

Abstract
Microglia play an important role in the development and maintenance of the central nervous system (CNS) under homeostatic conditions as well as during neurodegenerative diseases. Recent observations in human genomics and advances in genetic mouse models have provided insights into signaling pathways that control development, survival, proliferation and function of microglia. Alteration of these pathways contributes to the pathogenesis of CNS diseases. Here we review the current literature regarding the roles of these microglial pathways in both the normal and diseased brain and discuss areas that require further investigation.
Keywords: Microglia, IL-34, CSF-1, CSF-1R, TREM2, Alzheimer's, Neurodegeneration, Glioblastoma
1. Overview: signals that govern microglial survival and proliferation
Microglia are central nervous system (CNS) resident macrophages that perform many important functions during development and in maintaining homeostasis as well as in countering inflammatory conditions within the CNS [1,2]. Like other tissue macrophages, microglia develop during embryogenesis [3,4]. Primitive macrophages derived from early erythro-myeloid precursors in the embryonic yolk sac seed the developing brain as early as embryonic day 9.5 [5]. These primitive macrophages then proliferate, differentiate, and mature into microglia [3,4]. In steady state conditions, microglia are maintained in an autonomous fashion throughout life, with very little contribution from bone marrow-derived cells [3,4]. Several signals govern the development and maintenance of microglia (Fig. 1). First, like other macrophages, microglia require signals for survival and proliferation from colony stimulating factor-1 receptor (CSF-1R), which is triggered by either CSF-1 or interleukin-34 (IL-34), both of which are differentially expressed in specific regions of the prenatal and postnatal CNS [6–9]. Mice that are deficient in IL-34 or CSF-1 have fewer microglia in various regions of the brain [6,9,10]. Engagement of CSF-1R by CSF-1 or IL-34 results in oligomerization and trans-phosphorylation of CSF-1R, internalization of the ligand-receptor complex, as well as phosphorylation and activation of downstream cytoplasmic mediators that promote survival and proliferation [11,12]. Optimal CSF-1R signaling also requires additional signals, particularly from the DNAX activating protein of 12 kDa (DAP12), an adapter protein that contains cytosolic tyrosine-based activation motifs (ITAMs) [11,13,14]. Activation of DAP12 is achieved through association with an Ig superfamily cell surface receptor, triggering receptor expressed on myeloid cells 2 (TREM2), which detects phospholipids and lipoproteins [15]. Activation of the TREM2/DAP12 complex leads to the phosphorylation of the kinase Syk, which synergizes with CSF-1R signaling. Macrophages lacking DAP12 proliferate poorly and do not survive [14]. In addition, recent transcriptional profiling and epigenetic analyses revealed a requirement for TGF-β signaling [16,17]; TGF-β imprinting, combined with activation of the transcription factor PU.1, drives the expression of many microglial specific genes [16,17]. Interestingly, TGF-β deficiency results in a gradual decline in microglia numbers in the brain [16]. In this review, we discuss the impact of these survival signals on microglial function during normal homeostasis and their potential role during neurodegenerative diseases.
Fig. 1.
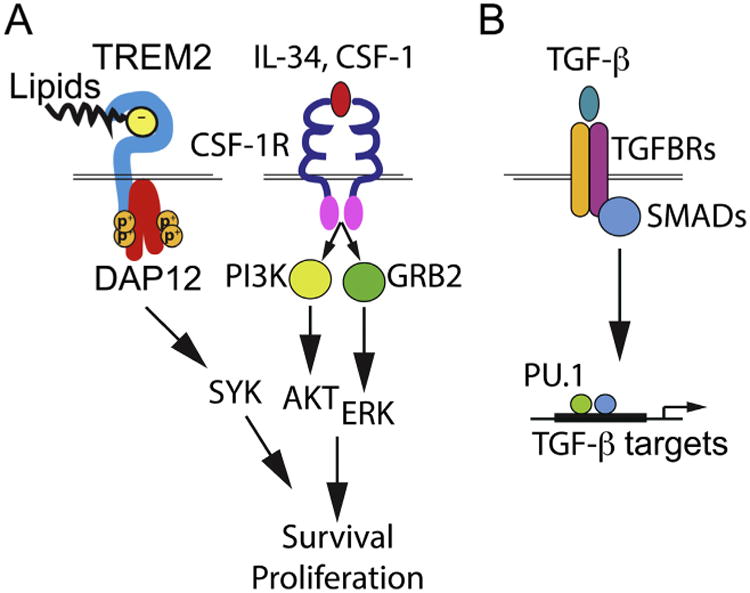
Signals that govern the development and maintenance of microglia.
The development and maintenance of microglia require several signals. (A) CSF-1 and its analog IL-34 trigger the activation of CSF-1R and the phosphorylation of AKT and ERK that promote the survival and proliferation of microglia. Optimized CSF-1R signaling requires TREM2, a lipid sensor. Through its adapter DAP12, activation of TREM2 leads to the phosphorylation of Syk. (B) In addition, recent transcriptional profiling and epigenetic analyses revealed a requirement for TGF-β signaling in microglia, resulting in the activation of SMADs. Activation and translocation of SMADs into the nuclei, combined with the transcription factor PU.1, drives the expression of many microglial specific genes.
