

Abstract
Previous data obtained in our laboratory suggested that there may be constitutive signaling through the myeloid differentiation primary response gene 88 (Myd88)-dependent signaling cascade in murine mammary carcinoma. Here, we extended these findings by showing that, in the absence of an added Toll-like receptor (TLR) agonist, the myddosome complex was preformed in 4T1 tumor cells, and that Myd88 influenced cytoplasmic extracellular signal–regulated kinase (Erk)1/Erk2 levels, nuclear levels of nuclear factor-kappaB (NFκB) and signal transducer and activator of transcription 5 (STAT5), tumor-derived chemokine (C–C motif) ligand 2 (CCL2) expression, and in vitro and in vivo tumor growth. In addition, RNA-sequencing revealed that Myd88-dependent signaling enhanced the expression of genes that could contribute to breast cancer progression and genes previously associated with poor outcome for patients with breast cancer, in addition to suppressing the expression of genes capable of inhibiting breast cancer progression. Yet, Myd88-dependent signaling in tumor cells also suppressed expression of genes that could contribute to tumor progression. Collectively, these data revealed a multifaceted role for Myd88-dependent signaling in murine mammary carcinoma.
Keywords: breast cancer, 4T1, Myd88, NFκB, RNA-Seq
Introduction
Myeloid differentiation primary response gene 88 (Myd88) is an adaptor protein for Toll-like receptor (TLR) signaling, which most TLRs utilize for signal transduction.1 Upon binding a TLR agonist, the Toll/interleukin (IL)-1R homology (TIR) domain of the TLR interacts with the C-terminal TIR domain of Myd88. The N-terminal death domain (DD) of Myd88 then associates with the DD of IL-1 receptor-associated kinase-2 (IRAK2) and IRAK4 molecules to form a functional myddosome complex composed of multiple molecules of Myd88, IRAK2, and IRAK4.2 Signaling culminates in nuclear translocation of several transcriptional factors, leading to expression of proinflammatory cytokines and chemokines.1
Inflammation in the context of cancer is a double-edged sword. While antitumor immunity has great potential for prevention, diagnosis, and treatment of patients with cancer, inflammation is also widely accepted as a contributing factor for cancer initiation, progression, and dissemination.3,4 This same dichotomy also applies to TLR signaling.5 As a result, it is not surprising to find a link between Myd88 and cancer progression. Notably, there have been several reports that expression of Myd88 may contribute to tumor progression. For instance, Rakoff-Nahoum and Medzhitov6 showed that Myd88 played a role in tumor development in mice with mutations in the adenomatous polyposis coli (APC) gene, perhaps by inducing expression of cyclooxygenase 2 (COX2), matrix metalloproteinases 7 and 10 (MMP7 and MMP10), IL-6, and tumor necrosis factor alpha (TNF-α), among other genes. Similar findings were obtained using a murine hepatocellular carcinoma (HCC) model, where diethylnitrosamine-induced HCC was shown to be related to IL-6 produced in a Myd88-dependent manner.7 Additionally, Chefetz et al,8 revealed that a Myd88-dependent pathway contributed to protumorigenic inflammation as well as stem cell renewal in an ovarian cancer model. Myd88 has also been shown to participate in signaling that culminates in recurrence after radiotherapy. Gao et al,9 reported that factors released during tumor regression triggered TLR9 signaling in myeloid cells, which could contribute to inflammation capable of promoting tumor recurrence.
Here, we were interested in the direct contribution of Myd88-dependent signaling in the tumor cells themselves. For this purpose, we used the 4T1 murine mammary carcinoma, which is often used as a model for stage IV disease in patients with breast cancer.10 Our data revealed that Myd88-dependent signaling affected 4T1 at several different levels, with changes evident in the cytoplasm (formation of myddosome complex and elevated extracellular signal–regulated kinase [Erk]1/Erk2 levels), nucleus (elevated levels of nuclear factor-kappaB [NFκB] and chemokine (C–C motif) ligand 2 [CCL2] transcription), and overall behavior (increased growth). Yet, these data represent a small picture of how Myd88-dependent signaling affects the tumor cells. RNA sequencing (RNA-Seq) analysis revealed several previously unrecognized ways in which Myd88-dependent signaling in the tumor cells themselves may contribute to breast cancer progression. For instance, we found that Myd88 contributed to expression of genes associated with breast cancer, such as Cdh3 and Adra2a,11,12 and genes associated with poor outcome for patients with breast cancer, such as Folr1 and Aldh1a3,13,14 in addition to suppressing the expression of genes that encode proteins capable of inhibiting breast cancer, such as Psd4 and Dmp1.15,16 Collectively, these data revealed several previously unrecognized ways in which Myd88 may contribute to tumor progression.
While it is clear that targeting Myd88-dependent signaling in tumor cells can significantly affect tumor growth, caution is necessary when blocking this pathway in murine mammary carcinoma because suppressing Myd88-dependent signaling in the tumor cells also contributed to an increase in expression of MMP9 and IL-1β genes, which are associated with disease progression.17,18 Overall, these data reveal a complex role for constitutive Myd88-dependent signaling in murine mammary carcinoma and suggest that the consequences of targeting Myd88 may not be as straightforward as previously thought.
Methods
Cells and mice
The 4T1 murine mammary carcinoma cells were maintained in complete RPMI (cRPMI) (RPMI 1640, Lonza, Walkersville, MD, USA) supplemented with 10% heat-inactivated fetal bovine serum (FBS; Lonza), glutamine (2 mM; Lonza), penicillin (100 U/mL; Lonza), streptomycin (100 µg/mL; Lonza), nonessential amino acids (Sigma), 2-mercaptoethanol (5 × 10−5 M; Sigma), and sodium pyruvate (1 mM; Lonza). All other cell lines used in this study were generated from 4T1. Constitutive Myd88low cells (consMydlow) expressing short hairpin RNA (shRNA) specific for Myd88 were generated with the pBLOCK-it DEST vector (Life Technologies, Carlsbad, CA, USA), as previously reported,19 and maintained in cRPMI with 800 µg/mL G418 (Life Technologies). 4T1 cells expressing lacZ under control of the same shRNA expression vector19 and maintained in 800 µg/mL G418 served as a control for the consMydlow cells. 4T1 cells expressing shRNA specific for Myd88 under a tetracycline-inducible promoter (inducible Myd88low cells, indMydlow) were generated using the BLOCK-it Inducible PolII miR RNAi Expression Vector Kit (Life Technologies) and maintained in 800 µg/mL G418 and 2.5 µM blasticidin (Life Technologies). Analysis of these cells in the absence of tetracycline served as a control for the indMydlow cells.
Six- to eight-week-old female BALB/c mice were bred on site and housed in a Thoren caging system (Thoren Caging Systems Inc, Hazelton, PA, USA). Food and water were provided ad libitum. All mice were used in accordance with a protocol approved by the Institutional Animal Care and Use Committee, following the guidelines for ethical conduct in care and use of animals.
Growth kinetics
To determine in vitro growth rates, tumor cells were plated at 1 × 104 cells per T25 cm2 flask for 24 hours and 48 hours, or in a T75 cm2 flask for 72 hours. Cells were trypsinized, washed, and counted at the specified time points. Each cell count was performed in duplicate. For studies with the Myd88 inhibitory peptide, 1 × 104 tumor cells were cultured in 24-well tissue culture plates with the Myd88-specific inhibitory peptide (MP) or a control peptide (CP) lacking the Myd88-binding domain (Novus Biologicals). For in vivo kinetics, after washing three times in Hanks’ balanced salt solution (HBSS), 5 × 104 cells in 100 µL HBSS were delivered subcutaneously to six- to eight-week-old female BALB/c mice in the left hind flank. Starting at day six, and every two-to-three days thereafter, tumors were measured using vernier calipers, and tumor volume was calculated (L × W2/2). Mice given the indMydlow cells were started on doxycycline (0.2–1.6 mg/mL) in drinking water (supplemented with 20% sucrose) one week before tumor delivery and maintained on doxycycline for the remainder of the experiment.
