

Abstract
Purpose of the Review
The pathogenesis of genetically complex granulomatous diseases, such as sarcoidosis and latent tuberculosis, remain largely unknown. With the recent advent of more powerful research tools, such as genome-wide expression platforms, comes the challenge of making sense of the enormous data sets so generated. This manuscript will provide demonstrations of how in silico (computer) analysis of large research data sets can lead to novel discoveries in the field of granulomatous lung disease.
Recent Findings
The application of in silico research tools has led to novel discoveries in the fields of non-infectious (e.g., sarcoidosis) and infectious granulomatous diseases. Computer models have identified novel disease mechanisms and can be used to perform “virtual” experiments rapidly and at low cost compared to conventional laboratory techniques.
Summary
Granulomatous lung diseases are extremely complex, involving dynamic interactions between multiple genes, cells and molecules. In silico interpretation of large data sets generated from new research platforms that are capable of comprehensively characterizing and quantifying pools of biological molecules promises to rapidly accelerate the rate of scientific discovery in the field of granulomatous lung disorders.
Keywords: Genomic, Systems Biology, Mathematical Model, Sarcoidosis
Introduction
Biological systems are infinitely complex in that their behavior is a manifestation of the net sum of dynamic interactions between the host and environment. The host and environment are co-dependent variables wherein the complex biological system of the host is strongly influenced by genetic and epigenetic factors; which, in turn, are driven by evolutionary pressures imposed by the environment. For example, the evolutionary pressure of malaria on humans in sub-Sahara Africa has selected for human genetic traits that would interrupt the lifecycle of the parasite, manifesting as thalassemias and sickle cell disease (1). Thus, accurate models of systems biology must account for the complex and dynamic relationships between host and environment, including individual host variability, such as the effects of genetics, age, gender, occupation, medications/drugs, nutrition, etc.
Fortunately, even the most intricate biological systems, such as the human body, have achieved a homeostatic balance that is regulated by a relatively small subset of cells, such as photoreceptor cells in the eyes, antigen sensing or regulatory immune cells, and molecules (e.g., receptors, enzymes, transcription factors). Biological systems are further confined by fundamental “laws of science”, such as the laws of thermodynamics (energy) and quantum mechanics (probability) which significantly simplifies model predictions. Nonetheless, classical reductionist research models focused on the functions of a single molecule or specific cell type are rarely adequate for the accurate representation of complex biological systems.
The field of “systems biology” has emerged as a new approach to scientific discovery that emerged to predict the behavior of living systems. Systems biology is founded upon the study of numerous interacting components of complex systems using mathematical equations to predict their behavior over time and in response to relevant experimental conditions (2).
In Silico Modeling to Interpret Complex ‘Omics Data
In silico or computer modeling refers to a recent shift from conventional “reductionist biology” to an “integrative biology” approach that considers how the components of complex biological systems engage with one another to produce a biological effect, such as the host immune response to a pathogen or malignant transformation of a cell caused by genetic variability or environmental exposures. The tools-of-the-trade include systems mathematics and computer simulations, and the goal is to produce “in silico organisms” that represent their in vivo counterparts (3). The components of an in silico biological model can be highly variable, but typically include molecular (e.g., genomic, cell signaling), biochemical (e.g., enzymatic) or physiological data. While the mathematics of in silico modeling is often complex (i.e., beyond the scope of this manuscript), the equations are necessarily constrained by the limits of biological behavior (i.e., what is possible, and what is not), allowing the mathematical equations to be simplified significantly. Using immune responses to environmental antigens in the context of granulomatous inflammation as an example, it is feasible to construct model constraints that effectively reduce the model components from millions of biological components (interactions among various immune cells, each of which has its own set of genes, enzymes, cytokines, chemokines, receptors, transcription factors, non-coding RNA, etc.) to produce a greatly simplified model that considers the most important determinants of granulomatous inflammation. In this regard, mathematical model creation is an iterative process whereby new input from conventional biochemical or genetic experiments (e.g., human, animal or in vitro cell experiments) informs the modification of in silico models to more closely match the biological system, including phenotypic variations of the disease. Such models have great potential for accelerating biological research based upon their interpretive (mechanistic) and predictive capacities, provided rapidly, and at low cost.