2. Rare hereditary neurodegenerative diseases resulting from loss of function in either the CSF-1R or TREM2/DAP12 signaling pathways
Recently, it has been appreciated that microglia play an essential role in the normal function of the CNS. Impaired microglia function has been reported to result from hemizygous loss of a functional CSF-1R or through homozygous loss of function mutations in either TREM2 or its signaling partner DAP12 [18–28]. Hemizygous loss of CSF-1R results in adult-onset leukoencephalopathy with axonal spheroids and pigmented glia (ALSP), whereas total loss of TREM2/DAP12 signaling causes polycystic lipomembranous osteodysplasia with sclerosing leukoencephalopathy, which is also known as Nasu-Hakola disease (NHD) [20–22,24–26,28].
In the CNS, CSF-1R signaling is important for the survival and maintenance of both neurons and microglia [29]. CSF-1R haploinsufficiency results in the autosomal dominant development of ALSP, which is characterized by adult-onset dementia with a progressive decline in motor skills accompanied by epilepsy [24,28]. In order to better understand the underlying biology of ALSP, a mouse model of ALSP was generated by deleting one copy of the Csf1r gene [29]. Aged Csf1r+/− mice have pronounced cognitive defects [29]. Histological examination revealed increased demyelination and neurodegeneration as well as fewer neurons in the brains of Csf1r+/− mice than in those of Csf1r+/+ mice [29]. Interestingly, although CSF-1R signaling is important for the survival of microglia, Csf1r+/− mice have elevated numbers of microglia throughout the brain [9,29]. Chitu et al. hypothesized that the additional microglia result from increased microglia activation/proliferation, which is promoted by inflammation triggered by the excess myelin debris in Csf1r+/− mice [29]. Further characterization of the Csf1r+/− mice will provide additional insight into ALSP.
In the CNS, TREM2 expression is limited to microglia [23,26,30,31]. Mutations resulting in the loss of TREM2 signaling, involving either TREM2 or its signaling adaptor DAP12, result in NHD, which is characterized by severe, early-onset, progressive dementia with bone involvement [20–22,25,26]. NHD patients typically succumb to disease before the 5th decade of life [20–22,26]. A hallmark of NHD is the demyelination of subcortical white matter [32]. Using a cuprizone-induced mouse model of oligodendrocyte toxicity and demyelination that recapitulates some of the pathological findings in individuals with NHD, we recently demonstrated that TREM2 promotes the microglial response to myelin debris [33]. Mice lacking TREM2 were unable to clear myelin debris and had more axonal dystrophy, fewer oligodendrocytes, and prolonged demyelination [33]. Additionally, microglia in Trem2-deficient mice did not expand as quickly as those in wild-type mice in response to cuprizone challenge [33]. However, the microglia population in Trem2-deficient mice was sustained for an extended period of time following cuprizone challenge, which correlated with the prolonged demyelination observed in these mice [33]. These results offer a mechanistic explanation for the role of microglia in a mouse model of NHD and provide insight into the function of microglia in the CNS.
3. Microglia play an important role in the development and maintenance of the CNS
Most functional studies of microglia have focused on the role of activated microglia in the inflamed or diseased brain. Until recently, the function of microglia in the normal resting CNS was unknown. However, two studies using two-photon microscopy revealed that microglia actively survey their surroundings in a healthy, resting CNS [34,35]. Microglia were observed extending and retracting their processes and interacting with astrocytes and neurons, which suggests that they are dynamic cells in the unperturbed CNS [34,35]. These studies supported the long-standing hypothesis that microglia may play an active role in maintaining CNS functions in steady-state.
The use of mouse models in recent studies has aided in elucidating the variety of roles that microglia play in the developing as well as the adult brain. In the developing brain, neurons make many productive and non-productive connections with surrounding cells. The “pruning” of non-productive connections is an important step in the development of a normally functioning brain. Recent evidence has indicated that microglia survey synaptic connections and engulf synapses during postnatal brain development [36–38]. The engulfment of synapses by microglia appears to depend on several signals. Following engagement with microglia, neurons transiently upregulate expression of CX3CL1, which interacts with CX3CR1 expressed on microglia [37]. A recent study found that microglial engulfment of synapses is dependent on expression of CX3CL1 by neurons [37]. A separate study showed that expression of the complement receptor 3 (CD11b/CD18) complex on microglia, along with deposition of complement component 3 on synapses, is also required for microglial engulfment of synapses [38]. Synaptic engulfment by microglia also appears to be influenced by the productivity of the synapse, as synapses that are active are less likely to be engulfed by microglia [38]. Finally, microglia do not appear to be important for brain development in the embryonic mouse, as Csf1r-deficient mice, which have significantly reduced numbers of microglia, appear to have normal brains at birth [36]. However, following birth, the brains of Csf1r-deficient mice develop enlarged ventricles and have more neurons and astrocytes but fewer oligodendrocytes than do brains of wild-type mice [36]. Csf1r-deficient mice generally do not survive until adulthood [36]. Taken together, these data indicate that unimpaired development and survival of microglia are essential for the development of normal neuronal function.
