

Supplemental Digital Content is available in the text.
Keywords: chemoreceptor, CXCR2, hypertension, inflammation, oxidant stress
Abstract
Background:
The recruitment of leukocytes to the vascular wall is a key step in hypertension development. Chemokine receptor CXCR2 mediates inflammatory cell chemotaxis in several diseases. However, the role of CXCR2 in hypertension development and the underlying mechanisms remain unknown.
Methods:
Angiotensin II (490 ng·kg-1·min-1) or deoxycorticosterone acetate (DOCA) salt–induced mouse hypertensive models in genetic ablation, pharmacologic inhibition of CXCR2, and adoptive bone marrow transfer mice were used to determine the role of CXCR2 in hypertension (measured by radiotelemetry and tail-cuff system), inflammation (verified by flow cytometry and quantitative real-time polymerase chain reaction [PCR] analysis), vascular remodeling (studied by haematoxylin and eosin and Masson’s trichrome staining), vascular dysfunction (assessed by aortic ring), and oxidative stress (indicated by nicotinamide adenine dinucleotide phosphate [NADPH] oxidase activity, dihydroethidium staining, and quantitative real-time PCR analysis). Moreover, the blood CXCR2+ cells in normotensive controls and hypertension patients were analyzed by flow cytometry.
Results:
Angiotensin II significantly upregulated the expression of CXCR2 mRNA and protein and increased the number of CD45+ CXCR2+ cells in mouse aorta (n=8 per group). Selective CXCR2 knockout (CXCR2-/-) or pharmacological inhibition of CXCR2 markedly reduced angiotensin II- or DOCA-salt-induced blood pressure elevation, aortic thickness and collagen deposition, accumulation of proinflammatory cells into the vascular wall, and expression of cytokines (n=8 per group). CXCR2 inhibition also ameliorated angiotensin II-induced vascular dysfunction and reduced vascular superoxide formation, NADPH activity, and expression of NADPH oxidase subunits (n=6 per group). Bone marrow reconstitution of wild-type mice with CXCR2-/- bone marrow cells also significantly abolished angiotensin II-induced responses (n=6 per group). It is important to note that CXCR2 blockade reversed established hypertension induced by angiotensin II or DOCA-salt challenge (n=10 per group). Furthermore, we demonstrated that CXCR2+ proinflammatory cells were higher in hypertensive patients (n=30) compared with normotensive individuals (n=20).
Conclusions:
Infiltration of CXCR2+ cells plays a pathogenic role in arterial hypertension and vascular dysfunction. Inhibition of CXCR2 pathway may represent a novel therapeutic approach to treat hypertension.
Editorial, see p 1369
Hypertension is the major risk factor for stroke, coronary heart disease, and chronic kidney disease. Despite extensive investigation for more than a century, the exact mechanisms responsible for essential hypertension remain unclear. Increasing evidence has established that inflammation plays a critical role in vascular dysfunction and hypertension.1–3 Various hypertensive stimuli, including angiotensin II and reactive oxygen species, are able to stimulate migration and infiltration of proinflammatory cells, including macrophages, neutrophils, and T cells, into the vasculature, kidney, and heart. Studies have demonstrated that T-cell activation promotes hypertension development in mice.4 T-cell costimulation by B7 ligand is also important for the development of experimental hypertension.5 It is interesting to note that recent evidence suggests that monocytes/macrophages rather than neutrophils are essential for the development of angiotensin II-induced hypertension and vascular dysfunction.6 However, the precise mechanisms by which hypertensive stimuli recruit these cells into the vasculature during hypertension have not been fully elucidated.
Numerous factors have been shown to stimulate leukocyte migration during the inflammatory response.7 Chemokines are low molecular weight proteins of the cytokine family, which activate G-protein-coupled receptors and induce the migration of neutrophils and monocytes/macrophages to the damaged vascular wall.7,8 Four major classes of chemokines have been identified: CXC, CC, C, and CX3C. The CXC ELR+ chemokines, including CXCL1, 2, 3 (GROα, β, and γ), 5, 6, 7, and 8, act through the chemokine receptor CXCR2 and are critical for neutrophil and mononuclear cell recruitment in the mouse.9 CXCR2 is of particular interest as accumulating evidence indicates a pivotal role for this receptor in tumor development and a variety of inflammatory disorders. For example, depletion of CXCR2 impairs neutrophil recruitment and protects against ischemia-reperfusion-induced cardiac injury.10 Neutralization of CXCR2 was also shown to inhibit neutrophil accumulation in the adventitia and suppress aortic rupture after acute aortic dissection.11 In addition, pharmacologic blockade of CXCR2 was shown to prevent inflammation and autoimmune responses in experimental models of arthritis,12,13 neuroinflammation,14 and CD4+ T cells-mediated hypersensitivity reactions in the lung.15
This study aimed to define the role of CXCR2 in the pathogenesis of hypertension and vascular dysfunction induced by hypertensive stimuli. Hypertension was studied in CXCR2 knockout (CXCR2-/-) mice, wild-type (WT) mice treated with CXCR2 inhibitor, and chimeric mice engineered to lack CXCR2 only in hematopoietic cells. Our results demonstrate that angiotensin II significantly increases the expression of CXCR2 on macrophages. Knockout or inhibition of CXCR2 markedly reduces macrophage infiltration into the vascular wall, limits activation of nicotinamide adenine dinucleotide phosphate (NADPH) oxidases, and attenuates arterial hypertension and vascular dysfunction. Reconstitution of CXCR2-/- mice with WT bone marrow cells reestablished hypertension, vascular remodeling, and oxidative stress in response to angiotensin II, whereas reconstitution of WT mice with CXCR2-/- bone marrow cells prevented hypertension, suggesting that hematopoietic cell CXCR2 expression plays a critical role in hypertension development.
Methods
Detailed methods are described in the online-only Data Supplement.
Animals and Experimental Protocols
WT mice (C57BL/6J, male) and CXCR2 knockout mice (CXCR2-/-, B6.129S2(C)-Cxcr2tm1Mwm/J) were purchased from Jackson Laboratory (Sacramento, CA). Mice at 8 to 12 weeks of age were used in an angiotensin II-induced hypertension model by subcutaneous infusion of angiotensin II (Sigma-Aldrich, St. Louis, MO) at a dose of 490 ng/kg/min or saline using osmotic mini-pumps (Alzet MODEL 1007D; DURECT, Cupertino, CA) for 14/28 days as described previously.16 Deoxycorticosterone acetate (DOCA)-salt-induced hypertension was also created as previously described.17 The selective CXCR2 antagonist SB265610 (Axon, Groningen, The Netherlands) was administered intraperitoneally (2 mg/g/d) beginning 1 day before angiotensin II/DOCA infusion and continued during angiotensin II/DOCA-salt treatment. To determine whether SB265610 could reverse established hypertension, SB265610 treatment was initiated 2 weeks after either angiotensin II infusion (for 28 days) or DOCA-salt challenge (for 21 days). All investigations were approved by the Animal Care and Use Committee of Dalian Medical University and conformed to the US National Institutes of Health Guide for the Care and Use of Laboratory Animals.
Blood Pressure Recordings
Blood pressure was measured by the tail-cuff system (SoftronBP-98A; Softron, Tokyo, Japan)16 and the telemetric blood pressure system (TA-PA11C10, Data Science International, Tilburg, The Netherlands).
Histopathology
Aorta sections were fixed in formalin, embedded in paraffin, sectioned (5 µm), and stained with haematoxylin and eosin and Masson’s trichrome reagent as described previously.16 Cryosections were stained with the dihydroethidine (1µM in PBS) for 30 minutes at 37°C. Green auto-fluorescence and red dihydroethidine fluorescence were detected using Nikon Labophot 2 microscope (Nikon, Tokyo, Japan).
Quantitative Real-Time PCR Analysis
Total mRNA was extracted by the Trizol Reagent method (Invitrogen, New York, NY), and the first strand of cDNA was synthesized with moloney murine leukemia virus reverse transcriptase (Promega, Southampton, UK). Quantitative real-time polymerase chain reaction (qPCR) was performed with an iCycler IQ system (Bio-Rad, CA) as described previously.16 The primer sequences are described in online-only Data Supplement Table SI.
Flow Cytometry
Inflammatory cells in aortic vessels and blood were analyzed by flow cytometry as previously described.6 Single-cell suspensions were treated with Fc block, washed, and stained with CD45 V500, CD3 PE-CF594, CD11b APC-Cy7, Ly6G PE-Cy7, Ly6C V450, F4/80 APC, and CXCR2 PE and their homologous isotype-matched negative controls (BD, Franklin Lakes, NJ). On the basis of a live gate, events were acquired on a Fortessa flow cytometer (BD) and analyzed.
Constitution of Bone Marrow Transplantation Chimeric Mice
We used bone marrow transplantation to produce chimeric mice to study the contribution of bone marrow-derived CXCR2-positive cells to hypertension as previously described.16
Vascular Relaxation Studies
The thoracic aorta was cut into 4-mm segments and gently mounted on force transducers (Power Laboratory, AD Instruments, Bella Vista, Australia) in organ chambers. After stimulation by noradrenaline, vascular responses to increasing concentrations of acetylcholine and sodium nitroprusside (SNP) were recorded.6
NADPH Oxidase Activity
Aortas were cut into pieces and homogenized by ultrasonic processor in PBS buffer and protein concentration was determined by BCA Protein Assay Kit (Life Technologies, Waltham, MA). The aorta lysate was mixed with 0.1 mM NADPH, 5μM lucigenin, PBS buffer, and water in 96-well plates and incubated in 37°C for 30 min. NADPH oxidase activity of the aortas was measured by photon emission from the chromogenic substrate lucigenin by luminometer at 450-nm wavelength. The data were converted to relative light units/min/mg of protein.
