

Abstract
Desmoplasia contributes to the aggressive behavior of pancreatic cancer. However, recent clinical trials testing several antifibrotic agents on pancreatic cancer have not shown clear efficacy. Therefore, further investigation of desmoplasia‐targeting antifibrotic agents by another mechanism is needed. Calpeptin, an inhibitor of calpains, suppressed fibroblast function and inhibited fibrosis. In this study, we investigated the anticancer effects of calpeptin on pancreatic cancer. We investigated whether calpeptin inhibited tumor progression using a mouse xenograft model. We used quantitative RT‐PCR to evaluate the expression of calpain‐1 and calpain‐2 mRNA in pancreatic cancer cells (PCCs) and pancreatic stellate cells (PSCs). We also undertook functional assays, including proliferation, migration, and invasion, to evaluate the inhibitory effects of calpeptin on PCCs and PSCs. Quantitative RT‐PCR indicated that PCCs and PSCs expressed calpain‐2 mRNA. Calpeptin reduced tumor volume (P = 0.0473) and tumor weight (P = 0.0471) and inhibited the tumor desmoplastic reaction (P < 0.001) in xenograft tumors in nude mice. Calpeptin also inhibited the biologic functions of PCCs and PSCs including proliferation (P = 0.017), migration (P = 0.027), and invasion (P = 0.035) in vitro. Furthermore, calpeptin reduced the migration of PCCs and PSCs by disrupting the cancer–stromal interaction (P = 0.0002). Our findings indicate that calpeptin is a promising antitumor agent for pancreatic cancer, due not only to its suppressive effect on PCCs and PSCs but also its disruption of the cancer–stromal interaction.
Keywords: Calpeptin, cancer–stromal interaction, desmoplasia, pancreatic cancer, pancreatic stellate cells
Pancreatic cancer is a lethal solid tumor, with a 5‐year survival of only 7% in the USA.1 The poor prognosis for pancreatic cancer is due to the difficulty of early detection, the presence of local invasion and distant metastasis by the time of diagnosis, and resistance to chemotherapy. Surgical resection is a unique therapeutic procedure that can cure pancreatic cancer, but unfortunately, <20% of pancreatic cancer patients can be subjects of surgical resection.2 The majority of pancreatic cancer patients are treated by chemotherapy, which can prolong a patient's life for a few months.3, 4, 5, 6
It has been reported that desmoplasia, a significant hyperplasia of fibrotic tissue, is one of the main contributors to the aggressive nature of pancreatic cancer and its resistance to chemotherapy.7, 8 Pancreatic stellate cells (PSCs) are the main components of pancreatic cancer stromal tissue and enhance proliferation and migration of pancreatic cancer cells.9 Desmoplasia is characterized by an excessive production of ECM, which can disturb the delivery of chemotherapeutic agents such as gemcitabine.7 Although clinical trials of antifibrotic agents such as IPI‐926, a Hedgehog pathway inhibitor, to treat pancreatic cancer have been carried out, none have been clearly effective as a treatment.10 Further investigation and strategies for targeting desmoplasia in various ways are required. Antifibrotic agents with an effect on desmoplasia by another mechanism might be effective in pancreatic cancer treatment.
Calpains are well‐conserved cysteine proteases that are expressed by mammalian cells.11 The calpain family is classified into subtypes according to structure. Calpain‐1 and calpain‐2 are classified as typical calpain family‐type proteins, which bind to a 30‐kDa regulatory small subunit (CAPNS1, also called calpain‐4).11 It has been reported that calpains are involved in apoptosis, cellular signaling, cell survival, and cytoskeletal remodeling.11, 12 In addition, it is known that calpains cleave focal adhesion kinase (FAK),13, 14, 15 IκB,16 and MYC,17 all of which are involved in the carcinogenesis of pancreatic cancer.18, 19, 20, 21
Calpeptin is a drug developed to specifically inhibit calpains to assess their physiological role.22 It was reported that calpeptin induced cell apoptosis and decreased migration in several cancers,15, 23 but there have been no reports describing the effects of calpeptin on pancreatic cancer. Furthermore, previous studies have reported that calpeptin has a suppressive effect on fibroblast function and inhibits pulmonary fibrosis,24, 25 leading us to hypothesize that calpeptin inhibited not only pancreatic cancer cells (PCCs) but also myofibroblast‐like PSCs, which have been reported to promote pancreatic cancer aggressiveness.26 The purpose of this study is to investigate the suppressive effects of calpeptin on PCCs and PSCs and to clarify its effects on pancreatic cancer.
Materials and Methods
Cell isolation and culture conditions
Human PSCs were isolated from fresh pancreatic surgical specimens using the outgrowth method described by Bachem et al.27, 28 The PSC cell type was confirmed by morphology (stellate‐like or spindle‐shaped cells) and by immunofluorescence staining for α‐smooth muscle actin (α‐SMA) and vimentin.28, 29, 30 The use of pancreatic cancer surgical specimens was approved by the Ethics Committee of Kyushu University (Fukuoka, Japan) and was carried out according to the Ethical Guidelines for Human Genome/Gene Research enacted by the Japanese Government and the Helsinki Declaration. We used 10 human PCC cell lines in this study: PANC‐1 (Institute of Physical and Chemical Research, Saitama, Japan), KP‐2 and KP‐3 (Japan Health Sciences Foundation, Tokyo, Japan), SUIT‐2 and MIA PaCa‐2 (Japan Health Science Research Resources Bank, Osaka, Japan), and AsPC‐1, CAPAN‐1, CAPAN‐2, Hs766T, and SW1990 (ATCC, Manassas, VA, USA), as well as normal human pancreatic epithelial cells (CS‐PE; Cell Systems, Applied Cell Biology Research Institute, Kirkland, WA, USA). The HPDE cell line was kindly provided by Dr. Ming‐Sound Tsao (University of Toronto, Toronto, ON, Canada). All of the cells were maintained as previously described.31
Immortalization of human PSCs
We synthesized double‐stranded DNA encoding the immortalizing genes hTERT and SV40 LargeT and cloned the DNA into the pLVSIN vector. Then we constructed lentiviral particles from this vector and transduced human PSCs, followed by G418 selection, to establish the immortalized PSCs (iPSCs).
