

Abstract
Human epidermal growth factor receptor 3 (HER3) expression in lung and breast cancers has a negative impact on survival. Patritumab, a human anti‐HER3 mAb, has shown anticancer activity in preclinical models. This study examined the safety and pharmacokinetics of patritumab in combination with trastuzumab and paclitaxel in patients with HER2‐overexpressing metastatic breast cancer. In this open‐label, multicenter, dose‐escalation, phase Ib study, patients received patritumab 9 or 18 mg/kg plus trastuzumab and paclitaxel at known tolerated doses. Safety and tolerability were assessed based on dose‐limiting toxicities and other non‐life threatening adverse events. The pharmacokinetic profile for patritumab was determined based on the target trough level. Clinical efficacy was evaluated based on the overall response rate and progression‐free survival. Six patients received patritumab 9 mg/kg and 12 received 18 mg/kg. The most common adverse events were diarrhea, alopecia, leukopenia, neutropenia, and maculopapular rash. No dose‐limiting toxicities were observed. The target trough serum concentration was achieved in all patients at a dose of 18 mg/kg. Overall response rate was 38.9% and median progression‐free survival was 274 days. In conclusion, patritumab plus trastuzumab and paclitaxel was tolerable and efficacious at both doses. We recommend the dose level of 18 mg/kg for future phase II studies. (Clinical trial registration: JapicCTI‐121772.)
Keywords: Breast cancer, HER3 protein, human, metastasis, patritumab, trastuzumab
Overall breast cancer mortality rates are declining in developed countries in Europe and North America. However, while there have been advances in supportive care and the development of active chemotherapy regimens and hormone therapies, there has been limited success in improving the survival of patients with metastatic disease, underscoring the need for additional treatment options.
The HER family tyrosine kinases are involved in the pathogenesis of a variety of human cancers. The family comprises HER1, HER2, HER3, and HER4, which are highly homologous.1
Human epidermal growth factor receptor 3, first cloned and sequenced approximately 25 years ago,2, 3 lacks tyrosine kinase activity and is not biologically active, but both HER1 and HER2 can reconstitute its extremely potent mitogenic activity through the formation of heterodimers. Heterodimers are more active than homodimers, with HER2/HER3 heterodimers being particularly active.1, 4 Advances in understanding the important role of HER3 have uncovered its potential as a therapeutic target.1 Moreover, high HER3 expression has a negative impact on survival in patients with lung and breast cancers.5, 6, 7
Patritumab (U3‐1287) is a fully human anti‐HER3 mAb that has reportedly shown anticancer activity in preclinical models.8, 9, 10 Phase I studies in Japan and the USA evaluated patritumab in patients with advanced solid tumors and showed that doses up to 20 mg/kg were well tolerated without DLTs.11, 12
Here, we describe the results of a phase I study of patritumab in combination with trastuzumab and paclitaxel in Japanese patients with HER2‐overexpressing MBC. The primary objectives of this study were to assess the safety and PK of patritumab and to determine the recommended dose for subsequent studies. Secondary objectives included evaluating the incidence of anti‐patritumab antibodies, a preliminary assessment of antitumor activity, and evaluating the potential of patritumab‐related biomarkers.
Materials and Methods
Assessments
Study end‐points were safety (AEs), PK (serum concentrations of patritumab and PK parameters), and efficacy (antitumor activity). Other end‐points included the presence of HAHAs and biomarker analysis.
Study design and intervention
This open‐label, multicenter, dose‐escalation, phase Ib study was carried out in Japan, and started on March 1, 2012.
Dose‐limiting toxicities for combined treatment with patritumab, trastuzumab, and paclitaxel were evaluated at two dose levels, where dose escalation followed a modified 3 + 3 design. The study comprised two regimens. The C trough of patritumab was 15 μg/mL, which was expected to sufficiently inhibit HER3 activation in humans based on the results of non‐clinical PK/PD analyses. Only the initial C trough before treatment in cycle 2 was lower than 15 μg/mL at the dose of 9 mg/kg every 3 weeks, and all C trough values in every cycle were twice as high as the target C trough at the dose of 18 mg/kg every 3 weeks by PK/PD simulation of the US phase I study.11 In another phase I study,12 the tolerability and safety of patritumab at doses of 9 and 18 mg/kg were evaluated in Japanese patients with advanced solid tumors. No DLTs were observed at the dose levels studied and the MTD was not reached. Therefore, we opted to use the same two dose levels (9 and 18 mg/kg) in this study. In regimen A, patients received patritumab 9 mg/kg (dose level 1) or 18 mg/kg (dose level 2), and trastuzumab 8 mg/kg for the initial dose, then 6 mg/kg for the second and subsequent doses, and paclitaxel 175 mg/m2. Drugs were given by i.v. infusion every 3 weeks. In regimen B, patients received the same dosing regimen of patritumab and trastuzumab, but paclitaxel was given at 80 mg/m2 once weekly for 2 consecutive weeks followed by a 1‐week rest period. In both regimens, one cycle was completed after 3 weeks and treatment was continued until patients met any of the discontinuation criteria. Treatment was started at level 1 with regimen A, with the first cycle considered the DLT evaluation period. After completing the DLT period at dose level 2 in regimen A, evaluation at dose level 1 of regimen B was started.
