

ABSTRACT
Translational GTPases (trGTPases) play key roles in facilitating protein synthesis on the ribosome. Despite the high degree of evolutionary conservation in the sequences of their GTP‐binding domains, the rates of GTP hydrolysis and nucleotide exchange vary broadly between different trGTPases. EF‐Tu, one of the best‐characterized model G proteins, evolved an exceptionally rapid and tightly regulated GTPase activity, which ensures rapid and accurate incorporation of amino acids into the nascent chain. Other trGTPases instead use the energy of GTP hydrolysis to promote movement or to ensure the forward commitment of translation reactions. Recent data suggest the GTPase mechanism of EF‐Tu and provide an insight in the catalysis of GTP hydrolysis by its unusual activator, the ribosome. Here we summarize these advances in understanding the functional cycle and the regulation of trGTPases, stimulated by the elucidation of their structures on the ribosome and the progress in dissecting the reaction mechanism of GTPases. © 2016 Wiley Periodicals, Inc. Biopolymers 105: 463–475, 2016.
Keywords: ribosome, translation, EF‐Tu, tRNA, decoding, GTP hydrolysis
INTRODUCTION
GTPases are molecular switches that alternate between two distinct conformations, active or inactive, depending on whether they are bound to GTP or GDP.1, 2, 3 The life cycle of a typical GTP‐binding protein involves interaction with guanine nucleotide exchange factors (GEFs) that regenerate the active form of the GTPase by facilitating the exchange of GDP for GTP (switch on signal) and GTPase activating proteins (GAPs) which trigger rapid GTP hydrolysis (switch off signal).4, 5 The structure of the GTP‐binding domain of all GTPases is evolutionarily conserved. Five conserved motifs, named G1 to G5, contact the nucleotide: the G1 motif, also known as P‐loop, holds the α‐ and β‐ phosphates of GTP. The G2 (switch I) and G3 (switch II) comprise flexible regions that have different structures in the GTP‐ or GDP‐conformation6; they make contacts with the γ phosphate and are parts of the coordination shell of a Mg2+ ion, an essential cofactor that helps to bind GTP and GDP. The switch regions are binding targets for the GAPs. Switch II also contains a key residue, often a glutamine or histidine, which contacts the catalytic water molecule and is thus essential for GTP hydrolysis. The G4 and G5 elements are responsible for binding the guanine base, thus ensuring substrate specificity.
One important family of the GTP binding proteins are translational GTPases (trGTPases),7 a group of factors that facilitate and control protein synthesis on the ribosome. Translation entails four distinct phases: initiation, elongation, termination, and recycling. Every phase comprises several checkpoints that allow the ribosome to control protein production and to achieve the optimal speed and fidelity of translation. These steps are regulated by trGTPases. In bacteria, trGTPase initiation factor (IF) 2 recruits the initiator fMet‐tRNAfMet to the P site of the small ribosomal subunit during initiation, thereby controlling correct reading frame selection. Translation elongation entails repetitive cycles of aminoacyl‐tRNA (aa‐tRNA) selection, peptide bond formation and movement of the ribosome on the next codon of the mRNA. A trGTPase elongation factor (EF) Tu (EF‐Tu) is responsible for the delivery to the A site of the ribosome of all elongator aminoacyl‐tRNAs (aa‐tRNAs), except for the Sec‐tRNASec which requires a specialized EF, SelB. After peptide bond formation EF‐G catalyzes translocation of the ribosome along the mRNA. The cycles of translation elongation continue until a stop codon is reached, which leads to the translation termination phase. The newly synthesized protein is released from the tRNA and the ribosome; release factor (RF) 3 is a GTPase involved in termination. Finally, during the recycling phase the ribosome complex is split into the ribosomal subunits with the help of EF‐G in order to start a new translation cycle. The GTPase activity of these factors is promoted by the ribosome which acts as a GAP.
Bacterial elongation and termination factors EF‐Tu, SelB, EF‐G, and RF3 have close homologs in eukaryotic organisms, eEF1A, EFsec, eEF2, and eRF3, respectively, which fulfill similar functions and also use the ribosome as a GAP. In contrast, the initiation phase is controlled differently in bacteria and eukaryotes. The initiator is delivered to the eukaryotic ribosome by eIF2, a trGTPase that has no similarity to bacterial IF2 and is activated by a specialized GAP, eIF5, rather than by the ribosome.8 The homolog of bacterial IF2, eIF5B, is a factor that facilitates joining of the ribosomal subunits at the end of the initiation phase9, 10; similarly to IF2, this GTPase is activated by the ribosome. All trGTPases share similar structural elements that extend beyond the nucleotide binding region. The G domain is usually followed by one or two β‐barrel domains7, 11 (Figure 1). For example, EF‐Tu, which has been a model trGTPase for more than 30 years ever since its structure was solved,12, 13, 14 consists of three domains: the GTP binding domain 1 (G domain) and the β‐barrel domains 2 and 3; their relative position is drastically different in the GTP‐ and GDP‐form. Many other trGTPases contain one or multiple additional domains that are usually unique for the given factor.
Figure 1.