In vivo experiments in mouse s.c. xenograft model
To analyze the effects of calpeptin on PCCs and PSCs in vivo, SW1990 cells (1 × 106) suspended in 100 μL DMEM and iPSCs (1 × 106) and SW1990 cells (1 × 106) in 100 μL DMEM were s.c. transplanted into the left and the right flank of 5‐week‐old female nude mice, respectively. One week after implantation, we randomly divided the mice into two groups (five mice/group) and i.p. administered 0.04 mg calpeptin diluted in DMSO in 0.2 mL PBS or 0.2 mL PBS containing the same amount of DMSO (controls) three times a week for 4 weeks. The mice were killed on day 35, and all of the xenograft tumors were excised and weighed. Tumor volume was calculated using the formula π/6 × (L × W × W), in which L is the largest tumor diameter and W is the smallest tumor diameter. All of the mouse experiments were approved by the Ethics Committee of Kyushu University.
Immunohistochemical procedures and measurements
Immunohistochemistry was carried out as described previously.32 The antibodies used for immunohistochemistry were: mouse monoclonal anti‐α‐SMA (1:500; Dako, Glostrup, Denmark), rabbit polyclonal anti‐periostin (sc‐67233, 1:500; Santa Cruz Biotechnology, Santa Cruz, CA, USA), mouse monoclonal anti‐cytokeratin 19 (sc‐376126, 1:500), and rabbit polyclonal anti‐PCNA (ab2426, 1:1000; both Abcam, Cambridge, UK). Cells were considered positive if the nucleus and/or cytoplasm were stained. The α‐SMA‐ and periostin‐positive areas and the proliferating cell nuclear antigen (PCNA)‐positive cell rate (PCNA index) were calculated in FIVE fields at a magnification of ×200 with a light microscope using ImageJ software (http://rsb.info.nih.gov/ij/) provided by the US National Institutes of Health.
Sirius red staining and measurements
Sections were cut to 4‐mm thickness from paraffin‐embedded material, deparaffinized in xylene, and rehydrated through a graded ethanol series. The sections were stained with Sirius red (Direct Red 80; Aldrich Chemical Co., Milwaukee, WI, USA) staining solution for 30 min, and the Sirius red‐positive area was measured in five fields at a magnification of ×100 with a light microscope using Adobe Photoshop CS (Adobe Systems Incorporated, San Jose, CA, USA).
Quantitative RT‐PCR
Quantitative RT‐PCR was carried out as described previously.33 We designed specific primers for α‐SMA, periostin, and fibroblast growth factor‐2 (FGF‐2) using Primer 3 software (http://primer3.sourceforge.net/). Specific primers for calpain‐2, collagen type I, fibronectin, transforming growth factor (TGF)‐β1, platelet‐derived growth factor (PDGF)‐A, PDGF‐B, connective tissue growth factor (CTGF), hepatocyte growth factor (HGF), vascular endothelial growth factor (VEGF), and GAPDH RNAs were purchased from Takara Bio Inc. (Tokyo, Japan). The expression level of each gene was normalized to that of GAPDH as an internal control and depicted as the ratio of target gene expression to GAPDH expression. All of the samples were run in triplicate, and each sample was analyzed at least twice. Gene expression levels were calculated using a standard curve constructed with total RNA from SW1990.
Cell viability assay
One thousand PCCs or PSCs per well were plated in triplicate into 96‐well plates with DMEM containing 10% FBS for 24 h. After cellular adhesion to the plates, the medium was replaced with fresh DMEM containing 10% FBS plus calpeptin at 0 (DMSO), 10, 20, 40, 60, or 80 μM (day 0). Cell viability was determined with a CellTiter‐Glo luminescent cell viability assay kit (Promega, Madison, WI, USA) according to the manufacturer's instructions on days 0–4 every 24 h.
Production of conditioned media from PCCs and PSCs
Conditioned media from PSCs and PCCs were produced using serum‐free DMEM to exclude the effects of growth factors present in serum. Subconfluent SUIT‐2 cells and iPSC cells were cultured in serum‐free DMEM for 24 or 48 h, and the supernatants were collected and designated PCC‐SN and PSC‐SN, respectively. In experiments designed to analyze the effects of calpeptin on PSCs or PCCs, subconfluent SUIT‐2 cells or iPSC cells were cultured in serum‐free DMEM containing 20 μM calpeptin for 24 or 48 h. To remove the calpeptin, 15 mL supernatant was separated with a centrifugal separator using filter units (Amicon Ultra‐15 Centrifugal Filter Units [Merck Millipore, Billerica, MA, USA]) at 4000 g for 40 min. The residue was diluted with serum‐free DMEM to a total of 15 mL. The conditioned media obtained by these processes was designated calpeptin‐treated PCC‐SN and calpeptin‐treated PSC‐SN and were used to stimulate PSCs and PCCs. Fifteen milliliters of PCC‐SN and PSC‐SN was also filtered as above and the residue was diluted with serum‐free DMEM to a total of 15 mL, as a comparison.
Cell migration and Matrigel invasion assay
The migration and invasion assays were carried out by counting the number of migrating or invading cells through uncoated or Matrigel‐coated Transwell chambers (BD Biosciences, Franklin Lakes, NJ, USA) as described previously,34 using calpeptin concentrations of 0 (DMSO), 1, 10, or 20 μM. To investigate the effects of calpeptin on the cancer–stromal interaction, we used the above‐mentioned supernatants PCC‐SN, PSC‐SN, calpeptin‐treated PCC‐SN, and calpeptin‐treated PSC‐SN in this assay. Ten percent FBS was added to the supernatant.
Silencing of calpain‐2 by siRNA
Subconfluent PCCs and PSCs were transfected with siRNA targeting calpain‐2 (siCalpain2‐1: sense, 5′‐CCAGCGAUACCUACAAGAA‐3′ and antisense, 5′‐UUCUUGUAGGUAUCGCUGG‐3′; and siCalpain2‐2: sense, 5′‐CGAGAAUACUGGAACAAUA‐3′ and antisense, 5′‐UAUUGUUCCAGUAUUCUCG‐3′) (Life Technologies, Carlsbad, CA, USA) by electroporation using the Nucleofector System (Amaxa Biosystems, Köln, Germany). To verify the specificity of the knockdown effects, we used non‐targeting siRNA as a control (Life Technologies). Calpain‐2‐silenced cells were used for the following assays 24 h post‐transfection.