Study Patients and Flow Cytometry Analysis
This investigation was performed with approval by the Institutional Ethics Committee of First Affiliated Hospital of Dalian Medical University. Thirty new-onset essential hypertensive patients (systolic blood pressure ≥140 mm Hg and diastolic blood pressure ≥90 mm Hg) and 20 normotensive individuals were included in the study. The baseline characteristics of normotensive control subjects and hypertensive patients are shown in Table 1. Blood samples were obtained from all participants in the recumbent position with a 21-gauge needle by antecubital venipuncture. Coagulation was blocked by K2EDTA. Blood was erythrocyte-depleted with RBC lysis buffer (BD) and incubated with CD45 Percp-cy5.5, CD13 APC, CD15 FITC, CD 64 V450, and CXCR2 PE (BD) as previously described.16 On the basis of a live gate, events were acquired on a FACS Canto IITM flow cytometer (BD) and analyzed.
Table 1.
Baseline Characteristics of Normotensive Control Subjects and Hypertensive Patients
Statistical Analysis
SPSS 16.0 was used to perform the statistical calculations. All data are expressed as mean±SE. First, the normality test (Shapiro-Wilk) was performed to determine whether the data were normally distributed with SPSS 16.0. If data were normally distributed (or in Gaussian distribution), the Student’s t test was used to test for the difference between 2 groups. If the data were not normally distributed, then the Mann-Whitney test was used to test for the difference between two groups. For acetylcholine or SNP-induced vasodilation tests in aortic rings, repeated-measures analysis of variance was used. If the analysis of variance demonstrated a significant effect, then post hoc comparisons were made pairwise with the Fisher least significant difference test. Multivariable logistic regression models were used to evaluate the association of human hypertension and blood CXCR2+ cell numbers (including CD45+CXCR2+ cells, CD45+CD13+CXCR2+ monocytes, CD45+CD13+CD15+CXCR2+ neutrophils, and CD45+CD13+CD64+CXCR2+ macrophages) while adjusting for sex, age, total cholesterol, low-density lipoprotein (LDL) cholesterol, high-density lipoprotein (HDL) cholesterol, and triglycerides. P<0.05 was considered statistically significant.
Results
Angiotensin II Induces CXCR2-Positive Myelomonocytes Infiltration in the Vascular Wall During Hypertension
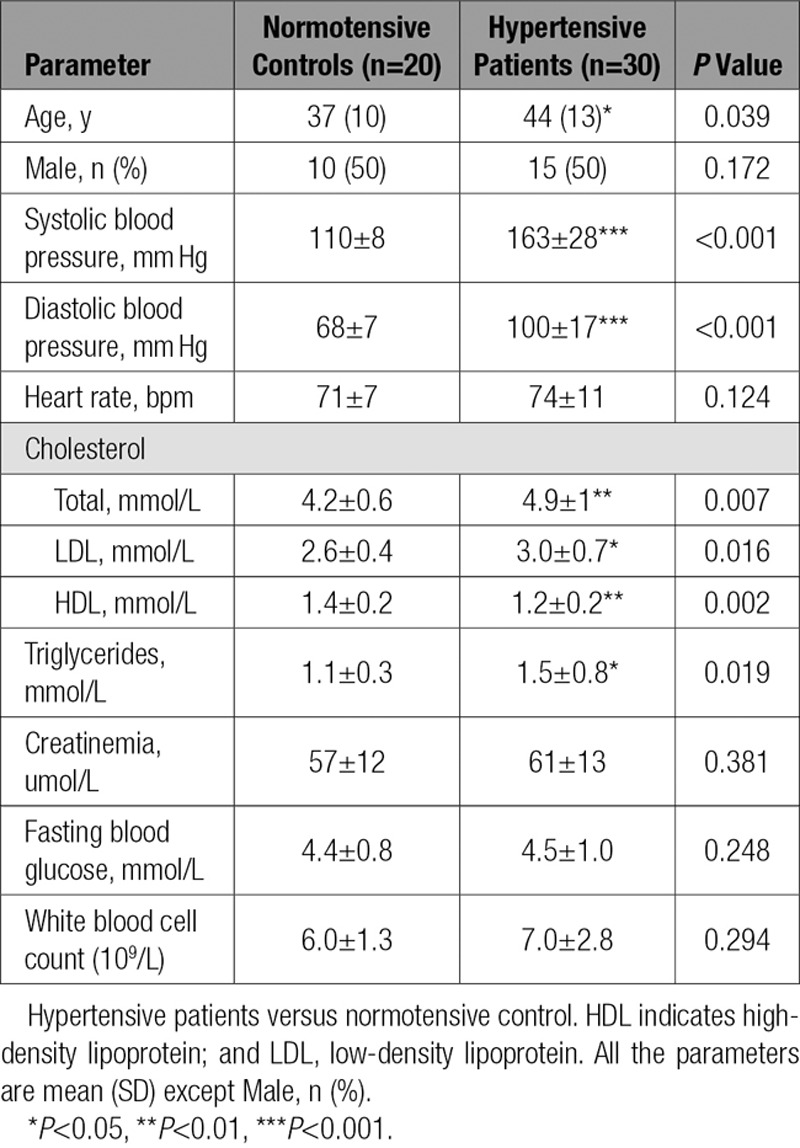
Chemokines are known to play a critical role in the infiltration of myelomonocytes.6 We first used a microarray assay to evaluate the chemokine gene expression profiles in the aortas of mice exposed to angiotensin II. We found that chemokine CXCL1 was the most highly expressed (13.9-fold vs saline-treated) in the aorta 1 day after angiotensin II treatment (Figure IA in the online-only Data Supplement). CXCL1 expression was further confirmed by qPCR analysis (Figure IB in the online-only Data Supplement). Because CXCL1 signals through the G protein-coupled receptor CXCR2, we analyzed the aortic infiltration of CXCR2+ cells by flow cytometry. Compared with saline infusion, angiotensin II infusion caused a significant increase of CD45+CXCR2+ cells in mice aortas (Figure 1A). Real-time qPCR showed that angiotensin II infusion significantly increased CXCR2 mRNA content in the aorta as well (Figure 1B). Immunohistochemical staining further demonstrated that angiotensin II infusion increased CXCR2 protein expression in aorta (Figure 1C).
Figure 1.
Angiotensin II promotes myeloid-derived CXCR2-positive cells. A, Flow cytometry analysis of CD45+CXCR2+ cells in aortic samples before and after angiotensin II infusion at day 1, 3, 7, and 14 (top). The percentage of CD45+CXCR2+ cells was shown as the histogram (bottom). B, Aortic mRNA level of CXCR2 was measured by quantitative real-time polymerase chain reaction (qPCR) after saline and angiotensin II infusion for 14 days. C, Immunohistochemical staining of the CXCR2-positive cells in aortic sections. Arrows indicate the CXCR2-positive cells. Scale bar, 50 μm. *P<0.05, ***P<0.001 versus saline, n=8 per group. A indicates adventitia; Ang II, angiotensin II; E, endothelium; M, media; and WT, wild-type.
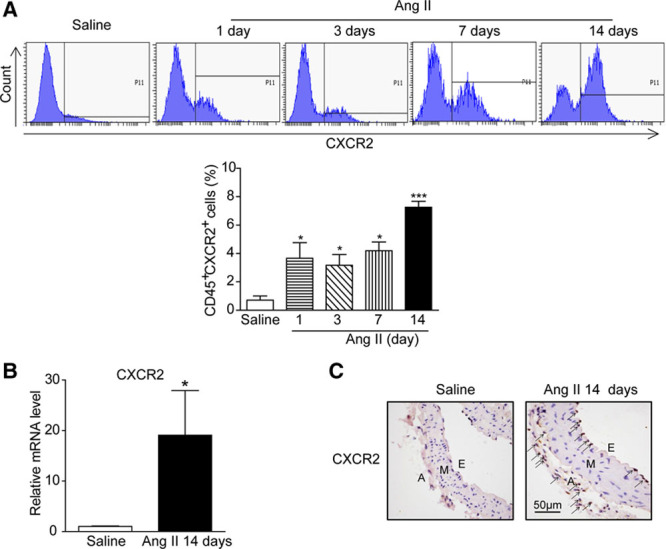
CXCR2 Knockout Abolished Angiotensin II-Induced Hypertension, Vascular Hypertrophy, Aortic Macrophage Infiltration, and Inflammatory Gene Expression
We next determined the effect of CXCR2 on blood pressure in CXCR2 knockout (CXCR2-/-) mice using both the noninvasive tail-cuff method (Figure II in the online-only Data Supplement) and invasive radio-telemetry (Figure 2A and 2B). To our surprise, angiotensin II-induced hypertension was almost totally abolished in CXCR2-/- mice compared with WT animals (Figure 2A and 2B). Radiotelemetry further confirmed that systolic pressure averaged 161±3.5 mm Hg in WT mice and 109±3.4 mm Hg in CXCR2-/- mice during the last week of angiotensin II infusion (Figure 2A and 2B). Likewise, diastolic pressure was reduced from 129±1.4 mm Hg to 95±0.7 mm Hg in CXCR2-/- mice (Figure 2B). Angiotensin II also causes aortic wall thickening and collagen deposition in WT animals, and this finding was significantly blunted in CXCR2-/- mice (Figure 2C and 2D). Moreover, angiotensin II-induced infiltration of CD45+ myelomonocytes, especially F4/80+ macrophages in the aorta, was markedly attenuated in CXCR2-/- mice. It is interesting to note that CD3+ T cells were also markedly decreased in CXCR2-/- mice compared with WT mice after angiotensin II infusion. Depletion of CXCR2 did not change the number of macrophages and T cells after saline infusion (Figure 2E). In addition, angiotensin II-induced expression of several proinflammatory genes and fibrotic markers (IL-1β, IL-6, TNF-α, CD68, VCAM, COX-2, α-SMA, collagen I, and collagen III) in aortic tissue was significantly reduced in CXCR2-/- mice (Figure 2F and 2G).