While these studies indicate that microglia are important for development of the CNS during a short period after birth, a potential role for microglia in maintaining the CNS throughout adulthood has also been explored. Using a CX3CR1CreER mouse, which allows for tamoxifen-inducible depletion of microglia in the adult brain, Parkhurst et al. examined the role of microglia in the adult CNS [39]. Microglia, through the production of brain-derived neurotrophic factor, were found to be important for maintaining synaptic plasticity, which has been linked to learning [39]. In addition, microglia have been implicated in synaptic transmission; several studies demonstrated that microglia regulate basal glutamatergic as well as GABAergic synaptic transmission [39–42]. As a result of microglia depletion later in life, these microglia-dependent signaling events do not occur and normal synaptic functions are compromised.
Because microglia play roles in synaptic pruning, maintenance of synaptic plasticity, as well as signaling between neurons, it was reasonable to suggest that they may also have an impact on higher functions, such as olfactory sensing and learning. Indeed, further examination of Csf1r-deficient mice revealed that they have a significantly impaired ability to find food, which is characteristic of compromised olfactory capacity [36]. Additionally, mice transiently depleted of microglia (CX3CR1CreER:R26iDTR/+ mice after tamoxifen administration) failed to adequately perform a number of tasks testing both motor skills and learning, including RotaRod training, freezing in response to a conditioned stimulus, and time spent interacting with a new object [39]. Taken together these data demonstrate that development and survival of microglia are essential for the development and maintenance of the CNS.
4. TREM2 variants in neurodegenerative diseases
The innate immune response to damage-associated signals is paramount for the early host response to both pathogens and sterile insults. The innate immune system relies primarily on both cell surface and intracellular receptors that recognize common motifs to trigger responses to insults. The TREM2/DAP12 heterodimer represents one example of a crucial innate immune receptor expressed by microglia [18,19,23,26,27,30,31].
Because of the dramatic early onset dementia and unique pathology observed in individuals with total loss of TREM2 signaling, it was surprising that recent genome wide association studies (GWAS) and more conventional sequencing studies found that heterozygousity for the rare TREM2 variant R47H is associated with a significantly increased odds ratio for the development of late-onset Alzheimer's disease (AD) [43–46]. Unlike the pathology in NHD, partial loss of TREM2 signaling resulted in individuals that are heterozygous for TREM2 R47H developing AD, which is characterized by deposition of amyloid-β (Aβ) plaques and hyper-phosphorylated tau tangles, which leads to the development of dementia [43–46]. We have recently demonstrated that, in a mouse model of AD, significantly more amyloid-β (Aβ) accumulated in the brains of TREM2-deficient than in those of TREM2-sufficient mice [27]. Accumulation of Aβ was strongly associated with an increase in neuronal loss [27]. We also determined that TREM2-deficient microglia were less responsive to amyloid plaques [27]. Furthermore, the microglia from TREM2-deficient animals were also much more dystrophic and more likely to undergo apoptosis than microglia from wild-type animals [27]. Using a reporter cell assay we determined that the R47H variant of TREM2 is less responsive to TREM2 activating phospholipid ligands than is wild-type TREM2 [27].
Interestingly, the rare TREM2 variant R47H does not contribute to an increased odds ratio for the development of AD in all populations that have been examined. GWAS studies performed on individuals of East Asian origin did not detect an increase in the risk of developing AD for those carrying the TREM2 R47H variant [47–51]. These findings most likely indicate that the development of AD is multifactorial and certain populations may possess factors that compensate for the partial loss of TREM2 function. Further investigation into the apparent lack of a role for the R47H TREM2 variant in the development of AD in these populations will improve our understanding of both TREM2 biology and the progression of AD.
Because the R47H variant of TREM2 was associated with a significantly increased chance for developing AD in certain populations, it was hypothesized that this TREM2-variant might also contribute to the development of other neurodegenerative diseases. A recent effort to quantify the contribution of the R47H variant to the development of Parkinson's disease (PD), frontotemporal dementia (FTD), amyotrophic lateral sclerosis (ALS), ischemic stroke, and progressive supranuclear palsy revealed that the R47H variant of TREM2 is associated with a statistically significant increase in the odds ratio for the development of both PD and FTD [52]. In contrast, another GWAS study found no correlation between the R47H variant of TREM2 and the development of FTD, PD, or ALS [53]. Finally, yet another study found a statistically significant increase in the odds ratio that R47H individuals are at risk for developing spontaneous ALS [54]. Therefore, it is currently unclear what, if any, role the R47H variant of TREM2 plays in the development of PD, FTD, and ALS [52–54]. Further characterization of the development of PD, FTD, or ALS in individuals harboring the R47H variant of TREM2 as well as the use of animal models to examine the role of TREM2 in models of PD, FTD, and ALS, will improve our understanding of the role of TREM2 in these neurodegenerative diseases.
5. Innate immune receptor in AD
Beyond TREM2, several other proteins that are expressed by microglia have also been implicated in the development and progression of AD including CD33 and complement receptor 1 (CR1). CD33 was identified as playing a role in the development of late-onset AD in GWAS studies [55,56]. Mechanistic studies into the role of CD33 in AD development were performed by analyzing samples obtained from individuals bearing either the common CD33 variant or one of the minor CD33 allele variants (rs3865444). Individuals with the CD33 rs3865444T variant had less CD33 expression on microglia and less detectable Aβ throughout their brains [57]. Additionally, CD33 expression inhibited the uptake of Aβ by microglia in vitro, while CD33-deficiency reduced brain levels of insoluble Aβ42 and amyloid plaque burden in APP(Swe)/PS1(ΔE9) mice [57]. Another study investigating the role of a different CD33 variant, rs3865444C, found that this variant is associated with increased CD33 expression [58]. Increased CD33 was associated with diminished Aβ internalization and more neuronal cell damage surrounding plaques as well as an increase in the overall number of activated microglia [58]. Taken together these studies indicate that CD33 expression inhibits phagocytosis of Aβ and that overex-pression of CD33 is detrimental while decreased CD33 is beneficial during AD development and progression [57,58].