Quantitative reverse transcriptase–polymerase chain reaction
Gene expression was analyzed by quantitative reverse transcriptase–polymerase chain reaction (qRT-PCR). For this purpose, RNA was isolated from 1 × 106 cells using the Aurum Total RNA Mini Kit (Bio-Rad Laboratories) according to manufacturer’s instructions. Briefly, the cells were lysed, RNA was bound to the column, treated with DNase, and following several washes, RNA was eluted and stored at −20°C. Complementary DNA (cDNA) was generated using the iScript cDNA synthesis kit (Bio-Rad Laboratories). For this purpose, 15 µL RNA, 4 µL 5 × iScript buffer and 1 µL iScript reverse transcriptase were combined in a 0.5 mL microcentrifuge tube and placed in a thermal cycler (MiniCycler; MJ Research Watertown). The reaction conditions consisted of 25°C for five minutes, 42°C for 30 minutes, and then 85°C for five minutes. All samples were stored at −20°C prior to PCR. An aliquot (0.5 µL) of cDNA was amplified in a reaction with 1 × iQ SYBR Green Supermix (Bio-Rad Laboratories) and 200 nM gene-specific primers. The reaction conditions consisted of 40 cycles of a two-step PCR reaction with 94°C for 10 seconds and 68°C for 30 seconds on an iQ5 Real Time PCR Detection System (Bio-Rad Laboratories). Gene-specific primers used included glyceraldehyde-3-phosphate dehydrogenase (GAPDH) forward 5′-cttccgtgttcctacccccaatgt-3′, GAPDH reverse 5′-gcctgcttcaccaccttcttgatgt-3′, Myd88 forward 5′-cctgaccccactcgcagtttgt-3′, Myd88 reverse 5′-tgcgcgacttcagctccttca-3′, CCL2 forward 5′-tcatgcttctgggcctgctgt-3′, and CCL2 reverse 5′-ctcattgggatcatcttgctggtg-3′. The primers were synthesized by Integrated DNA Technologies and analyzed for specificity with the National Center for Biotechnology Information’s Basic Local Alignment Search Tool (BLAST) program. Standard curves were used to examine efficiency and reproducibility of each reaction, and melt curves were used to validate amplification of single products. The housekeeping gene GAPDH was used to establish normalized expression (ΔΔCT).
Western blot and coimmunoprecipitation
To analyze protein expression, 5 × 106 cells were washed three times with ice-cold phosphate-buffered saline (PBS), resuspended in 150 µL of buffer A (10 mM Hepes, 10 mM KCl, 0.1 mM EDTA, and 0.1 mM EGTA [all from Sigma]), supplemented with the protease inhibitors aprotinin, leupeptin, chymostatin, and pefabloc (Roche Molecular Biochemicals) and placed on ice. Following 15 minutes’ incubation, 10 µL of 10% Nonidet P-40 (Sigma) was added. The samples were vortexed for 10 seconds and centrifuged at 15,000 × g at 4°C for 1 minute. NuPAGE LDS sample buffer (Life Technologies) was added, and the supernatants containing the cytoplasmic proteins were stored at −20°C. The pellets were washed one time with buffer A, then resuspended in 75 µL buffer B (25% glycerol, 20 mM Hepes, 0.4 M NaCl, 1 mM EDTA, and 1 mM EGTA [all from Sigma]) supplemented with the protease inhibitors aprotinin, leupeptin, chymostatin, and pefabloc, and then sonicated for 30 seconds. After 10 minutes’ centrifugation at 15,000 × g, the supernatants containing the nuclear proteins were transferred to new tubes, NuPAGE LDS sample buffer was added, and the samples were stored at −20°C. For coimmunoprecipitation, proteins were isolated from 2 × 107 cells and the Immunocruz IP/WB Optima System (Santa Cruz Biotechnology) was used. For analysis using sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS–PAGE), gels (12.5%; Life Technologies) were loaded with 15 µL of proteins, electrophoresed, and transferred to polyvinylidene difluoride membranes (Life Technologies). The membranes were blocked at room temperature in PBS with 5% powdered milk (Carnation) and 0.05% Tween 20 (Sigma) for two hours. Primary antibodies (10 µg) specific for Myd88, IRAK2, IRAK4, Erk1/2, pErk1/2, p50, p52, p65, RelB, cRel, STAT5, and activating transcription factor (ATF2) (Santa Cruz Biotechnology) were added, and the blots were incubated at room temperature for one hour, followed by overnight incubation at 4°C. After washing four times with blocking buffer, a horseradish peroxidase-conjugated secondary antibody (Santa Cruz Biotechnology) was added, and the blots were incubated for two hours at room temperature. Following four washes, proteins were visualized by enhanced chemiluminesence on an Alpha Innotech Gel Documentation System (Alpha Innotech Corp). Densitometry data were normalized to actin and nuclear histone 3 levels, which showed similar patterns.
Viability and cell cycle analysis
The impact of Myd88 levels on cell viability was determined using trypan blue exclusion following study of the in vitro growth kinetics. To determine whether Myd88 affected the progression of cells through the cell cycle, propidium iodide (PI) staining was used. To synchronize the cell cycle, 1 × 105 cells were cultured in T75 cm2 tissue culture flasks in cRPMI minus FBS. After 24 hours, the medium was replaced with cRPMI, and the cells were harvested 48 hours later for cell cycle analysis. Following a wash with 10 mL cold PBS, the cells were resuspended in 200 µL cold PBS and then slowly added to 4 mL cold 70% ethanol while vortexing. Following 90 minutes’ incubation on ice, the cells were centrifuged at 450 × g, resuspended in 500 µL PI/RNase solution (BD Biosciences), and sent to Hershey Medical Center (Hershey, PA, USA) for analysis.
RNA-Seq
For RNA-Seq analysis, RNA was isolated from consMydlow, indMydlow, and the appropriate controls for each line using the Aurum Total RNA Mini Kit (Bio-Rad Laboratories) according to manufacturer’s instructions. After quantifying the RNA and checking the purity, RNA was shipped to GENEWIZ (South Plainfield, NJ, USA) for next-generation sequencing and RNA-Seq analysis. Following next-generation sequencing, the Fastq files were used to trim the ends to remove low-quality bases. After mapping to the mouse genome and calculating hit counts and expression values (reads per kilobase of exon per million) for each gene, differential analysis was conducted using CLC Genomics to identify genes that were up- or downregulated when Myd88 was inhibited in the tumor cells. The number of reads for consMydlow, control for consMydlow, indMydlow, and control for indMydlow were 72,624,237, 95,204,904, 86,891,625, and 92,649,213, respectively. Mean Q Scores for all these groups were 35, and the percentage of Q Scores ≥30 was >90.