Figure 2.
Depletion of CXCR2 prevents angiotensin II-induced hypertension, vascular inflammation, and fibrosis. A, Representative original systolic blood pressure recordings in WT and CXCR2-/- mice 3 days before angiotensin II treatment (baseline) and during the last 3 days of angiotensin II infusion (Ang II) by telemetric blood pressure measurements. B, Average systolic (top) and diastolic (bottom) blood pressure of WT and CXCR2-/- mice before and after angiotensin II treatment obtained by telemetry. ##P<0.01 versus WT + Ang II; n=8 per group. C, H&E staining of thoracic aorta (top) and the wall thickness of each group were analyzed (bottom). Scale bar, 50 μm. D, Masson staining of thoracic aorta (top) and the percentage of fibrotic area were analyzed (bottom). Scale bar, 50 μm. E, Flow cytometry analysis of CD45+ cells, CD45+CD11b+F4/80+ macrophages, and CD45+CD3+ T cells in aortic lysates (left). The histograms indicate the percentage of gated cells in the total cells (right). F, qPCR analysis of IL-1β, IL-6, TNF-α, CD68, VCAM-1, and COX-2 mRNA expression levels in the aorta. G, qPCR analysis of α-SMA, collagen I, and collagen III mRNA expression levels in the aorta. *P<0.05 versus WT + saline, #P<0.05 versus WT + Ang II; n=8 per group. α-SMA indicates α-smooth muscle actin; A, adventitia; Ang II, angiotensin II; COX-2, cyclooxygenase-2; E, endothelium; H&E, hematoxylin and eosin; IL-1β, interleukin-1β; IL-6, interleukin-6; M, media; SSC, side scatter; TNF-α, tumor necrosis factor-α; VCAM, vascular cell adhesion molecule-1; and WT, wild-type.
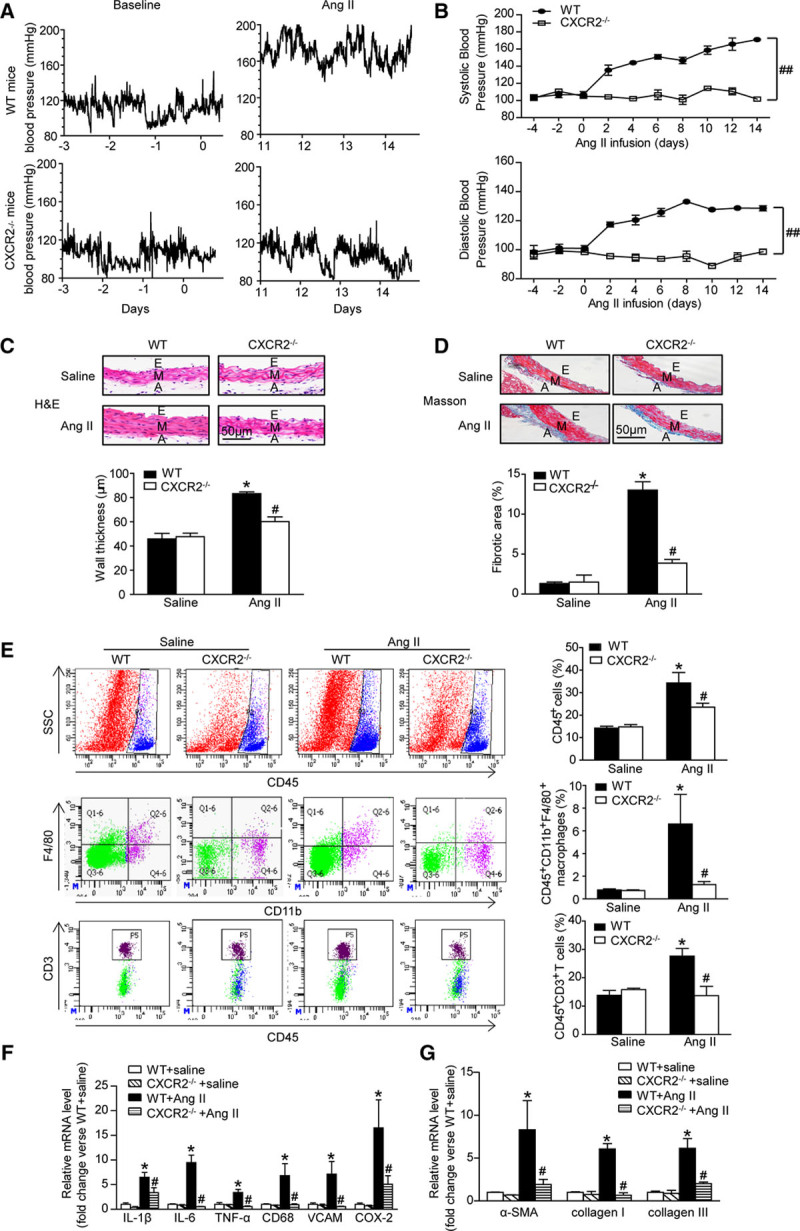
Pharmacological Inhibition of CXCR2 Profoundly Suppressed Angiotensin II-Induced Hypertension, Vascular Remodeling, and Inflammation
To further assess the role of CXCR2 in hypertension, we performed additional experiments in WT mice treated with CXCR2 inhibitor SB265610. Consistent with the results obtained from CXCR2-/- mice, treatment with SB265610 fully attenuated angiotensin II-induced hypertension (Figure 3A). Furthermore, angiotensin II induced aortic wall thickness, collagen deposition, and accumulation of CD45+ myeloid-derived inflammatory cells, including macrophages and T cells, and expression of proinflammatory cytokines and fibrotic markers (IL-1β, IL-6, TNF-α, CD68, VCAM, COX-2, α-SMA, collagen I, and collagen III) in aortic tissue was also significantly attenuated in SB265610-treated mice (Figure 3B through 3F). These results suggest that CXCR2 contributes to angiotensin II-induced hypertension and vascular injury.
Figure 3.
The CXCR2 inhibitor SB265610 alleviates hypertension, vascular inflammation, and fibrosis after angiotensin II infusion. A, Systolic blood pressure was measured by the noninvasive tail-cuff method in vehicle or SB265610-treated mice before (C) and after angiotensin II treatment (T, 490ng/kg/min). B, H&E staining of thoracic aorta (left) and the wall thickness of each group were analyzed (right). Scale bar, 50 μm. C, Masson staining of thoracic aorta (left) and the percentage of fibrotic area were analyzed (right). Scale bar, 50 μm. D, Flow cytometry analysis of CD45+ cells, CD45+CD11b+F4/80+ macrophages, and CD45+CD3+ T cells in aortic lysates (left). The histograms indicate the percentage of gated cells (right). E, qPCR analysis of IL-1β, IL-6, TNF-α, CD68, VCAM-1, and COX-2 mRNA expression levels in the aorta. F, qPCR analysis of α-SMA, collagen I, and collagen III mRNA expression levels in the aorta. *P<0.05, **P<0.01 versus vehicle + saline, #P<0.05, ##P<0.01 versus vehicle + Ang II; n=8 per group. α-SMA indicates α-smooth muscle actin; A, adventitia; A, after Ang II treatment; Ang II, angiotensin II; C, before Ang II treatment; COX-2, cyclooxygenase-2; E, endothelium; H&E, hematoxylin and eosin; IL-1β: interleukin-1β; IL-6, interleukin-6; M, media; TNF-α, tumor necrosis factor-α; VCAM, vascular cell adhesion molecule-1; and WT, wild-type.
Inhibition of CXCR2-Attenuated Angiotensin II-Induced Vascular Dysfunction and Oxidative Stress
To examine how CXCR2 inhibition ameliorated vascular dysfunction, we measured ex vivo vascular function in WT, CXCR2-/-, or SB265610-treated mice treated with angiotensin II. Intact aortas were isolated from WT and CXCR2-/- or SB265610-treated mice 14 days after saline or angiotensin II infusion, and concentration-relaxation curves in response to acetylcholine or SNP were examined. Consistent with previous observations,6,18–20 chronic angiotensin II infusion significantly impaired endothelium-dependent vasodilatation to acetylcholine compared with saline-treated control, whereas these effects were markedly preserved in CXCR2-/- and SB265610-treated mice. Angiotensin II infusion only mildly affected endothelium-independent vasodilatation to SNP in WT mice, and there was no statistically significant difference between angiotensin II-treated mice and saline control. Acetylcholine and SNP-induced vasodilatation was not different between WT and CXCR2-/- mice under control conditions (treated with saline) (Figure 4A and 4B). These results indicate that blocking CXCR2 function prevented angiotensin II-induced endothelial dysfunction.
Figure 4.