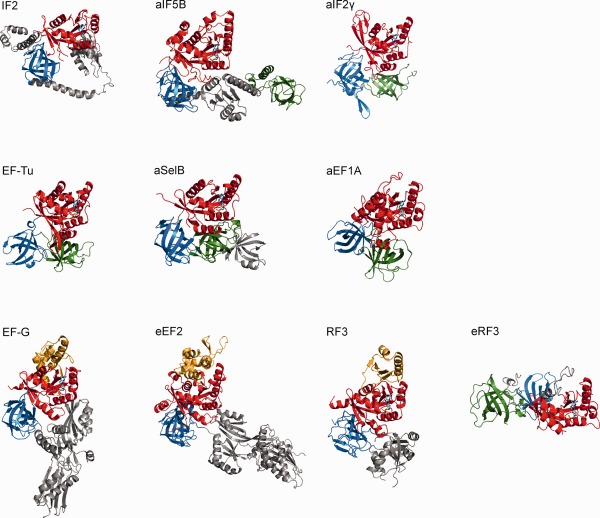
Structural overview of trGTPases. Crystal structures of trGTPases (PDB IDs: IF2 (4KJZ); aIF5B (1G7S); aIF2γ (1KK3); EF‐Tu (1EFC); aSelB (4AC9); aEF1A (1SKQ); EF‐G (2EFG); eEF2 (1N0U); RF3 (2HSE); eRF3 (1R5N)) in the complex with GDP (except for eEF2, which is shown in the apo‐form) were aligned on the P‐loop and GDP to highlight the structural conservation of the nucleotide binding (red) and the adjacent β‐barrel domains (blue and green, respectively). The G’ extension is represented in orange, the GDP molecule in cyan. Additional domains specific for each trGTPase are represented in gray.
Except for eIF2, which binds to the small ribosomal subunit and is released upon subunits joining, trGTPases bind to the ribosome at the so‐called GTPase associated center (GAC) located on the large ribosomal subunit (Figure 2), which comprises the sarcin‐ricin loop (SRL) of 23S rRNA (residues 2653–2667, E. coli numbering is used throughout the text), as well as ribosomal proteins L10 and L7/12 (P0 and P1/P2 in eukaryotes, respectively). L10 and the multiple copies of L7/12 (4, 6, or even 8 copies depending on the organism15) form the ribosomal L12 stalk that recruits all trGTPases to the ribosome16 and appears to accelerate GTP hydrolysis, at least of EF‐Tu and EF‐G.17, 18 Upon docking onto the GAC, the nucleotide pocket of trGTPases is positioned at the interface with the SRL; recent structures of EF‐Tu and EF‐G trapped on the ribosome in the pre‐hydrolysis state show the conserved histidine (His84 in EF‐Tu) in the switch II contacting the phosphate group of base A2662.19, 20, 21 This review summarizes our current knowledge on the trGTPases and presents the recent advances in understanding the GTPase activity of EF‐Tu. Because the structure of the nucleotide binding pocket is conserved in all trGTPases, this mechanism may be common for most translation factors whose GTPase activity is facilitated by the ribosome.
Figure 2.

trGTPases bind to the GAC center of the ribosome. Structure of the complex EF‐Tu·GDPCP·aa‐tRNA trapped on the programmed ribosome before GTP hydrolysis.20 In (a), the ribosome elements constituting the GAC are highlighted. The color code for EF‐Tu is the same as in Figure 1; the A/T aa‐tRNA is shown in magenta, the P‐site tRNA in purple, the E‐site tRNA in dark purple. (b) Close‐up view of the nucleotide binding pocket of EF‐Tu: the residues important for catalysis of GTP hydrolysis are shown as sticks; the catalytic water and the Mg2+ ion are shown in red and green, respectively (PDB: 2XQD and 2XQE).
NUCLEOTIDE BINDING AND EXCHANGE
With the exception of EF‐Tu, trGTPases, including IF2, SelB, EF‐G and their eukaryotic homologs typically bind GTP with a similar or higher affinity than GDP (Table 1). Nucleotide exchange occurs spontaneously due to the higher concentration of GTP in the cell. In contrast, EF‐Tu has a much higher affinity for GDP than for GTP.22, 23 Because the dissociation rate of GDP from EF‐Tu is very slow compared to the rate of protein elongation, which would delay recycling of EF‐Tu during elongation, formation of the active EF‐Tu‐GTP requires a GEF, EF‐Ts, which increases the GDP dissociation rate by more than four orders of magnitude.23 This feature is conserved also in eEF1A, which requires eEF1B as a GEF,24 and eIF2 which uses eIF2B to catalyze nucleotide exchange. Given the structural conservation of trGTPases, it is not immediately clear why would some of them need a GEF and others not. However, because EF‐Tu, eEF1A, and eIF2 have to control the delivery of aa‐tRNAs to their respective matching (cognate) codon, a stringent switch‐like response might provide a tighter quality control. Furthermore, the need for a GEF correlates with the structural differences between the GTP‐ and GDP‐bound forms of a trGTPase. For example, in the GTP conformation the three domains of EF‐Tu/eEF1A are compacted by interactions of the domain 1 with domains 2 and 3; this structure allows EF‐Tu to bind aa‐tRNA to form a complex EF‐Tu–GTP–aa‐tRNA.25, 26 The high affinity of binding ensures that at the cellular concentrations of EF‐Tu and aa‐tRNA,27, 28 virtually all the aa‐tRNA is bound in the ternary complex. The conformation switch triggered by GTP hydrolysis propagates from the nucleotide pocket to the rest of the protein, inducing a large rotation of domain 1 with respect to domains 2 and 313, 29; this “open” conformation is incompatible with aa‐tRNA binding. In contracts to EF‐Tu, EF‐G, IF2/eIF5B and SelB apparently do not undergo such dramatic GTP/GDP conformational switches; rather, the rearrangements are local and involve domain interfaces.30, 31, 32
Table 1.