Results
The myddosome complex is preformed in 4T1 tumor cells
We previously reported that in vitro growth of 4T1 could be decreased by treatment with blocking antibodies specific for TLR2, TLR4, or the receptor for advanced glycation end products, each of which is capable of signaling in a Myd88-dependent manner,20 and that targeting Myd88 affected CCL2 expression.19 These data suggested that there may be constitutive signaling through the Myd88 signaling cascade. Because TLR signaling leads to formation of the myddosome complex by bringing together Myd88, IRAK2, and IRAK4,2 we used coimmunoprecipitation to determine whether the myddosome complex was preformed in the 4T1 tumor cells. The data showed that upon immunoprecipitation of Myd88, both IRAK2 and IRAK4 coprecipitated (Fig. 1). Likewise, immunoprecipitating IRAK2 led to coprecipitation of Myd88 and IRAK4, and immunoprecipitating IRAK4 led to coprecipitation of Myd88 and IRAK2 (Fig. 1). These data support the contention that the myddosome complex was preformed in 4T1 tumor cells and therefore may be capable of constitutive Myd88-dependent signaling.
Figure 1.
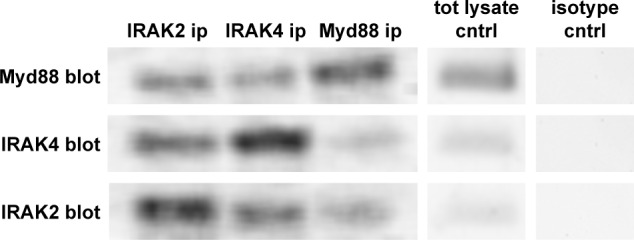
The myddosome complex is preformed in 4T1 murine mammary carcinoma. Antibodies specific for Myd88, IRAK2, or IRAK4 were used for immunoprecipitation (ip), and then Western blots were used to screen for association with the other components of the myddosome complex using Myd88, IRAK2, or IRAK4 antibodies (blot). The data represent one of three separate experiments. Baseline levels of Myd88, IRAK4, and IRAK2 (tot lysate cntrl) and isotype controls are also shown.
Decreasing Myd88 levels affect cytoplasmic Erk1/Erk2 levels and nuclear levels of NFκB and STAT5
Downstream of Myd88 lie several signaling pathways, including the Ras/mitogen-activated protein kinase and NFκB pathways. To begin to examine constitutive Myd88-dependent signaling in the tumor cells, we looked at some of these downstream effects, including cytoplasmic levels of Erk1/Erk2 and nuclear levels of several transcriptional factors. Examining the cytoplasmic levels of Erk1/Erk2 revealed that the tumor cells with decreased levels of Myd88 also exhibited decreased levels of Erk1 and Erk2. Both consMydlow and indMydlow cells showed a significant decrease in Erk1 and Erk2 levels (Fig. 2A and B). Phosphorylated Erk1/Erk2 levels were also decreased in the consMydlow and indMydlow cells, although the decrease was only significant for phosphorylated Erk2 in one of the cell lines (indMydlow; Fig. 2A and B). Next, to investigate other signaling pathways downstream of Myd88, we examined several transcriptional factors including NFκB, STAT5, and ATF2. The data revealed that decreasing Myd88 levels affected the nuclear levels of these transcriptional factors, with a significant decrease in the levels of NFκB p50, NFκB p52, and STAT5 in both consMydlow and indMydlow cells (Fig. 2C and D). Because modulating Myd88 levels influenced downstream signaling, these data support the contention that there was constitutive Myd88-dependent signaling in 4T1 tumor cells.
Figure 2.

Constitutive Myd88-dependent signaling contributes to Erk1/Erk2 levels and nuclear levels of NFκB and STAT5. Cytoplasmic (A, B) and nuclear (C, D) proteins were isolated from 4T1 cells expressing shRNA specific for Myd88 (consMydlow), a cell line expressing shRNA specific for Myd88 under a tetracycline-inducible promoter and treated with tetracycline (indMydlow), or the appropriate controls (cntrl). A representative blot and densitometry data from the average and standard error of at least three separate experiments are shown. Where indicated (*), P < 0.05 using Student’s t-test relative to control.
Tumor-derived CCL2 is produced in a Myd88-dependent manner
If constitutive Myd88-dependent signaling contributed to elevated levels of transcriptional factors in the nucleus, then changes in gene expression should also be evident. We previously reported that CCL2 was produced in a Myd88-dependent manner.19 Here, we validated and extended these findings using three different methods to target Myd88 in the tumor cells. For this purpose, we examined CCL2 gene expression using cells with reduced Myd88 levels as well as a Myd88-specific inhibitory peptide (MP) that would affect the function, but not the Myd88 protein or RNA expression. As expected, the consMydlow cells exhibited reduced Myd88 and CCL2 levels (Fig. 3A), and the indMydlow cells exhibited a dose-dependent decrease in CCL2, which paralleled the decrease in Myd88 upon treatment with tetracycline (Fig. 3B). 4T1 cells treated with MP also exhibited decreased CCL2 expression in a time- and dose-dependent manner (Fig. 3C and D). Collectively, these data revealed a direct correlation between Myd88-dependent signaling and CCL2 gene expression in the tumor cells.
Figure 3.

CCL2 expression is regulated by constitutive Myd88-dependent signaling. (A) Myd88 was decreased in a stable cell line expressing shRNA specific for Myd88 (consMydlow) and assessed for Myd88 and CCL2 expression by qRT-PCR relative to a cell line expressing shRNA specific for lacZ as a control (cntrl). (B) Myd88 and CCL2 expressions were assessed by qRT-PCR in a cell line expressing shRNA specific for Myd88 under a tetracycline-inducible promoter upon no treatment (cntrl) or treatment with different doses of tetracycline (indMydlow) for 48 hours. (C) 4T1 cells were treated with 100 µM control peptide (cntrl) or Myd88-specific inhibitory peptide for 24–72 hours and examined for CCL2 expression by qRT-PCR. (D) 4T1 cells were treated with different doses of the control peptide (CP) and Myd88-specific inhibitory peptide (MP) for 72 hours and examined for CCL2 expression by qRT-PCR. Data were normalized to GAPDH and expressed relative to the control. All experiments were repeated at least three times, and where indicated (*), P < 0.05 using Student’s t-test relative to control.
Constitutive Myd88-dependent signaling contributes to tumor growth, but not viability or cell cycle progression
To further examine the biological effects of Myd88-dependent signaling in the tumor cells, we next examined in vitro and in vivo growth using all three methods to inhibit Myd88-dependent signaling. The stable cell line expressing shRNA specific for Myd88 (consMydlow) exhibited a significant reduction in the in vitro and in vivo growth (Fig. 4A and B). Likewise, the indMydlow line exhibited a significant reduction in the in vitro growth when treated with tetracycline and in the in vivo growth when the mice were treated with doxycycline (Fig. 4C and D). We examined several different levels of tetracycline in vitro (0.25 to 2 µg/mL) and doxycycline in vivo (0.2 to 1.6 mg/mL) and found 2 µg/mL and 0.2 mg/mL had the most significant and reproducible effects respectively. 4T1 cells with normal Myd88 levels, but treated with MP also showed a significant reduction in the in vitro and in vivo growth (Fig. 4E and F). We examined several different doses of the inhibitory peptide (50 µM, 100 µM, 200 µM, and 1 mM) and found that 100 µM delivered into the tumor cells had the most significant and reproducible effects in vivo. Thus, in addition to modulating gene expression, decreasing Myd88-dependent signaling significantly reduced in vitro and in vivo growth of the tumor cells.
Figure 4.

Constitutive Myd88-dependent signaling contributes to tumor growth. In vitro and in vivo growth were examined over time using a stable cell line expressing shRNA specific for Myd88 (consMydlow; A, B). Growth was also examined for a cell line expressing shRNA specific for Myd88 under a tetracycline-inducible promoter upon treatment with 2 µg/mL tetracycline in vitro (indMydlow, C) or with 0.2 mg/mL doxycycline in vivo (indMydlow, D). 4T1 cells treated with 100 µM of a Myd88-specific inhibitory peptide (MP) was also followed for growth over time in vitro (E) and in vivo (F). All data are presented with the appropriate controls (cntrl) and represent the average and standard error of at least three separate experiments. For in vitro growth, where indicated (*), P < 0.05 using Student’s t-test relative to control at the 72-hour time point. For in vivo studies, at least five mice per group were used for each experiment, and where indicated (*), P < 0.05 using two-way ANOVA.