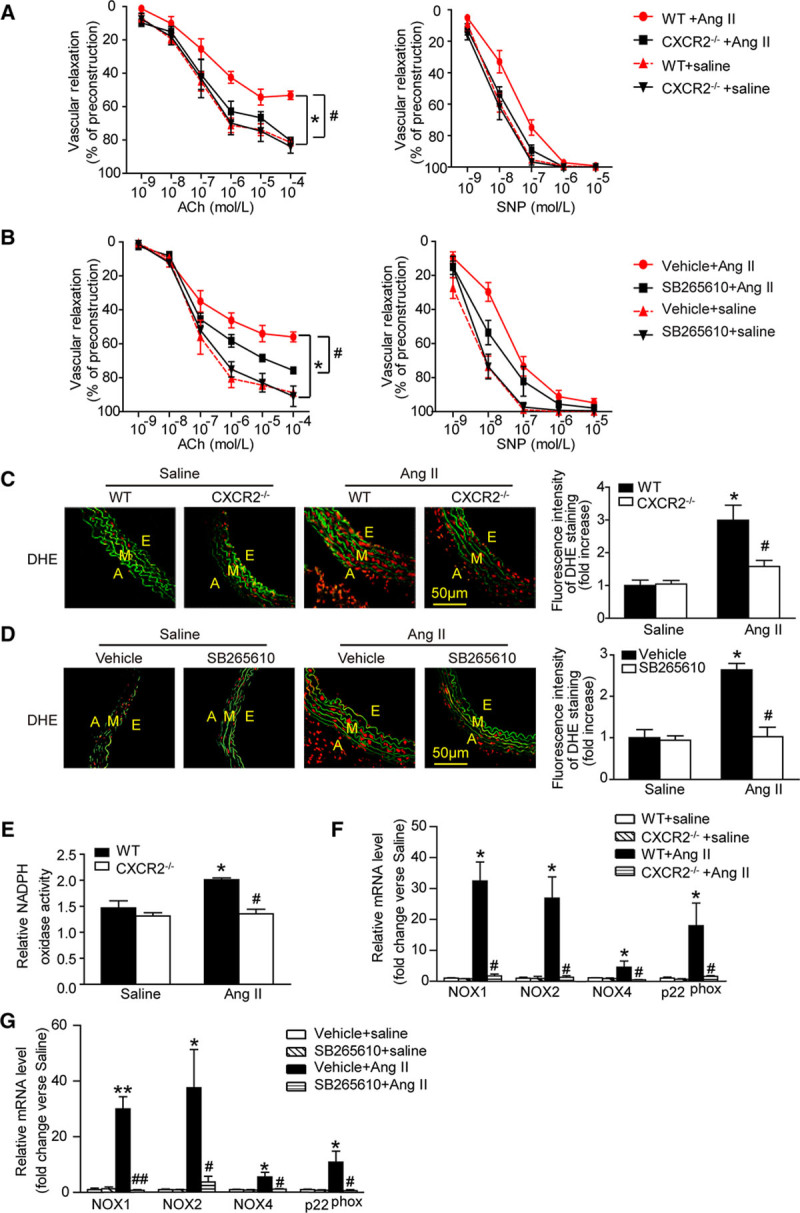
Inhibition or genetic ablation of CXCR2 improves vascular function and reduces oxidative stress in response to Angiotensin II. A and B, Concentration-response curves of endothelium-dependent (acetylcholine, left) and nonendothelium-dependent (SNP, right) relaxation. C and D, DHE staining of the aortic segments (left) and the DHE fluorescence intensity of each group were analyzed (right). Red fluorescence reflects the superoxide formation, whereas green fluorescence shows the laminae. Scale bar, 50 μm. E, Aortic NADPH oxidase activity was measured. F and G, qPCR analysis of NOX1, NOX2, NOX4, and p22phox mRNA expression. *P<0.05, **P<0.01 versus WT/vehicle + saline, #P<0.05, ##P<0.01 versus WT + Ang II; n=6 per group. A indicates adventitia; Ach, acetylcholine; Ang II, angiotensin II; DHE, dihydroethidine; E, endothelium; M, media; NADPH, nicotinamide adenine dinucleotide phosphate; NOX, NADPH oxidase; SNP, sodium nitroprusside; and WT, wild-type.
Increased vascular tone is thought to be mediated by an increase in vascular superoxide production.21,22 We found that angiotensin II significantly increased aortic superoxide (as indicated by dihydroethidine staining), and this effect was abrogated in CXCR2-/- or SB265610-treated mice (Figure 4C and 4D). Moreover, the increase of superoxide formation was accompanied by increased aortic NADPH oxidase activity in both groups after angiotensin II infusion (Figure 4E). qPCR analysis demonstrated that aortic NADPH oxidase NOX1, NOX2, NOX4 catalytic subunits, and p22phox mRNA levels were significantly elevated by angiotensin II treatment in WT mice, but this increase was markedly attenuated in aortas from CXCR2-/- mice or in WT mice treated with SB265610 (Figure 4F and 4G). Thus, CXCR2 drives angiotensin II-induced increases in aortic NADPH oxidase activity and superoxide formation, which likely contribute to vascular dysfunction.
CXCR2 Knockout Prevents DOCA-Salt-Induced Hypertension
To determine whether the reduced hypertensive response in CXCR2-/- or SB265610-treated mice was specific to angiotensin II, we also examined a mouse model of DOCA-salt-induced hypertension. Similar to the angiotensin II study, DOCA-salt-induced hypertension was markedly attenuated in CXCR2-/- mice compared with WT mice (Figure 5A). Likewise, DOCA-salt-induced increases in aortic wall thickness and fibrosis, superoxide production (red dihydroethidine-derived fluorescence), and mRNA expression of proinflammatory cytokines (IL-1β, IL-6, TNF-α, CD68, VCAM, and iNOS) and NADPH oxidase subunits (NOX1, NOX2, NOX4, and p22phox) were markedly reduced in CXCR2-/- mice (Figure 5B through 5F).
Figure 5.
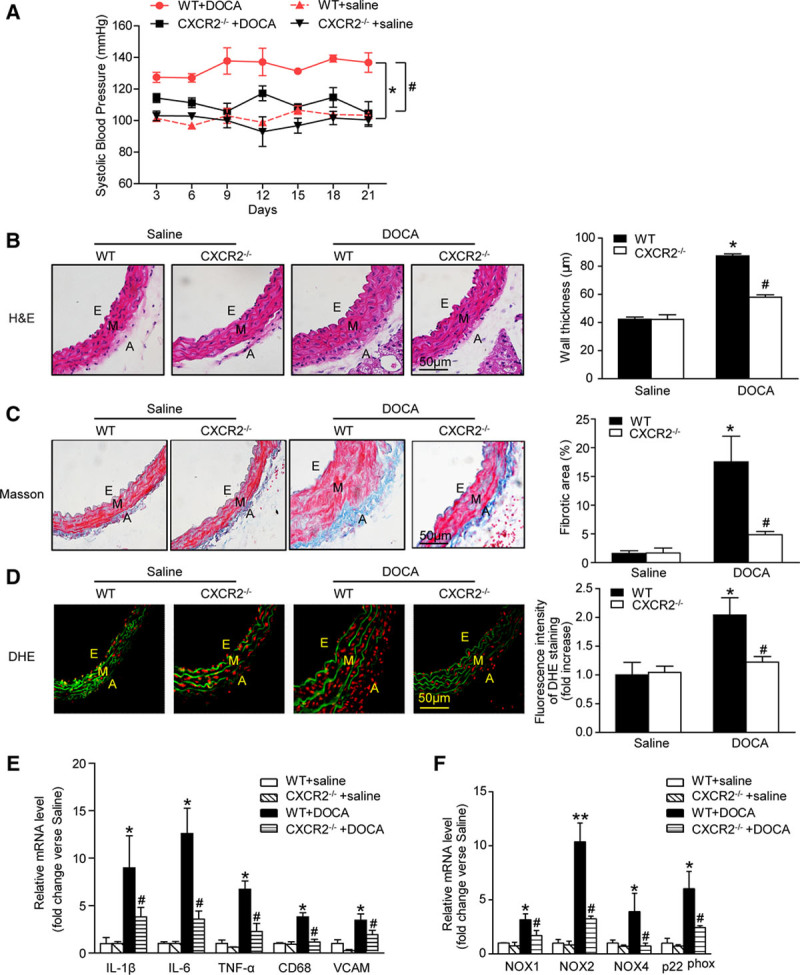
Depletion of CXCR2 attenuates DOCA-salt-induced hypertension. A, Systolic blood pressure was measured by the tail-cuff method of each group. B and C, H&E and Masson staining of thoracic aorta (left), wall thickness, and percentage of fibrotic area of each group were analyzed (right). Scale bar, 50 μm. D, DHE staining of the aortic segments (left) and the DHE fluorescence intensity of each group were analyzed (right). Scale bar, 50 μm. E and F, IL-1β, IL-6, TNF-α, CD68, VCAM-1, NOX1, NOX2, NOX4, and p22phox mRNA expression levels were measured by qPCR. *P<0.05, **P<0.01 versus WT + saline; #P<0.05 versus WT + DOCA; n=8 per group. A indicates adventitia; DHE, dihydroethidine; DOCA, desoxycorticosterone acetate; E, endothelium; H&E, hematoxylin; IL-1β: interleukin-1β; IL-6, interleukin-6; M, media; NOX, nicotinamide adenine dinucleotide phosphate oxidase; TNF-α, tumor necrosis factor-α; VCAM, vascular cell adhesion molecule-1; and WT, wild-type.
Selective Deletion of CXCR2 on Bone Marrow–Derived Cells Attenuated Angiotensin II-Induced Hypertension, Inflammation, Vascular Dysfunction, and Oxidative Stress
Because CXCR2 is expressed on macrophages and neutrophils and on nonmyeloid cells such as fibroblasts, smooth muscle cells, and endothelial cells,23–28 we specifically examined the role of myeloid cell CXCR2 expression in the development of hypertension and vascular dysfunction. We generated chimeric mice by transplanting bone marrow (BM)–derived cells from WT or CXCR2-/- mice into lethally irradiated WT and CXCR2-/- mice, respectively. Four weeks later, these mice were infused with either angiotensin II or vehicle for 2 weeks. WT mice transplanted with CXCR2-/- BM showed significantly reduced systolic pressure, aortic thickening, collagen deposition, and F4/80+ macrophages infiltration into the adventitia, and reduced mRNA expression of proinflammatory cytokines and fibrotic markers (IL-6, IL-1β, TNF-α, CD68, VCAM, COX-2, α-SMA, collagen I, and collagen III) compared with WT mice transplanted with WT BM. In contrast, transplantation of WT BM into CXCR2-/- mice completely restored these pathologic features of hypertension. CXCR2-/- mice transplanted with CXCR2-/- BM responded the same as WT mice transplanted with CXCR2-/- BM (Figure 6A through 6D, 6H, Figure IIIA and IV in the online-only Data Supplement).
Figure 6.