In order to better appreciate the challenges of reducing complex biological data into a more simplified form, it is useful to consider how mathematical modeling interfaces with huge biological data sets that are generated from high-throughput ‘omics research. Based upon the premise that changes in the relative expression of proteins, genes, and lipids are reflective of the biological processes that contribute to disease, demands for systems biology expertise have rapidly expanded. Recent innovations in the field of biological research provide for the simultaneous measurement of multiple parameters from a single sample. For instance, using mass spectroscopy technology the relative expression of hundreds of proteins can be determined from less than a milligram of tissue. More remarkably, nanogram quantities of DNA or RNA can be amplified, identified (based upon conserved nucleotide sequence), and quantified (relative expression) to yield a readout consisting of tens of thousands of genes, and millions of RNA transcripts. The cumulative data so generated from protein, gene, and transcript expression analyses are designated as the “proteome”, “genome”, and “transcriptome”, respectively. The rapid evolution of proteomic, genomic and other ‘omics research techniques facilitated the generation of massive amounts of data, thereby creating challenges relating to the objective statistical analysis and subsequent interpretation of the data.
A thorough description of the complex algorithms applied to ‘omics research are beyond the scope of this manuscript [the interested reader is encouraged to read the expert review by Joyce and Palsson (4)], however, “Machine learning” techniques are among the most widely-used approaches to address this problem (5). The algorithms used in machine learning include applications of graph theory and clustering, and can be applied in ways that incorporate existing information or ideas (supervised), that largely ignore preconceived ideas (unsupervised) or somewhere in between those two extremes (semi-supervised). Machine learning techniques are valuable for identifying molecular patterns that correspond with disease pathogenesis and for detecting biomarkers that distinguish distinct disease phenotypes.
Among the earliest applications of systems biology to advance our understanding of granulomatous disease compared tissue gene expression in two distinct leprosy populations performed by Bleharski and colleagues in 2003 (6). Like sarcoidosis and TB, leprosy presents as a clinical and immunological spectrum of disease. With the use of gene expression profiling, it was shown that gene expression correlates with and accurately classifies the clinical form of the disease. Genes belonging to the leukocyte immunoglobulin-like receptor (LIR) family, including LIR-7, were significantly up-regulated in lesions of lepromatous patients suffering from the disseminated form of the infection. In functional studies, LIR-7 protein suppressed innate host defense mechanisms by shifting monocyte production from interleukin-12 toward interleukin-10 and by blocking antimicrobial activity triggered by Toll-like receptors. This study was the first to show that gene expression profiles may be useful in defining clinical forms of disease and providing novel insights into the regulation of immune responses in the context of granulomatous disorders (7). The Bleharski study has inspired a number of subsequent studies that have further investigated the role of LIR-7 (a.k.a., LILRA2) in the pathogenesis of altered immunity in the context of infectious diseases (7,8).
Gene expression profiling has since been applied to study sarcoidosis disease pathogenesis using a systems biology approach. Crouser and colleagues compared lung tissue gene expression in sarcoidosis patients to disease-free controls of sarcoidosis using an unsupervised approach. Namely, using a well-curated database representing the known functions of the gene product (e.g., transcription factor, enzyme, receptor, etc.) and considering the known interactions among the gene products (e.g., genes known to be regulated by specific transcription factors, receptors known to activate signaling pathways), an unbiased mathematic approach (i.e., having no a priori hypothesis as to how the genes interact) was employed to statistically determine the molecular processes, represented as gene “networks”, that differentiate sarcoidosis lung disease from normal lung. Based upon a statistical analysis that is roughly based upon the number of differentially expressed genes conforming to each gene network so identified, a molecular pathway that is regulated by the transcription factor signal transducer and activator of transcription 1 (STAT1) emerged as being statistically most probable (i.e., least likely to be explained by “chance”). Furthermore, the two most highly expressed transcripts, MMP12 and ADAMDEC1, were included in this network (9), but were not previously incriminated in the pathogenesis of sarcoidosis. Molecules of this class (metalloproteinases) are integral to the function of cells participating in granulomatous inflammation (10), and it is interesting to note that the expression of ADAMDEC1 was most prominent in cells located adjacent to the stroma, presumably immature antigen-producing cells (11), whereas MMP12 (encoding a macrophage elastase enzyme) gene expression was highest in macrophages located within the granulomas. Gene and protein expression of MMP12 and ADAMDEC1 were significantly higher in BAL samples from patients with clinically active sarcoidosis compared to those with inactive disease (9), further supporting the likelihood that these molecules are integral to sarcoidosis pathogenesis. Subsequent investigations have shown that STAT1-regulated genes are highly expressed in extra-pulmonary sarcoidosis tissues, leading the investigators to hypothesize that STAT1 may be therapeutic target (12). Thus, the “machine learning” or computer generated interpretation of large disease-specific “omics” data can be used to generate new hypotheses relating to disease mechanisms and treatments.