Statistical analysis
For the in vitro experiments, values are expressed as the mean values ± SD. Comparisons between groups were carried out using the one‐way anova test followed by the Tukey–Kramer multiple comparisons test. All of the experiments were repeated independently three times, and all of the statistical analyses were undertaken using JMP 11 software (SAS Institute, Cary, NC, USA).
Results
Calpeptin inhibited s.c. tumor growth in vivo in mice
First, we examined the effects of calpeptin on PCCs and PSCs in vivo. In the control group, co‐implanted PCC and PSC tumor growth was much greater than PCCs alone (P < 0.001, Fig. 1a,b). The tumors consisting of PCCs co‐implanted with PSCs contained marked desmoplasia with larger Sirius red‐, αSMA‐, and periostin‐positive areas than tumors containing PCCs alone (P < 0.05, Fig. 1c–f). The growth of co‐implanted tumors was suppressed by calpeptin treatment compared with the control group (Fig. 1a,b). Calpeptin also reduced the Sirius red‐, α‐SMA‐ and periostin‐positive area in tumors consisting of PCCs co‐implanted with PSCs (P < 0.001, Fig. 1c–f). Although the tumor growth of PCCs alone was also suppressed by calpeptin, the difference was not significant. We also investigated the cell proliferative status of xenograft tumors. The PCNA index of co‐implantation tumors was higher than that of PCCs alone, and this value decreased after calpeptin treatment (Fig. 1c,g).
Figure 1.
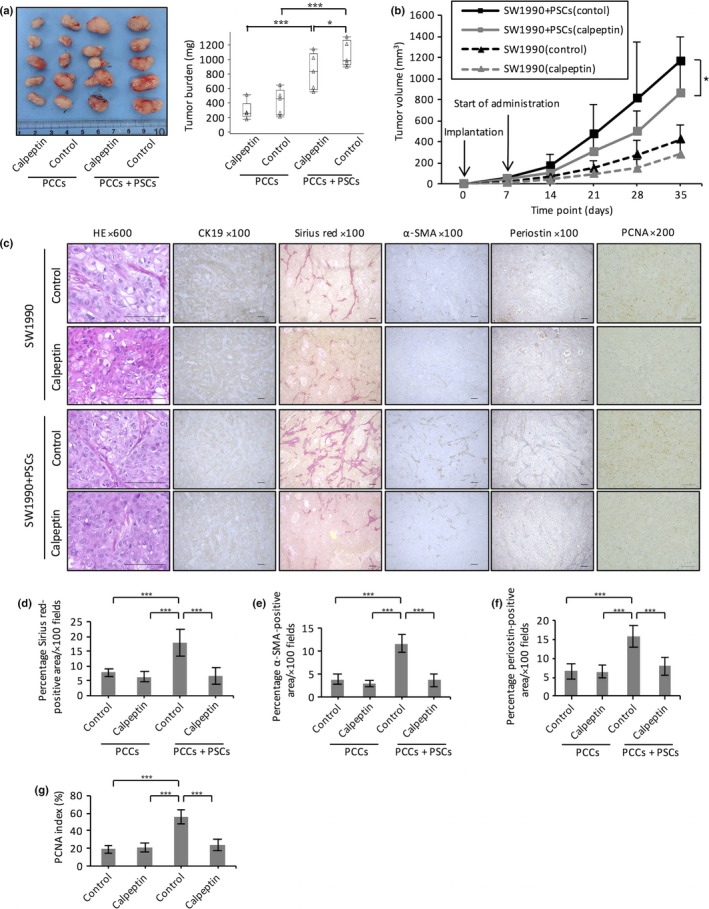
Effect of the calpain inhibitor calpeptin on s.c. xenograft tumors. Six‐week‐old nude mice were injected with SW1990 cells and immortalized pancreatic stellate cells (iPSCs) in the right flank and SW1990 cells only in the left flank. Calpeptin or DMSO was given i.p. three times a week during the experiment. On day 35, the mice (n = 5, each experiment) were killed. (a) Images of xenograft tumors formed 5 weeks after the injection of pancreatic cancer cells (PCCs) only or PCCs and pancreatic stellate cells (PSCs). The right panel charts the xenograft tumor burden. Calpeptin decreased xenograft tumor weight in the co‐implantation groups. (b) Time course of tumor growth. (c) Histological evaluation of desmoplastic activity and cell proliferative status in PCCs and PCCs co‐implanted with PSCs with or without calpeptin treatment. Calpeptin treatment reduced interstitial tumor tissues in the co‐implantation group as assessed by Sirius red staining (c, d), immunohistochemistry of α‐smooth muscle actin (α‐SMA) (c, e), and periostin (c, f). (c, g) Calpeptin decreased the proliferating cell nuclear antigen (PCNA) index in tumors containing SW1990 cells co‐implanted with PSCs. Data are presented as the mean ± SD. *P < 0.05, ***P < 0.001. Scale bar = 100 μm.
Calpain‐2 mRNA expression in pancreatic cancer cells and PSCs
We investigated the expression level of calpain‐2 mRNA in 10 pancreatic cancer cell lines, three primary PSC cultures, and normal pancreatic ductal epithelial cells (HPDE), all of which expressed calpain‐2 mRNA (Fig. 2). AsPC‐1 cells showed the highest expression of calpain‐2, followed by PANC‐1, Hs766T, CAPAN‐2, and the others. HPDE cells expressed a relatively high level of calpain‐2 mRNA. All of the examined primary PSC cultures also expressed calpain‐2 mRNA, but at a lower level. CS‐PE cells did not express detectable levels of calpain‐2 mRNA. Calpain‐1 mRNA expression was not detected in any of these cell lines (data not shown). These data showed that both PCCs and PSCs expressed calpain‐2 mRNA, suggesting that calpeptin affected both PCCs and PSCs and possibly explaining the suppression of xenograft growth in nude mice.
