1RBS-phaC1, pCUP3-PphaC1dRBS-phaC1 and pCUP3-PphaC1d50RBS-phaC1 were constructed as follows. The DNA fragment containing PphaC1, the following potential RBS, and the phaC1 gene was obtained by PCR with PphaC1F/phaC1R primers and C. necator H16 genomic DNA as a template. This fragment was digested by EcoRI and SpeI, and then cloned into the MunI and SpeI site of pCUP3 to yield pCUP3-PphaC1RBS-phaC1. Next, the DNA fragment containing PphaC1 and the RBS disruptant (dRBS) was obtained by PCR with PphaC1F/dRBSPphaC1R primers and C. necator H16 genomic DNA as a template. The phaC1 gene fragment was amplified using dRBSphaC1F/phaC1R primers and C. necator H16 genomic DNA as a template. These PphaC1dRBS and phaC1 fragments were joined by fusion PCR with PphaC1F/phaC1R, and then digested by EcoRI and SpeI. This fragment was then cloned into the MunI and SpeI site of pCUP3 to yield pCUP3-PphaC1dRBS-phaC1.
The resulting plasmid vectors were transformed into the recombinant C. necator strain H16/ds containing a disrupted phaC1 gene. The transformation was performed by electroporation as described previously [17].
The lacZ expression plasmids pCUP3-RBS-lacZ, pCUP3-PlacUV5RBS-lacZ, pCUP3-PtrcRBS-lacZ, and pCUP3-PtrpRBS-lacZ were constructed as follows. The DNA fragment containing potential RBS of phaC1 and lacZ gene was obtained by PCR with MunRBSlacZF/lacZR primers and E. coli HB101 genomic DNA as a template. The fragment was digested by MunI and SpeI, and cloned into the same site of pCUP3 to yield pCUP3-RBS-lacZ. Next, the PlacUV5 was amplified by PCR with lacF/lacUV5R primers and E. coli HB101 genomic DNA as a template. The PlacUV5 fragment was digested with MunI, and then ligated into the pCUP3-RBS-lacZ digested with the same enzyme to generate pCUP3-PlacUV5RBS-lacZ.
Similarly, the Ptrc and Ptrp fragments, which were amplified by PCR using pKK388-1 (Clontech) as a template with trcF/trcR or trcF/trpR primers, were digested with MunI and ligated into MunI-digested pCUP3-RBS-lacZ to generate pCUP3-PtrcRBS-lacZ and pCUP3-PtrpRBS-lacZ, respectively.
Subsequently, the plasmid pCUP3-PphaC1RBS-lacZ was constructed as follows. The DNA fragment containing PphaC1 and the following potential RBS was obtained by PCR with PphaC1F/RBSR primers and C. necator H16 genomic DNA as a template. The lacZ gene fragment was amplified using RBSlacZF/lacZR and E. coli HB101 genomic DNA as a template. These PphaC1RBS and lacZ fragments were joined by fusion PCR with PphaC1F/lacZR, and then digested by EcoRI and SpeI. The digested fragment was then cloned into the MunI and SpeI site of pCUP3 to yield pCUP3-PphaC1RBS-lacZ.
Plasmids pCUP3-dRBS-lacZ, pCUP3-PlacUV5dRBS-lacZ, pCUP3-PtrcdRBS-lacZ, pCUP3-PtrpdRBS-lacZ, and pCUP3-PphaC1dRBS-lacZ were all constructed in the same manner as described above, except in the use of primers, where MundRBSlacZF, dRBSPphaC1R, and dRBSlacZF were used instead of MunRBSlacZF, RBSR, and RBSlacZF, respectively.
These plasmid vectors were transformed via electroporation into the recombinant C. necator strain 005dZ126, which habors the PHA synthase gene derived form A. caviae FA440.
Construction of chromosomal mutants for the monomer composition modulated PHBHHx production
The oligonucleotides used for the constructions of plasmids for chromosomal recombination are listed in Additional file 1: Table S2. The mutant strain CnUA was constructed as follows. The fragments of C. necator DNA corresponding to the regions immediately upstream of the phaJ4a open reading frame (ORF) and potential RBS-fused phaJ4a ORF were amplified by PCR with J4aUF/J4aUR or RBSJ4aF/J4aR primers and C. necator H16 genomic DNA as a template. Furthermore, the fragment containing PlacUV5 and potential RBS was amplified by PCR with the J4aUlacUV5F/RBSR primers and pCUP3-PlacUV5RBS-lacZ as a template. These three fragments were combined and amplified with the primers J4aUF/J4aR to produce the amplicon containing the sequence upstream of phaJ4a, PlacUV5, potential RBS of phaC1, and the phaJ4a ORF. This amplicon was digested with SmiI and cloned into the same site of pNS2X-sacB [17] to create the plasmid pNS2X-sacB + PlacUV5RBS-J4a. The chromosomal mutant of C. necator was created by homologous recombination as described previously [17]. The plasmid pNS2X-sacB + PlacUV5RBS-J4a was introduced into the C. necator H16 derivative strain 005dZ126, to generate mutant CnUA by conjugation from the donor strain E. coli S17-1. Accuracy of the resulting recombination was confirmed via PCR and DNA sequencing.