Differentiation of sarcoidosis from infectious lung diseases is a critical diagnostic consideration given that the treatments typically administered for sarcoidosis (immune suppressants) are dramatically different (i.e., antibiotics for infections). The utility of genomic approaches for distinguishing sarcoidosis from infections has been challenged by recent publications showing minimal discernable differences between the two clinical conditions in terms of the signal derived from blood cells (13). As yet unpublished data comparing gene expression of lung tissue from sarcoidosis patients (the same cohort described above) to lung tissue from patients with confirmed granulomatous infections (i.e., histologically and/or tissue cultures) showed patterns that were similar in many respects (Figure 1a). However, two molecular pathways distinguished sarcoidosis from granulomatous infection in the lungs, and these pathways have implications for distinct disease mechanisms. Sarcoidosis was associated with higher expression of a specific MHC Class II complex, HLA-DRB1 (Figure 1b), a molecule well-known to influence the pathogenesis of sarcoidosis, presumably through engagement of as yet unidentified environmental antigens (14,15). Furthermore, this pathway was predicted to be regulated to some degree by a transcription factors encoded by PAX3, which has recently been incriminated in the pathogenesis of COPD using a similar gene expression analysis platform (16), and SIX1, which is thought to be instrumental during lung development (17). Fungal infection was distinguished from sarcoidosis by higher expression of transcripts regulated by the “B cell complex” (Figure 1c), which has implications for B cell development and maturation (18), and B cell antigen presentation to CD4+ T cells via MHC class II receptors (19).
Figure 1. Comparison of lung gene expression profiles in patients with sarcoidosis and granulomatous fungal infections.

Gene expression was compared in lung tissue derived from sarcoidosis (n=6), infectious granulomatous disease (n=6; 4 atypical mycobacterium, 2 histoplasmosis), and disease-free controls (n=6) using the Affymetrix Human U133 Plus 2.0 gene array platform using fold difference of >2, p<0.005, and false discovery rate of 3% as the criterion. Differentially expressed genes were then analyzed by Ingenuity Pathway Analysis [Ingenuity Systems (a QIAGEN company)]. Panel a: principal component analysis shows similar gene expression profiles in sarcoidosis (green) and infection (blue), which were distinct from controls (red). Panel b: expression of a gene network that is predicted to be regulated by transcription factors PAX3 and SIX1 were higher (darker shades of red = higher expression) in sarcoidosis compared to infection. Panel c: expression of genes regulated by the “B Cell Receptor” was reduced in sarcoidosis compared to infection.
In Silico Epigenetics Models and the “Interactome”
The vast majority of the human genome is classified as “non-coding” in that DNA does not encode a protein, tRNA or other functional molecule, and for many years the non-coding DNA was considered to be useless. This concept has dramatically changed over the past several decades during which a number of functional small RNA transcripts have been discovered to arise from non-coding DNA, the function of which is to (in most cases) suppress the translation of mRNA by binding to complementary sequences in the 3’ untranslated region (UTR), thereby negatively regulating mRNA translation (20). The most studied of the non-coding RNA are “micro-RNA” (miRNA), so called because they are only 21–25 nucleotides in length. The suppression of mRNA is targeted to some degree in that each miRNA has different affinity for each mRNA, and in most situations the net expression of any given mRNA is regulated by several miRNA simultaneously (20). Computer models have been developed to mathematically predict the most likely mRNA targets of any set of miRNA provided, which has implications for gene transcription (21). Indeed, miRNA is used experimentally to suppress mRNA transcription.