To begin to delineate a possible reason for the impact on growth, we next evaluated viability and cell cycle progression. Surprisingly, targeting Myd88 levels did not affect the viability of the tumor cells or the ability of the cells to progress through the cell cycle. Viability of all cells was similar at 72 hours, indicating that decreased growth was not attributable to cell death. consMydlow, indMydlow, and cells treated with MP exhibited 96%, 99%, and 94% viability compared to the controls, which showed 97%, 99%, and 96% viability, respectively (Fig. 5A). Although the percentage of cells in the G1, S, and G2 phases of the cell cycle were slightly different between the controls and the cells with reduced levels of Myd88, there were no consistent differences, indicating that Myd88 levels did not affect progression through the cell cycle (Fig. 5B). For instance, compared to their respective controls, more consMyd88low cells were in the S phase and fewer in the G1 and G2 phases of the cell cycle, while fewer indMydlow cells were in the S phase and more were in the G1 and G2 phases of the cell cycle (Fig. 5B). These data suggested that factors other than viability or cell cycle progression influenced growth of the tumor cells when Myd88 was targeted.
Figure 5.

Myd88 does not directly affect tumor cell viability or cell cycle progression.
Notes: (A) Following 72 hours of in vitro growth, consMydlow cells and indMydlow cells treated with 2 µg/mL tetracycline or 4T1 cells treated with 100 µM of a Myd88-specific inhibitory peptide (MP) were assessed for viability by trypan blue exclusion. (B) Following 72 hours of in vitro growth, the same cell lines were assessed for cell cycle progression. Comparisons were made to each of the appropriate controls (cntrl). All data represent the average and standard error of at least three separate experiments.
RNA-Seq analysis revealed a multifaceted role for Myd88-dependent signaling in the tumor cells
Finally, we used RNA-Seq in order to gain better understanding of how modulating Myd88 levels in the tumor cells affected cell signaling and growth. The number of reads for each sample ranged from 72 million to 95 million. For consMydlow cells, there were 3,955 differentially expressed genes (more than or less than a twofold change in gene expression) relative to its control, with 2,379 upregulated and 1,576 downregulated genes. For the indMydlow cells, there were 1,969 differentially expressed genes relative to its control, with 652 upregulated and 1,317 downregulated genes. We identified a total of 116 genes that were jointly upregulated and 137 genes that were jointly downregulated in both lines with reduced Myd88 levels.
The downregulated genes represent those that were normally expressed downstream of the Myd88-dependent signaling cascade in the tumor cells. Among others, we found 39 genes associated with cancer, 11 genes involved in adhesion, nine genes encoding solute carriers, and three genes involved in lipid metabolism. Downregulation of some of these genes upon inhibition of Myd88-dependent signaling in the tumor cells may help explain the decreased growth of the consMydlow and indMydlow lines. Table 1 shows some of the downregulated genes that are associated with breast cancer. Among these are genes associated with metastasis (Cdh3, Aldh1a3, Lgals4, and Vldlr), proliferation (Fgd5 and Rab3b), and poor outcome for patients with breast cancer (Folr1, Anxa8, and Tspyl5) (Table 1).11,13,14,21–26 Some of these genes have also been associated with other types of cancer, such as hepatocellular carcinoma (Dnajc6 and Lgals4), colorectal cancer (Lgals4), and ovarian cancer (Cdh3 and Folr1).27–31
Table 1.
Genes expressed in a Myd88-dependent manner in 4T1 cells.
| aconsMydlow | bindMydlow | cRELEVANT ASSOCIATED FUNCTION(S) | REFS | |
|---|---|---|---|---|
| Krt14 | −6.76 | −8.26 | Associated with a migratory phenotype in breast cancer. | 55 |
| Dnajc6 | −6.28 | −7.56 | Associated with progression of hepatocellular carcinoma. | 27 |
| Adra2a | −3.27 | −4.32 | Associated with breast cancer proliferation and tumor growth. | 12 |
| Fam83c | −2.48 | −3.88 | Associated with mammary epithelial cell transformation. | 56 |
| Vipr1 | −2.50 | −3.14 | Expressed by breast cancer. | 63 |
| Cdh3 | −8.87 | −2.07 | Associated with breast cancer invasion and metastasis. | 11 |
| Slco5a1 | −2.08 | −7.31 | Associated with the uptake of hormones during breast cancer development. | 57 |
| Fgd5 | −2.29 | −3.92 | Amplified in breast cancer and associated with cell proliferation. | 23 |
| Rab3b | −3.55 | −2.09 | Associated with breast cancer proliferation and invasion. | 24 |
| Folr1 | −2.04 | −3.31 | Expressed by double and triple negative breast cancers and associated with poor outcome for patients with breast cancer. | 13 |
| Aldh1a3 | −2.05 | −3.33 | Correlated with tumor stage for patients with triple negative breast cancer. | 14 |
| Ccdc170 | −2.24 | −2.64 | Associated with genetic rearrangements in ER+ breast cancer. | 64 |
| Lgals4 | −2.66 | −2.13 | Considered an angiogenesis and metastasis factor. | 21 |
| Anxa8 | −2.59 | −2.13 | Predicts poor survival for patients with breast cancer. | 25 |
| Tspyl5 | −2.18 | −2.12 | Predictor of poor outcome in patients with breast cancer. | 26 |
| Vldlr | −2.07 | −2.85 | Associated with metastasis in breast cancer. | 22 |
| Ppp1r16b | −2.02 | −2.39 | Contributes to angiogenesis by decreasing PTEN. | 65 |
Notes:
Relative downregulation of genes in consMydlow cells relative to control.
Relative downregulation of genes in indMydlow cells relative to control.
Select functions that have been attributed to the downregulated genes.
The upregulated genes included those that were normally repressed by Myd88-dependent signaling in the tumor cells. Among others, we found 18 genes that could function as tumor suppressors or help fight cancer, three genes involved in adhesion, one gene that encodes a solute carrier, and seven genes involved in lipid metabolism. Upregulation of some of these genes upon inhibition of Myd88-dependent signaling in the tumor cells may help explain the decreased growth of the consMydlow and indMydlow lines. Among this cohort of upregulated genes, we found several that have been reported to function as tumor suppressors (Arl11 and Dmp1) and proapoptotic factors (Dapk2 and Usp17la), in addition to a couple of genes that have been associated with inhibition of breast cancer (Psd4 and Idi1) and few involved in initiation of adaptive immune responses (Sectm1a, Nlrp10, and Rnf125) (Table 2).15,16,32–38
Table 2.