The role of CR1 in microglia appears to be neuroprotective but in some respects can enhance neurodegeneration [59]. A copy number variant (CNV) of CR1 that results in the overproduction of a larger CR1 variant known as CR1-s has been found to be strongly associated with the development of AD [60]. The mechanism by which the production of CR1-s is linked to neurodegeneration in AD is still unclear. It has been proposed that because CR1-s has additional binding sites for C3b/C4b the production of CR1-s is directly linked to enhanced complement activation and inflammation which in turn drives the development of AD [60,61]. Additional work to determine the mechanisms by which both CD33 and CR1 contribute to the development of AD is warranted as an understanding of these mechanisms may directly lead to more effective therapeutic approaches for AD.
6. CSF-1R blockade in glioblastoma multiforme tumors
Glioblastoma multiforme (GBM) tumors are the most common primary brain tumor. GBMs represent approximately 45% of all malignant primary brain tumors and are one of the most aggressive and difficult to treat, with a mean 5 year survival rate of ∼3.3% [62,63]. Current treatment for GBM is limited to resection of the tumor mass when possible and irradiation; in most cases treatment is not efficacious [62]. Recent characterization of GBM tumors has determined that several cell populations that do not originate in the tumor are present within the tumor mass [64]. The primary immune cells that have been identified as part of GBM tumors are tumor-associated macrophages/microglia (TAMs) [65]. The origin of the TAMs is currently controversial. A study by Badie and Schartner suggested that the origin of the TAMs was mixed with microglia representing the TAMs on the outside border of the tumor while the tumor infiltrating TAMs originated from the monocyte pool [66]. However, recent studies have suggested that resident microglia are the primary contributor to the TAM pool with little contribution from monocytes [67]. In GBMs, TAMs comprise up to 1% of the total cells in the tumor mass [65]. TAMs are known to have severely compromised immune function, with a decreased capacity for cytokine secretion and a hypoactive TLR response [65]. As TAMs have been implicated in the maintenance and progression of several different types of tumors, including GBMs, and TAMs have been associated with higher tumor grade and poor prognosis, the regulation and perturbation of TAMs represents an interesting therapeutic approach for the treatment of GBMs [67–71].
A role for CSF-1R signaling in the survival of TAMs during the development of GBMs has recently been appreciated; Pyonteck et al. demonstrated that blockade of CSF-1R signaling significantly reduced the number of TAMs in a mouse model of GBM [70]. They also found that, in the context of CSF-1R blockade, TAM survival became dependent on the secretion of granulocyte-macrophage colony stimulating factor (GM-CSF) and interferon-γ (IFN-γ) by the glioma [70]. GM-CSF and IFN-γ signaling not only promoted TAM survival but also influenced the polarization of the TAMs [70]. TAMs stimulated with GM-CSF and IFN-γ in the absence of CSF-1R signaling developed a less M2-like phenotype and were less efficient at promoting the growth and survival of the tumor [70]. Further insight into the effect of CSF-1R blockade on TAMs can be gained from examination of CSF-1R blockade in other tumor models. CSF-1R blockade in combination with radiotherapy was found to significantly improve the efficacy of radiotherapy treatment in a prostate cancer model [71]. Taken together, these data indicate that the disruption of normal CSF-1R signaling, either alone or in combination with radiotherapy, may dramatically affect TAMs and may be a more effective therapeutic approach for the treatment of GBMs in the future.
7. Concluding remarks
The roles of microglia in normal health and disease pathogenesis are increasingly appreciated. It has become clear that microglia play an essential part in the development and maintenance of the CNS and that perturbation of normal microglia functions can contribute to the development of neurodegenerative diseases i.e. NHD and AD. Future work focused on signaling pathways that modulate microglial activation, survival and proliferation will allow for further insights into the pathogenesis of disease of the CNS and may provide new and more effective therapeutics for the treatment of diseases of the CNS including AD and GBM.
Acknowledgments
T.K.U. is supported by NIH 5T32CA009547-30. Y.W. is supported by the Lilly Innovation Fellowship Award (Eli Lilly and Company). M.C. is supported by the Knight Alzheimer's Disease Research Center pilot grant P50 AG005681-30, the Cure Alzheimer's Fund and NIH 1RF1AG051485-01.