The same construction strategy was used when preparing the mutant strains, CnUDA, CnUB, CnTRCDA, CnTRCB, CnTRPA, CnTRPDA, CnTRPB, CnCA, CnCB, and CnCDB. The DNA fragments used for construction of pNS2X-sacB + PlacUV5dRBS-J4a were generated using J4aUF/J4aUR, dRBSJ4aF/J4aR, and J4aUlacUV5F/dRBSR primers with C. necator H16 genomic DNA and pCUP3-PlacUV5dRBS-lacZ as templates. The J4bUF/J4bUR, RBSJ4bF/J4bR, and J4bUlacUV5F/RBSR primers with C. necator H16 genomic DNA and pCUP3-PlacUV5RBS-lacZ as templates were used to generate pNS2X-sacB + PlacUV5RBS-J4b; the J4aUF/J4aUR, dRBSJ4aF/J4aR primers and J4aUtrcF/dRBSR with C. necator H16 genomic DNA and pCUP3-PtrcdRBS-lacZ as templates were used to generate pNS2X-sacB + PtrcdRBS-J4a; the J4bUF/J4bUR, RBSJ4bF/J4bR, and J4bUtrcF/RBStrcR primers with C. necator H16 genomic DNA and pKK388-1 as templates were used to generate pNS2X-sacB + PtrcRBS-J4b; the J4aUF/J4aUR, RBSJ4aF/J4aR, and J4aUtrcF/RBSR primers with C. necator H16 genomic DNA and pCUP3-PtrpRBS-lacZ as templates were used to generate pNS2X-sacB + PtrpRBS-J4a; the J4aUF/J4aUR, dRBSJ4aF/J4aR, and J4aUtrcF/dRBSR primers with C. necator H16 genomic DNA and pCUP3-PtrpdRBS-lacZ as templates were used to generate pNS2X-sacB + PtrpdRBS-J4a; the J4bUF/J4bUR, RBSJ4bF/J4bR, and J4bUtrcF/RBSR primers with C. necator H16 genomic DNA and pCUP3-PtrpRBS-lacZ as templates were used to generate pNS2X-sacB + PtrpRBS-J4b; the J4aUF/J4aUR, RBSJ4aF/J4aR, and J4aUPphaC1F/RBSR primers with C. necator H16 genomic DNA and pCUP3-PphaC1RBS-lacZ a templates were used to generate pNS2X-sacB + PphaC1RBS-J4a; the J4bUF/J4bUR, RBSJ4bF/J4bR, and J4bUPphaC1F/RBSR primers with C. necator H16 genomic DNA and pCUP3-PphaC1RBS-lacZ as templates were used to generate pNS2X-sacB + PphaC1RBS-J4b; the J4bUF/J4bUR, dRBSJ4bF/J4bR, and J4bUPphaC1F/dRBSR primers with C. necator H16 genomic DNA and pCUP3-PphaC1dRBS-lacZ as templates were used to generate pNS2X-sacB + PphaC1dRBS-J4b.
Enzyme activity assay
β-Galactosidase activity was measured using cell lysates prepared with B-PER Bacterial Protein Extraction Reagent (Pierce Biotechnology, Rockford, IL) and the chromogenic substrate chlorophenol red-β-d-galactopyranoside (CPRG) as previously described [28]. B-PER reagent with protease inhibitors (Halt Protease Inhibitor Cocktail, EDTA-free, Thermo Scientific) was added to the recombinant C. necator cell pellets, and after incubation, cell debris was removed by centrifugation. Protein concentrations in the lysates were measured using Takara BCA Protein Assay Kit (Takara Bio Inc., Shiga, Japan). β-Galactosidase assay and the calculation of specific activities were performed according to the manufacturer’s instructions [β-galactosidase assay (CPRG), G-Biosciences, St. Louis, MO].