Dose‐limiting toxicities were evaluated at each dose level for the first three patients. If no DLTs occurred at level 1, the dose was increased to level 2. If there were zero or one DLTs at level 2, an additional three patients were recruited and safety was evaluated in the six patients. The MTD was defined as the highest dose level with an observed incidence of DLT in <33% of patients. Dose escalation was not carried out in the same patient. At least three patients were aimed to be included for each dose level, requiring 9–18 patients for regimen A and 9–18 patients for regimen B.
Patients
Patients were those with histologically or cytologically confirmed invasive carcinoma of the breast with metastatic disease. Confirmation of HER2‐positive status was by FISH or IHC. This study enrolled patients whose disease had progressed after at least one prior course of chemotherapy. Patients were eligible if they had progressed after receiving trastuzumab and/or paclitaxel. If chemotherapy was only provided as postoperative adjuvant therapy, patients could be documented as relapsed within 1 year after the completion of the adjuvant therapy including trastuzumab. Other inclusion criteria included: women aged ≥20 years and ≤75 years; ECOG PS 0 to 1; adequate hematologic, hepatic, and renal function; life expectancy of ≥3 months; no brain metastasis with clinical symptoms or requiring treatment; and recovered (≤grade 1) from adverse drug reaction, excluding alopecia, or prior treatment for MBC.
Exclusion criteria included: prior anti‐HER3 therapy; a history of interstitial lung disease; total prior anthracycline dose >360 mg/m2; active synchronous cancer; decreased LVEF <55% at screening or a decrease of LVEF by 10% versus the baseline value during treatment with trastuzumab to reach <55%; pregnancy/nursing; and cardiovascular or cerebrovascular comorbidities.
Background information was obtained from patients, including age, hormone receptor status, HER2 status, medical history, complications and treatment for breast cancer, height, weight, vital signs, echocardiography, and tumor findings.
Ethical considerations
All participants provided written informed consent. The study was carried out in compliance with the ethical principles of the Declaration of Helsinki. The study was approved by the Institutional Review Boards at each participating site.
Safety assessments
Adverse event grading was in accordance with the Common Terminology Criteria for Adverse Events version 4.0 (National Cancer Institute, National Institutes of Health, U.S. Department of Health and Human Services) throughout the treatment period until 30 days after the last dose was given. Safety evaluations were based on a medical review of AEs and the results of clinical laboratory tests, vital sign measurements, 12‐lead electrocardiograms, physical examination, ECOG PS, LVEF, and X‐ray/CT scans. The presence of anti‐patritumab antibodies was assessed every two treatment cycles and measured by an ECL immunoassay. Dose‐limiting toxicities were defined as follows: febrile neutropenia (>38.3°C, neutrophil count < 1000/μL) or grade 4 neutropenia persisting for >7 days; grade 4 thrombocytopenia or grade 3 thrombocytopenia requiring blood transfusion; uncontrollable grade 3 or worse fatigue, anorexia, nausea, vomiting, skin disorders (eruptions, urticaria) or diarrhea despite maximal supportive treatment; reduction of LVEF by >15% compared with baseline value or a decrease to <45%; and grade 3 or worse toxicities except for the above four definitions. Fever without neutropenia, grade 3 anemia, lymphopenia, transient electrolyte abnormality, and transient laboratory abnormality not requiring treatment and without clinical symptoms did not qualify as DLTs. The MTD was defined as the highest dose level with an observed incidence of DLT in <33% of patients in the first cycle.
Pharmacokinetic analysis of patritumab
On day 1 of the first cycle, 4‐mL blood samples were collected prior to and 15 min after the end of the initial infusion and 4 and 7 h after initiation of the infusion. Subsequent samples were collected on day 2 (24 h after the start of the infusion), day 4 (72 h after the start of the infusion), day 8, day 15, and day 22 (the end of cycle 1). In cycles 2 and 3, samples were taken prior to and 15 min after the infusion, and at day 22. Serum concentrations of U3‐1287 were determined by ELISA. Pharmacokinetic parameters were calculated by non‐compartmental analysis using WinNonlin software (version 6.2; Certara GK, Tokyo, Japan).