Suppression of gene expression downstream of mRNA gene expression, such as is regulated by miRNA, is referred to as “epigenetic” gene regulation. Epigenetics explains phenotypic variation among people with nearly identical genetics (e.g., siblings), and can be influenced by environmental factors, such as smoking, stress or diet. Epigenetic factors are known to contribute to cancer risk (22) and to chronic inflammatory conditions, including inflammation in the context of granulomatous lung infections (23).
The first attempt to interpret the epigenome in sarcoidosis was recently performed in my laboratory, and provides interesting insights into the potential importance of miRNA in this disease. MiRNA array technology was used to quantify hundreds of known miRNA in peripheral blood mononuclear cells (PBMCs) from patients with active sarcoidosis, compared to disease-free volunteers. There were dramatic differences in miRNA expression in these groups, and a subset of the highly differentially expressed miRNA was further validated in other sarcoidosis tissues (lung, lymph nodes). These “highly expressed in sarcoidosis” miRNA were then analyzed using online access to computer programs with the ability to predict mRNA targets of the miRNA in the data set (i.e., common mRNA targets of all miRNA in the sample). The results of these investigations were very interesting in that the predicted miRNA targets were predicted to suppress TGFβ and WNT signaling pathways, which regulate Th2 immune responses (24). Presumably, these epigenetic mechanisms favor polarization away from Th2 immune responses towards the typical Th1 immune response that is characteristic of sarcoidosis and other granulomatous disorders.
Another variation on this theme, which requires yet another level of computer model complexity, is to model the interaction between highly expressed genes (mRNA) with highly expressed miRNA in the same biological sample. This type of analysis falls into the category of an “interactome” wherein the sum of multiple molecular interactions, reflected by two or more ‘omics platforms, are considered in the context of a clinical phenotype (e.g., disease). An example of the predicted interactions between differentially expressed genes and differentially expressed miRNA is shown in Figure 2. It is interesting to note that these molecular interactions represent many of the known stages of granuloma formation, including antigen presentation, T cell and macrophage activation, and, as previously shown in human pulmonary sarcoidosis genomic studies, STAT1 signaling (9,12). Moreover, the pathway is predicted to regulate the production of cytokines (TNFα, IFNγ) and other byproducts of activated immune cells (neopterin) that are differentially expressed in the lungs of patients with pulmonary sarcoidosis (25)
Figure 2. Predicted interactions between genes and microRNA (miRNA) in pulmonary sarcoidosis.
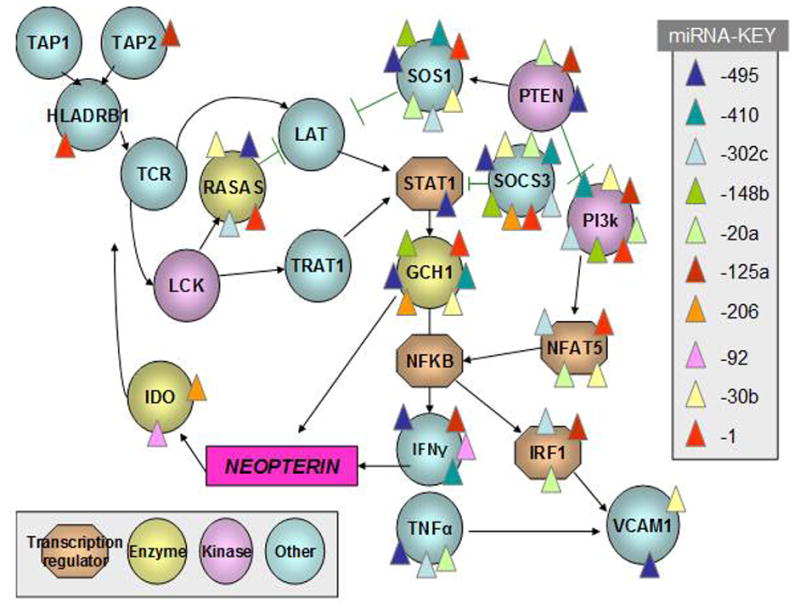
Genes that were differentially expressed in lung tissues of patients with pulmonary sarcoidosis compared to controls (as described in Figure 1) are depicted according to the function of their protein products. The predicted targets of differentially expressed miRNA transcripts, which were identified in pulmonary sarcoidosis in a previously published report (23) were then analyzed using curated online miRNA target prediction resources (microRNA.org, targetscan.org). The predicted interactions among gene products are represented by black arrows, and the predicted interactions of specific miRNA with specific genes are represented by color-coded triangles. The sum total of these interactions is designated the “interactome”.