Figure 2.
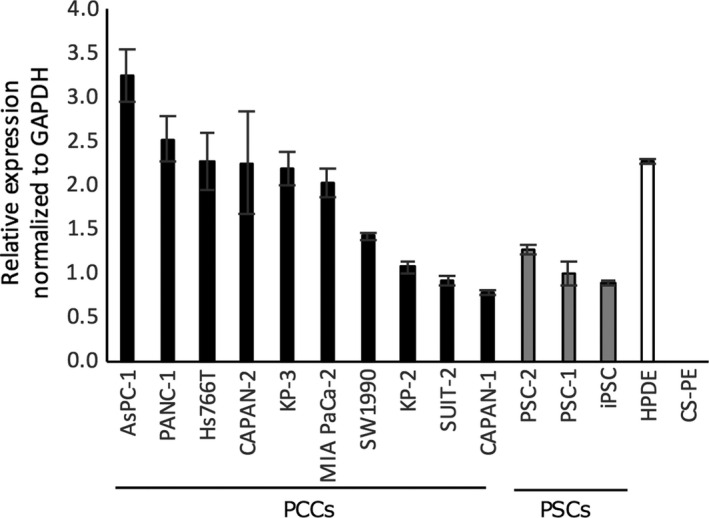
Calpain‐2 mRNA expression levels in 10 pancreatic cancer cell lines (PCCs), three pancreatic stellate cell lines (PSCs), and HPDE and CS‐PE normal pancreatic ductal epithelial cells. Calpain‐2 mRNA expression was normalized to GAPDH RNA expression.
Calpeptin inhibited proliferation, migration, and invasion of pancreatic cancer cells
Next, we examined the effects of calpeptin on the functions of PCCs. Calpeptin suppressed the proliferation of SW1990 cells at high doses (>20 μM), whereas calpeptin did not suppress proliferation at low doses (1 or 10 μM; P < 0.05, Fig. 3a). The IC50 value was 74.2 μM. Calpeptin similarly suppressed the proliferation of SUIT‐2 cells (data not shown) and inhibited the migration (P < 0.05, Fig. 3b,c) and invasion (P < 0.05, Fig. 3d,e) of PCCs. To investigate the specific role of calpain‐2 in this mechanism, we used siRNAs targeting calpain‐2 and investigated the effect of silencing of calpain‐2 on proliferation, migration, and invasion of PCCs. As shown in Figure 3, silencing of calpain‐2 decreased the proliferation, migration, and invasion of SW1990 cells (Fig. 3f,g) and SUIT‐2 cells (data not shown).
Figure 3.
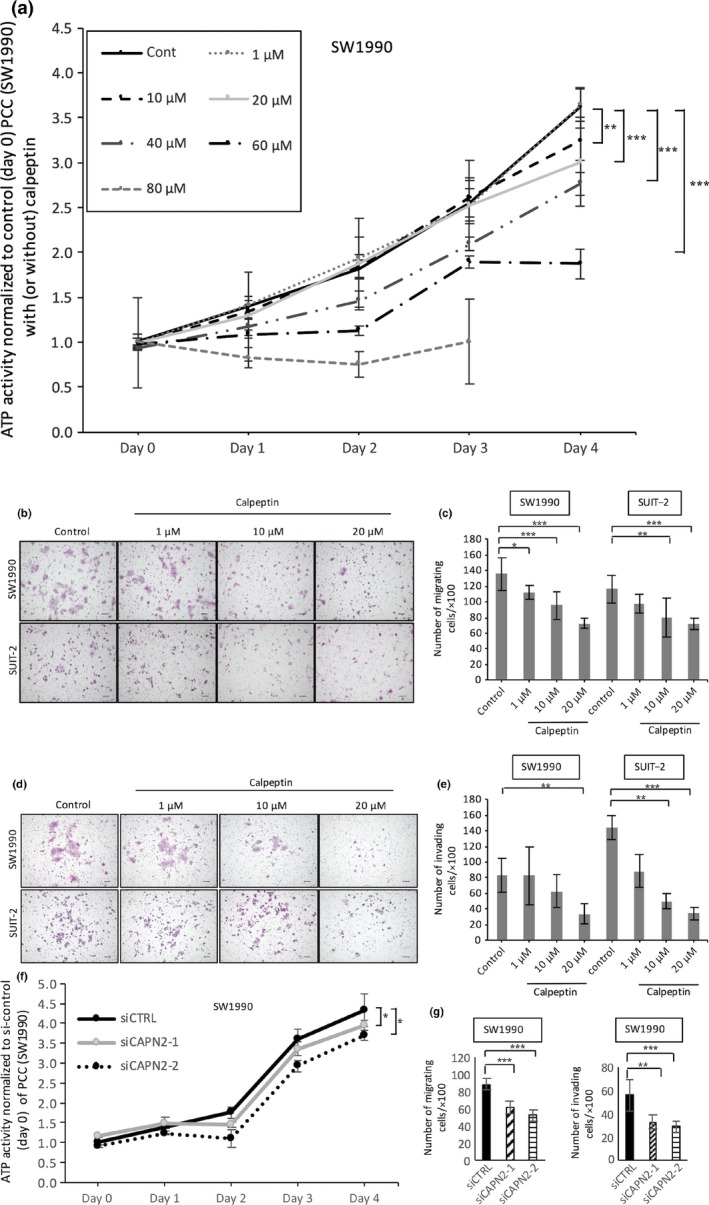
Effect of calpeptin on proliferation, migration, and invasion of pancreatic cancer cells (PCCs). (a) Cell growth of PCCs was assessed using an ATP‐based assay with or without calpeptin (0, 10, 20, 40, 60, or 80 μM). Representative images of migrating (b) and invading (d) PCCs are shown. Calpeptin dose‐dependently decreased the migration (c) and invasion (e) of PCCs. Silencing of calpain‐2 decreased proliferation (f), and migration and invasion (g) of SW1990 cells. Data are presented as the mean ± SD. *P < 0.05, **P < 0.01, ***P < 0.001. Cells were stained with HE; original magnification, ×100. Scale bar = 100 μm.