References
- 1.Gomez Perdiguero E, Schulz C, Geissmann F. Development and homeostasis of resident myeloid cells: the case of the microglia. Glia. 2013;61:112–120. doi: 10.1002/glia.22393. [DOI] [PubMed] [Google Scholar]
- 2.Shemer A, Erny D, Jung S, Prinz M. Microglia plasticity during health and disease: an immunological perspective. Trends Immunol. 2015;36:614–624. doi: 10.1016/j.it.2015.08.003. [DOI] [PubMed] [Google Scholar]
- 3.Ginhoux F, Prinz M. Origin of microglia: current concepts and past controversies. Cold Spring Harb Perspect Biol. 2015;7 doi: 10.1101/cshperspect.a020537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Waisman A, Ginhoux F, Greter M, Bruttger J. Homeostasis of microglia in the adult brain: review of novel microglia depletion systems. Trends Immunol. 2015;36:625–636. doi: 10.1016/j.it.2015.08.005. [DOI] [PubMed] [Google Scholar]
- 5.Kierdorf K, Erny D, Goldmann T, Sander V, Schulz C, Perdiguero EG, et al. Microglia emerge from erythromyeloid precursors via Pu.1- and Irf8-dependent pathways. Nat Neurosci. 2013;16:273–280. doi: 10.1038/nn.3318. [DOI] [PubMed] [Google Scholar]
- 6.Greter M, Lelios I, Pelczar P, Hoeffel G, Price J, Leboeuf M, et al. Stroma-derived interleukin-34 controls the development and maintenance of langerhans cells and the maintenance of microglia. Immunity. 2012;37:1050–1060. doi: 10.1016/j.immuni.2012.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Nandi S, Gokhan S, Dai XM, Wei S, Enikolopov G, Lin H, et al. The CSF-1 receptor ligands IL-34 and CSF-1 exhibit distinct developmental brain expression patterns and regulate neural progenitor cell maintenance and maturation. Dev Biol. 2012;367:100–113. doi: 10.1016/j.ydbio.2012.03.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Stanley ER, Chitu V. CSF-1 receptor signaling in myeloid cells. Cold Spring Harb Perspect Biol. 2014;6 doi: 10.1101/cshperspect.a021857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wang Y, Szretter KJ, Vermi W, Gilfillan S, Rossini C, Cella M, et al. IL-34 is a tissue-restricted ligand of CSF1R required for the development of langerhans cells and microglia. Nat Immunol. 2012;13:753–760. doi: 10.1038/ni.2360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wegiel J, Wisniewski HM, Dziewiatkowski J, Tarnawski M, Kozielski R, Trenkner E, et al. Reduced number and altered morphology of microglial cells in colony stimulating factor-1-deficient osteopetrotic op/op mice. Brain Res. 1998;804:135–139. doi: 10.1016/s0006-8993(98)00618-0. [DOI] [PubMed] [Google Scholar]
- 11.Tos M, Trojaborg N, Thomsen J. The contralateral ear after translabyrinthine removal of acoustic neuromas: is there a drill-noise generated hearing loss. J Laryngol Otol. 1989;103:845–849. doi: 10.1017/s0022215100110278. [DOI] [PubMed] [Google Scholar]
- 12.Yeung YG, Wang Y, Einstein DB, Lee PS, Stanley ER. Colony-stimulating factor-1 stimulates the formation of multimeric cytosolic complexes of signaling proteins and cytoskeletal components in macrophages. J Biol Chem. 1998;273:17128–17137. doi: 10.1074/jbc.273.27.17128. [DOI] [PubMed] [Google Scholar]
- 13.Lanier LL, Corliss BC, Wu J, Leong C, Phillips JH. Immunoreceptor DAP12 bearing a tyrosine-based activation motif is involved in activating NK cells. Nature. 1998;391:703–707. doi: 10.1038/35642. [DOI] [PubMed] [Google Scholar]
- 14.Otero K, Turnbull IR, Poliani PL, Vermi W, Cerutti E, Aoshi T, et al. Macrophage colony-stimulating factor induces the proliferation and survival of macrophages via a pathway involving DAP12 and beta-catenin. Nat Immunol. 2009;10:734–743. doi: 10.1038/ni.1744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bouchon A, Hernandez-Munain C, Cella M, Colonna M. A DAP12-mediated pathway regulates expression of CC chemokine receptor 7 and maturation of human dendritic cells. J Exp Med. 2001;194:1111–1122. doi: 10.1084/jem.194.8.1111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Butovsky O, Jedrychowski MP, Moore CS, Cialic R, Lanser AJ, Gabriely G, et al. Identification of a unique TGF-beta-dependent molecular and functional signature in microglia. Nat Neurosci. 2014;17:131–143. doi: 10.1038/nn.3599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gosselin D, Link VM, Romanoski CE, Fonseca GJ, Eichenfield DZ, Spann NJ, et al. Environment drives selection and function of enhancers controlling tissue-specific macrophage identities. Cell. 2014;159:1327–1340. doi: 10.1016/j.cell.2014.11.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Atagi Y, Liu CC, Painter MM, Chen XF, Verbeeck C, Zheng H, et al. Apolipoprotein E is aligand for triggering receptor expressed on myeloid cells 2 (TREM2) J Biol Chem. 2015 doi: 10.1074/jbc.M115.679043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Bailey CC, DeVaux LB, Farzan M. The triggering receptor expressed on myeloid cells 2 binds apolipoprotein E. J Biol Chem. 2015 doi: 10.1074/jbc.M115.677286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Guerreiro R, Bilgic B, Guven G, Bras J, Rohrer J, Lohmann E, et al. Novel compound heterozygous mutation in TREM2 found in a Turkish frontotemporal dementia-like family. Neurobiol Aging. 