Dynamic Math Models as an Experimental Platform for Granulomatous Disease Research
In silico models applied to human data, such as discussed previously, generally reflect a snapshot image of the disease. The static nature of such models is a major research limitation in that the evolution of granulomas is a dynamic process, including initiation, accumulation, maintenance, and resolution phases, which are altered in the context of human diseases such as sarcoidosis. In lieu of animal models that accurately model sustained pulmonary and systemic granuloma formation in human sarcoidosis or latent Mycobacterium tuberculosis infection, mathematical models of granulomatous disease may provide a practical alternative.
As with ‘omics models that are based upon well-curated knowledge platforms, math models rely upon accurate characterization of established molecular and cellular interactions represented by an interactive molecular network. The interaction of each network component with others is characterized as reinforcing or suppressing of other components based upon peer-reviewed scientific evidence. Each component of the model network is then represented by complex differential equations to account for all known interactions (e.g., activation, inhibition, degradation, proliferation) with other components of the network (e.g., CD4+ T cell activity is promoted by antigen presenting cells, and is suppressed by regulatory T cells), which are solved simultaneously and reevaluated over time to determine how the components of the network change over time. During the construction of such mathematical models, it often becomes apparent that some model components are more influential than others in terms of changing the final configuration (steady state) that is achieved when all of the equations are solved on the computer. These sensitive system components most often possess the capacity to amplify a biological signal (e.g., cell receptor, transcription factor, enzyme, cytokine) and, as such, become molecules of interest for future mechanistic or therapeutic research (26).
Mathematical models are only as good as the model assumptions, and, as such, it is best to generate the models from actual human data. An example of how mathematical modeling can be applied to granulomatous disorders is provided by a recent study by Hao and colleagues (27). Based upon the known interactions of a critical granuloma components (immune cells, chemokines, cytokines) (Figure 3a) and related quantitative data derived from human sarcoidosis tissues, it is shown that the mathematical model closely replicates the molecular profile of human pulmonary sarcoidosis (Figure 3b). Moreover, perturbations of the model equating with the effects of various “treatments” or “genetic variation” (e.g., anti-TNFα drugs or the effects of genetic or epigenetic factors that influence TNFα activity) are shown to equate with a change in steady state conditions that influence the predicted burden of granulomatous disease (Figure 3c) (27). As is the case with computer models previously described, it is important to emphasize that computer simulations require validation using more conventional research approaches (e.g., randomized controlled clinical trials) and may require modification as new information becomes available (e.g., newly discovered molecular interactions).
Figure 3. A mathematical model of pulmonary sarcoidosis.

Panel a: a basic schematic network of sarcoidosis: arrowhead means production or activation, block head means inhibition, and diamond means chemoattraction. Panel b: comparison of math model simulations of chemokine and cytokine profiles in sarcoidosis with actual human sarcoidosis clinical data [from Ref (8)], showing nearly identical results. Panel c: math model predictions of TNFα concentrations in sarcoidosis tissues over time, and matching granuloma size simulations (wherein “R” refers to granuloma radius), showing the effects of “anti-TNFα treatment” rendered 15 weeks after the onset of sarcoidosis. Suppression of TNFα is shown to create a new steady state associated with reduced granuloma radius around week 20. (Reproduced with permission from Hao W, Crouser ED, Freidman A. Mathematical model of sarcoidosis. Proc Natl Acad Sci USA 2014;111:16065-16070.).