Calpeptin also inhibited proliferation, migration, and invasion of PSCs
We examined whether calpeptin inhibits the functions of PSCs and observed that, similar to PCCs, calpeptin dose‐dependently suppressed the proliferation of PSCs (P < 0.05, Fig. 4a). The IC50 value was 62.1 μM. Similarly, calpeptin inhibited the migration (P < 0.05, Fig. 4b,c) and invasion (P < 0.05, Fig. 4d,e) of PSCs. The number of migrating or invading PSCs was decreased at doses over 10 μM (P < 0.05, Fig. 4c,e). Furthermore, silencing of calpain‐2 decreased proliferation, migration, and invasion of iPSC cells (Fig. 4f,g) and PSC‐1 cells (data not shown).
Figure 4.
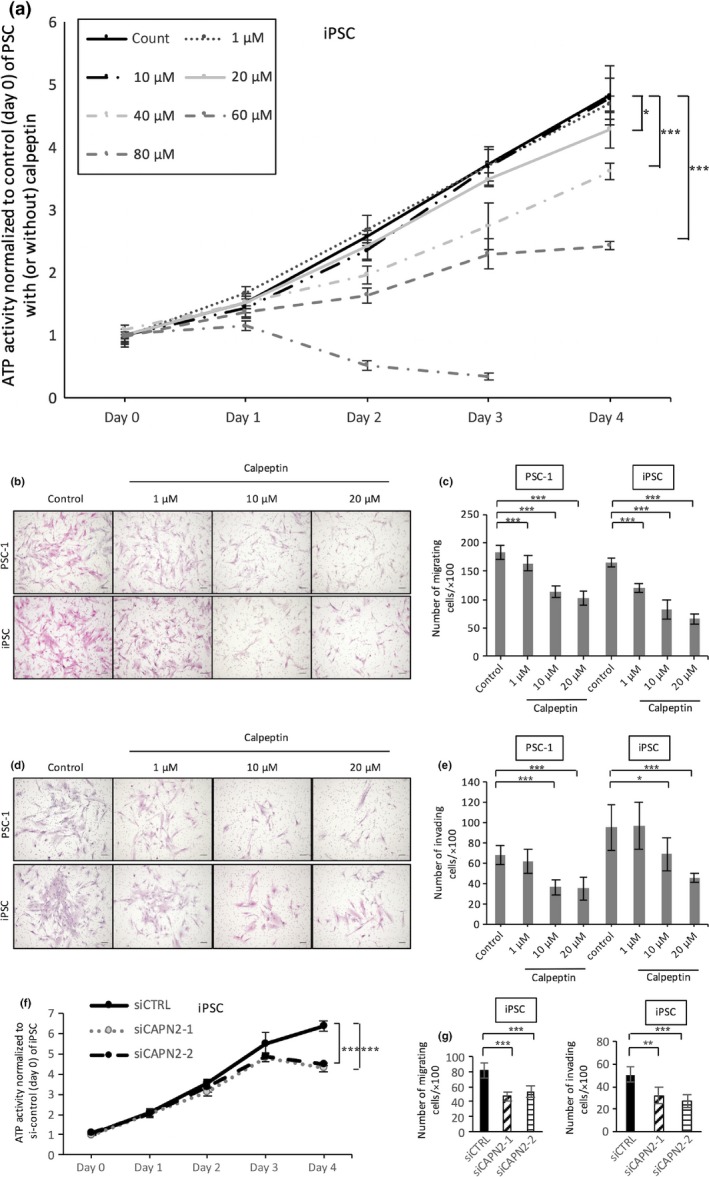
Effect of calpeptin on proliferation, migration, and invasion of pancreatic stellate cells (PSCs). (a) Cell growth of PSCs was assessed with an ATP‐based assay with or without calpeptin (0, 10, 20, 40, 60, or 80 μM). Representative images of migrating (b) and invading (d) PSCs are shown. Calpeptin dose‐dependently decreased the migration (c) and invasion (e) of PSCs. Silencing of calpain‐2 decreased proliferation (f) and migration and invasion (g) of immortalized PSC (iPSC) cells. Data are presented as the mean ± SD. *P < 0.05, **P < 0.01, ***P < 0.001. Cells were stained with HE; original magnification, ×100. Scale bar = 100 μm.
Calpeptin inhibited cancer–stromal interactions between PCCs and PSCs
It was reported that the migration of PCCs is enhanced by cancer–stromal interactions with PSCs.9 Because the results of the in vivo experiments suggested that calpeptin might inhibit the cancer–stromal interactions between PCCs and PSCs, we investigated the effects of calpeptin on this interaction using supernatant derived from PCC and PSC culture (PCC‐SN and PSC‐SN). It was shown that PSC‐SN increased the migration of PCCs (P < 0.05, Fig. 5a,b), and the increase in PCC migration by PSC‐SN was prevented by calpeptin‐treated PSC‐SN (P < 0.05, Fig. 5a,b). Similarly, PCC‐SN significantly increased the migration of PSCs, which was reversed with calpeptin‐treated PCC‐SN (P < 0.05, Fig. 5c,d). To identify the molecules involved in the calpeptin‐induced suppression of cancer–stromal interactions, we investigated the expression of the major known factors related with cancer–stromal interaction by qRT‐PCR. As shown in Figure 5, treatment with calpeptin inhibited the mRNA levels of PDGF‐A, PDGF‐B, and CTGF in SUIT‐2 cells (Fig. 5e) and PDGF‐B, epidermal growth factor (EGF), collagen 1A1, periostin, and α‐SMA in PSC‐1 cells (Fig. 5f).
Figure 5.