Efficacy assessments
Tumor response was assessed in accordance with Response Evaluation Criteria in Solid Tumors version 1.1.13 Tumor lesions and lymph nodes were measured using MRI, CT, or other imaging techniques, and were categorized as measurable or non‐measurable. Measurable lesions were those measuring >10 mm in diameter by CT or caliper measurement (>20 mm for chest X‐ray). Lymph nodes ≥15 mm by CT were regarded as pathologically enlarged and measurable. Target lesions were observed during follow‐up using the same method as that used for baseline assessment. The diameters of the target lesions (longest for tumor lesions, short axis for nodal lesions), any disappearance or progression of non‐target lesions, and any appearance of new lesions were recorded. The antitumor effect was evaluated in terms of response rate (CR + PR), disease control rate (CR + PR + stable disease), and PFS.
Other assessments
To evaluate HAHAs, blood samples were collected on days 1 and 21 of cycle 1, and day 21 of every subsequent two cycles. The HAHAs were measured by ECL immunoassay. Levels of soluble HER3 and HRG in serum were measured on days 1, 8, 15, and 21 of cycle 1, and day 21 of every two subsequent cycles. Soluble HER3 and soluble HRG levels were measured by ELISA using the following kits: soluble HER3, human ErbB3/Her3 DuoSet [DY348] (R&D Systems, Minneapolis, MN, USA); and soluble HRG, human NRG1‐β1/HRG1‐β1 DuoSet [DY377] (R&D Systems). Tissue biomarkers were assessed in participants who had given informed consent for biomarker research using formalin‐fixed, paraffin‐embedded tumor samples collected prior to enrolment. Expression of HER2, HER3, phosphorylated HER3, HRG, phosphatase and tensin homolog, and EGFR was detected by IHC. Immunohistochemistry was carried out in accordance with Mosaic Laboratories' (Lake Forest, CA, USA) standard operating procedures and validated protocols. The following antibodies were used: HercepTest, rabbit polyclonal HER2 antibody (Dako, Carpinteria, CA, USA); HER3, mouse clone 5A12 antibody (Santa Cruz Biotechnology, Santa Cruz, CA, USA); pHER3, rabbit clone 21D3 antibody (Cell Signaling Technology, Danvers, MA, USA); HRG, rabbit polyclonal antibody (Santa Cruz Biotechnology); phosphatase and tensin homolog, rabbit clone 138G6 antibody (Cell Signaling Technology); and EGFR, mouse clone 31G7 antibody (Invitrogen, Camarillo, CA, USA)]. HER2 and HER3 gene amplifications were detected by fluorescence in situ hybridization, and gene mutations of PIK3CA and EGFR were identified by next‐generation sequencing. Fluorescence in situ hybridization was carried out in accordance with Mosaic Laboratories' standard operating procedures and validated protocols, using the following kits and probes: HER2, PathVysion HER2 DNA Probe Kit (Abbott Molecular, Des Plaines, IL, USA); and HER3, ZytoLight SPEC HER3/CEN12 dual color FISH probe (ZytoVision, Bremerhaven, Germany). HER2/CEP17 ratios ≥2.2 were defined as amplified, according to cut‐off points established in a study by the Cancer and Leukemia Group B (CALGB 8869), subsequent comparative studies, and the American Society of Clinical Oncology/College of American Pathologists HER‐2 Guideline Recommendations. HER3/CEN12 ratios ≥2.0 were defined as amplified. Next Generation Sequencing assays were developed and validated at LabCorp Clinical Trials (Seattle, WA, USA) using Illumina's (San Diego, CA, USA) MiSeq Next Generation Sequencing platform.
Statistical analyses
The ORR and disease control rate were calculated with 95% confidence intervals. Additionally, waterfall and Kaplan–Meier plots were provided for the best overall response and PFS, respectively. All statistical analyses were carried out using SAS system version 9.2 (SAS Institute Inc., Cary, NC, USA). In this study, no significance level was established because no hypothesis test was undertaken.
Results
Patient disposition and characteristics
Patient characteristics are shown in Table 1. All patients had received prior trastuzumab and 12 patients (66.7%) had received prior paclitaxel. A total of 18 patients (mean age, 59 years) were enrolled in both dose levels (9 and 18 mg/kg) of regimens A and B. As of April 1, 2015, the data cut‐off, four patients were continuing treatment. Reasons for withdrawal included progressive disease (n = 8), AEs (n = 4), and withdrawal by the patient (n = 2). A CR was achieved in one patient and a CR in the target lesion was achieved in another patient; these patients chose to discontinue treatment as they thought the therapy was sufficiently effective.
Table 1.