Genes repressed by Myd88-dependent signaling in 4T1 cells.
| aconsMydlow | bindMydlow | cRELEVANT ASSOCIATED FUNCTION(S) | REFS | |
|---|---|---|---|---|
| Slfn2 | 352.58 | 3.89 | Considered a negative regulator of growth. | 58 |
| Sectm1a | 6.49 | 11.34 | May function as a T cell co-stimulatory ligand. | 36 |
| Dapk2 | 6.80 | 3.53 | Can function as a pro-apoptotic protein kinase. | 33 |
| Psd4 | 4.97 | 3.44 | Can function as an antagonist for breast cancer. | 15 |
| Nlrp10 | 6.78 | 2.90 | Associated with dendritic cells and innate and adaptive immunity. | 37 |
| Rnf125 | 3.25 | 2.96 | Positive regulator of T cell activation. | 38 |
| Idi1 | 2.21 | 3.20 | Associated with breast cancer growth inhibition. | 35 |
| Creb3l3 | 5.98 | 2.12 | Growth suppressor for hepatocellular carcinoma. | 59 |
| Arl11 | 4.18 | 2.12 | May function as a tumor suppressor in lung cancer. | 32 |
| Dmp1 | 2.16 | 2.69 | May be considered a tumor suppressor in breast cancer. | 16 |
| Gng7 | 2.06 | 2.34 | Candidate tumor suppressor in Hodgkin lymphoma. | 60 |
| Usp17la | 2.06 | 2.00 | Can function as a pro-apoptotic factor and growth suppressor. | 34 |
Notes:
Relative upregulation of genes in consMydlow cells relative to control.
Relative upregulation of genes in indMydlow cells relative to control.
Select functions that have been attributed to the upregulated genes.
However, not all of the RNA-Seq data could help to explain the decreased tumor growth evident when Myd88 was inhibited. For instance, in addition to the genes mentioned above, there was upregulation in the expression of some genes that would be expected to favor tumor growth, such as Ptn, Tslp, Il1b, Mmp9, MMP13, Fos, and Hsd17b7, and downregulation of a few genes that would be expected to favor tumor growth, such as Psd3 and Tshz2 (Table 3).17,18,39–45 Collectively, these data revealed a multifaceted role for constitutive Myd88-dependent signaling in murine mammary carcinoma.
Table 3.
Additional genes influenced by Myd88-dependent signaling in 4T1 cells.
| aconsMydlow | bindMydlow | cRELEVANT ASSOCIATED FUNCTION(S) | REFS | |
|---|---|---|---|---|
| Ptn | 3.33 | 22.17 | May contribute to angiogenesis in breast cancer. | 39 |
| Tslp | 15.61 | 3.10 | Correlated with growth and metastasis of 4T1 tumors. | 40 |
| Mmp13 | 3.55 | 2.45 | Associated with breast cancer invasion and metastasis. | 41 |
| Il1b | 8.27 | 2.20 | Associated with breast cancer progression. | 18 |
| Fos | 2.55 | 2.10 | May contribute to tumor growth. | 42 |
| Mmp9 | 2.99 | 2.07 | Associated with breast cancer metastasis. | 17 |
| Hsd17b7 | 2.06 | 2.15 | Associated with estradiol synthesis in breast cancer. | 43 |
| Psd3 | −15.15 | −2.81 | Breast cancer candidate metastasis suppressor gene. | 44 |
| Tshz2 | −2.57 | −4.07 | May function as a tumor suppressor in breast and prostate cancer. | 45 |
Notes:
Relative up- or downregulated genes in consMydlow cells relative to control.
Relative up- or downregulated genes in indMydlow cells relative to control.
Select functions that have been attributed to the genes.
Discussion
We focused here on the Myd88-dependent signaling cascade exclusively in the tumor cells themselves. Previously, we described how 4T1 responded to TLR agonist treatment46,47 and showed that inhibiting TLR4 or Myd88 could affect the growth of tumor cells.19 In this study, we showed that Myd88-dependent signaling was present in 4T1 in the absence of TLR agonist treatment and that targeting this pathway was sufficient to inhibit in vitro and in vivo growth.
One of the first components of the Myd88-dependent signaling cascade is formation of the myddosome complex, and the presence of this complex in the tumor cells without addition of TLR agonists suggested constitutive signaling through Myd88. We also found evidence of Myd88-dependent signaling downstream of the myddosome complex, with altered Erk1/Erk2 protein levels. These data partially agree with another study that examined the role of Myd88 in tumor cells. Coste et al48 reported that Myd88 contributed to proliferation and cell cycle progression through the Ras/Erk signaling pathway. They found that blocking Myd88 decreased the number of cells in the S phase of the cell cycle, suggesting fewer cells were escaping G1.48 Although we also found a relationship between Myd88, growth, and Erk levels in the tumor cells, we did not find an alteration in cell cycle progression. One possible explanation for altered in vitro and in vivo growth in absence of a change in viability or cell cycle control is that there could be a change in Myd88-driven paracrine growth factors, which results in slowing of the cell cycle rather than causing disruption in progression through one of the checkpoints. It also appears as though a certain level of autocrine signaling through Myd88 is necessary for growth of the tumor cells because we have been unable to generate a tumor cell line that completely lacks Myd88-dependent signaling. Indeed, this was one of the main reasons we generated the tetracycline-inducible cell line.
For downstream signaling, we focused on transcriptional factors. Although there are several transcriptional factors downstream of the Myd88 signaling cascade, we investigated the ones that could drive CCL2 expression. Our data showed that Myd88-dependent signaling influenced nuclear levels of NFkB p50, p52, p65, cRel, RelB, STAT5, and ATF2. In addition to these, we looked at other CCL2 transcriptional factors such as ETS-1, which was present at below-detection levels, and C/EPB, which was not consistently modulated by Myd88 (data not shown). Nevertheless, the NFκB, STAT5, and ATF2 data provided additional evidence for constitutive Myd88-dependent signaling in 4T1 cells and helps explain the direct relationship between Myd88 and CCL2 expression. These data are particularly interesting considering the well-established relationship between breast cancer progression and CCL2.49–51 Our interest in CCL2 began with the finding that 4T1 constitutively produced this chemokine, that tumor-bearing mice possessed T-cells with desensitized CCL2 receptors, and that exposure to tumor-derived or recombinant CCL2 could negatively influence T-cell effector function.52,53 Perhaps more importantly, CCL2 participates in recruiting suppressor cells such as tumor-associated macrophages and myeloid-derived suppressor cells.54 Thus, there are several different ways through which Myd88-driven CCL2 expression could contribute to tumor progression in vivo.
In order to gain insight into the additional mechanisms by which Myd88-dependent signaling could contribute to tumor progression, we turned to RNA-Seq. To reduce the likelihood of identifying genes unique to a particular cell line and to increase the likelihood of identifying genes related to Myd88-dependent signaling, we focused on genes that were up- or downregulated in both the consMydlow and indMydlow lines. The downregulated genes represent those normally expressed in a Myd88-dependent manner by the tumor cells. We were particularly interested in genes that may contribute to tumor progression. Of the 137 genes that were jointly downregulated in both lines, we found 39 genes that were associated with cancer, and most of the genes shown in Table 1 have been known to be associated with breast cancer. In addition to the genes mentioned herein, we also found Krt14 (which has been shown to be associated with a migratory phenotype in breast cancer)55 Fam83c (which has been shown to be associated with mammary epithelial cell transformation),56 and Slco5a1, which may be associated with the uptake of hormones during breast cancer development.57
Genes upregulated during Myd88 suppression represent those normally suppressed in a Myd88-dependent manner by the tumor cells. We were particularly interested in genes that may help slow tumor progression if expression was increased. Of the 116 genes that were jointly upregulated in both lines, we found 12 genes that may fulfill this criteria. In addition to the genes mentioned, we found Slfn2 (which may be a negative regulator of growth),58 Creb3l3 (which may function as a growth suppressor for hepatocellular carcinoma),59 and Gng7 (which may be considered a candidate tumor suppressor in Hodgkin’s lymphoma.60
Although we are yet to decide which genes to pursue further, one approach is to rely upon the analysis conducted by the Database for Annotation, Visualization and Integrated Discovery (DAVID). Annotation clusters from the DAVID analysis of upregulated genes included several recurring terms, most notably cholesterol, sterol, and lipid biosynthesis, while downregulated genes included terms such as transmembrane, glycoprotein, and glycosylation. These data suggest that in murine mammary carcinoma, Myd88-dependent signaling may normally suppress genes related to cholesterol biosynthesis while upregulating the expression of genes that contribute to glycosylation and localization of transmembrane proteins. Thus, on the whole, the data support the contention that Myd88-dependent signaling in the tumor cell itself plays an important role in tumor progression.