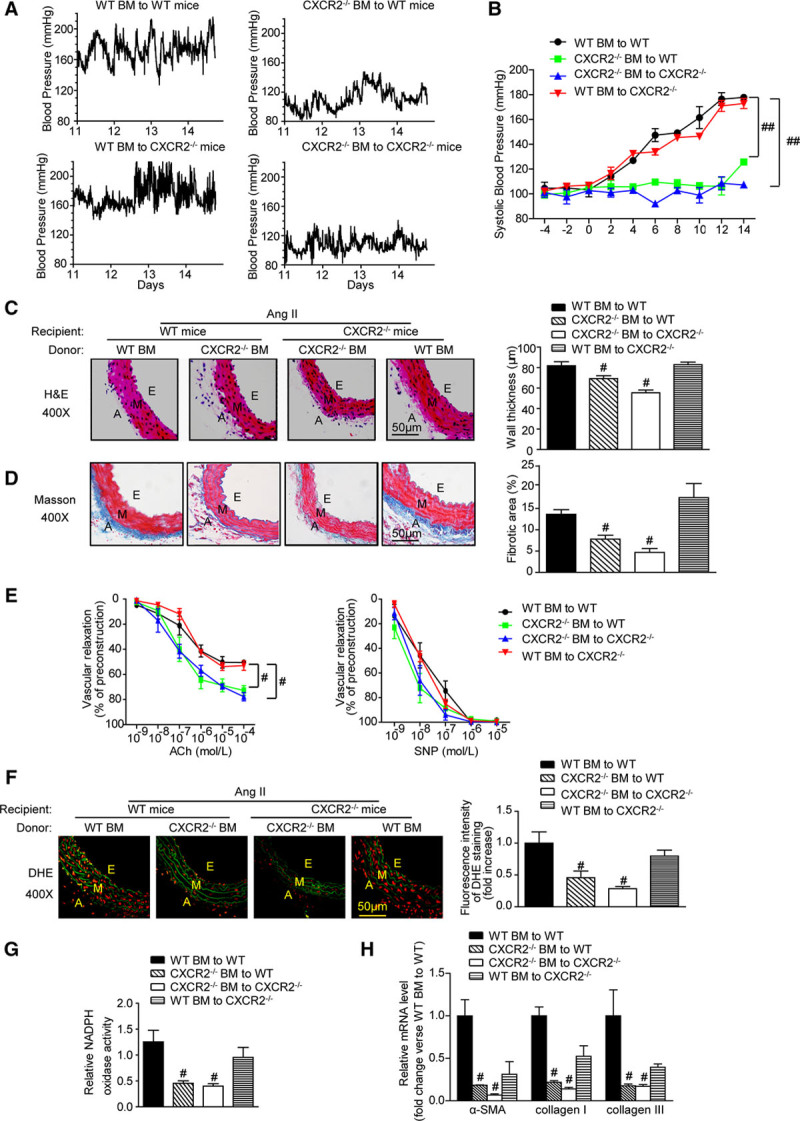
CXCR2 deficiency in bone marrow-derived cells prevents angiotensin II-induced hypertension, vascular inflammation, fibrosis, dysfunction, and oxidative stress. A, Representative telemetric trace recording of systolic blood pressure of each group during the last 3 days of angiotensin II infusion. B, Average systolic blood pressure of each group before and after angiotensin II treatment obtained by telemetry. C and D, H&E and Masson staining of thoracic aorta (left), wall thickness, and percentage of fibrotic area of each group were analyzed (right). Scale bar, 50 μm. E, Concentration-response curves of endothelium acetylcholine-dependent and nonendothelium (SNP)-dependent relaxation. F, DHE staining of the aortic segments (left) and the DHE fluorescence intensity of each group were analyzed (right). Scale bar, 50 μm. G, Aortic NADPH oxidase activity was measured. H, qPCR analysis of α-SMA, collagen I, and collagen III mRNA expression levels in the aorta. #P<0.05, ##P<0.01 versus WT BM to WT; n=6 per group. α-SMA indicates α-smooth muscle actin; A, adventitia; Ach, acetylcholine; BM, bone marrow; DHE, dihydroethidine; E, endothelium; H&E, hematoxylin and eosin; M, media; NADPH oxidase, nicotinamide adenine dinucleotide phosphate oxidase; and SNP, sodium nitroprusside.
The effect of angiotensin II infusion on vascular dysfunction was also examined in BM-transplanted mice. Angiotensin II-induced impairment of endothelium-dependent vasodilatation was much greater in WT mice transplanted with WT BM than in WT mice transplanted with CXCR2-/- BM, but there was no significant difference of endothelium-independent vasodilation between each group (Figure 6E). Likewise, angiotensin II-induced aortic superoxide production (as indicated by dihydroethidine staining), NADPH oxidase activity, and expression of NOX2, NOX4, and p22phox were markedly attenuated in WT mice transplanted with CXCR2-/- BM cells. CXCR2-/- mice transplanted with CXCR2-/- BM responded the same as WT mice transplanted with CXCR2-/- BM (Figure 6F and 6G, Figure IIIB in the online-only Data Supplement). Overall, these findings demonstrate that inhibition of myeloid cell CXCR2 activity prevents angiotensin II-induced hypertension, superoxide production, vascular remodeling, and dysfunction.
CXCR2 Inhibitor Reverses Established Hypertension
We next tested whether inhibition of CXCR2 can reverse preestablished hypertension in mice induced by angiotensin II or DOCA-salt. Consistent with previous and present data,5 angiotensin II infusion or DOCA-salt treatment for 2 weeks elevated blood pressure to 173±5.9 mm Hg or 131±4.5 mm Hg, respectively, compared with saline control (97±6.9 mm Hg). Mice were then divided into two groups: one received intraperitoneal injection of inhibitor SB265610 (2 mg/kg/d per mouse) for an additional 2 weeks, and another group received the vehicle only. Treatment of mice with SB265610 significantly lowered blood pressure in mice induced by either angiotensin II (Figure 7A) or DOCA-salt (Figure 7B) within 1 week of treatment. These results indicate that blockade of CXCR2 lowers hypertension and reduces preestablished hypertension.
Figure 7.
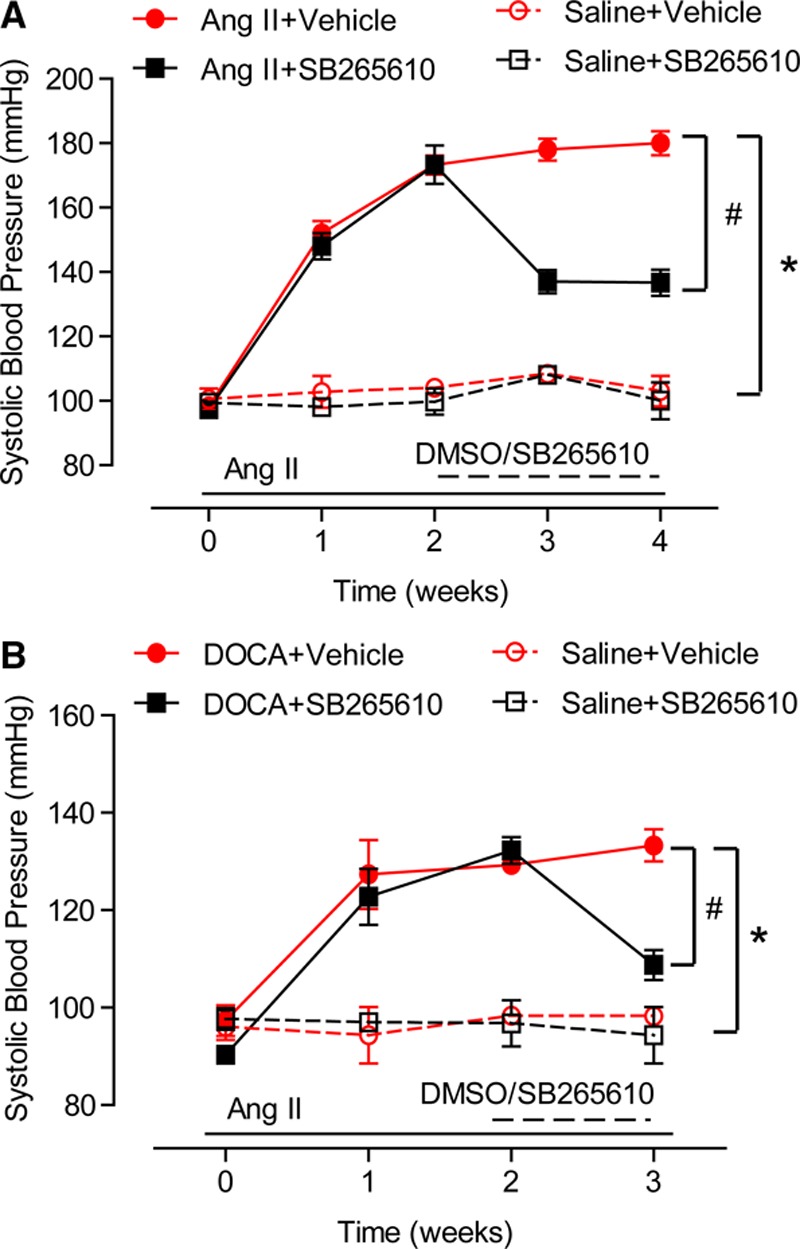
Treatment with SB265610 reverses angiotensin II- and DOCA-salt–induced hypertension. A and B, After 2 weeks of saline, angiotensin II, or DOCA-salt treatment, mice were given either vehicle or SB265610 for the remaining 1 or 2 weeks of saline, angiotensin II, or DOCA-salt treatment. The systolic blood pressure was recorded by the tail-cuff method every week. *P<0.05 versus saline + vehicle, #P<0.05 versus Ang II/DOCA + SB265610; n=10 per group.