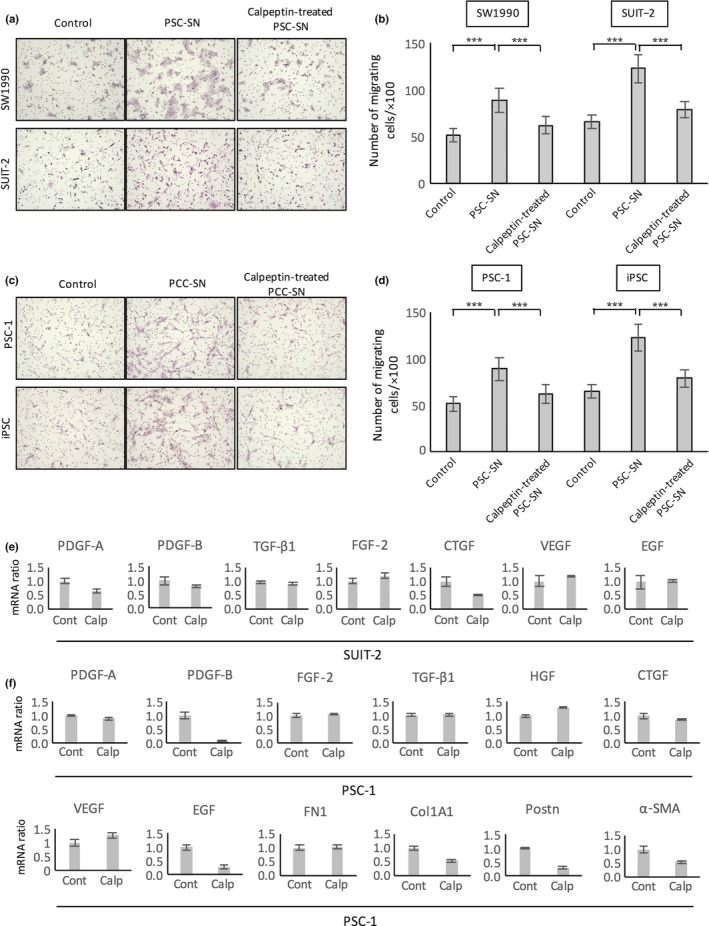
Effect of calpeptin on the cancer–stromal interaction between pancreatic cancer cells (PCCs) and pancreatic stellate cells (PSCs). Representative images of migrating PCCs (a) and PSCs (c). (b) PSC supernatant (PSC‐SN) significantly enhanced the migration of PCCs. Calpeptin‐treated PSC‐SN prevented the activation of PCCs stimulated by PSC‐SN. (d) PCC supernatant (PCC‐SN) significantly enhanced the migration of PSCs. Calpeptin‐treated PCC‐SN prevented the activation of PSCs stimulated by PCC‐SN. Effects of calpeptin on gene expression levels of growth factors and ECM proteins associated with cancer–stromal interactions. PCCs (e) and PSCs (f) were untreated (Cont) or treated with calpeptin (Calp). mRNA expression levels were normalized to GAPDH expression and are presented as the fold change in gene expression relative to control PCCs and PSCs. Data are presented as the mean ± SD. ***P < 0.001. Cells were stained with HE; original magnification, ×100. α‐SMA, α‐smooth muscle actin; COL1A1, collagen 1A1; CTGF, connective tissue growth factor; EGF, epidermal growth factor; FGF‐2, fibroblast growth factor‐2; FN1, fibronectin 1; HGF, hepatocyte growth factor; PDGF, platelet‐derived growth factor; POSTN, periostin; TGF‐β1, transforming growth factor‐β1; VEGF, vascular endothelial growth factor.
Discussion
In this study, we showed the anticancer effects of calpeptin on pancreatic cancer with an in vivo xenograft model. In tumors containing both PCCs and PSCs, calpeptin significantly reduced the tumor volume, tumor weight, and number of stromal cells in the tumors. We evaluated the amount of stroma by staining with Sirius red (a dye used for collagen staining), α‐SMA (a marker of activated PSCs) and periostin, which is predominantly expressed in collagen‐rich fibrous connective tissues in several organs, where it regulates collagen fibrillogenesis.35 Several studies reported that stromal cells such as PSCs promote pancreatic cancer aggressiveness26, 36 and that the inhibition of stromal cells leads to the suppression of tumor growth.34, 37 These studies support our results indicating that calpeptin inhibited not only xenograft tumor growth but also the desmoplastic reaction. However, although calpeptin reduced tumor volume in both groups, the inhibitory effect of calpeptin on tumors was more pronounced in the co‐implantation group, suggesting that calpeptin prevented tumor growth by an inhibitory effect on not only PCCs but also on the cancer–stromal interaction.
In addition to in vivo studies, we observed that calpeptin has inhibitory effects on the proliferation, migration, and invasion of PCCs and PSCs in vitro and that PCCs and PSCs expressed only calpain‐2, not calpain‐1. Therefore, these data suggest that calpeptin inhibited the proliferation of PCCs and PSCs by inhibiting the calpain‐2 catalytic subunit, which is regulated by the binding and dissociation of CAPNS1.38, 39, 40, 41 Dai et al.42 observed that downregulation of CAPNS1 in hepatocellular carcinoma cells suppressed the proliferation, migration, and invasion of the cells and showed that CAPNS1 contributes to the growth and metastasis of hepatocellular carcinoma by activating the FAK–Src signaling pathway. The expression of FAK is increased in a variety of human malignant tumors, including pancreatic cancer.18, 43 It has also been shown that MMP2 plays a crucial role in the growth and metastasis of HCC through activation of the FAK–Src signaling pathway by CAPNS1.42 Sundaramoorthy et al.15 reported that activated calpain cleaves FAK in colon cancer cells, leading to increased migration of the cells. Mataga et al.23 reported that calpeptin induced the apoptosis of breast cancer cells and increased the mRNA level of ARHI, a tumor suppressor gene. Furthermore, it was reported that suppression of calpains inhibited phosphorylation in many regulatory signaling pathways including AKT, a serine/threonine‐specific protein kinase that plays a key role in multiple cellular processes such as apoptosis, cell proliferation, transcription, and cell migration.11 In this study, we did not examine changes in ARHI expression or assess the inhibitory effect of calpeptin on the Akt or FAK–Src signaling pathways. Therefore, to investigate whether calpeptin affects these pathways in pancreatic cancer, additional examination is required.