One concern with respect to targeting Myd88 is the increased expression of Mmp9, Mmp13, and Il1b, as well as the decreased expression of Psd3 and Tshz2, which could exacerbate tumor growth.17,18,41,44,45 However, because inhibiting Myd88 in the tumor cells resulted in decreased tumor growth, it is likely that slower tumor growth was associated with one or more of the genes indicated in Tables 1 and 2 and that the effects of these genes outweigh the effects of genes that could exacerbate growth such as those indicated in Table 3.
Finally, in this study, we did not address what may be contributing to the Myd88-dependent signaling in the tumor cells. While mutations in Myd88 can drive TLR signaling and cancer,61 we found no mutations in Myd88 in the tumor cells (data not shown); yet, we did previously report on a possible autocrine mechanism of signaling. Tumor cells are known to secrete a range of damage-associated molecular pattern molecules (DAMPs), which are capable of mediating effects in a TLR-dependent manner.62 We found that high mobility group box 1 protein (HMGB1) was produced by 4T1 in a Myd88-dependent manner and that neutralizing extracellular HMGB1 resulted in 50% decrease in growth of the tumor cells.20 Though there are probably numerous other factors present in the serum in vivo and in the tissue culture medium in vitro, these data support the contention that there is at least one tumor-derived DAMP capable of mediating the effects in a TLR-dependent autocrine manner in the tumor cells.
Collectively, we believe the focus of this study is important because it concentrates on TLR-driven inflammation in a mouse model for stage IV breast cancer. Several previously unrecognized consequences of Myd88-dependent signaling have been revealed, and results from this study provide important information about how Myd88-dependent signaling in the tumor cells themselves may contribute to tumor progression. Ultimately, the main message from this study is that the consequence of Myd88-dependent signaling in cancer is not only very important but more complicated than is currently understood. While this study has generated more questions than it answered, we feel our results are important to recognize and justify further investigations due to the increasing interest in using TLR agonists in a tumor setting. While a few seminal papers6–8 have reported some now-well-established roles for Myd88-dependent signaling in a tumor setting, neither the importance nor the consequences of autocrine Myd88-dependent signaling in breast cancer cells themselves have been sufficiently delineated.
Acknowledgments
We thank Eric Ho for his assistance and advice regarding the RNA-Seq analysis.
Footnotes
ACADEMIC EDITOR: Goberdhan P. Dimri, Editor in Chief
PEER REVIEW: Two peer reviewers contributed to the peer review report. Reviewers’ reports totaled 607 words, excluding any confidential comments to the academic editor.
FUNDING: This work was supported by the National Institutes of Health (R15CA137858 to RAK) and by a grant from the Howard Hughes Medical Institute to Lafayette College under the Precollege and Undergraduate Science Education Program. The Department of Biology and Lafayette College Excel Scholar program also supported this work. The authors confirm that the funder had no influence over the study design, content of the article, or selection of this journal.
COMPETING INTERESTS: ASeifert could not be contacted to provide a statement regarding any potential conflicts of interest. Other authors disclose no potential conflicts of interest.
Paper subject to independent expert single-blind peer review. All editorial decisions made by independent academic editor. Upon submission manuscript was subject to anti-plagiarism scanning. Prior to publication all authors have given signed confirmation of agreement to article publication and compliance with all applicable ethical and legal requirements, including the accuracy of author and contributor information, disclosure of competing interests and funding sources, compliance with ethical requirements relating to human and animal study participants, and compliance with any copyright requirements of third parties. This journal is a member of the Committee on Publication Ethics (COPE).
Author Contributions
Conceived and designed the experiments: MJH, KCT, RAK. Analyzed the data: MJH, TP, JMR, KCT, RAK. Wrote the first draft of the manuscript: RAK. Contributed to the writing of the manuscript: MJH, JMR, KCT, RAK. Agree with manuscript results and conclusions: MJH, AS, KYB, VAP, TP, AS, JMR, KCT, ASE, MAC, RAK. Jointly developed the structure and arguments for the paper: MJH, RAK. Made critical revisions and approved final version: MJH, JMR, KCT, RAK. ASeifert could not be contacted to give approval of the final version for publication. All other authors reviewed and approved of the final manuscript.
REFERENCES
- 1.Brown J, Wang H, Hajishengallis GN, Martin M. TLR-signaling networks: an integration of adaptor molecules, kinases, and cross-talk. J Dent Res. 2011;90:417–427. doi: 10.1177/0022034510381264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Lin SC, Lo YC, Wu H. Helical assembly in the Myd88-IRAK4-IRAK2 complex in TLR/IL-1R signaling. Nature. 2010;465:885–890. doi: 10.1038/nature09121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Elinav E, Nowarski R, Thaiss CA, Hu B, Jin C, Flavell RA. Inflammation-induced cancer: crosstalk between tumours, immune cells and microorganisms. Nat Rev Cancer. 2013;13:759–771. doi: 10.1038/nrc3611. [DOI] [PubMed] [Google Scholar]
- 4.Helmy KY, Patel SA, Nahas GR, Rameshwar P. Cancer immunotherapy: accomplishments to date and future promise. Ther Deliv. 2013;4:1307–1320. doi: 10.4155/tde.13.88. [DOI] [PubMed] [Google Scholar]
- 5.Yu L, Wang L, Chen S. Dual character of toll-like receptor signaling: protumorigenic affects and anti-tumor functions. Biochim Biophys Acta. 2013;1835:144–154. doi: 10.1016/j.bbcan.2012.10.006. [DOI] [PubMed] [Google Scholar]
- 6.Rakoff-Nahoum S, Medzhitov R. Regulation of spontaneous intestinal tumorigenesis through adaptor protein Myd88. Science. 2007;317:124–127. doi: 10.1126/science.1140488. [DOI] [PubMed] [Google Scholar]
- 7.Naugler WE, Sakurai T, Kim S, et al. Gender disparity in liver cancer due to sex differences in Myd88-dependent IL-6 production. Science. 2007;317:121–124. doi: 10.1126/science.1140485. [DOI] [PubMed] [Google Scholar]
- 8.Chefetz I, Alvero AB, Holmberg JC, et al. TLR2 enhances ovarian cancer stem cell self-renewal and promotes tumor repair and recurrence. Cell Cycle. 2013;12:511–521. doi: 10.4161/cc.23406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Gao C, Kozlowska A, Nechaev S, et al. TLR9 signaling in the tumor microenvironment initiates cancer recurrence after radiotherapy. Cancer Res. 2013;73:7211–7221. doi: 10.1158/0008-5472.CAN-13-1314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Alakson CJ, Miller FR. Selective events in the metastatic process defined by analysis of the sequential dissemination of subpopulations of a mouse mammary tumor. Cancer Res. 1992;52:1399–1405. [PubMed] [Google Scholar]