The next frontier of granulomatous disease research has to further consider the dynamic interaction of the host with the environment, as relates to the course of disease. In this regard, new evidence is emerging to support the notion that occupational exposures can adversely influence the clinical course of sarcoidosis (28), and there is strong evidence showing that the interaction between host immune components and infections, such as TB, are associated with dynamic biological adjustments by both the host and pathogen that dictate disease phenotypes, ranging from complete eradication of infection to latent infection or fulminant infection. As such, the mathematical representation of the host and pathogen become interdependent and are critical when considering disease mechanisms or potential treatments. A very recent attempt to create an in silico model representing the complexity of host-pathogen interactions in the context of human TB was reported by Hao et al. (29). The selection of host and pathogen model components, including their functions and the expected magnitude of the effect (represented by a rate constant) were derived from the available literature and with guidance from an established expert in the field of TB host-pathogen immunology. The results of the model dynamics were expressed in terms of the “total bacterial load”, reflecting the degree to which the host immune response controlled mycobacterial growth over time. The results were interesting in that pro-inflammatory mediators such as TNFα and IFNβ, often presumed to be most important variables regulating the growth of mycobacteria in humans, were not deemed to be most critical for suppressing TB growth. Instead, the transition from a M1 to M2 macrophage phenotype was shown to depend upon IL-10, a potent inhibitor of pro-inflammatory pathways in the context of granulomatous infectious (30,31). Furthermore, suppression of IL-10 activity (e.g., using anti-IL-10 antibody treatment) was predicted to delay the M1 (favoring inflammation) to M2 (favoring repair) transition, thereby decreasing the bacterial burden, in a dose-dependent fashion. Additional information about the adaptive responses of TB pathogens to IL-10 treatment is required to validate these results and to would potentially identify new mechanisms by which the pathogens are capable of escaping the host immune response. New discoveries of this sort would be incorporated into a revised version of the in silico model. And so goes, until the model is shown to sufficiently model the human condition in terms of predicting the superimposed effects of human variability (e.g., genetic and environmental factors) or model perturbations (e.g., proposed treatments).
Conclusion; The Future of Mathematical Modeling
With the advent of ‘omics research and a shift towards attempts to comprehend complex molecular interactions, or systems biology, mathematical or in silico modeling has emerged as an essential research tool. Just as computers have led to innovations in the fields of communication, aerospace, automobile performance, and almost every other technology-dependent field, the next generation of health science researchers will increasingly rely on mathematical and computer models to understand the biological complexities of human health and disease.
In the future, mathematical and computer models may replace much of the time-consuming and expensive pre-clinical research that is conducted on animals in the laboratory setting or in humans leading to more rapid progress towards understanding and treating human diseases. In the context of sarcoidosis, the anticipated expansion of knowledge relating to the Genomic Research in Alpha-1 Antitrypsin Deficiency and Sarcoidosis (GRADS) study, including genomic information from diseased humans and environmental factors influencing their immune environment (the microbiome) (32), will provide the substrate for more advanced in silico models, and related insights into dynamic disease mechanisms and novel treatments. The success of the GRADS program will accentuate the transition from reliance on conventional, hypothesis-driven research towards discoveries based upon systems biology research platforms and related computer models.
Key Points.
‘Omics research platforms are capable of comprehensively identifying and quantifying numerous molecules in a biological sample, and in silico models have been used to hypothesize how these molecules are likely to interact in the context of granulomatous lung disease.
In silico models are only as good as the quality of the data that is used to develop them such that the performance of in silico simulations requires validation using more conventional experimental approaches.
Well-designed in silico models promise to accelerate the pace while reducing the costs of research to advance our understanding and to improve the treatment of granulomatous lung diseases
Acknowledgments
The author would like to thank Dr. Avner Friedman for his assistance with the manuscript.
Financial Support and Sponsorship:
This work was supported, in part, by the National Institutes for Health, U.S.A. [HL077466 (EDC)].
Footnotes
Conflicts of Interest:
Dr. Crouser has received honoraria from Beckman Coulter, Inc. and grants from National Institutes of Health (HL123586, HL126399).