2013;34:2890. e2891–e2895. doi: 10.1016/j.neurobiolaging.2013.06.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Guerreiro RJ, Lohmann E, Bras JM, Gibbs JR, Rohrer JD, Gurunlian N, et al. Using exome sequencing to reveal mutations in TREM2 presenting as a frontotemporal dementia-like syndrome without bone involvement. JAMA Neurol. 2013;70:78–84. doi: 10.1001/jamaneurol.2013.579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kleinberger G, Yamanishi Y, Suarez-Calvet M, Czirr E, Lohmann E, Cuyvers E, et al. TREM2 mutations implicated in neurodegeneration impair cell surface transport and phagocytosis. Sci Transl Med. 2014;6:243ra286. doi: 10.1126/scitranslmed.3009093. [DOI] [PubMed] [Google Scholar]
- 23.Kiialainen A, Hovanes K, Paloneva J, Kopra O, Peltonen L. Dap12 and Trem2, molecules involved in innate immunity and neurodegeneration, are co-expressed in the CNS. Neurobiol Dis. 2005;18:314–322. doi: 10.1016/j.nbd.2004.09.007. [DOI] [PubMed] [Google Scholar]
- 24.Nicholson AM, Baker MC, Finch NA, Rutherford NJ, Wider C, Graff-Radford NR, et al. CSF1R mutations link POLD and HDLS as a single disease entity. Neurology. 2013;80:1033–1040. doi: 10.1212/WNL.0b013e31828726a7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Numasawa Y, Yamaura C, Ishihara S, Shintani S, Yamazaki M, Tabunoki H, et al. Nasu-Hakola disease with a splicing mutation of TREM2 in a Japanese family. Eur J Neurol. 2011;18:1179–1183. doi: 10.1111/j.1468-1331.2010.03311.x. [DOI] [PubMed] [Google Scholar]
- 26.Paloneva J, Manninen T, Christman G, Hovanes K, Mandelin J, Adolfsson R, et al. Mutations in two genes encoding different subunits of a receptor signaling complex result in an identical disease phenotype. Am J Hum Genet. 2002;71:656–662. doi: 10.1086/342259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wang Y, Cella M, Mallinson K, Ulrich JD, Young KL, Robinette ML, et al. TREM2 lipid sensing sustains the microglial response in an Alzheimer's disease model. Cell. 2015;160:1061–1071. doi: 10.1016/j.cell.2015.01.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wider C, Van Gerpen JA, De Armond S, Shuster EA, Dickson DW, Wszolek ZK. Leukoencephalopathy with spheroids (HDLS) and pigmentary leukodystrophy (POLD): a single entity. Neurology. 2009;72:1953–1959. doi: 10.1212/WNL.0b013e3181a826c0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Chitu V, Gokhan S, Gulinello M, Branch CA, Patil M, Basu R, et al. Phenotypic characterization of a Csf1r haploinsufficient mouse model of adult-onset leukodystrophy with axonal spheroids and pigmented glia (ALSP) Neurobiol Dis. 2015;74:219–228. doi: 10.1016/j.nbd.2014.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Schmid CD, Sautkulis LN, Danielson PE, Cooper J, Hasel KW, Hilbush BS, et al. Heterogeneous expression of the triggering receptor expressed on myeloid cells-2 on adult murine microglia. J Neurochem. 2002;83:1309–1320. doi: 10.1046/j.1471-4159.2002.01243.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Thrash JC, Torbett BE, Carson MJ. Developmental regulation of TREM2 and DAP12 expression in the murine CNS: implications for Nasu-Hakola disease. Neurochem Res. 2009;34:38–45. doi: 10.1007/s11064-008-9657-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Verloes A, Maquet P, Sadzot B, Vivario M, Thiry A, Franck G. Nasu-Hakola syndrome: polycystic lipomembranous osteodysplasia with sclerosing leucoencephalopathy and presenile dementia. J Med Genet. 1997;34:753–757. doi: 10.1136/jmg.34.9.753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Poliani PL, Wang Y, Fontana E, Robinette ML, Yamanishi Y, Gilfillan S, et al. TREM2 sustains microglial expansion during aging and response to demyelination. J Clin Invest. 2015;125:2161–2170. doi: 10.1172/JCI77983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Davalos D, Grutzendler J, Yang G, Kim JV, Zuo Y, Jung S, et al. ATP mediates rapid microglial response to local brain injury in vivo. Nat Neurosci. 2005;8:752–758. doi: 10.1038/nn1472. [DOI] [PubMed] [Google Scholar]
- 35.Nimmerjahn A, Kirchhoff F, Helmchen F. Resting microglial cells are highly dynamic surveillants of brain parenchyma in vivo. Science (New York, NY) 2005;308:1314–1318. doi: 10.1126/science.1110647. [DOI] [PubMed] [Google Scholar]