In this study, calpeptin not only suppressed the activities of PCCs and PSCs but also inhibited the cancer–stromal interactions between PCCs and PSCs. It was reported that PCCs stimulate PSCs through the secretion of growth factors known to induce PSC activation such as transforming growth factor‐β1, PDGF, FGF, and vascular endothelial growth factor.9 In addition, PSCs stimulate PCCs through the secretion of mitogenic factors such as PDGF, FGF, EGF, and insulin‐like growth factor‐1 (IGF‐1).9 We investigated several candidates suppressing cancer–stromal interactions. Calpeptin inhibited mRNA expression of PDGF‐A, PDGF‐B, and CTGF in SUIT‐2 cells, and PDGF‐B, EGF, collagen 1A1, periostin, and α‐SMA in PSCs. Inhibition of these molecules in PCCs and PSCs may be involved in the inhibitory effect of calpeptin on xenograft tumor growth.
The present data suggest that calpeptin might be effective as a pancreatic cancer treatment by inhibiting the activity of PCCs and PSCs and disrupting cancer–stromal interactions. However, calpeptin is not currently available for clinical use. Although in‐depth investigation on the cellular mechanisms of calpeptin in pancreatic cancer is necessary before clinical application, a new therapeutic strategy using calpeptin might improve the prognosis of patients with pancreatic cancer.
Disclosure Statement
The authors have no conflict of interest.
Acknowledgments
This study was supported by the Japan Society for the Promotion of Science KAKENHI (Grant Nos. 25462117, 26293305, 26462066, 26462063, 26462062, 25462118, 25461024, 25293285, and 15H04933) and Scientific Research on Innovative Areas (Grant No. 26108010).
Cancer Sci 107 (2016) 1443–1452
Funding Information
Japan Society for the Promotion of Science (25462117, 26293305, 26462066, 26462063, 26462062, 25462118, 25461024, 25293285, 15H04933); Scientific Research on Innovative Areas (26108010)
Contributor Information
Yoshihiro Miyasaka, Email: yoshi-m@surg1.med.kyushu-u.ac.jp.
Kenoki Ohuchida, Email: kenoki@surg1.med.kyushu-u.ac.jp.
References
- 1. Siegel RL, Miller KD, Jemal A. Cancer statistics, 2015. CA Cancer J Clin 2015; 65: 5–29. [DOI] [PubMed] [Google Scholar]
- 2. Hidalgo M. Pancreatic cancer. N Engl J Med 2010; 362: 1605–17. [DOI] [PubMed] [Google Scholar]
- 3. Burris HA 3rd, Moore MJ, Andersen J et al Improvements in survival and clinical benefit with gemcitabine as first‐line therapy for patients with advanced pancreas cancer: a randomized trial. J Clin Oncol 1997; 15: 2403–13. [DOI] [PubMed] [Google Scholar]
- 4. Moorcraft SY, Khan K, Peckitt C et al FOLFIRINOX for locally advanced or metastatic pancreatic ductal adenocarcinoma: the Royal Marsden experience. Clin Colorectal Cancer 2014; 13: 232–8. [DOI] [PubMed] [Google Scholar]
- 5. Conroy T, Desseigne F, Ychou M et al FOLFIRINOX versus gemcitabine for metastatic pancreatic cancer. N Engl J Med 2011; 364: 1817–25. [DOI] [PubMed] [Google Scholar]
- 6. Von Hoff DD, Ervin T, Arena FP et al Increased survival in pancreatic cancer with nab‐paclitaxel plus gemcitabine. N Engl J Med 2013; 369: 1691–703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Minchinton AI, Tannock IF. Drug penetration in solid tumours. Nat Rev Cancer 2006; 6: 583–92. [DOI] [PubMed] [Google Scholar]
- 8. Netti PA, Berk DA, Swartz MA, Grodzinsky AJ, Jain RK. Role of extracellular matrix assembly in interstitial transport in solid tumors. Cancer Res 2000; 60: 2497–503. [PubMed] [Google Scholar]
- 9. Li X, Ma Q, Xu Q, Duan W, Lei J, Wu E. Targeting the cancer‐stroma interaction: a potential approach for pancreatic cancer treatment. Curr Pharm Des 2012; 18: 2404–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Amakye D, Jagani Z, Dorsch M. Unraveling the therapeutic potential of the Hedgehog pathway in cancer. Nat Med 2013; 19: 1410–22. [DOI] [PubMed] [Google Scholar]
- 11. Storr SJ, Carragher NO, Frame MC, Parr T, Martin SG. The calpain system and cancer. Nat Rev Cancer 2011; 11: 364–74. [DOI] [PubMed] [Google Scholar]
- 12. Potter DA, Tirnauer JS, Janssen R et al Calpain regulates actin remodeling during cell spreading. J Cell Biol 1998; 141: 647–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Carragher NO, Fincham VJ, Riley D, Frame MC. Cleavage of focal adhesion kinase by different proteases during SRC‐regulated transformation and apoptosis. Distinct roles for calpain and caspases. J Biol Chem 2001; 276: 4270–5. [DOI] [PubMed] [Google Scholar]
- 14. Chan KT, Bennin DA, Huttenlocher A. Regulation of adhesion dynamics by calpain‐mediated proteolysis of focal adhesion kinase (FAK). J Biol Chem 2010; 285: 11418–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Sundaramoorthy P, Sim JJ, Jang YS et al Modulation of intracellular calcium levels by calcium lactate affects colon cancer cell motility through calcium‐dependent calpain. PLoS ONE 2015; 10: e0116984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Pianetti S, Arsura M, Romieu‐Mourez R, Coffey RJ, Sonenshein GE. Her‐2/neu overexpression induces NF‐kappaB via a PI3‐kinase/Akt pathway involving calpain‐mediated degradation of IkappaB‐alpha that can be inhibited by the tumor suppressor PTEN. Oncogene 2001; 20: 1287–99. [DOI] [PubMed] [Google Scholar]
- 17. Conacci‐Sorrell M, Ngouenet C, Eisenman RN. Myc‐nick: a cytoplasmic cleavage product of Myc that promotes alpha‐tubulin acetylation and cell differentiation. Cell 2010; 142: 480–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Duxbury MS, Ito H, Zinner MJ, Ashley SW, Whang EE. Focal adhesion kinase gene silencing promotes anoikis and suppresses metastasis of human pancreatic adenocarcinoma cells. Surgery 2004; 135: 555–62. [DOI] [PubMed] [Google Scholar]