PHA synthase activities were determined by the 5,5′-dithiobis(2-nitrobenzoic acid) (DTNB) method as previously described [29]. Protein concentrations were measured using the Quick Start Bradford Protein Assay (Bio-Rad Laboratories, Hercules, CA, USA).
Production of PHBHHx by high-cell-density fed-batch culture
A 5-L jar fermenter (B.E. Marubishi, Co., Ltd.) containing 2 L MB medium [17] was used for high-cell-density fed-batch fermentation. The seed culture was grown over-night at 30 °C in a 500-mL flask containing 100 mL of MBM medium prior to being transferred to the jar fermenter. The jar fermenter operating conditions were as follows: agitation at 500 rpm and aeration rate of 3 L/min, and pH controlled between 6.7 and 6.8 using a 14% aqueous solution of ammonium hydroxide. Cultures were grown at 30 °C for 68 h in a fed-batch process. Palm kernel oil was fed as a carbon source. The feeding rate was 5 g/h from 0 to 24 h, and thereafter, 10 g/h.
Analysis of dry cell weight and PHA
After culturing, the cells were harvested by centrifugation, washed, and vacuum dried. The dry cell weight was then measured. The cellular content and monomer composition of PHA were determined by gas chromatography (GC) of samples prepared from dry cells as previously described [27].
Differential scanning calorimetry measurements were performed with the EXSTAR 6000 (DSC 6220, SII Nano Technology Inc., Tokyo, Japan). PHA samples were prepared by extraction from dry cells using chloroform and subsequently precipitation with methanol. The analysis was carried out using the thermal conditions as described previously [30]. Samples were first heated at a rate of 10 °C/min to 200 °C and maintained at that temperature for 2 min. Then, they were cooled at a rate of 10 °C/min to −50 °C and maintained at that temperature for 2 min. Subsequently, a second heating was performed at the same rate to 200 °C. The crystallinity was calculated according as
is the enthalpy of melting per gram of 100% crystalline 146 J/g [31] and is the measured enthalpy of melting for PHB or PHBHHx.
Authors’ contributions
HA and KM started the study. HA carried out the experiments, analyzed the data, and wrote the manuscript. KM contributed general advice, supervised the study, and edited the manuscript. Both authors read and approved the final manuscript.
Acknowledgements
We would like to thank Mr R. Koyama for valuable discussions about the DSC results.
Competing interests
The authors declare that they have no competing interests.
Availability of data and material
The datasets supporting the conclusions of this article are included within the article and its additional file.
Funding
This work was fully funded by Kaneka Corporation.
Abbreviations
- PHA
polyhydroxyalkanoate
- PHB
poly[(R)-3-hydroxybutyrate]
- PHBHHx
poly[(R)-3-hydroxybutyrate-co-3-hydroxyhexanoate]
- 3HHx
(R)-3-hydroxyhexanoate
- RBS
ribosome binding site
- ORF
open reading frame
- DSC
differential scanning calorimetry
Additional file
Additional file 1: Table S1. Plasmids for chromosomal recombination. Table S2. Oligonucleotides used in this study.
Contributor Information
Hisashi Arikawa, Phone: +81-79-445-2423, Email: Hisashi.Arikawa@kaneka.co.jp.
Keiji Matsumoto, Email: Keiji.Matsumoto@kaneka.co.jp.