Demographics and baseline characteristics of patients with metastatic breast cancer
| Characteristic | Dosage group | Total (n = 18) | |
|---|---|---|---|
| 9 mg/kg (n = 6) | 18 mg/kg (n = 12) | ||
| Age, years, median (range) | 56.0 (34–69) | 59.0 (39–64) | 59.0 (34–69) |
| ECOG PS | |||
| 0 | 5 (83.3) | 8 (66.7) | 13 (72.2) |
| 1 | 1 (16.7) | 4 (33.3) | 5 (27.8) |
| ER‐positive | 2 (33.3) | 3 (25.0) | 5 (27.8) |
| PgR‐positive | 1 (16.7) | 2 (16.7) | 3 (16.7) |
| HER2 IHC/FISH‐positive | 6 (100.0) | 12 (100.0) | 18 (100.0) |
| Number of prior regimens,† median (range) | 2 (1–9) | 3 (0§–6) | 2 (0§–9) |
| Prior trastuzumab | 6 (100.0) | 12 (100.0) | 18 (100.0) |
| Prior paclitaxel | 5 (83.3) | 7 (58.3) | 12 (66.7) |
All values are n (%) unless stated otherwise. †Included hormone therapy. §One patient relapsed during adjuvant therapy including trastuzumab. In this protocol, if chemotherapy was only provided as postoperative adjuvant therapy, patients could be documented as relapsed within 1 year after the completion of the adjuvant therapy including trastuzumab. ECOG, Eastern Cooperative Oncology Group; ER, estrogen receptor; HER2, human epidermal growth factor receptor 2; IHC, immunohistochemistry; PgR, progesterone receptor; PS, performance status.
Safety assessments
The most common AEs (≥50% of all patients) were diarrhea, alopecia, leukopenia, neutropenia, and maculopapular rash (Table 2). One patient experienced serious AEs (cataract and sinusitis), although these were not assessed as being related to the study drugs. Common Terminology Criteria for Adverse Events grade ≥3 AEs that occurred during the study were neutropenia (n = 5), leukopenia (n = 3), cataract (n = 1), neuropathy (n = 1), and granulocytopenia (n = 1). No DLT was observed. The AEs that led to treatment discontinuation were: sinusitis in one patient; peripheral neuropathy, arthralgia, and malaise in one patient; peripheral neuropathy and diarrhea in one patient; and decreased LVEF in one patient. The reduction in LVEF, from 55% at baseline to 42% after two cycles of treatment, was marginal in terms of the discontinuation criterion of LVEF reduction to <45%. This LVEF decrease was not considered a DLT because the study protocol defined the first cycle as the DLT evaluation period. No safety risks were identified from laboratory tests, vital sign measurements, 12‐lead electrocardiograms, physical examination, ECOG PS, LVEF, or X‐ray/CT scans.
Table 2.
Adverse events observed in five or more patients (27.8%) during a phase 1b study of patritumab plus trastuzumab and paclitaxel for human epidermal growth factor receptor 2‐overexpressing metastatic breast cancer
| Preferred term | Dosage group, all grades | Dosage group, grade ≥3 | Overall all grades (n = 18) | Overall grade ≥3 (n = 18) | ||
|---|---|---|---|---|---|---|
| 9 mg/kg (n = 6) | 18 mg/kg (n = 12) | 9 mg/kg (n = 6) | 18 mg/kg (n = 12) | |||
| Diarrhea | 4 (66.7) | 12 (100.0) | 0 (0.0) | 0 (0.0) | 16 (88.9) | 0 (0.0) |
| Alopecia | 4 (66.7) | 9 (75.0) | 0 (0.0) | 0 (0.0) | 13 (72.2) | 0 (0.0) |
| Leukopenia | 5 (83.3) | 9 (75.0) | 1 (16.7) | 2 (16.7) | 14 (77.8) | 3 (16.7) |
| Neutropenia | 4 (66.7) | 8 (66.7) | 2 (33.3) | 3 (25.0) | 12 (66.7) | 5 (27.3) |
| Maculopapular rash | 5 (83.3) | 4 (33.3) | 0 (0.0) | 0 (0.0) | 9 (50.0) | 0 (0.0) |
| Peripheral sensory neuropathy | 2 (33.3) | 6 (50.0) | 0 (0.0) | 0 (0.0) | 8 (44.4) | 0 (0.0) |
| Nausea | 3 (50.0) | 5 (41.7) | 0 (0.0) | 0 (0.0) | 8 (44.4) | 0 (0.0) |
| Upper respiratory infection | 2 (33.3) | 4 (33.3) | 0 (0.0) | 0 (0.0) | 6 (33.3) | 0 (0.0) |
| Neuropathy peripheral | 2 (33.3) | 4 (33.3) | 0 (0.0) | 1 (8.3) | 6 (33.3) | 1 (5.6) |
| Myalgia | 2 (33.3) | 4 (33.3) | 0 (0.0) | 0 (0.0) | 6 (33.3) | 0 (0.0) |
| Malaise | 1 (16.7) | 5 (41.7) | 0 (0.0) | 0 (0.0) | 6 (33.3) | 0 (0.0) |
| Decreased appetite | 1 (16.7) | 4 (33.3) | 0 (0.0) | 0 (0.0) | 5 (27.8) | 0 (0.0) |
| Arthralgia | 1 (16.7) | 4 (33.3) | 0 (0.0) | 0 (0.0) | 5 (27.8) | 0 (0.0) |
All values are n (%). Coded using MedDRA version 17.0 (MedDRA MSSO, McLean, VA, USA).