Binding of Different trGTPases to GTP and GDP
| GTP | GDP | GEF | T °C | GTPase | Ref | |||||
|---|---|---|---|---|---|---|---|---|---|---|
| trGTPase | k on (M−1 s−1) | k off (s−1) | K d (M) | k on (M−1 s−1) | k off (s−1) | K d (M) | k GTP s−1 | |||
| IF2 | 4 × 105 | 15 |
3.8 × 10−5‐
7.1 × 10−6 |
1.4 × 10−6 | − | 20° | 33 | 128,129 | ||
| EF‐Tu | 5.0 × 105 | 0.03 | 6.0 × 10−8 | 2 × 106 | 0.002 | 1 × 10−9 | EF‐Ts | 20° | >500 | 130 |
| EF‐G | 5.8 × 105 | 13 | 2.2 × 10−5 | 4.0 × 10−5 | − | 37° | 250 | 45 | ||
| SelB | 1.7 × 105 | 0.1 | 6 × 10−7 | 1.8 × 106 | 15 | 8.3 × 10−6 | − | 20° | 131,132 | |
| RF3 | 1.2 × 109 | 24a | 2.0 × 10−8 | 2.7 × 107 | 0.13 | 5 × 10−9 | − | 37° | 0.15 | 41 |
| eIF2 | >5.8 × 105 | > 0.1 | 1.7 × 10−6 | 1.6 × 105 | 0.2 | 2.0 × 10−8 | eIF2B | 26° | 119 | |
| eIF5B | 1.5 × 106 | 22 | 1.5 × 10−5 | 4.3 × 106 | 9.6 | 2.3 × 10−6 | − | 25° | 133 | |
| eEF1A | 9 × 104 | 0.1 | 1.1 × 10−6 | 3.3 × 105 | 0.13 | 4 × 10−4 | eEF1B | 20° | 24 | |
| eEFSec | 1.1 × 10−7 | 3.3 × 10−7 | − | 30° | 134 | |||||
| eEF2 | 3 × 10‐6 | 2.5 × 10‐6 | 25° | 135 | ||||||
| eRF3 | 7 × 105 | 13 | 2.3 × 10−5 | 3.1 × 106 | 2.4 | 8 × 10−7 | − | 25° | 136 | |
Measured with GDPNP.
Surprisingly, some trGTPases can adopt different conformations independent of the nucleotide‐bound form. Crystal structures of isolated EF‐G captured the factor in an elongated conformation with domain arrangement and interactions similar in the GTP‐ and GDP‐bound form,31 whereas a recent single molecule FRET study suggested that in solution EF‐G–GTP may adopt a compact conformation with a quite different domain arrangement compared to the elongated structure observed in the crystal.33 The compact form may bind to the ribosome and rearrange into the elongated structure upon GTP hydrolysis and tRNA translocation.34 The same seems to apply to IF2/eIF5B as well: in free IF2 the relative orientation of the GTP‐binding domain and the domain analogous to domain 2 of EF‐Tu remains the same regardless of the nucleotide bound; the difference pertains to the orientation of the IF2‐specific domain C1. The structure of IF2 changes drastically upon binding to the ribosome, similarly to EF‐G.35, 36, 37
Despite the different structures of EF‐Ts, eEF1B, and eIF2B (and other GEFs), the mechanism of nucleotide exchange is conserved among trGTPases and other GTPases families such as Ras: in every case the GEF introduces a conformational change of the nucleotide pocket, thereby disturbing the coordination shell of the Mg2+ ion38, 39 and initiating GDP release. Nucleotide exchange in EF‐Tu proceeds through formation of a complex EF‐Tu–GTP–EF‐Ts. In the complex, the binding of Mg2+ ion at the nucleotide binding pocket is destabilized; removal of the Mg2+ ion alone reduces the affinity for GDP by two orders of magnitude.23 After GDP release, GTP enters to form the EF‐Tu–GTP–EF‐Ts complex. Finally, the accommodation of Mg2+ ion is required for the transition to the conformation from which EF‐Ts is released40 (Figure 3).
Figure 3.

Nucleotide exchange mechanism in EF‐Tu. Schematic of the nucleotide exchange pathway catalyzed by EF‐Ts. Binding of EF‐Ts to EF‐Tu–GDP causes dissociation of the Mg2+ ion, which initiates GDP dissociation. GTP is then loaded due to its higher cellular concentration compared to GDP. The change in the conformation into the GTP‐bound form occurs upon rebinding of Mg2+ and causes the dissociation of EF‐Ts.23, 40
Curiously, the ribosome itself can accelerate nucleotide exchange on RF3. Initial reports suggested that (i) the binding affinity of RF3 for GDP is much higher than for GTP and (ii) the ribosome in a state that is committed to peptide release can increase its nucleotide exchange rate by at least 100‐fold.41, 42 The following work confirmed that the ribosome in complex with RF1 or RF2 (the factors that promote peptide release during termination) can accelerate nucleotide exchange in RF3, even though the intrinsic dissociation rate of the RF3–GDP complex is high enough to support in vivo termination rates.41 However, the different binding affinity of RF3 for GTP and GDP turned out to be smaller than initially estimated.41, 43 Thus, in the cell RF3 is predominantly in the GTP‐bound form. Apparently, rapid nucleotide exchange in RF3 on the ribosome may act as a backup mechanism for the rare cases when RF3–GDP happens to bind to the termination complex.