- 11.Paredes J, Figueiredo J, Albergaria A, et al. Epithelial E- and P-cadherins: role and clinical significance in cancer. Biochim Biophys Acta. 2012;1826:297–311. doi: 10.1016/j.bbcan.2012.05.002. [DOI] [PubMed] [Google Scholar]
- 12.Pérez Piñero C, Bruzzone A, Sarappa MG, Castillo LF, Lüthy IA. Involvement of α2- and β2-adrenoceptors on breast cancer cell proliferation and tumour growth regulation. Br J Pharmacol. 2012;166:721–736. doi: 10.1111/j.1476-5381.2011.01791.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zhang Z, Wang J, Tacha DE, et al. Folate receptor α associated with triple-negative breast cancer and poor prognosis. Arch Pathol Lab Med. 2014;138:890–895. doi: 10.5858/arpa.2013-0309-OA. [DOI] [PubMed] [Google Scholar]
- 14.Opdenaker LM, Arnold KM, Pohlig RT, Padmanabhan JS, Flynn DC, Sims-Mourtada J. Immunohistochemical analysis of aldehyde dehydrogenase isoforms and their association with estrogen-receptor status and disease progression in breast cancer. Breast Cancer. 2014;6:205–209. doi: 10.2147/BCTT.S73674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zangari J, Partisani M, Bertucci F, et al. EFA6B antagonizes breast cancer. Cancer Res. 2014;74:5493–5506. doi: 10.1158/0008-5472.CAN-14-0298. [DOI] [PubMed] [Google Scholar]
- 16.Fry EA, Taneja P, Maglic D, Zhu S, Sui G, Inoue K. Dmp1α inhibits HER2/neu-induced mammary tumorigenesis. PLoS One. 2013;8(10):e77870. doi: 10.1371/journal.pone.0077870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Yousef EM, Tahir MR, St-Pierre Y, Gaboury LA. MMP-9 expression varies according to molecular subtypes of breast cancer. BMC Cancer. 2014;14:609. doi: 10.1186/1471-2407-14-609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Goldberg JE, Schwertfeger KL. Proinflammatory cytokines in breast cancer: mechanisms of action and potential targets for therapeutics. Curr Drug Targets. 2010;11:1133–1146. doi: 10.2174/138945010792006799. [DOI] [PubMed] [Google Scholar]
- 19.Egunsola AT, Zawislak CL, Akuffo AA, et al. Growth, metastasis, and expression of CCL2 and CCL5 by murine mammary carcinomas are dependent upon Myd88. Cell Immunol. 2012;272:220–229. doi: 10.1016/j.cellimm.2011.10.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Chalmers SA, Eidelman AS, Ewer JC, et al. A role for HMGB1, HSP60 and Myd88 in growth of murine mammary carcinoma in vitro. Cell Immunol. 2013;282:136–145. doi: 10.1016/j.cellimm.2013.04.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Chen C, Duckworth CA, Fu B, Pritchard DM, Rhodes JM, Yu LG. Circulating galectins −2, −4 and −8 in cancer patients make important contributions to the increased circulation of several cytokines and chemokines that promote angiogenesis and metastasis. Br J Cancer. 2014;110:741–752. doi: 10.1038/bjc.2013.793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.He L, Lu Y, Wang P, Zhang J, Yin C, Qu S. Up-regulated expression of type II very low density lipoprotein receptor correlates with cancer metastasis and has a potential link to β-catenin in different cancers. BMC Cancer. 2010;10:601. doi: 10.1186/1471-2407-10-601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Gatza ML, Silva GO, Parker JS, Fan C, Perou CM. An integrated genomics approach identifies drivers of proliferation in luminal-subtype human breast cancer. Nat Genet. 2014;46:1051–1059. doi: 10.1038/ng.3073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ye F, Tang H, Liu Q, et al. miR-200b as a prognostic factor in breast cancer targets multiple members of RAB family. J Transl Med. 2014;12:17. doi: 10.1186/1479-5876-12-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Stein T, Price KN, Morris JS, et al. Annexin A8 is up-regulated during mouse mammary gland involution and predicts poor survival in breast cancer. Clin Cancer Res. 2005;11:6872–6879. doi: 10.1158/1078-0432.CCR-05-0547. [DOI] [PubMed] [Google Scholar]
- 26.Epping MT, Meijer LA, Krijgsman O, Bos JL, Pandolfi PP, Bernards R. TSPYL5 suppresses p53 levels and function by physical interaction with USP7. Nat Cell Biol. 2011;13:102–108. doi: 10.1038/ncb2142. [DOI] [PubMed] [Google Scholar]
- 27.Yang T, Li XN, Li XG, Li M, Gao PZ. DNAJC6 promotes hepatocellular carcinoma progression through induction of epithelial-mesenchymal transition. Biochem Biophys Res Commun. 2014;455:298–304. doi: 10.1016/j.bbrc.2014.11.011. [DOI] [PubMed] [Google Scholar]
- 28.Cai Z, Zeng Y, Xu B, et al. Galectin-4 serves as a prognostic biomarker for the early recurrence/metastasis of hepatocellular carcinoma. Cancer Sci. 2014;105:1510–1517. doi: 10.1111/cas.12536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Watanabe M, Takemasa I, Kaneko N, et al. Clinical significance of circulating galectins as colorectal cancer markers. Oncol Rep. 2011;25:1217–1226. doi: 10.3892/or.2011.1198. [DOI] [PubMed] [Google Scholar]
- 30.Ko SY, Naora H. HOXA9 promotes homotypic and heterotypic cell interactions that facilitate ovarian cancer dissemination via its induction of P-cadherin. Mol Cancer. 2014;13:170. doi: 10.1186/1476-4598-13-170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Leung F, Dimitromanolakis A, Kobayashi H, Diamandis EP, Kulasingam V. Folate-receptor 1 (FOLR1) protein is elevated in the serum of ovarian cancer patients. Clin Biochem. 2013;46:1462–1468. doi: 10.1016/j.clinbiochem.2013.03.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Yendamuri S, Trapasso F, Calin GA. ARLTS1—a novel tumor suppressor gene. Cancer Lett. 2008;264:11–20. doi: 10.1016/j.canlet.2008.02.021. [DOI] [PubMed] [Google Scholar]
- 33.Michie AM, McCaig AM, Nakagawa R, Vukovic M. Death-associated protein kinase (DAPK) and signal transduction: regulation in cancer. FEBS J. 2010;277:74–80. doi: 10.1111/j.1742-4658.2009.07414.x. [DOI] [PubMed] [Google Scholar]
- 34.Ramakrishna S, Suresh B, Baek KH. Biological functions of hyaluronan and cytokine-inducible deubiquitinating enzymes. Biochim Biophys Acta. 2015;1855:83–91. doi: 10.1016/j.bbcan.2014.11.006. [DOI] [PubMed] [Google Scholar]
- 35.Huang L, Pardee AB. Suberoylanilide hydroxamic acid as a potential therapeutic agent for human breast cancer treatment. Mol Med. 2000;6:849–866. [PMC free article] [PubMed] [Google Scholar]
- 36.Howie D, Garcia Rueda H, Brown MH, Waldmann H. Secreted and transmembrane 1A is a novel co-stimulatory ligand. PLoS One. 2013;8(9):e73610. doi: 10.1371/journal.pone.0073610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Eisenbarth SC, Williams A, Colegio OR, et al. NLRP10 is a NOD-like receptor essential to initiate adaptive immunity by dendritic cells. Nature. 2012;484:510–513. doi: 10.1038/nature11012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Zhao H, Li CC, Pardo J, et al. A novel E3 ubiquitin ligase TRAC-1 positively regulates T cell activation. J Immunol. 2005;174:5288–5297. doi: 10.4049/jimmunol.174.9.5288. [DOI] [PubMed] [Google Scholar]