CXCR2+ Cells Are Increased in Human Hypertension
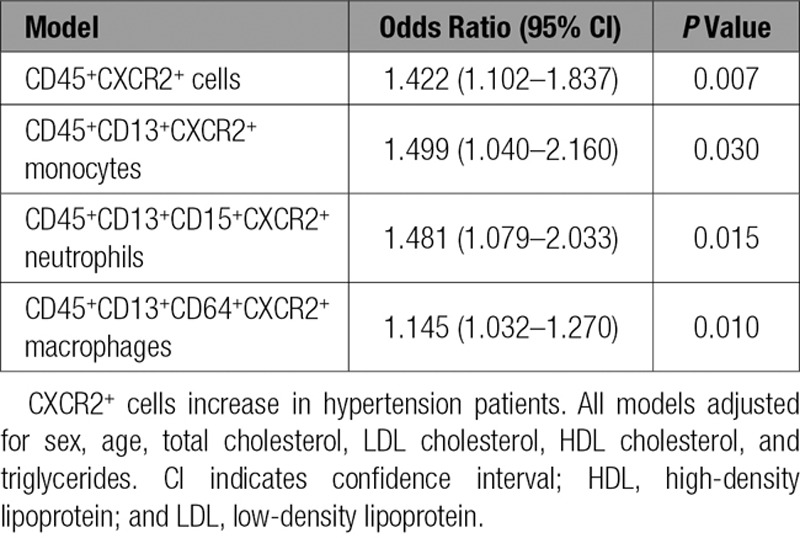
To determine whether CXCR2+ macrophages are important in human hypertension, we analyzed blood CXCR2+ cells in patients (30 with hypertension and 20 normotensive individuals, Table 1) with flow cytometry. We found that hypertensive patients were older and had higher total cholesterol, LDL cholesterol, and triglyceride and lower HDL cholesterol concentrations in serum compared with normotensive control (Table 1). Flow cytometry showed that CXCR2 was mainly expressed in CD45+CD13+ monocytes, CD45+CD13+CD15+ neutrophils, and CD45+CD13+CD64+ macrophages (Figure IC in the online-only Data Supplement). The numbers of circulating CD45+CXCR2+ inflammatory cells, CD45+CD13+CXCR2+ monocytes, CD45+CD13+CD15+CXCR2+ neutrophils, and CD45+CD13+CD64+CXCR2+ macrophages in hypertensive patients were significantly higher than in normotensive individuals (Figure 8). We then evaluated the association of blood CXCR2+ cell numbers and hypertension with multivariable logistic regression models. After adjusting for sex, age, total cholesterol, LDL cholesterol, HDL cholesterol, and triglycerides, a statistically significant association was found between the numbers of CXCR2+ cells, including CD45+ cells (odds ratio [OR] 1.422), monocytes (OR 1.499), neutrophils (OR 1.481), macrophages (OR 1.145), and hypertension (Table 2).
Figure 8.
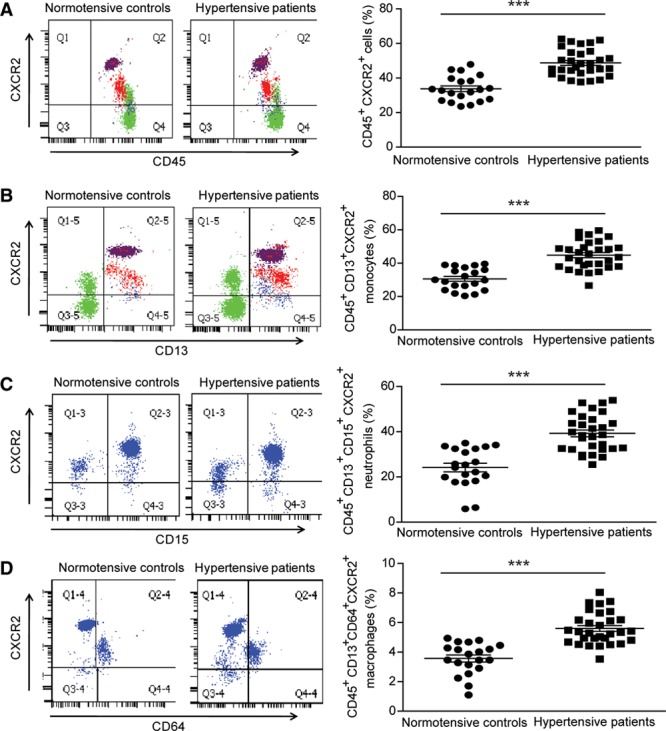
Blood CXCR2+ cells are increased in hypertensive patients. A–D, Flow cytometric analysis of circulating CD45+CXCR2+ inflammatory cells (A), CD45+CD13+CXCR2+ monocytes (B), CD45+CD13+CD15+CXCR2+ neutrophils (C), and CD45+CD13+CD64+CXCR2+ macrophages (D) in both hypertensive patients (n=30) and normotensive individuals (n=20). ***P<0.001 hypertensive patients versus normotensive control.
Table 2.
Results of the Multivariable Logistic Regression Models of CXCR2+ Cells on Hypertension
Discussion
In this study, we demonstrated that inhibition of chemokine receptor CXCR2 activity by genetic deletion or pharmacologic CXCR2 inhibitor-attenuated hypertension development in mice. CXCR2 disruption reduced angiotensin II or DOCA-salt-induced aortic infiltration of macrophages, proinflammatory cytokine expression, and vascular superoxide production and prevented development of vascular endothelial dysfunction. In addition, adoptive transfer of BM-derived cells from WT mice into CXCR2-/- mice reestablished angiotensin II-induced hypertension, vascular dysfunction, inflammation, fibrosis, and oxidative stress, indicating a specific involvement of leukocyte CXCR2 expression in the development of hypertension. It is important to note that CXCR2 blockade also reversed established angiotensin II or DOCA-salt-induced hypertension. This study thus provides novel evidence supporting a strong causative role of CXCR2+ leukocytes in the pathogenesis of arterial hypertension (Figure V in the online-only Data Supplement). Inhibition of CXCR2 activation may therefore be a promising approach for treatment of hypertension and vascular dysfunction.
Previous studies demonstrated that hypertensive stimuli cause vascular recruitment and activation of proinflammatory cells such as macrophages and T lymphocytes, which play a critical role in the pathogenesis of hypertension.6,16,29 An unanswered question is how hypertensive stimuli such as angiotensin II and high salt promote the infiltration of these cells into the vascular wall in the early stages of hypertension. Emerging evidence indicates that several chemokines (MCP-1, IL-8, CCL5, and CX3CL1) and their receptors (CCR2, CXCR1, CCR5, and CX3CR1) play an important role in the development of hypertension.8 Chemokine-receptor pathways strongly influence the migration of monocytes/macrophages, production of reactive oxygen species and nitric oxide, endothelial dysfunction, and smooth muscle cell proliferation.8 Chemokine receptor CXCR2 is expressed on multiple immune cell types and is essential for recruitment of neutrophils at inflammation sites. In the present study, our data revealed that ablation or inhibition of CXCR2 activity reduced vascular endothelial dysfunction, attenuated vascular oxidative stress, decreased the extent of vascular medial hypertrophy and collagen deposition (Figure 2 through 5), and, most important, nearly abolished the increase in systolic blood pressure induced by angiotensin II or DOCA-salt. In addition, the number of vascular macrophages and T lymphocytes, and the expression of proinflammatory cytokines, were markedly reduced by knockout of CXCR2 or inhibitor SB265610 (Figure 2E and 3D), demonstrating a critical role for CXCR2 in vascular infiltration of inflammatory cells, oxidative stress, and hypertension.
Inflammatory cells, including monocytes/macrophages, neutrophils, and T lymphocytes, exert differential effects in experimental model of hypertension, vascular inflammation, and dysfunction.30 For example, infiltration of neutrophils is important to initially activate or fully stimulate monocytes/macrophages in the vessel wall but has no effect on arterial hypertension induced by angiotensin II. 6 T cells but not B cells have been reported to promote vascular dysfunction and hypertension in mice after angiotensin II infusion or DOCA-salt stress.31 It is important to note that monocytes/macrophages but not neutrophils were shown to be responsible for angiotensin II-induced vascular dysfunction and hypertension.6 In this study, our flow cytometry measurements showed that CXCR2 was highly expressed in macrophages and neutrophils but almost undetectable in T lymphocytes (Figure IC in the online-only Data Supplement). Furthermore, depletion or inhibition of CXCR2 effectively reduced blood pressure elevation and infiltration of F4/80+ macrophages in aorta in response to angiotensin II (Figure 2 and 3). Although adoptive transfer of CXCR2-/- BM into WT mice markedly attenuated hypertension, vascular dysfunction, and oxidative stress, transfer of WT BM into CXCR2-/- mice reestablished these effects in response to angiotensin II infusion (Figure 6). These findings demonstrate that CXCR2+ macrophages mediate angiotensin II- or DOCA-salt-induced arterial hypertension and vascular dysfunction.