References
- 1.Anderson AJ, Haywood GW, Dawes EA. Biosynthesis and composition of bacterial poly (hydroxyalkanoates) Int J Biol Macromol. 1990;12:102–105. doi: 10.1016/0141-8130(90)90060-N. [DOI] [PubMed] [Google Scholar]
- 2.Akiyama M, Tsuge T, Doi Y. Environmental life cycle composition of polyhydroxyalkanoates produced from renewable carbon resources by bacterial fermentation. Polym Degrad Stab. 2003;80:183–194. doi: 10.1016/S0141-3910(02)00400-7. [DOI] [Google Scholar]
- 3.Ryu HW, Hahn SK, Chang YK, Chang HN. Production of poly (3-hydroxybutyrate) by high cell density fed-batch culture of Alcaligenes eutrophus with phosphate limitation. Biotechnol Bioeng. 1997;55:28–32. doi: 10.1002/(SICI)1097-0290(19970705)55:1<28::AID-BIT4>3.0.CO;2-Z. [DOI] [PubMed] [Google Scholar]
- 4.Shimamura E, Kasuya K, Kobayashi G, Shiotani T, Shima Y, Doi Y. Physical properties and biodegradability of microbial poly(3-hydroxybutyrate-co-3-hydroxyhexanoate) Macromolecules. 1994;27:878–880. doi: 10.1021/ma00081a041. [DOI] [Google Scholar]
- 5.Doi Y, Kitamura S, Abe H. Microbial synthesis and characterization of poly(3-hydroxybutyrate-co-hydroxyhexanoate) Macromolecules. 1995;28:4822–4828. doi: 10.1021/ma00118a007. [DOI] [Google Scholar]
- 6.Srinivasan S, Barnard GC, Gerngross TU. A novel high-cell-density protein expression system based on Ralstonia eutropha. Appl Environ Microbiol. 2002;68:5925–5932. doi: 10.1128/AEM.68.12.5925-5932.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Srinivasan S, Barnard GC, Gerngross TU. Production of recombinant proteins using multiple-copy gene integration in high-cell-density fermentations of Ralstonia eutropha. Biotechnol Bioeng. 2003;84:114–120. doi: 10.1002/bit.10756. [DOI] [PubMed] [Google Scholar]
- 8.Lütte S, Pohlmann A, Zaychikov E, Schwartz E, Becher JR, Heumann H, Friedrich B. Autotrophic production of stable-isotope-labeled arginine in Ralstonia eutropha strain H16. Appl Environ Microbiol. 2012;78:7884–7890. doi: 10.1128/AEM.01972-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lu J, Brigham CJ, Gai CS, Sinskey AJ. Studies on the production of branched-chain alcohols in engineered Ralstonia eutropha. Appl Microbiol Biotechnol. 2012;96:283–297. doi: 10.1007/s00253-012-4320-9. [DOI] [PubMed] [Google Scholar]
- 10.Müller J, MacEachran D, Burd H, Sathitsuksanoh N, Bi C, Yeh YC, Lee TS, Hillson NJ, Chhabra SR, Singer SW, Beller HR. Engineering of Ralstonia eutropha H16 for autotrophic and heterotrophic production of methyl ketones. Appl Environ Microbiol. 2013;79:4433–4439. doi: 10.1128/AEM.00973-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Chen JS, Colón B, Dusel B, Ziesack M, Way JC, Torella JP. Production of fatty acids in Ralstonia eutropha H16 by engineering β-oxidation and carbon storage. PeerJ. 2015;3:e1468. doi: 10.7717/peerj.1468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Barnard GC, Henderson GE, Srinivasan S, Gerngross TU. High level recombinant protein expression in Ralstonia eutropha using T7 RNA polymerase based amplification. Protein Expr Purif. 2004;38:264–271. doi: 10.1016/j.pep.2004.09.001. [DOI] [PubMed] [Google Scholar]
- 13.Bi C, Su P, Müller J, Yeh YC, Chhabra SR, Beller HR, Singer SW, Hillson NJ. Development of a broad-host synthetic biology toolbox for Ralstonia eutropha and its application to engineering hydrocarbon biofuel production. Microb Cell Fact. 2013;12:107. doi: 10.1186/1475-2859-12-107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Fukui T, Suzuki M, Tsuge T, Nakamura S. Microbial synthesis of poly((R)-3-hydroxybutyrate-co-3-hydroxypropionate) from unrelated carbon sources by engineered Cupriavidus necator. Biomacromolecules. 2009;10:700–706. doi: 10.1021/bm801391j. [DOI] [PubMed] [Google Scholar]
- 15.Fukui T, Ohsawa K, Mifune J, Orita I, Nakamura S. Evaluation of promoters for gene expression in polyhydroxyalkanoate-producing Cupriavidus necator H16. Appl Microbiol Biotechnol. 2011;89:1527–1536. doi: 10.1007/s00253-011-3100-2. [DOI] [PubMed] [Google Scholar]
- 16.Delamarre SC, Batt CA. Comparative study of promoters for the production of polyhydroxyalkanoates in recombinant strains of Wautersia eutropha. Appl Microbiol Biotechnol. 2006;71:668–679. doi: 10.1007/s00253-005-0217-1. [DOI] [PubMed] [Google Scholar]