Pharmacokinetic assessments
The PK parameters of patritumab are summarized in Table 3. For the 9‐ and 18‐mg/kg dose groups, the mean areas under the plasma concentration–time curve from 0 to 21 days were 1110 and 2600 μg/day/mL, the maximum plasma drug concentrations were 199 and 446 μg/mL, C trough values were 14.9 and 39.0 μg/mL, and the terminal half‐lives were 7.95 and 7.82 days, respectively. The target C trough (15 μg/mL), which was expected to sufficiently inhibit HER3 activation in humans based on the results of non‐clinical PK/PD analyses, was achieved in all patients at a dose of 18 mg/kg. The PK parameters in this study were similar to those of phase I studies of patritumab monotherapy in Japan and the USA (Fig. 1).11, 12
Table 3.
Serum patritumab pharmacokinetic parameters in patients with human epidermal growth factor receptor 2‐overexpressing metastatic breast cancer
| Parameter | Dosage group | |
|---|---|---|
| 9 mg/kg (n = 6) | 18 mg/kg (n = 12) | |
| AUC0–21 days, μg·day/mL | 1110 ± 153 | 2600 ± 661 |
| C max, μg/mL | 199 ± 24.9 | 446 ± 92.7 |
| C trough, μg/mL | 14.9 ± 2.73 | 39.0 ± 16.1 |
| CL, mL/day/kg | 7.11 ± 1.04 | 6.31 ± 1.70 |
| V ss, mL/kg | 67.7 ± 11.2 | 61.1 ± 12.5 |
| t 1/2, days | 7.95 ± 1.24 | 7.82 ± 1.70 |
All values are mean ± SD. AUC0–21 days, area under the plasma concentration–time curve from 0 to 21 days; CL, apparent total body clearance of the drug from plasma; C max, maximum plasma drug concentration; C trough, trough concentration; t 1/2, elimination half‐life; V ss, apparent volume of distribution at steady state.
Figure 1.
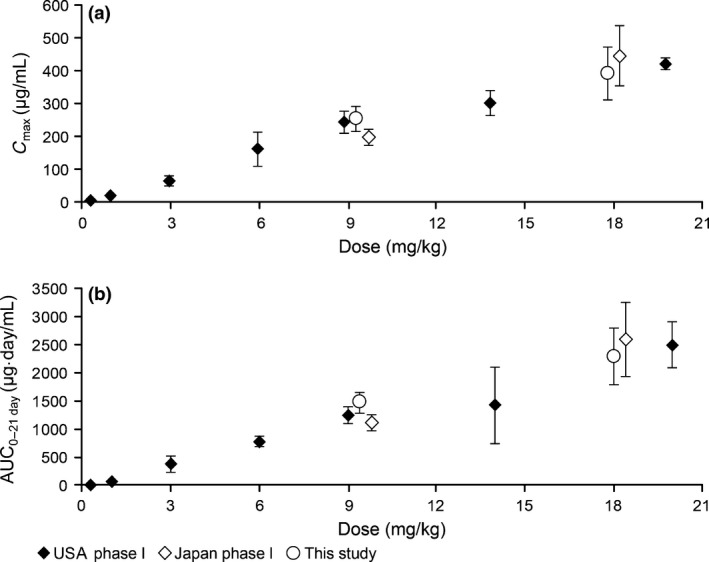
Comparison of maximum plasma drug concentration (C max) (a) and area under the plasma concentration–time curve (AUC) (b) in phase I studies of patritumab monotherapy in Japan and the USA and the present study.
Efficacy assessments
Two CRs (one for each patritumab dose level) and five PRs (two and three patients receiving 9 and 18 mg/kg doses, respectively) were observed and the ORR was 38.9% (95% confidence interval, 20.3–61.4%). Among the 13 patients with target lesions, tumor shrinkage was observed in 10 patients including four with disappearance of the target lesion (Fig. 2). The median PFS was 274 days and four patients are still progression‐free (Fig. 3). The two longest durations of PFS obtained from ongoing patients at dose level 1 in regimen A were almost 3 years.
Figure 2.