Early studies demonstrated that angiotensin II-induced hypertension and vascular dysfunction are associated with increased vascular superoxide production.21 NADPH oxidase isoforms NOX1-5 are the major source of superoxide in vessels.32,33 NOX1 is expressed mainly in vascular smooth muscle cells; NOX2 in endothelial cells, cardiomyocytes, and some VSMCs; NOX4 in endothelial cells, VSMCs, and other cells; and NOX5 in human endothelial cells.34 The various NOX isoforms are believed to play important roles in vascular remodeling and dysfunction. Here we found that angiotensin II-induced production of superoxide, NADPH oxidase activity, and the mRNA levels of NADPH oxidase subunits NOX1, NOX2, NOX4, and p22phox were markedly abrogated in CXCR2-/- or SB265610-treated mice (Figure 4E through 4G). Proinflammatory cells are believed to be the main source of superoxide in the vascular wall.35 As shown in our study, inhibition or ablation of CXCR2 markedly reduced the number of CD45+ cells, particularly macrophages, which infiltrated the vascular wall after angiotensin II infusion. Together these data suggest that inhibition or ablation of CXCR2 attenuates hypertension by reducing inflammatory cell infiltration and subsequent vascular oxidative stress. CXCR2 has been implicated in a number of immune-mediated inflammatory diseases.36 Although several CXCR2-specific inhibitors have been developed as anti-inflammatory agents, there are currently no clinically approved CXCR2 inhibitors. Recently, agents that inhibit CXCR2 activation, such as SB265610, reparixin, and CX797, have been developed and examined in various animal disease models, including autoimmune diseases, ischemia-reperfusion injury, diabetes, lung injury, and vascular injury.11,15,37–39 It is interesting to note that comparison of these inhibitors shows a distinct mechanism of action on CXCR2. For example, CX797 inhibits IL-8-mediated β-arrestin-2 recruitment and cell migration,36 whereas SB265610 inhibits CXCR2 function by binding to an allosteric site.40 Recently, reparixin was used to effectively decrease systolic blood pressure in spontaneously hypertensive rats.41 Our results also demonstrated that SB265610 markedly inhibited vascular inflammation and hypertension induced by angiotensin II. It is important to note that SB265610 treatment reversed established hypertension in our model (Figure 7), suggesting that CXCR2 may be a new therapeutic target for treatment of arterial hypertension.
Conclusions
Our study demonstrated for the first time that hypertension induced by various stimuli, including angiotensin II, mineralocorticoids, and high salt, requires CXCR2 activity, which is essential for vascular infiltration of macrophages, dysfunction, and oxidative stress (Figure V in the online-only Data Supplement). These findings may be clinically relevant because CXCR2-positive cells were also markedly elevated in hypertensive patients. Therefore, selective blockade of proinflammatory CXCR2+ cells, especially macrophages, might represent a novel therapeutic target for arterial hypertension treatment.
Acknowledgments
The authors gratefully thank Professor Yu Huang (School of Biomedical Sciences, The Chinese University of Hong Kong) and Professor Li Ma (School of Public Health, Dalian Medical University).
Sources of Funding
This work was supported by grants from the National Basic Research Program of China (973 program: No. 2012CB517802 to Dr Li), the State Key Program of National Natural Science Foundation of China (81630009 to Dr Li), the Youth Science Fund Program of National Natural Science Foundation of China (81500303 to Dr Wang), the International S&T Cooperation Program of China (2014DFA31930 to Drs Yang and Xia), and the Chang Jiang Scholar Program of China (to Dr Li).
Disclosures
None.
Supplementary Material
Footnotes
Sources of Funding, see page 1366
The online-only Data Supplement is available with this article at http://circ.ahajournals.org/lookup/suppl/doi:10.1161/CIRCULATIONAHA.115.020754/-/DC1.
Circulation is available at http://circ.ahajournals.org.
Clinical Perspective
What Is New?
The recruitment of leukocytes to the vascular wall is an early step to the pathological process of hypertension. Chemokines and their respective receptors play a critical role in mediating inflammatory cells chemotaxis in cardiovascular diseases. Therefore, identifying specific therapeutic targets in the chemokine system is crucial for the development of new therapeutic strategies against hypertension.
In this study, we provide experimental evidence that CXCR2+ macrophages are mediators of hypertension, vascular collagen deposition, inflammation, vascular dysfunction, and oxidative stress in angiotensin II- or DOCA-salt-induced animal models. Moreover, CXCR2+ proinflammatory cells were higher in hypertensive patients compared with normotensive individuals.
What Are the Clinical Implications?
The findings that CXCR2 inhibition prevents and reverses hypertension and vascular dysfunction in response to multiple hypertensive stimuli increase our understanding of the mechanisms involved in CXCR2 action and the potential clinical use of CXCR2 inhibitor for the treatment of hypertension.
Thus, selective inhibition of CXCR2 signal may be a novel therapeutic approach to prevent and cure hypertension and vascular remodeling in patients.
References
- 1.Rodríguez-Iturbe B, Pons H, Quiroz Y, Lanaspa MA, Johnson RJ. Autoimmunity in the pathogenesis of hypertension. Nat Rev Nephrol. 2014;10:56–62. doi: 10.1038/nrneph.2013.248. doi: 10.1038/nrneph.2013.248. [DOI] [PubMed] [Google Scholar]
- 2.Harrison DG, Guzik TJ, Lob HE, Madhur MS, Marvar PJ, Thabet SR, Vinh A, Weyand CM. Inflammation, immunity, and hypertension. Hypertension. 2011;57:132–140. doi: 10.1161/HYPERTENSIONAHA.110.163576. doi: 10.1161/HYPERTENSIONAHA.110.163576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Harrison DG. The immune system in hypertension. Trans Am Clin Climatol Assoc. 2014;125:130–38. [PMC free article] [PubMed] [Google Scholar]
- 4.Guzik TJ, Hoch NE, Brown KA, McCann LA, Rahman A, Dikalov S, Goronzy J, Weyand C, Harrison DG. Role of the T cell in the genesis of angiotensin II induced hypertension and vascular dysfunction. J Exp Med. 2007;204:2449–2460. doi: 10.1084/jem.20070657. doi: 10.1084/jem.20070657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Vinh A, Chen W, Blinder Y, Weiss D, Taylor WR, Goronzy JJ, Weyand CM, Harrison DG, Guzik TJ. Inhibition and genetic ablation of the B7/CD28 T-cell costimulation axis prevents experimental hypertension. Circulation. 2010;122:2529–2537. doi: 10.1161/CIRCULATIONAHA.109.930446. doi: 10.1161/CIRCULATIONAHA.109.930446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wenzel P, Knorr M, Kossmann S, Stratmann J, Hausding M, Schuhmacher S, Karbach SH, Schwenk M, Yogev N, Schulz E, Oelze M, Grabbe S, Jonuleit H, Becker C, Daiber A, Waisman A, Münzel T. Lysozyme M-positive monocytes mediate angiotensin II-induced arterial hypertension and vascular dysfunction. Circulation. 2011;124:1370–1381. doi: 10.1161/CIRCULATIONAHA.111.034470. doi: 10.1161/CIRCULATIONAHA.111.034470. [DOI] [PubMed] [Google Scholar]
- 7.Moser B, Wolf M, Walz A, Loetscher P. Chemokines: multiple levels of leukocyte migration control. Trends Immunol. 2004;25:75–84. doi: 10.1016/j.it.2003.12.005. doi: 10.1016/j.it.2003.12.005. [DOI] [PubMed] [Google Scholar]
- 8.Martynowicz H, Janus A, Nowacki D, Mazur G. The role of chemokines in hypertension. Adv Clin Exp Med. 2014;23:319–325. doi: 10.17219/acem/37123. [DOI] [PubMed] [Google Scholar]
- 9.Olson TS, Ley K. Chemokines and chemokine receptors in leukocyte trafficking. Am J Physiol Regul Integr Comp Physiol. 2002;283:R7–28. doi: 10.1152/ajpregu.00738.2001. doi: 10.1152/ajpregu.00738.2001. [DOI] [PubMed] [Google Scholar]
- 10.Tarzami ST, Miao W, Mani K, Lopez L, Factor SM, Berman JW, Kitsis RN. Opposing effects mediated by the chemokine receptor CXCR2 on myocardial ischemia-reperfusion injury: recruitment of potentially damaging neutrophils and direct myocardial protection. Circulation. 2003;108:2387–2392. doi: 10.1161/01.CIR.0000093192.72099.9A. doi: 10.1161/01.CIR.0000093192.72099.9A. [DOI] [PubMed] [Google Scholar]
- 11.Anzai A, Shimoda M, Endo J, Kohno T, Katsumata Y, Matsuhashi T, Yamamoto T, Ito K, Yan X, Shirakawa K, Shimizu-Hirota R, Yamada Y, Ueha S, Shinmura K, Okada Y, Fukuda K, Sano M. Adventitial CXCL1/G-CSF expression in response to acute aortic dissection triggers local neutrophil recruitment and activation leading to aortic rupture. Circ Res. 2015;116:612–623. doi: 10.1161/CIRCRESAHA.116.304918. doi: 10.1161/CIRCRESAHA.116.304918. [DOI] [PubMed] [Google Scholar]
- 12.Coelho FM, Pinho V, Amaral FA, Sachs D, Costa VV, Rodrigues DH, Vieira AT, Silva TA, Souza DG, Bertini R, Teixeira AL, Teixeira MM. The chemokine receptors CXCR1/CXCR2 modulate antigen-induced arthritis by regulating adhesion of neutrophils to the synovial microvasculature. Arthritis Rheum. 2008;58:2329–2337. doi: 10.1002/art.23622. doi: 10.1002/art.23622. [DOI] [PubMed] [Google Scholar]