- 17.Sato S, Fujiki T, Matsumoto K. Construction of a stable plasmid vector for industrial production of poly(3-hydroxybutyrate-co-3-hydroxyhexanoate) by a recombinant Cupriavidus necator H16 strain. J Biosci Bioeng. 2013;116:677–681. doi: 10.1016/j.jbiosc.2013.05.026. [DOI] [PubMed] [Google Scholar]
- 18.Gruber S, Hagen J, Schwab H, Koefinger P. Versatile and stable vectors for efficient gene expression in Ralstonia eutropha H16. J Biotechnol. 2014;186:74–82. doi: 10.1016/j.jbiotec.2014.06.030. [DOI] [PubMed] [Google Scholar]
- 19.Schubert P, Krüger N, Steinbüchel A. Molecular analysis of the Alcaligenes eutrophus poly(3-hydroxybutyrate) biosynthetic operon: identification of the N terminus of poly(3-hydroxybutyrate) synthase and identification of the promoter. J Bacteriol. 1991;173:168–175. doi: 10.1128/jb.173.1.168-175.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Fukui T, Shiomi N, Doi Y. Expression and characterization of (R)-specific enoyl coenzyme A hydratase involved in polyhydroxyalkanoate biosynthesis by Aeromonas caviae. J Bacteriol. 1998;180:667–673. doi: 10.1128/jb.180.3.667-673.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Tsuge T, Fukui T, Matsusaki H, Taguchi S, Kobayashi G, Ishizaki A, Doi Y. Molecular cloning of two (R)-specific enoyl-CoA hydratase genes from Pseudomonas aeruginosa and their use for polyhydroxyalkanoate synthesis. FEMS Microbiol Lett. 1999;184:193–198. doi: 10.1111/j.1574-6968.2000.tb09013.x. [DOI] [PubMed] [Google Scholar]
- 22.Park SJ, Lee SY. Identification and characterization of a new enoyl coenzyme A hydratase involved in biosynthesis of medium-chain-length polyhydroxyalkanoates in recombinant Escherichia coli. J Bacteriol. 2003;185:5391–5397. doi: 10.1128/JB.185.18.5391-5397.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kawashima Y, Cheng W, Mifune J, Orita I, Nakamura S, Fukui T. Characterization and functional analyses of R-specific enoyl coenzyme A hydratases in polyhydroxyalkanoate-producing Ralstonia eutropha. Appl Environ Microbiol. 2012;78:493–502. doi: 10.1128/AEM.06937-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Shimizu R, Chou K, Orita I, Suzuki Y, Nakamura S, Fukui T. Detection of phase-dependent transcriptomic changes and Rubisco-mediated CO2 fixation into poly (3-hydroxybutyrate) under heterotrophic condition in Ralstonia eutropha H16 based on RNA-seq and gene deletion analyses. BMC Microbiol. 2013;13:169. doi: 10.1186/1471-2180-13-169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Liu J, Turnbough CL., Jr Identification of the Shine-Dalgarno sequence required for expression and translational control of the pyrC gene in Escherichia coli K-12. J Bacteriol. 1994;176:2513–2516. doi: 10.1128/jb.176.9.2513-2516.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Mousaviouna P, Halleyb PJ, Doherty WOS. Thermophysical properties and rheology of PHB/lignin blends. Ind Crop Prod. 2013;50:270–275. doi: 10.1016/j.indcrop.2013.07.026. [DOI] [Google Scholar]
- 27.Arikawa H, Sato S, Fujiki T, Matsumoto K. A study on the relation between poly(3-hydroxybutyrate) depolymerases or oligomer hydrolases and molecular weight of polyhydroxyalkanoates accumulating in Cupriavidus necator H16. J Biotechnol. 2016;227:94–102. doi: 10.1016/j.jbiotec.2016.04.004. [DOI] [PubMed] [Google Scholar]
- 28.Hughes RA, Ellington AD. Rational design of an orthogonal tryptophanyl nonsense suppressor tRNA. Nucleic Acids Res. 2010;38:6813–6830. doi: 10.1093/nar/gkq521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hyakutake M, Tomizawa S, Mizuno K, Abe H, Tsugea T. Alcoholytic cleavage of polyhydroxyalkanoate chains by class IV synthases induced by endogenous and exogenous ethanol. Appl Environ Microbiol. 2014;80:1421–1429. doi: 10.1128/AEM.03576-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Wen X, Lu X, Peng Q, Zhu F, Zheng N. Crystallization behaviors and morphology of biodegradable poly (3-hydroxybutyrate-co-4-hydroxybutyrate) J Therm Anal Calorim. 2012;109:959–966. doi: 10.1007/s10973-011-1768-2. [DOI] [Google Scholar]
- 31.Avella M, Martuscelli E, Raimo M. Properties of blends and composites based on poly (3-hydroxy) butyrate (PHB) and poly (3-hydroxybutyrate-hydroxyvalerate) (PHBV) copolymers. J Mater Sci. 2000;35:523–545. doi: 10.1023/A:1004740522751. [DOI] [Google Scholar]