- 36.Erblich B, Zhu L, Etgen AM, Dobrenis K, Pollard JW. Absence of colony stimulation factor–1 receptor results in loss of microglia, disrupted brain development and olfactory deficits. PLoS One. 2011;6:e26317. doi: 10.1371/journal.pone.0026317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Paolicelli RC, Bolasco G, Pagani F, Maggi L, Scianni M, Panzanelli P, et al. Synaptic pruning by microglia is necessary for normal brain development. Science (New York, NY) 2011;333:1456–1458. doi: 10.1126/science.1202529. [DOI] [PubMed] [Google Scholar]
- 38.Schafer DP, Lehrman EK, Kautzman AG, Koyama R, Mardinly AR, Yamasaki R, et al. Microglia sculpt postnatal neural circuits in an activity and complement-dependent manner. Neuron. 2012;74:691–705. doi: 10.1016/j.neuron.2012.03.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Parkhurst CN, Yang G, Ninan I, Savas JN, Yates JR, 3rd, Lafaille JJ, et al. Microglia promote learning-dependent synapse formation through brain-derived neurotrophic factor. Cell. 2013;155:1596–1609. doi: 10.1016/j.cell.2013.11.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Coull JA, Beggs S, Boudreau D, Boivin D, Tsuda M, Inoue K, et al. BDNF from microglia causes the shift in neuronal anion gradient underlying neuropathic pain. Nature. 2005;438:1017–1021. doi: 10.1038/nature04223. [DOI] [PubMed] [Google Scholar]
- 41.Pascual O, Ben Achour S, Rostaing P, Triller A, Bessis A. Microglia activation triggers astrocyte-mediated modulation of excitatory neurotransmission. Proc Natl Acad Sci U S A. 2012;109:E197–E205. doi: 10.1073/pnas.1111098109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Tsuda M, Shigemoto-Mogami Y, Koizumi S, Mizokoshi A, Kohsaka S, Salter MW, et al. P2X4 receptors induced in spinal microglia gate tactile allodynia after nerve injury. Nature. 2003;424:778–783. doi: 10.1038/nature01786. [DOI] [PubMed] [Google Scholar]
- 43.Guerreiro R, Wojtas A, Bras J, Carrasquillo M, Rogaeva E, Majounie E, et al. TREM2 variants in Alzheimer's disease. N Engl J Med. 2013;368:117–127. doi: 10.1056/NEJMoa1211851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Jonsson T, Stefansson H, Steinberg S, Jonsdottir I, Jonsson PV, Snaedal J, et al. Variant of TREM2 associated with the risk of Alzheimer's disease. N Engl J Med. 2013;368:107–116. doi: 10.1056/NEJMoa1211103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Korvatska O, Leverenz JB, Jayadev S, McMillan P, Kurtz I, Guo X, et al. R47H variant of TREM2 associated with Alzheimer disease in a large late-onset family: clinical, genetic, and neuropathological study. JAMA Neurol. 2015;72:920–927. doi: 10.1001/jamaneurol.2015.0979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Rosenthal SL, Bamne MN, Wang X, Berman S, Snitz BE, Klunk WE, et al. More evidence for association of a rare TREM2 mutation (R47H) with Alzheimer's disease risk. Neurobiol Aging. 2015;36:2443. e2421–e2446. doi: 10.1016/j.neurobiolaging.2015.04.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Huang M, Wang D, Xu Z, Xu Y, Xu X, Ma Y, et al. Lack of genetic association between TREM2 and Alzheimer's disease in East Asian population: a systematic review and meta-analysis. Am J Alzheimers Dis Other Dement. 2015 doi: 10.1177/1533317515577128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Jiao B, Liu X, Tang B, Hou L, Zhou L, Zhang F, et al. Investigation of TREM2, PLD3, and UNC5C variants in patients with Alzheimer's disease from mainland. China Neurobiol Aging. 2014;35:2422. e2429–e2422. e2411. doi: 10.1016/j.neurobiolaging.2014.04.025. [DOI] [PubMed] [Google Scholar]
- 49.Ma J, Zhou Y, Xu J, Liu X, Wang Y, Deng Y, et al. Association study of TREM2 polymorphism rs75932628 with late-onset Alzheimer's disease in Chinese Han population. Neurol Res. 2014;36:894–896. doi: 10.1179/1743132814Y.0000000376. [DOI] [PubMed] [Google Scholar]
- 50.Miyashita A, Wen Y, Kitamura N, Matsubara E, Kawarabayashi T, Shoji M, et al. Lack of genetic association between TREM2 and late-onset Alzheimer's disease in a Japanese population. J Alzheimers Dis. 2014;41:1031–1038. doi: 10.3233/JAD-140225. [DOI] [PubMed] [Google Scholar]
- 51.Yu JT, Jiang T, Wang YL, Wang HF, Zhang W, Hu N, et al. Triggering receptor expressed on myeloid cells 2 variant is rare in late-onset Alzheimer's disease in Han Chinese individuals. Neurobiol Aging. 2014;35:937. e931–e933. doi: 10.1016/j.neurobiolaging.2013.10.075. [DOI] [PubMed] [Google Scholar]