- 19. Storr SJ, Zaitoun AM, Arora A et al Calpain system protein expression in carcinomas of the pancreas, bile duct and ampulla. BMC Cancer 2012; 12: 511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Zhang Z, Rigas B. NF‐kappaB, inflammation and pancreatic carcinogenesis: NF‐kappaB as a chemoprevention target (review). Int J Oncol 2006; 29: 185–92. [PubMed] [Google Scholar]
- 21. Hessmann E, Schneider G, Ellenrieder V, Siveke JT. MYC in pancreatic cancer: novel mechanistic insights and their translation into therapeutic strategies. Oncogene 2016; 35: 1609–18. [DOI] [PubMed] [Google Scholar]
- 22. Tsujinaka T, Kajiwara Y, Kambayashi J et al Synthesis of a new cell penetrating calpain inhibitor (calpeptin). Biochem Biophys Res Commun 1988; 153: 1201–8. [DOI] [PubMed] [Google Scholar]
- 23. Mataga MA, Rosenthal S, Heerboth S et al Anti‐breast cancer effects of histone deacetylase inhibitors and calpain inhibitor. Anticancer Res 2012; 32: 2523–9. [PubMed] [Google Scholar]
- 24. Tabata C, Tabata R, Nakano T. The calpain inhibitor calpeptin prevents bleomycin‐induced pulmonary fibrosis in mice. Clin Exp Immunol 2010; 162: 560–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Nassar D, Letavernier E, Baud L, Aractingi S, Khosrotehrani K. Calpain activity is essential in skin wound healing and contributes to scar formation. PLoS ONE 2012; 7: e37084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Neesse A, Michl P, Frese KK et al Stromal biology and therapy in pancreatic cancer. Gut 2011; 60: 861–8. [DOI] [PubMed] [Google Scholar]
- 27. Bachem MG, Schneider E, Gross H et al Identification, culture, and characterization of pancreatic stellate cells in rats and humans. Gastroenterology 1998; 115: 421–32. [DOI] [PubMed] [Google Scholar]
- 28. Bachem MG, Schunemann M, Ramadani M et al Pancreatic carcinoma cells induce fibrosis by stimulating proliferation and matrix synthesis of stellate cells. Gastroenterology 2005; 128: 907–21. [DOI] [PubMed] [Google Scholar]
- 29. Hwang RF, Moore T, Arumugam T et al Cancer‐associated stromal fibroblasts promote pancreatic tumor progression. Cancer Res 2008; 68: 918–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Ikenaga N, Ohuchida K, Mizumoto K et al CD10+ pancreatic stellate cells enhance the progression of pancreatic cancer. Gastroenterology 2010; 139: 1041–51, 51 e1–8. [DOI] [PubMed] [Google Scholar]
- 31. Ohuchida K, Mizumoto K, Murakami M et al Radiation to stromal fibroblasts increases invasiveness of pancreatic cancer cells through tumor‐stromal interactions. Cancer Res 2004; 64: 3215–22. [DOI] [PubMed] [Google Scholar]
- 32. Mizuuchi Y, Aishima S, Ohuchida K et al Anterior gradient 2 downregulation in a subset of pancreatic ductal adenocarcinoma is a prognostic factor indicative of epithelial‐mesenchymal transition. Lab Invest 2015; 95: 193–206. [DOI] [PubMed] [Google Scholar]
- 33. Fujiwara K, Ohuchida K, Sada M et al CD166/ALCAM expression is characteristic of tumorigenicity and invasive and migratory activities of pancreatic cancer cells. PLoS ONE 2014; 9: e107247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Kozono S, Ohuchida K, Eguchi D et al Pirfenidone inhibits pancreatic cancer desmoplasia by regulating stellate cells. Cancer Res 2013; 73: 2345–56. [DOI] [PubMed] [Google Scholar]
- 35. Norris RA, Damon B, Mironov V et al Periostin regulates collagen fibrillogenesis and the biomechanical properties of connective tissues. J Cell Biochem 2007; 101: 695–711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Vonlaufen A, Joshi S, Qu C et al Pancreatic stellate cells: partners in crime with pancreatic cancer cells. Cancer Res 2008; 68: 2085–93. [DOI] [PubMed] [Google Scholar]
- 37. Olive KP, Jacobetz MA, Davidson CJ et al Inhibition of Hedgehog signaling enhances delivery of chemotherapy in a mouse model of pancreatic cancer. Science 2009; 324: 1457–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Sasaki T, Yoshimura N, Kikuchi T et al Similarity and dissimilarity in subunit structures of calpains I and II from various sources as demonstrated by immunological cross‐reactivity. J Biochem 1983; 94: 2055–61. [DOI] [PubMed] [Google Scholar]
- 39. Yumoto N, Kikuchi T, Sasaki T, Murachi T. Comparison of tryptic peptides from the heavy and light subunits of calpain I and calpain II by high performance liquid chromatography. J Biochem 1984; 96: 1531–7. [DOI] [PubMed] [Google Scholar]
- 40. Inomata M, Nomoto M, Hayashi M, Nakamura M, Imahori K, Kawashima S. Comparison of low and high calcium requiring forms of the calcium‐activated neutral protease (CANP) from rabbit skeletal muscle. J Biochem 1984; 95: 1661–70. [DOI] [PubMed] [Google Scholar]
- 41. Kawasaki H, Imajoh S, Kawashima S, Hayashi H, Suzuki K. The small subunits of calcium dependent proteases with different calcium sensitivities are identical. J Biochem 1986; 99: 1525–32. [DOI] [PubMed] [Google Scholar]
- 42. Dai Z, Zhou SL, Zhou ZJ et al Capn4 contributes to tumour growth and metastasis of hepatocellular carcinoma by activation of the FAK‐Src signalling pathways. J Pathol 2014; 234: 316–28. [DOI] [PubMed] [Google Scholar]
- 43. Owens LV, Xu L, Craven RJ et al Overexpression of the focal adhesion kinase (p125FAK) in invasive human tumors. Cancer Res 1995; 55: 2752–5. [PubMed] [Google Scholar]