References Cited
- 1.Mangano VD, Modiano D. An evolutionary perspective of how infection drives human genome diversity: the case of malaria. Curr Opin Immunol. 2014;30:39–47. doi: 10.1016/j.coi.2014.06.004. [DOI] [PubMed] [Google Scholar]
- 2.Kitano H. Computational systems biology. Nature. 2002;420:206–210. doi: 10.1038/nature01254. [DOI] [PubMed] [Google Scholar]
- 3.Palsson B. The challenges of in silico biology. Nature Biotech. 2000;18:1147–1150. doi: 10.1038/81125. [DOI] [PubMed] [Google Scholar]
- 4.Joyce AR, Palsson BO. The model organism as a system: integrating ‘omics’ data sets. Nature Reviews Molecular Cell Biology. 2006;7:198–210. doi: 10.1038/nrm1857. [DOI] [PubMed] [Google Scholar]
- 5**.Libbrecht MW, Noble WS. Machine learning applications in genetics and genomics. Nature Reviews. 2015;16:321–32. doi: 10.1038/nrg3920. (An outstanding review of the concept of machine learning, which forms a foundation for ‘omics-based research) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bleharski JR, Li H, Meinken LH, et al. Use of genetic profiling in leprosy to discriminate clinical forms of the disease. Science. 2003;301(5639):1527–1530. doi: 10.1126/science.1087785. [DOI] [PubMed] [Google Scholar]
- 7.Lu HK, Mitchell A, Endoh Y, et al. LILRA2 selectively modulates LPS-mediated cytokine production and inhibits phagocytosis by monocytes. PLoS One. 2012;7:e33478. doi: 10.1371/journal.pone.0033478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lee DJ, Sieling PA, Ochoa MT, et al. LILRA2 activation inhibits dendritic cell differentiation and antigen presentation to T cells. J Immunol. 2007;179:8128–8136. doi: 10.4049/jimmunol.179.12.8128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Crouser ED, Culver DA, Knox KS, et al. Gene expression profiling identifies MMP-12 and ADAMDEC1 as potential pathogenic mediators of pulmonary sarcoidosis. Am J Respir Crit Care Med. 2009;179:929–938. doi: 10.1164/rccm.200803-490OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Salgame P. MMPs in tuberculosis: granuloma creators and tissue destroyers. J Clin Invest. 2011;121:1686–1688. doi: 10.1172/JCI57423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Fritsche J, Muller A, Hausmann M, Rogler G, Andreesen R, Kreutz M. Inverse regulation of the ADAM-family members, decysin and MADDAM/ADAM19 during monocyte differentiation. Immunology. 2003;110:450–457. doi: 10.1111/j.1365-2567.2003.01754.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Rosenbaum JT, Braziel RM, Crouser ED, et al. Hypothesis: sarcoidosis is a STAT1-mediated disease. Clin Immunol. 2009;132:174–183. doi: 10.1016/j.clim.2009.04.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Maertzdorf J, Weiner J, 3rd, Mollenkopf HJ, et al. Common patterns and disease-related signatures in tuberculosis and sarcoidosis. Proc Natl Acad Sci USA. 2012;109:7853–7858. doi: 10.1073/pnas.1121072109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Grunewald J, Kaiser Y, Ostadkarampour M, et al. T-cell receptor-HLA-DRB1 associations suggest specific antigens in pulmonary sarcoidosis. Eur Respir J. 2016;47:898–909. doi: 10.1183/13993003.01209-2015. [DOI] [PubMed] [Google Scholar]
- 15.Levin AM, Adrianto I, Datta I, et al. Association of HLA-DRB1 with sarcoidosis susceptibility and progression in African Americans. Am J Respir Cell Mol Biol. 2015;53:206–216. doi: 10.1165/rcmb.2014-0227OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Acquaah-Mensah GK, Malhotra D, Vulimiri M, McDermott JE, Biswal S. Suppression expression of T-Box transcription factors is involved in senescence in chronic obstructive pulmonary disease. PLoS Comput Biol. 2012;8(7):e1002597. doi: 10.1371/journal.pcbi.1002597. [DOI] [PMC free article] [PubMed] [Google Scholar] [Research Misconduct Found]