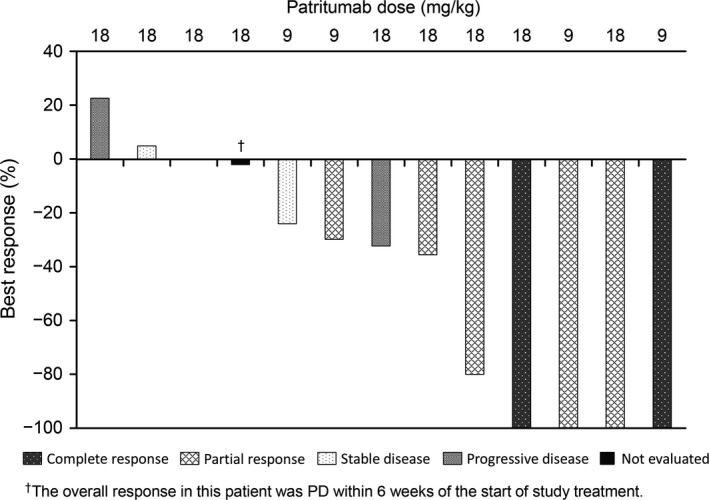
Waterfall plot showing best response to patritumab 9 or 18 mg/kg plus trastuzumab and paclitaxel in patients with human epidermal growth factor receptor 2‐overexpressing metastatic breast cancer. PD, progressive disease.
Figure 3.
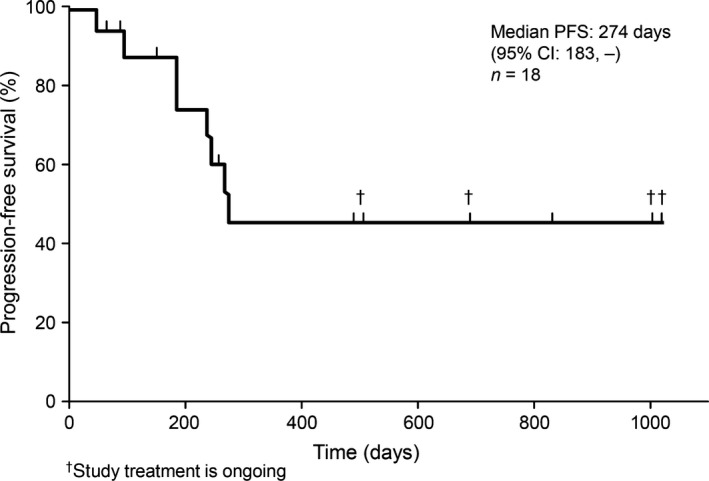
Kaplan–Meier curve of progression‐free survival (PFS) in patients with human epidermal growth factor receptor 2‐overexpressing metastatic breast cancer treated with patritumab 9 or 18 mg/kg plus trastuzumab and paclitaxel. The upper limit of the 95% confidence interval for the PFS could not be determined. CI, confidence interval.
Other assessments
All patients were negative for HAHAs during ≥4 cycles. Serum biomarker assessment was carried out for all 18 patients and tissue biomarker assessment was carried out for 10. The findings of the biomarker assessments showed no significant relationships between the serum and tissue biomarkers assessed and the efficacy and safety outcomes.
Discussion
This phase Ib study showed that patritumab in combination with trastuzumab and paclitaxel was well tolerated up to 18 mg/kg in HER2‐overexpressing MBC patients. Trastuzumab and paclitaxel were given at approved tolerated doses for both treatment regimens; thus, pharmacokinetics, safety, and efficacy were not evaluated for the two treatment regimens because the objective of this study was to determine the safety and efficacy of patritumab. Two patients had a best response of CR and five patients had a best response of PR. In later‐line‐treated (≥3rd‐line) patients (n = 11), CR was observed in one patient and PR was observed in three patients. Although there were some differences in ORR between earlier‐line‐ and later‐line‐treated patients, no definite conclusion can be drawn based on the results of this study, in which an assessment of efficacy was not the primary objective. No DLTs were reported at any dose level, and the tested doses did not reach the MTD. The efficacy and safety profile did not correlate with the patritumab dose level. The PK parameters in this study were similar to those of phase I studies of patritumab monotherapy in patients with advanced solid tumors carried out in Japan and the USA.11, 12 In the Japanese study, patients received 9 or 18 mg/kg patritumab, and this was well tolerated up to 18 mg/kg without DLTs.12 In the US dose‐escalation study, the maximum dose given was 20 mg/kg. Similar to the present study, no DLTs were reported and the data supported a dosage of 9–20 mg/kg every 2–3 weeks.11
All patients in this study received prior trastuzumab, and 12 patients had received prior paclitaxel (median number of prior treatment regimens, 2 [range, 0–9]; recurrences within 1 year after trastuzumab‐containing adjuvant therapy were observed in treatment‐naïve patients). The CLEOPATRA Study Group examined the combined use of pertuzumab and trastuzumab plus docetaxel compared with trastuzumab plus docetaxel.14, 15, 16 Pertuzumab, an anti‐HER2 humanized mAb that inhibits receptor dimerization, was associated with significantly prolonged PFS and overall survival in these studies. Verma et al.17 examined the use of T‐DM1 compared with the HER2 inhibitor lapatinib plus capecitabine in patients who had previously received trastuzumab plus a taxane. T‐DM1 significantly prolonged PFS and overall survival and showed less toxicity compared with lapatinib plus capecitabine. Consequently, pertuzumab and T‐DM1 have become standard first‐ and second‐line treatments for HER2‐positive MBC on the basis of these clinical trials.14, 15, 16, 17 It is a limitation that neither T‐DM1 nor pertuzumab were used as pretreatments in the patients included in this study.