Contrary to RF3, GTP binding to IF2 and EF‐G is stabilized by the ribosome.44, 45 This may reflect structuring of the switch regions of the factors or closing of the nucleotide binding pocket upon the interaction with the ribosome.19, 36, 46, 47 Notably, these interactions seem different for RF3, whose G domain binds to the ribosome in a markedly rotated conformation compared to EF‐Tu and EF‐G, which impedes the interaction of the crucial histidine (H92 in RF3) with the SRL.48 Thus, the ribosome may change the nucleotide binding properties and the GTPase activity of trGTPases depending on the details of their docking to the SRL.
MULTIPLE PATHWAYS OF GTP HYDROLYSIS
Upon binding to their GAP, the ribosome, trGTPases rapidly hydrolyze GTP, which is required to maintain high speed of translation. EF‐Tu and EF‐G have evolved exceptionally rapid GTPase activities in the presence of their cognate ribosome complex, with estimated rates at 37°C of >500 s−1 49, 50, 51 and 250 s−1,52 respectively. Several models have been proposed for the mechanism of GTP hydrolysis by EF‐Tu and other GTPases based on structural, theoretical and biochemical studies.20, 53, 54, 55, 56, 57, 58, 59, 60, 61, 62 In general, the reaction proceeds through a nucleophilic attack of a water molecule on the γ‐phosphate of GTP. For the reaction to become efficient, the water molecule has to be activated. In principle, this could be achieved by employing a general base that abstracts a proton and thus increases the nucleophilicity of the attacking group (Figure 4a). Alternatively, the GTP molecule itself may initiate the reaction by abstracting a proton from the water molecule, in a “substrate assisted” mechanism60, 63 (Figure 4b). Depending on the presence or absence of a stable intermediate in the reaction coordinate, fully associative (AN + DN), fully dissociative (DN + AN) or concerted, SN2 (ANDN) pathways are possible. Furthermore, the extent of bond cleavage and bond formation determines the structure of the transition state (TS) – associative or dissociative – and its symmetry (Figure 4c). The mechanistic pathway of the cleavage reaction itself is controversial; the evidence supporting each of the proposed models for GTP hydrolysis in solution and on several enzymes has been extensively reviewed elsewhere54, 62, 64, 65, 66, 67, 68 and is thus not discussed here. It is however important to keep in mind that in an associative‐like TS, bond formation dominates over bond cleavage, and thus the negative charge is expected to accumulate on the γ phosphate, while in a dissociative‐like TS, where bond cleavage dominates over bond formation, the charge would mainly build up on the β phosphate, and in particular on the β‐γ bridging oxygen of the nucleotide. The distinct charge accumulation has important consequences for enzyme catalysis, as the GTPase would have to use different residues in the switch regions and the P‐loop element to stabilize the TS.
Figure 4.

Models for GTP hydrolysis of EF‐Tu on the ribosome. (a) General base mechanism; (b) substrate assisted pathway; (c) Two‐dimensional More O'Ferral‐Jencks plot for an ANDN reaction. The reaction coordinate proceeds from the lower left (ground state, GS) to the upper right corner (product state, PS). Bond cleavage and bond formation occur along the x and y axes, respectively. The TS can be located anywhere in the area, indicating that GTP hydrolysis can proceed via a continuum of reaction pathways. The farthermost cases of AN + DN and DN + AN will have additional intermediates in the upper left and lower right corners, respectively. Adapted from Ref. 65.
Biochemical studies of EF‐Tu guided by structures and comparisons to other GTPases identified two key residues that are required for its GTPase activity on the ribosome, His8469, 70, 71, 72, 73, 74, 75, 76, 77 and Asp21.73, 78 His84 in the switch II is conserved in all trGTPases. Mutation of His84 to alanine (His84Ala) completely abolishes the acceleration of GTP hydrolysis by the ribosome.71, 73 Due to the neutral pKa of histidine in solution and its position close to the catalytic water molecule, as seen from the structure of the ribosome‐bound EF‐Tu in the pre‐hydrolysis state20, 79, 80 (Figure 2b), His84 was proposed to act as a general base.14, 20, 59, 72 This model was initially supported by the finding that substitution of His84 with a glutamine (His84Gln), another good proton acceptor, does not completely abolish the GTPase activity of EF‐Tu.69, 70 However, biochemical data showed that hydrolysis of GTP and of the slowly hydrolysable analog GTPγS on the ribosome is not pH‐dependent over the neutral range, where the side chain of His84 is expected to ionize71, 73; thus, the general base mechanism seems unlikely at least in the presence of the ribosome.55 Computation analysis of the relative charges in the vicinity of the nucleotide in the EF‐Tu–ribosome complex suggested, in fact, that the pKa of the side chain of His84 would be significantly upshifted (>9) due to the presence of the negatively charged nucleotide and the interactions with the SRL of 23S rRNA.53, 56 GTP would also repel the activated hydroxide ion, thereby increasing the overall activation energy of the general base reaction.56 Also mutation of EF‐Tu Asp21 to alanine (Asp21Ala) reduces the GTPase rate by three orders of magnitude.73 As with His84 mutations, the chemistry itself is impaired, rather than the preceding conformational rearrangement that leads to the GTPase activation. The inhibitory effect of Asp21 replacements resembles that of His84 mutants and suggests that Asp21 also contributes to the chemistry step and thus can be regarded as an additional catalytic residue.