- 17.El-Hashash AH, Al Alam D, Turcatel G, Rogers O, Li X, Bellusci S, Warburton D. Six1 transcription factor is critical for coordination of epithelial, mesenchymal and vascular morphogenesis in the mammalian lung. Dev Biol. 2011;353(3):242–258. doi: 10.1016/j.ydbio.2011.02.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wang LD, Clark MR. B-cell antigen-receptor signaling in lymphocyte development. Immunology. 2003;110:411–420. doi: 10.1111/j.1365-2567.2003.01756.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kim YM, Pan JYJ, Korbel GA, Peperzak V, Boes M, Ploegh HL. Monovalent ligation of the B cell receptor induces receptor activation but fails to promote antigen presentation. Proc Natl Acad Sci USA. 2006;103:3327–3332. doi: 10.1073/pnas.0511315103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20*.Hayes J, Peruzzi PP, Lawler S. MicroRNAs in cancer: biomarkers, functions and therapy. Trends in Molecular Medicine. 2014;20:460–469. doi: 10.1016/j.molmed.2014.06.005. (This is an excellent review of microRNA biology, and provides insight into epigenetic regulation of disease, which is a relatively new and rapidly expanding area of research.) [DOI] [PubMed] [Google Scholar]
- 21.Li Y, Zhang Z. Computational biology in microRNA. Wiley Interdiscip Rev RNA. 2015;6:435–452. doi: 10.1002/wrna.1286. [DOI] [PubMed] [Google Scholar]
- 22.Iorio MV, Croce CM. Causes and consequences of microRNA dsyregulation. Cancer J. 2012;18:215–222. doi: 10.1097/PPO.0b013e318250c001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Liu Y, Li H, Xiao T, Lu Q. Epigentics in immune-mediated pulmonary diseases. Clin Rev Allergy Immunol. 2013;45:314–330. doi: 10.1007/s12016-013-8398-3. [DOI] [PubMed] [Google Scholar]
- 24.Crouser ED, Julian MW, Crawford M, et al. Differential expression of microRNA and predicted targets in pulmonary sarcoidosis. Biochem Biophys Res Commun. 2012;417:886–891. doi: 10.1016/j.bbrc.2011.12.068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Prior C, Knight, Herold M, Ott G, Spiteri MA. Pulmonary sarcoidosis: patterns of cytokine release in vitro. Eur Respir J. 1996;9(1):47–53. doi: 10.1183/09031936.96.09010047. [DOI] [PubMed] [Google Scholar]
- 26.Edelman LB, Eddy JA, Price ND. In silico models of cancer. Wiley Interdiscip Rev Syst Biol Med. 2010;2:438–459. doi: 10.1002/wsbm.75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27**.Hao W, Crouser ED, Freidman A. Mathematical model of sarcoidosis. Proc Natl Acad Sci USA. 2014;111:16065–16070. doi: 10.1073/pnas.1417789111. (This study is the best example of how in silico models can be used to rapidly and inexpensively test new hypotheses using a computer, and without relying on conventional, more expensive and time-consuming laboratory techniques.) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Liu H, Patel D, Welch A, et al. Association between occupational exposures and sarcoidosis: an analysis from death certificates in the United States, 1988–1999. Chest. 2016 Jan 30; doi: 10.1016/j.chest.2016.01.020. Pil:S0012-3692(16)00514-6. Epub ahead of print. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hao W, Schlesinger LS, Friedman A. Modeling granulomas in response to infection in the lung. PLoS One. 2016;11(3):e0148738. doi: 10.1371/journal.pone.0148738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Abdalla AE, Lambert N, Duan X, Xie J. Interleukin-10 family and tuberculosis: an old story renewed. Int J Biol Sci. 2016;12(6):710–717. doi: 10.7150/ijbs.13881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Sadhu S, Kahaitan BK, Joshi B, Sengupta U, Nautiyal AK, Mitra DK. Reciprocity between regulatory T cells and Th17 cells: Relevance to polarized immunity in leprosy. PLoS Negl Trop Dis. 2016;10(1):e0004338. doi: 10.1371/journal.pntd.0004338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Moller DR, Koth LL, Maier LA, et al. Rationale and design of the Genomic Research in Alpha-1 Antitrypsin Deficiency and Sarcoidosis (GRADS) study. Sarcoidosis Protocol. Ann Am Thorac Soc. 2015;12(10):1561–1571. doi: 10.1513/AnnalsATS.201503-172OT. [DOI] [PMC free article] [PubMed] [Google Scholar]