- 39.Lynn KD, Roland CL, Brekken RA. VEGF and pleiotrophin modulate the immune profile of breast cancer. Cancers (Basel) 2010;2:970–988. doi: 10.3390/cancers2020970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Erdmann RB, Gartner JG, Leonard WJ, Ellison CA. Lack of functional TSLP receptors mitigates Th2 polarization and the establishment and growth of 4T1 primary breast tumours but has different effects on tumour quantities in the lung and brain. Scand J Immunol. 2013;78:408–418. doi: 10.1111/sji.12106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Rizki A, Weaver VM, Lee SY, et al. A human breast cell model of preinvasive to invasive transition. Cancer Res. 2008;68:1378–1387. doi: 10.1158/0008-5472.CAN-07-2225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Milde-Langosch K. The Fos family of transcription factors and their role in tumourigenesis. Eur J Cancer. 2005;41:2449–2461. doi: 10.1016/j.ejca.2005.08.008. [DOI] [PubMed] [Google Scholar]
- 43.Shehu A, Albarracin C, Devi YS, et al. The stimulation of HSD17B7 expression by estradiol provides a powerful feed-forward mechanism for estradiol biosynthesis in breast cancer cells. Mol Endocrinol. 2011;25:754–766. doi: 10.1210/me.2010-0261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Thomassen M, Tan Q, Kruse TA. Gene expression meta-analysis identifies chromosomal regions and candidate genes involved in breast cancer metastasis. Breast Cancer Res Treat. 2009;113:239–249. doi: 10.1007/s10549-008-9927-2. [DOI] [PubMed] [Google Scholar]
- 45.Yamamoto M, Cid E, Bru S, Yamamoto F. Rare and frequent promoter methylation, respectively, of TSHZ2 and 3 genes that are both downregulated in expression in breast and prostate cancers. PLoS One. 2011;6:3.e17149. doi: 10.1371/journal.pone.0017149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Nair P, O’Donnell CM, Janasek K, et al. Lipopolysaccharide treated mammary carcinomas secrete proinflammatory chemokines and exhibit reduced growth rates in vivo, but not in vitro. Immunol Invest. 2009;38:730–748. doi: 10.3109/08820130903177810. [DOI] [PubMed] [Google Scholar]
- 47.Palha De, Sousa C, Blum CM, Sgroe EP, Crespo AM, Kurt RA. Murine mammary carcinoma cells and CD11c+ dendritic cells elicit distinct responses to lipopolysaccharide and exhibit differential expression of genes required for TLR4 signaling. Cell Immunol. 2010;266:67–75. doi: 10.1016/j.cellimm.2010.08.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Coste I, Le Corf K, Kfoury A, et al. Dual function of MyD88 in RAS signaling and inflammation, leading to mouse and human cell transformation. J Clin Invest. 2010;120:3663–3667. doi: 10.1172/JCI42771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Saji H, Koike M, Yamori T, et al. Significant correlation of monocyte chemoattractant protein-1 expression with neovascularization and progression of breast carcinoma. Cancer. 2001;92:1085–1091. doi: 10.1002/1097-0142(20010901)92:5<1085::aid-cncr1424>3.0.co;2-k. [DOI] [PubMed] [Google Scholar]
- 50.Lebrecht A, Grimm C, Lantzsch T, et al. Monocyte chemoattractant protein-1 serum levels in patients with breast cancer. Tumour Biol. 2004;25:14–17. doi: 10.1159/000077718. [DOI] [PubMed] [Google Scholar]
- 51.Ueno T, Toi M, Saji H, et al. Significance of macrophage chemoattractant protein-1 in macrophage recruitment, angiogenesis, and survival in human breast cancer. Clin Cancer Res. 2000;6:3282–3289. [PubMed] [Google Scholar]
- 52.Kurt RA, Baher A, Wisner KP, Tackitt S, Urba WJ. Chemokine receptor desensitization in tumor-bearing mice. Cell Immunol. 2001;207:81–88. doi: 10.1006/cimm.2000.1754. [DOI] [PubMed] [Google Scholar]
- 53.Vitiello PF, Shainheit MG, Allison EM, Adler EP, Kurt RA. Impact of tumor-derived CCL-2 on T cell effector function. Immunol Lett. 2004;91:239–245. doi: 10.1016/j.imlet.2003.12.009. [DOI] [PubMed] [Google Scholar]
- 54.Ben-Baruch A. The tumor-promoting flow of cells into, within and out of the tumor site: regulation by the inflammatory axis of TNFα and chemokines. Cancer Microenviron. 2012;5:151–164. doi: 10.1007/s12307-011-0094-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Sizemore GM, Sizemore ST, Seachrist DD, Keri RA. GABA(A) receptor pi (GABRP) stimulates basal-like breast cancer cell migration through activation of extracellular-regulated kinase 1/2 (ERK1/2) J Biol Chem. 2014;289:24102–24113. doi: 10.1074/jbc.M114.593582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Cipriano R, Miskimen KL, Bryson BL, Foy CR, Bartel CA, Jackson MW. Conserved oncogenic behavior of the FAM83 family regulates MAPK signaling in human cancer. Mol Cancer Res. 2014;12:1156–1165. doi: 10.1158/1541-7786.MCR-13-0289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Kindla J, Rau TT, Jung R, et al. Expression and localization of the uptake transporters OATP2B1, OATP3A1 and OATP5A1 in non-malignant and malignant breast tissue. Cancer Biol Ther. 2011;11:584–591. doi: 10.4161/cbt.11.6.14533. [DOI] [PubMed] [Google Scholar]
- 58.Katsoulidis E, Carayol N, Woodard J, et al. Role of Schlafen 2 (SLFN2) in the generation of interferon alpha-induced growth inhibitory responses. J Biol Chem. 2009;284:25051–25064. doi: 10.1074/jbc.M109.030445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Chin KT, Zhou HJ, Wong CM, et al. The liver-enriched transcription factor CREB-H is a growth suppressor protein underexpressed in hepatocellular carcinoma. Nucleic Acids Res. 2005;33:1859–1873. doi: 10.1093/nar/gki332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Giefing M, Arnemann J, Martin-Subero JI, et al. Identification of candidate tumour suppressor gene loci for Hodgkin and Reed-Sternberg cells by characterisation of homozygous deletions in classical Hodgkin lymphoma cell lines. Br J Haematol. 2008;142:916–924. doi: 10.1111/j.1365-2141.2008.07262.x. [DOI] [PubMed] [Google Scholar]
- 61.Wang JQ, Jeelall WS, Ferguson LL, Horikawa K. Toll-like receptors and cancer: Myd88 mutation and inflammation. Front Immunol. 2014;5:1–10. doi: 10.3389/fimmu.2014.00367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Srikrishna G, Freeze HH. Endogenous damage-associated molecular pattern molecules at the crossroads of inflammation and cancer. Neoplasia. 2009;11:615–628. doi: 10.1593/neo.09284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Thakur ML, Zhang K, Berger A, et al. VPAC1 receptors for imaging breast cancer: a feasibility study. J Nucl Med. 2013;54:1019–1025. doi: 10.2967/jnumed.112.114876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Veeraraghavan J, Tan Y, Cao XX, et al. Recurrent ESR1-CCDC170 rearrangements in an aggressive subset of oestrogen receptor-positive breast cancers. Nat Commun. 2014;5:4577. doi: 10.1038/ncomms5577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Obeidat M, Li L, Ballermann BJ. TIMAP promotes angiogenesis by suppressing PTEN-mediated Akt inhibition in human glomerular endothelial cells. Am J Physiol Renal Physiol. 2014;307:F623–F633. doi: 10.1152/ajprenal.00070.2014. [DOI] [PubMed] [Google Scholar]