No patient developed HAHAs during the present study. The findings of the biomarker assessments showed no significant relationships between the serum and tissue HRG levels and the efficacy and safety outcomes. However, a phase II study previously reported a correlation between high HRG mRNA expression in tumor tissue and PFS in patients with non‐small‐cell lung cancer when treated with patritumab and erlotinib compared with erlotinib alone, suggesting the potential of HRG as a biomarker.18 These results warrant the investigation of HRG mRNA as a biomarker in subsequent studies of HER2‐overexpressing MBC.
Regarding the exploratory variables examined, the lack of any significant relationships between serum and tissue biomarkers and efficacy and safety outcomes will need to be confirmed in a larger number of patients. In particular, tissue biomarker assessment was only performed in 10 patients in this study and archived tissue samples were used; thus, further larger scale studies are required to explore these preliminary findings.
In the present study, patritumab in combination with trastuzumab and paclitaxel was shown to be tolerable, and the favorable efficacy was encouraging in patients with HER2‐overexpressing MBC. We recommend 18 mg/kg patritumab as the dose level for future phase II studies based on safety, efficacy, and PK parameters.
Disclosure Statement
H.M. has received honoraria from Eisai, Daiichi Sankyo Co., Ltd., Taiho Pharmaceutical, Ono Pharmaceutical, Boehringer Ingelheim, Chugai Japan, Novartis Healthcare A/S, and Asia & Emerging Markets Innovative Medicine of AstraZeneca R&D, and research funding from Daiichi Sankyo Co., Ltd., Eisai, Nippon Kayaku, Pfizer, Sanofi, Chugai Japan, and Novartis Healthcare A/S. T.S. received honoraria from Daiichi Sankyo Co., Ltd. K.A. has received honoraria from Eisai, Otsuka, Ono Pharmaceutical, Sanofi, Daiichi Sankyo Co., Ltd., Taiho Pharmaceutical, Becton Dickinson, Nihon Medi‐Physics, Chugai Japan, and Asia & Emerging Markets Innovative Medicine of AstraZeneca R&D. Y.N. has received honoraria from Daiichi Sankyo Co., Ltd. N.M. has received honoraria and/or research grants from Janssen Pharmaceutical K.K., Sanofi K.K., Taiho Pharmaceutical Co., Ltd., Bayer Yakuhin, Ltd, and AstraZeneca K.K. Fumikata Hara has received research funding from Eisai and Chugai Japan. T.Y. and S.O. are employees of Daiichi Sankyo Co., Ltd. Y.S. has received honoraria from Eisai, Taiho Pharmaceutical, Merck Serono, and Chugai Japan, and research funding from Taiho Pharmaceutical, Eli Lilly and Company, Bristol‐Myers Squibb, Boehringer Ingelheim, and Chugai Japan. T.S., S.U., and S.T. have no conflict of interest.
Abbreviations
- AE
adverse event
- CR
complete response
- CT
computed tomography
- Ctrough
trough concentration
- DLT
dose‐limiting toxicities
- ECOG
Eastern Cooperative Oncology Group
- EGFR
epidermal growth factor receptor
- HAHA
human anti‐human antibody
- HER
human epidermal growth factor receptor
- HRG
heregulin
- IHC
immunohistochemistry
- LVEF
left ventricular ejection fraction
- MBC
metastatic breast cancer
- MTD
maximum tolerated dose
- ORR
objective response rate
- PD
pharmacodynamic
- PFS
progression‐free survival
- PK
pharmacokinetic
- PR
partial response
- PS
performance status
- T‐DM1
trastuzumab emtansine
Acknowledgments
Funding for this study and manuscript development was provided by Daiichi Sankyo Co., Ltd. The authors would like to thank Helen Roberton and Dr Sarah Williams for providing medical writing support.
Cancer Sci 107 (2016) 1465–1470
Funding Information
Daiichi Sankyo Co., Ltd.