- 52.Rayaprolu S, Mullen B, Baker M, Lynch T, Finger E, Seeley WW, et al. TREM2 in neurodegeneration: evidence for association of the p.R47H variant with frontotemporal dementia and Parkinson's disease. Mol Neurodegener. 2013;8:19. doi: 10.1186/1750-1326-8-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Lill CM, Rengmark A, Pihlstrom L, Fogh I, Shatunov A, Sleiman PM, et al. The role of TREM2 R47H as a risk factor for Alzheimer's disease, frontotemporal lobar degeneration, amyotrophic lateral sclerosis, and Parkinson's disease. Alzheimers Dement. 2015 doi: 10.1016/j.jalz.2014.12.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Cady J, Koval ED, Benitez BA, Zaidman C, Jockel-Balsarotti J, Allred P, et al. TREM2 variant p.R47H as a risk factor for sporadic amyotrophic lateral sclerosis. JAMA Neurol. 2014;71:449–453. doi: 10.1001/jamaneurol.2013.6237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Hollingworth P, Harold D, Sims R, Gerrish A, Lambert JC, Carrasquillo MM, et al. Common variants at ABCA7, MS4A6A/MS4A4E, EPHA1, CD33 and CD2AP are associated with Alzheimer's disease. Nat Genet. 2011;43:429–435. doi: 10.1038/ng.803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Naj AC, Jun G, Beecham GW, Wang LS, Vardarajan BN, Buros J, et al. Common variants at MS4A4/MS4A6E, CD2AP, CD33 and EPHA1 are associated with late-onset Alzheimer's disease. Nat Genet. 2011;43:436–441. doi: 10.1038/ng.801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Griciuc A, Serrano-Pozo A, Parrado AR, Lesinski AN, Asselin CN, Mullin K, et al. Alzheimer's disease risk gene CD33 inhibits microglial uptake of amyloid beta. Neuron. 2013;78:631–643. doi: 10.1016/j.neuron.2013.04.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Bradshaw EM, Chibnik LB, Keenan BT, Ottoboni L, Raj T, Tang A, et al. CD33 Alzheimer's disease locus: altered monocyte function and amyloid biology. Nat Neurosci. 2013;16:848–850. doi: 10.1038/nn.3435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Crehan H, Hardy J, Pocock J. Blockage of CR1 prevents activation of rodent microglia. Neurobiol Dis. 2013;54:139–149. doi: 10.1016/j.nbd.2013.02.003. [DOI] [PubMed] [Google Scholar]
- 60.Brouwers N, Van Cauwenberghe C, Engelborghs S, Lambert JC, Bettens K, Le Bastard N, et al. Alzheimer risk associated with a copy number variation in the complement receptor 1 increasing C3b/C4b binding sites. Mol Psychiatry. 2012;17:223–233. doi: 10.1038/mp.2011.24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Hazrati LN, Van Cauwenberghe C, Brooks PL, Brouwers N, Ghani M, Sato C, et al. Genetic association of CR1 with Alzheimer's disease: a tentative disease mechanism. Neurobiol Aging. 2012;33:2949. e2945–e2949. e2912. doi: 10.1016/j.neurobiolaging.2012.07.001. [DOI] [PubMed] [Google Scholar]
- 62.Stupp R, Mason WP, van den Bent MJ, Weller M, Fisher B, Taphoorn MJ, et al. Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. N Engl J Med. 2005;352:987–996. doi: 10.1056/NEJMoa043330. [DOI] [PubMed] [Google Scholar]
- 63.Thakkar JP, Dolecek TA, Horbinski C, Ostrom QT, Lightner DD, Barnholtz-Sloan JS, et al. Epidemiologic and molecular prognostic review of glioblastoma. Cancer Epidemiol Biomarkers Prev. 2014;23:1985–1996. doi: 10.1158/1055-9965.EPI-14-0275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Charles NA, Holland EC, Gilbertson R, Glass R, Kettenmann H. The brain tumor microenvironment. Glia. 2012;60:502–514. doi: 10.1002/glia.21264. [DOI] [PubMed] [Google Scholar]
- 65.Hussain SF, Yang D, Suki D, Aldape K, Grimm E, Heimberger AB. The role of human glioma-infiltrating microglia/macrophages in mediating antitumor immune responses. Neuro Oncol. 2006;8:261–279. doi: 10.1215/15228517-2006-008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Badie B, Schartner JM. Flow cytometric characterization of tumor-associated macrophages in experimental gliomas. Neurosurgery. 2000;46:957–961. doi: 10.1097/00006123-200004000-00035. discussion 961–962. [DOI] [PubMed] [Google Scholar]
- 67.Coniglio SJ, Eugenin E, Dobrenis K, Stanley ER, West BL, Symons MH, et al. Microglial stimulation of glioblastoma invasion involves epidermal growth factor receptor (EGFR) and colony stimulating factor 1 receptor (CSF-1R) signaling. Mol Med. 2012;18:519–527. doi: 10.2119/molmed.2011.00217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Engler JR, Robinson AE, Smirnov I, Hodgson JG, Berger MS, Gupta N, et al. Increased microglia/macrophage gene expression in a subset of adult and pediatric astrocytomas. PLoS One. 2012;7:e43339. doi: 10.1371/journal.pone.0043339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Komohara Y, Ohnishi K, Kuratsu J, Takeya M. Possible involvement of the M2 anti-inflammatory macrophage phenotype in growth of human gliomas. J Pathol. 2008;216:15–24. doi: 10.1002/path.2370. [DOI] [PubMed] [Google Scholar]
- 70.Pyonteck SM, Akkari L, Schuhmacher AJ, Bowman RL, Sevenich L, Quail DF, et al. CSF-1R inhibition alters macrophage polarization and blocks glioma progression. Nat Med. 2013;19:1264–1272. doi: 10.1038/nm.3337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Xu J, Escamilla J, Mok S, David J, Priceman S, West B, et al. CSF1R signaling blockade stanches tumor-infiltrating myeloid cells and improves the efficacy of radiotherapy in prostate cancer. Cancer Res. 2013;73:2782–2794. doi: 10.1158/0008-5472.CAN-12-3981. [DOI] [PMC free article] [PubMed] [Google Scholar]