Given that the general base mechanism—as originally proposed—appears unlikely, the reaction may instead proceed via the substrate‐assisted mechanism which has been originally proposed for the Ras GTPase based on linear free energy relationships, and later by theoretical simulation of the reaction in Ras and EF‐Tu.53, 56, 60, 81, 82 According to this model, His84 does not participate in catalysis directly, but contributes to precise positioning of the water molecule with respect to the γ phosphate (also referred to as an “allosteric” function of His8453, 54). Comparison of the structures of free and ribosome‐bound ternary complex EF‐Tu–GDP(N/C)P–aa‐tRNA suggests that the active site of EF‐Tu does not require large conformational changes to reach the activated state: in fact, only a ∼90° rotation of the side chain of His84 is sufficient to induce a GTPase‐prone structure.20, 83 This structural rearrangement occurs only upon interaction of His84 with the negatively charged phosphate of A2662 in the SRL, which results in the large pKa increase mentioned above. The structure of the GTPase TS of EF‐Tu is difficult to deduce based on biochemical evidence alone. However, the lack of kinetic solvent isotope effect (KSIE)73 suggests that proton transfer does not occur at the rate‐limiting step, which speaks in favor of a concerted pathway, where proton abstraction and hydroxide attack are expected to occur concomitantly. As such, the lack of KSIE might also be compatible with a dissociative mechanism, where phosphate cleavage is expected to occur at the rate‐limiting step. However, the large contribution of His84 to catalysis is indicative of a charge accumulation on the γ phosphate. It seems, thus, that the concerted, substrate‐assisted catalysis is the most likely mechanism for GTP hydrolysis in the presence of the ribosome.
THE INTRINSIC GTPASE OF EF‐TU
Similarly to many other GTPases, EF‐Tu possesses a very low intrinsic GTPase activity in the absence of its GAP, with a rate in the order of 10−5 s−1.73 Although probably irrelevant for its biological function, the intrinsic activity of EF‐Tu has been extensively studied in the past using a range of biochemical techniques, as it provides an insight into the basic mechanism of catalysis. Monovalent and divalent ions stimulate the spontaneous GTP hydrolysis,84, 85 which raised the interesting possibility that EF‐Tu might act as a potassium‐activated GTPase,78 such as MnmE and FeoB.86, 87 In these GTPases, a conserved Asn residue in the P‐loop is responsible of coordinating the catalytic K+ ion. Noteworthy, the structurally related and invariant Asp21 is found in the same position in EF‐Tu. This residue is conserved among trGTPases, with the notable exception of the eIF2 γ‐subunit, which is, so far, the only known trGTPase that is not activated by the ribosome, but by another protein factor.8 The recent crystal structure of eIF5B and a careful analysis of previously determined structures of the archaeal aEF1A identified a monovalent ion bound between the conserved Asp and an invariant Gly residue in the switch II region.78 Mutations of Asp21 in eIF5B and EF‐Tu abolish the K+ simulation, indicating that Asp21 EF‐Tu mutants are not able to coordinate the monovalent ion. However, the mutant proteins still show a measurable GTPase activity,73, 78 which contrasts with the idea that EF‐Tu might behave as a metal ion‐activated GTPase. Furthermore, in the presence of the ribosome, no stimulation by K+ ions was observed,73 in agreement with the notion that the K+ ion is easily exchanged by a water molecule or a Mg2+ ion upon binding to the ribosome, as suggested by structural and theoretical studies.54, 58
Recently, we analyzed the GTPase activity of three variants of EF‐Tu carrying the His84Ala, His84Gln or His84Arg mutations.73 The mutations impair the GTPase activity of EF‐Tu stimulated by the ribosome. However, in the absence of the ribosome the mutants hydrolyze GTP with the same rate as wild‐type EF‐Tu.73 Taking into account the structure of free EF‐Tu, which shows the catalytic histidine shielded from the water molecule by the “hydrophobic gate” constituted by Val20 and Ile61,14 the lack of effect of His84 replacements might indicate that GTP hydrolysis on and off the ribosome follows different mechanisms. A dissociative‐like mechanism would be more likely in the free protein, with His84 pointing to the solvent and the charged Asp21 being neutralized by the K+ ion. This would ultimately explain why we and others failed to identify a single catalytic residue responsible for the intrinsic GTPase, as the negative charge developing on the β‐γ bridging oxygen would be stabilized efficiently by the multiple main‐chain interactions with the P‐loop residues and the side chain of Lys24, a charged residue in the vicinity of β‐ and γ‐phosphates, as already suggested for the intrinsic activity of Ras based on Fourier transform infrared spectroscopy data.88
THE RIBOSOME AS A GAP
The weak GTPase activity of EF‐Tu is dramatically stimulated by binding to the ribosome. The maximum stimulation occurs when the aa‐tRNA in the EF‐Tu–GTP–aa‐tRNA complex is cognate (e.g., fully matching) to the codon in the A site, which increases the GTPase rate by about 6 orders of magnitude.49, 50, 51 In comparison, when the codon is near‐ or non‐cognate to the aa‐tRNA, the extent of the GTPase activation by the ribosome is much smaller, 2 to 4 orders of magnitude. The programmed ribosome thus acts as a GAP for EF‐Tu and the question is how the ribosome contributes to catalysis. For members of other families of GTP binding proteins, solving the structures of several GTPase–GAP complexes in the presence of TS analogs, such as aluminum tri‐ or tetra‐fluorides, allowed elucidation of the distinct mechanisms of GAP activation.4, 5 Unfortunately, EF‐Tu does not bind such fluorine compounds89, 90, 91; instead, the TS has to be inferred from the structures of complexes trapped with ground state analogs. In the case of small GTPases such as Ras, the GAP introduces an arginine residue—known as “arginine finger” (Arg finger)—directly into the nucleotide binding pocket, where it stabilizes the charges developing on the β‐γ bridging oxygen of GTP (Figure 5a). For this interaction to occur, the presence of two adjacent glycine residues, Gly12 and Gly13, in the P‐loop is required, as any other side chain would sterically hinder the Arg finger binding92; in fact, mutations of either Gly12 or Gly13 abolish GTPase activation.93 While Gly12 is conserved in all known G proteins, Gly13 of Ras is replaced by Asp21 in EF‐Tu and other trGTPases (Figure 5b), making an approach of an external Arg finger unlikely. Furthermore, an exhaustive search for potential Arg residues in the vicinity of the nucleotide in the trGTPases or the ribosome, e.g., in the ribosomal protein L7/12, showed that the Arg finger mechanism is not likely for EF‐Tu and EF‐G.17, 18
Figure 5.