References
- 1. Campbell MR, Amin D, Moasser MM. HER3 comes of age: new insights into its functions and role in signaling, tumor biology, and cancer therapy. Clin Cancer Res 2010; 16: 1373–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Gullick WJ. The c‐erbB3/HER3 receptor in human cancer. Cancer Surv 1996; 27: 339–49. [PubMed] [Google Scholar]
- 3. Yarden Y, Sliwkowski MX. Untangling the ErbB signalling network. Nat Rev Mol Cell Biol 2001; 2: 127–37. [DOI] [PubMed] [Google Scholar]
- 4. Pinkas‐Kramarski R, Soussan L, Waterman H et al Diversification of Neu differentiation factor and epidermal growth factor signaling by combinatorial receptor interactions. EMBO J 1996; 15: 2452–67. [PMC free article] [PubMed] [Google Scholar]
- 5. Bièche I, Onody P, Tozlu S, Driouch K, Vidaud M, Lidereau R. Prognostic value of ERBB family mRNA expression in breast carcinomas. Int J Cancer 2003; 106: 758–65. [DOI] [PubMed] [Google Scholar]
- 6. Müller‐Tidow C, Diederichs S, Bulk E et al Identification of metastasis‐associated receptor tyrosine kinases in non‐small cell lung cancer. Cancer Res 2005; 65: 1778–82. [DOI] [PubMed] [Google Scholar]
- 7. Yi ES, Harclerode D, Gondo M et al High c‐erbB‐3 protein expression is associated with shorter survival in advanced non‐small cell lung carcinomas. Mod Pathol 1997; 10: 142–8. [PubMed] [Google Scholar]
- 8. Freeman D, Ogbagabriel S, Schneider M, Radinsky R, Hettman T. U3‐1287 (AMG 888), a fully human anti‐HER3 mAb, demonstrates in vitro and in vivo efficacy in NSCLC models. Mol Cancer Ther 2009; 8 (12 Suppl): B171 (abstract). [Google Scholar]
- 9. Hettman T, Schneider M, Blum S et al U3‐1287 (AMG 888), a fully human anti‐HER3 mAb, demonstrates preclinical efficacy in HER2 + and HER2− breast cancer models. Mol Cancer Ther 2009; 8 (12 Suppl): B161 (abstract). [Google Scholar]
- 10. Lum P, Perez Ruixo JJ, Ogbagabriel S et al Identifying first in human (FIH) doses and schedule of U3‐1287 (AMG 888), a fully human anti‐HER3 mAb, based on preclinical pharmacokinetic (PK), pharmacodynamic (PD) and efficacy data. Mol Cancer Ther 2009; 8 (12 Suppl): B167 (abstract). [Google Scholar]
- 11. LoRusso P, Jänne PA, Oliveira M et al Phase I study of U3‐1287, a fully human anti‐HER3 monoclonal antibody, in patients with advanced solid tumors. Clin Cancer Res 2013; 19: 3078–87. [DOI] [PubMed] [Google Scholar]
- 12. Wakui H, Yamamoto N, Nakamichi S et al Phase 1 and dose‐finding study of patritumab (U3‐1287), a human monoclonal antibody targeting HER3, in Japanese patients with advanced solid tumors. Cancer Chemother Pharmacol 2014; 73: 511–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Eisenhauer EA, Therasse P, Bogaerts J et al New response evaluation criteria in solid tumours: revised RECIST guideline (version 1.1). Eur J Cancer 2009; 45: 228–47. Translated into Japanese by the Japanese Clinical Oncology Group (2014) Steering committee. Available from: http://www.jcog.jp/doctor/tool/RECISTv11J_20100810.pdf (accessed 14 October 2015). [DOI] [PubMed] [Google Scholar]
- 14. Baselga J, Cortés J, Kim SB et al Pertuzumab plus trastuzumab plus docetaxel for metastatic breast cancer. N Engl J Med 2012; 366: 109–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Swain SM, Kim SB, Cortés J et al Pertuzumab, trastuzumab, and docetaxel for HER2‐positive metastatic breast cancer (CLEOPATRA study): overall survival results from a randomised, double‐blind, placebo‐controlled, phase 3 study. Lancet Oncol 2013; 14: 461–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Swain SM, Baselga J, Kim SB et al Pertuzumab, trastuzumab, and docetaxel in HER2‐positive metastatic breast cancer. N Engl J Med 2015; 372: 724–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Verma S, Miles D, Gianni L et al Trastuzumab emtansine for HER2‐positive advanced breast cancer. N Engl J Med 2012; 367: 1783–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Mendell J, Freeman DJ, Feng W et al Clinical translation and validation of a predictive biomarker for patritumab, an anti‐human epidermal growth factor receptor 3 (HER3) monoclonal antibody, in patients with advanced non‐small cell lung cancer. EBioMedicine 2015; 2: 264–71. [DOI] [PMC free article] [PubMed] [Google Scholar]