- 13.Barsante MM, Cunha TM, Allegretti M, Cattani F, Policani F, Bizzarri C, Tafuri WL, Poole S, Cunha FQ, Bertini R, Teixeira MM. Blockade of the chemokine receptor CXCR2 ameliorates adjuvant-induced arthritis in rats. Br J Pharmacol. 2008;153:992–1002. doi: 10.1038/sj.bjp.0707462. doi: 10.1038/sj.bjp.0707462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Veenstra M, Ransohoff RM. Chemokine receptor CXCR2: physiology regulator and neuroinflammation controller? J Neuroimmunol. 2012;246:1–9. doi: 10.1016/j.jneuroim.2012.02.016. doi: 10.1016/j.jneuroim.2012.02.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Konrad FM, Reutershan J. CXCR2 in acute lung injury. Mediators Inflamm. 2012;2012:740987. doi: 10.1155/2012/740987. doi: 10.1155/2012/740987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wang L, Li YL, Zhang CC, Cui W, Wang X, Xia Y, Du J, Li HH. Inhibition of Toll-like receptor 2 reduces cardiac fibrosis by attenuating macrophage-mediated inflammation. Cardiovasc Res. 2014;101:383–392. doi: 10.1093/cvr/cvt258. doi: 10.1093/cvr/cvt258. [DOI] [PubMed] [Google Scholar]
- 17.Du YH, Guan YY, Alp NJ, Channon KM, Chen AF. Endothelium-specific GTP cyclohydrolase I overexpression attenuates blood pressure progression in salt-sensitive low-renin hypertension. Circulation. 2008;117:1045–1054. doi: 10.1161/CIRCULATIONAHA.107.748236. doi: 10.1161/CIRCULATIONAHA.107.748236. [DOI] [PubMed] [Google Scholar]
- 18.Madhur MS, Lob HE, McCann LA, Iwakura Y, Blinder Y, Guzik TJ, Harrison DG. Interleukin 17 promotes angiotensin II-induced hypertension and vascular dysfunction. Hypertension. 2010;55:500–507. doi: 10.1161/HYPERTENSIONAHA.109.145094. doi: 10.1161/HYPERTENSIONAHA.109.145094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Matsuno K, Yamada H, Iwata K, Jin D, Katsuyama M, Matsuki M, Takai S, Yamanishi K, Miyazaki M, Matsubara H, Yabe-Nishimura C. Nox1 is involved in angiotensin II-mediated hypertension: a study in Nox1-deficient mice. Circulation. 2005;112:2677–2685. doi: 10.1161/CIRCULATIONAHA.105.573709. doi: 10.1161/CIRCULATIONAHA.105.573709. [DOI] [PubMed] [Google Scholar]
- 20.Liu L, Liu J, Tian XY, Wong WT, Lau CW, Xu A, Xu G, Ng CF, Yao X, Gao Y, Huang Y. Uncoupling protein-2 mediates DPP-4 inhibitor-induced restoration of endothelial function in hypertension through reducing oxidative stress. Antioxid Redox Signal. 2014;21:1571–1581. doi: 10.1089/ars.2013.5519. doi: 10.1089/ars.2013.5519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Rajagopalan S, Kurz S, Münzel T, Tarpey M, Freeman BA, Griendling KK, Harrison DG. Angiotensin II-mediated hypertension in the rat increases vascular superoxide production via membrane NADH/NADPH oxidase activation. Contribution to alterations of vasomotor tone. J Clin Invest. 1996;97:1916–1923. doi: 10.1172/JCI118623. doi: 10.1172/JCI118623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Guzik TJ, West NE, Black E, McDonald D, Ratnatunga C, Pillai R, Channon KM. Vascular superoxide production by NAD(P)H oxidase: association with endothelial dysfunction and clinical risk factors. Circ Res. 2000;86:E85–E90. doi: 10.1161/01.res.86.9.e85. [DOI] [PubMed] [Google Scholar]
- 23.Reutershan J, Morris MA, Burcin TL, Smith DF, Chang D, Saprito MS, Ley K. Critical role of endothelial CXCR2 in LPS-induced neutrophil migration into the lung. J Clin Invest. 2006;116:695–702. doi: 10.1172/JCI27009. doi: 10.1172/JCI27009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Schraufstatter IU, Chung J, Burger M. IL-8 activates endothelial cell CXCR1 and CXCR2 through Rho and Rac signaling pathways. Am J Physiol Lung Cell Mol Physiol. 2001;280:L1094–L1103. doi: 10.1152/ajplung.2001.280.6.L1094. [DOI] [PubMed] [Google Scholar]
- 25.Miyake M, Goodison S, Urquidi V, Gomes Giacoia E, Rosser CJ. Expression of CXCL1 in human endothelial cells induces angiogenesis through the CXCR2 receptor and the ERK1/2 and EGF pathways. Lab Invest. 2013;93:768–778. doi: 10.1038/labinvest.2013.71. doi: 10.1038/labinvest.2013.71. [DOI] [PubMed] [Google Scholar]
- 26.Hastings NE, Feaver RE, Lee MY, Wamhoff BR, Blackman BR. Human IL-8 regulates smooth muscle cell VCAM-1 expression in response to endothelial cells exposed to atheroprone flow. Arterioscler Thromb Vasc Biol. 2009;29:725–731. doi: 10.1161/ATVBAHA.109.184382. doi: 10.1161/ATVBAHA.109.184382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Frydelund-Larsen L, Penkowa M, Akerstrom T, Zankari A, Nielsen S, Pedersen BK. Exercise induces interleukin-8 receptor (CXCR2) expression in human skeletal muscle. Exp Physiol. 2007;92:233–240. doi: 10.1113/expphysiol.2006.034769. doi: 10.1113/expphysiol.2006.034769. [DOI] [PubMed] [Google Scholar]
- 28.Rigamonti E, Fontaine C, Lefebvre B, Duhem C, Lefebvre P, Marx N, Staels B, Chinetti-Gbaguidi G. Induction of CXCR2 receptor by peroxisome proliferator-activated receptor gamma in human macrophages. Arterioscler Thromb Vasc Biol. 2008;28:932–939. doi: 10.1161/ATVBAHA.107.161679. doi: 10.1161/ATVBAHA.107.161679. [DOI] [PubMed] [Google Scholar]
- 29.Hilgers KF. Monocytes/macrophages in hypertension. J Hypertens. 2002;20:593–596. doi: 10.1097/00004872-200204000-00010. [DOI] [PubMed] [Google Scholar]
- 30.Morton J, Coles B, Wright K, Gallimore A, Morrow JD, Terry ES, Anning PB, Morgan BP, Dioszeghy V, Kühn H, Chaitidis P, Hobbs AJ, Jones SA, O’Donnell VB. Circulating neutrophils maintain physiological blood pressure by suppressing bacteria and IFNgamma-dependent iNOS expression in the vasculature of healthy mice. Blood. 2008;111:5187–5194. doi: 10.1182/blood-2007-10-117283. doi: 10.1182/blood-2007-10-117283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Singh MV, Chapleau MW, Harwani SC, Abboud FM. The immune system and hypertension. Immunol Res. 2014;59:243–253. doi: 10.1007/s12026-014-8548-6. doi: 10.1007/s12026-014-8548-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Griendling KK, Sorescu D, Lassègue B, Ushio-Fukai M. Modulation of protein kinase activity and gene expression by reactive oxygen species and their role in vascular physiology and pathophysiology. Arterioscler Thromb Vasc Biol. 2000;20:2175–2183. doi: 10.1161/01.atv.20.10.2175. [DOI] [PubMed] [Google Scholar]
- 33.Seshiah PN, Weber DS, Rocic P, Valppu L, Taniyama Y, Griendling KK. Angiotensin II stimulation of NAD(P)H oxidase activity: upstream mediators. Circ Res. 2002;91:406–413. doi: 10.1161/01.res.0000033523.08033.16. [DOI] [PubMed] [Google Scholar]
- 34.Anilkumar N, Weber R, Zhang M, Brewer A, Shah AM. Nox4 and nox2 NADPH oxidases mediate distinct cellular redox signaling responses to agonist stimulation. Arterioscler Thromb Vasc Biol. 2008;28:1347–1354. doi: 10.1161/ATVBAHA.108.164277. doi: 10.1161/ATVBAHA.108.164277. [DOI] [PubMed] [Google Scholar]
- 35.Mittal M, Siddiqui MR, Tran K, Reddy SP, Malik AB. Reactive oxygen species in inflammation and tissue injury. Antioxid Redox Signal. 2014;20:1126–1167. doi: 10.1089/ars.2012.5149. doi: 10.1089/ars.2012.5149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ha H, Neamati N. Pyrimidine-based compounds modulate CXCR2-mediated signaling and receptor turnover. Mol Pharm. 2014;11:2431–2441. doi: 10.1021/mp500180e. doi: 10.1021/mp500180e. [DOI] [PubMed] [Google Scholar]
- 37.Opfermann P, Derhaschnig U, Felli A, Wenisch J, Santer D, Zuckermann A, Dworschak M, Jilma B, Steinlechner B. A pilot study on reparixin, a CXCR1/2 antagonist, to assess safety and efficacy in attenuating ischaemia-reperfusion injury and inflammation after on-pump coronary artery bypass graft surgery. Clin Exp Immunol. 2015;180:131–142. doi: 10.1111/cei.12488. doi: 10.1111/cei.12488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Citro A, Valle A, Cantarelli E, Mercalli A, Pellegrini S, Liberati D, Daffonchio L, Kastsiuchenka O, Ruffini PA, Battaglia M, Allegretti M, Piemonti L. CXCR1/2 inhibition blocks and reverses type 1 diabetes in mice. Diabetes. 2015;64:1329–1340. doi: 10.2337/db14-0443. doi: 10.2337/db14-0443. [DOI] [PubMed] [Google Scholar]
- 39.Zarbock A, Allegretti M, Ley K. Therapeutic inhibition of CXCR2 by Reparixin attenuates acute lung injury in mice. Br J Pharmacol. 2008;155:357–364. doi: 10.1038/bjp.2008.270. doi: 10.1038/bjp.2008.270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Bradley ME, Bond ME, Manini J, Brown Z, Charlton SJ. SB265610 is an allosteric, inverse agonist at the human CXCR2 receptor. Br J Pharmacol. 2009;158:328–338. doi: 10.1111/j.1476-5381.2009.00182.x. doi: 10.1111/j.1476-5381.2009.00182.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kim HY, Choi JH, Kang YJ, Park SY, Choi HC, Kim HS. Reparixin, an inhibitor of CXCR1 and CXCR2 receptor activation, attenuates blood pressure and hypertension-related mediators expression in spontaneously hypertensive rats. Biol Pharm Bull. 2011;34:120–127. doi: 10.1248/bpb.34.120. [DOI] [PubMed] [Google Scholar]