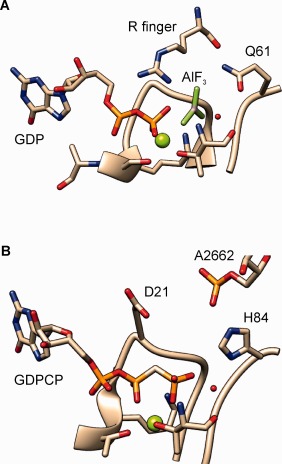
The active site of Ras and EF‐Tu in complex with the GAP. (a) Structure of the transition state of GTP hydrolysis in H‐Ras in complex with the RasGAP (PDB: 1WQ1) showing the Arg‐finger reaching the β‐γ bridging oxygen of the nucleotide; (b) Structure of the active site of EF‐Tu bound to the programmed ribosome in the pre‐hydrolysis state (PDB: 2XQD and 2XQE). The nucleophilic water molecule is highlighted in red, the Mg2+ in green.
Comparison of the structures of the activated Ras–RasGAP complex92 and EF‐Tu bound to the ribosome in the presence of mRNA and cognate aa‐tRNA20, 94 shows that, although the ribosome does not introduce an Arg finger into the active site of EF‐Tu, it does contact the nucleotide pocket via the SRL of the large ribosomal subunit. In particular, the interaction of the phosphate group of A2662 with the side chain of His84 is likely responsible for the stabilization of the rotated conformation of His84 towards the nucleotide in the activated state.20 This is in agreement with the results of atomic mutagenesis studies, where replacement of one of the two nonbridging phosphate oxygens of A2662 with a methyl group resulted in a complete loss of GTPase stimulation of EF‐G by the ribosome.95 Also Asp21 might take part in the stabilization of the TS by SRL,58 which would explain why this residue is present in trGTPases, but not in Ras‐like GTPases. Recent high‐resolution structures of the ribosome‐bound EF‐G show the side chain of the respective Asp residue coordinating a Mg2+ ion close to the crucial A2662, indicating that the GTPase‐activated state might also involve a rearrangement of Asp21.19, 21 Molecular dynamics and free energy calculations suggested that the opposite charge of the side chain of Asp21 and His84 would effectively “push” the negative charge towards the γ phosphate of GTP.58 Taken together, these results show that the ribosome accelerates GTP hydrolysis by EF‐Tu by arranging the catalytic site in a productive way. This finding is supported by the large entropic contribution to the overall activation energy of the GTPase reaction in the presence of the ribosome,50, 51, 96 which may result from the favorable positioning of the reactive groups, electrostatic effects or shielding from the bulk water.97 In this context, the contribution of L7/12, which accelerates GTP hydrolysis by EF‐Tu by 2 orders of magnitude,18 remains unknown.
THE GTPASE CYCLE AND EF‐TU FUNCTION
The function of EF‐Tu is to deliver aa‐tRNA to the ribosome in response to the codon exposed in the A site. The ribosome must select an aa‐tRNA matching the given codon from a large pool of different aa‐tRNAs. The process of aa‐tRNA selection on the ribosome entails two steps irreversibly separated by GTP hydrolysis by EF‐Tu (Figure 6). The recruitment of the ternary complex to the translating ribosome is mediated by the L12 stalk.16 The initial binding of the ternary complex to the ribosome is reversible and independent of the mRNA.98 Aa‐tRNA samples the codon presented in the A site by spontaneous fluctuations in the tRNA structure that allow to present the anticodon opposite the codon in the A site of the ribosome.99, 100 Most mismatching ternary complexes (non‐cognate) containing 2 or 3 unpaired anticodon bases are rejected at this step. When the anticodon of aa‐tRNA encounters a cognate A‐site codon, which allows for the formation of a codon‐anticodon duplex with a Watson‐Crick geometry (codon recognition), the universally conserved rRNA residues A1492, A1493 and G530 rearrange to interact with the minor groove of the codon‐anticodon helix.49, 101, 102 With the anticodon in the A site and the acceptor arm still bound to EF‐Tu (A/T state), the aa‐tRNA is now strained into a distorted conformation, which is essential to trigger a series of rearrangements that result in the docking of the nucleotide pocket of EF‐Tu onto the GAC (reviewed in Ref. 103). The GTPase is then rapidly activated and GTP is cleaved,49, 104 forming an EF‐Tu–GTP–Pi intermediate.105 The rate of GTP hydrolysis is different for the cognate, near‐cognate and non‐cognate ternary complexes, which allows for the discrimination against non‐matching tRNAs.102, 106, 107, 108, 109, 110 The steps prior to and including GTP hydrolysis constitute the initial selection step of decoding.102, 106, 107 Pi release initiates the switch to the GDP‐form; in this conformation EF‐Tu has a much lower affinity for both the ribosome and aa‐tRNA, and thus dissociates leaving the aa‐tRNA free to accommodate into the peptidyl transferase center (PTC), where peptide bond formation takes place. Recent smFRET studies indicate that EF‐Tu dissociates from the GAC prior to aa‐tRNA release,111 which would lead to the immediate drop‐off of EF‐Tu–GDP from the ribosome after the contact with aa‐tRNA is lost. The accommodation is rapid and efficient for the cognate aa‐tRNA. In contrast, if a non‐matching aa‐tRNA happens to escape the initial selection screen, it is rejected upon accommodation, before peptide bond formation can take place; this second selection step is called proofreading. The selection process relies on the kinetic discrimination of incorrect aa‐tRNAs: cognate ternary complexes induce rapid GTPase activation and accommodation, which are the rate‐limiting steps for the two irreversible reactions of GTP hydrolysis and peptide bond formation, respectively.106, 112, 113 The intrinsic selectivity each of initial selection and proofreading is not high, but the combination of two subsequent selection steps increases the fidelity to about 1 wrong amino acid incorporated every 400 codons at in vitro conditions.106 The same set of rate constants, with only small adjustments for the cellular conditions, account for both the rate and fidelity of decoding in vivo.51
Figure 6.
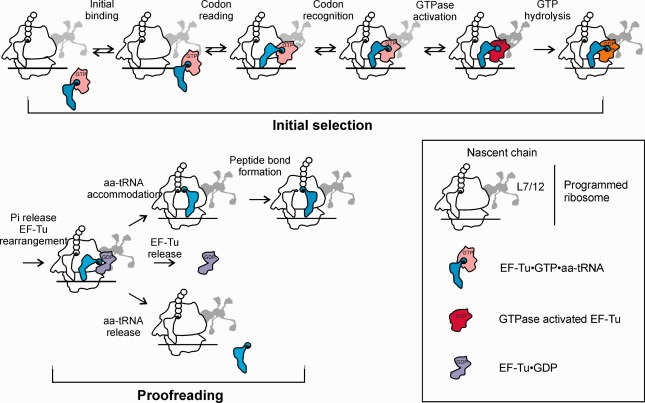
EF‐Tu function during decoding on the ribosome. EF‐Tu‐dependent selection of cognate aa‐tRNA and rejection of aa‐tRNAs that do not match the A‐site codon is achieved in two selection steps separated by GTP hydrolysis by EF‐Tu. Shown and individual kinetically‐resolved steps identified by rapid kinetics and smFRET approaches. Adapted from Ref. 112.
EF‐Tu is one of the fastest known GTPases. This is somewhat surprising, because if GTP hydrolysis is too fast, the discrimination potential cannot be used in full, a phenomenon known as a trade‐off between speed and accuracy.112, 113 At the two extremes, decoding can be very fast, but not accurate, or very accurate, but infinitely slow. The minimum theoretical error frequency of initial selection (at zero processivity) was estimated in the range of 1 incorrect amino acid incorporated in every 1200 − 80,000 decoding events depending on the type of mismatch, mismatch position in the codon, and tRNA isoacceptor type.114 These calculations are based on fidelity measurements at decreasing concentrations of Mg2+, which has a caveat that the activity of the ribosome may be irreversibly lost at particularly low Mg2+ concentrations.113 It thus remains to be seen whether this approach to estimate the maximum selectivity yields values that faithfully reflect in vivo conditions. However, as the actual rate of decoding is rather high, it is clear that this maximum discriminatory potential cannot be used in full, and the most important question is how much of this intrinsic selectivity is lost in favor of speed. The evolutionary pressure for rapid translation—at a cost of a moderate fidelity—may explain the high GTPase activity of EF‐Tu.112
One remaining question is the exact mechanism of the GTPase activation. The model suggesting the propagation of conformational rearrangements initiated by tRNA distortions to the nucleotide binding pocket of EF‐Tu is based on structures of the complex stalled by the antibiotic kirromycin in the post‐hydrolysis state.79, 115 Although we argued—based on fluorescence measurements using mant‐GTP derivatives—that the GTPase state persists in the kirromycin‐stalled complexes over a time span from milliseconds to hours,104 it is difficult to prove that the same state is maintained upon crystallization (over many days). The sequence of events (conformational changes in EF‐Tu followed by GTPase activation vs. GTPase activation resulting in rearrangements in EF‐Tu) cannot be inferred from a crystal structure. Furthermore, the distorted structure of aa‐tRNA is formed not only with the cognate, but also with a near‐cognate codon116, 117, 118; yet, the GTPase rates are different by orders of magnitude.106 The tRNA is crucial for GTPase activation of EF‐Tu108, 119; nevertheless, tRNA distortion appears to be a necessary, but not sufficient condition for the GTPase activation.
GTPASE CYCLES OF OTHER TRANSLATIONAL GTPASES
Structural, biochemical, and computational studies indicate that GTP hydrolysis by EF‐Tu in the presence of programmed ribosomes occurs due to the specific contribution of residues in the switch II and P‐loop regions (see above). Because these regions are highly conserved among trGTPases and because mutations of the conserved histidine abolish GTP hydrolysis of EF‐G,74 IF2,75, 76 and RF3 (unpublished data from our group), it is likely that all trGTPases use a similar mechanism of GTP hydrolysis. As for the function of Asp21 in other trGTPases, additional biochemical data are required; a recent study of the archaeal aIF2γ suggests that the residue corresponding to Asp21 in EF‐Tu contributes to catalysis, although apparently to a lesser extent than in EF‐Tu.77 One important question that still remains to be answered is whether GTPase activation by the ribosome involves the same conformational changes in different trGTPases. Despite the high conservation of the residues involved in the reaction, the rate of GTP hydrolysis differs greatly among trGTPases, ranging from about 0.3 s−1 for RF3 to >500 s−1 for EF‐Tu, which might indicate that the rate‐limiting GTPase activation follows distinct pathways (Table 1).
For all trGTPases, GTP hydrolysis promotes dissociation of the factor from the ribosome, thereby acting as a conventional GTPase switch. However, there are also additional roles for GTP hydrolysis in function of trGTPases on the ribosome, which are specific for every factor. eEF1A and SelB may function in a similar way as EF‐Tu, that is GTP hydrolysis is activated in response to the correct codon‐anticodon recognition, which allows the factors to release their cargo aa‐tRNA into the ribosome. Also eIF2 uses GTP hydrolysis and Pi release as sensors for recognition of the correct initiation codon.120 However, EF‐G utilizes the energy of GTP hydrolysis in a different way. EF‐G hydrolyses GTP almost immediately after binding to the ribosome, but remains in a GDP–Pi form; Pi is released concomitantly with the tRNA–mRNA translocation.52, 121, 122 GTP hydrolysis by EF‐G accelerates translocation by inducing an open (“unlocked”) ribosome conformation and prevents backward movements by acting as a door stopper,74, 122, 123, 124 in addition to its conventional role in promoting EF‐G dissociation.125 GTP hydrolysis by IF2 is induced upon docking of the large ribosomal subunit onto the 30S initiation complex containing IF1, IF3, IF2 and fMet‐tRNAfMet.126, 127 It promotes irreversible release of fMet‐tRNAfMet from IF2 and the subsequent dissociation of IF2 and IF1 from the ribosome.128 Also in this case the release of the Pi is delayed relative to GTP hydrolysis and occurs at a later stage together with the dissociation of the tRNA from IF2.127, 128 Finally, RF3 appears to be a very slow GTPase, even in the presence of its cognate termination complex41; the biological significance of this phenomenon remains unclear. In summary, trGTPases represent a group of proteins that have a common GTPase mechanism but function not only as switches, but also as mediators that link the energy of GTP hydrolysis to promoting reactions on the ribosome.
CONCLUSIONS AND PERSPECTIVES
Although the nucleotide binding pocket of the trGTPases is highly conserved, they have very different nucleotide binding properties and GTPase rates. While we start to understand the GTPase mechanism of EF‐Tu, the question arises as to what determines the variations in nucleotide binding and hydrolysis. Furthermore, it is still unclear how correct codon‐anticodon interaction activates the GTPase of EF‐Tu: is it the distortion of aa‐tRNA induced by the codon‐anticodon recognition at the decoding site which results in a long‐range effect at the nucleotide binding site of EF‐Tu, or is it the docking of the factor at the SRL? Why does in some cases Pi release, rather than GTP hydrolysis, act as a regulator of the events on the ribosome? How are the rearrangements caused by GTP hydrolysis and Pi release coupled to the conformational changes of the ribosome? It is important to solve these questions, as trGTPases are key components of protein synthesis and translational control. Understanding the mechanism of trGTPases will require not only concerted efforts of biochemical, biophysical, structural, and theoretical work, but also fresh ideas and new technologies.
This article was originally published online as an accepted preprint. The “Published Online” date corresponds to the preprint version. You can request a copy of any preprints from the past two calendar years by emailing the Biopolymers editorial office at biopolymers@wiley.com.
REFERENCES
- 1. Bourne, H. R. ; Sanders, D. A. ; McCormick, F. Nature 1991, 349, 117–127. [DOI] [PubMed] [Google Scholar]
- 2. Wittinghofer, A. ; Vetter, I. R. Annual Rev Biochem 2011, 80, 943–971. [DOI] [PubMed] [Google Scholar]
- 3. Hilgenfeld, R. Curr Opin Struct Biol 1995, 5, 810–817. [DOI] [PubMed] [Google Scholar]
- 4. Cherfils, J. ; Zeghouf, M. Physiol Rev 2013, 93, 269–309. [DOI] [PubMed] [Google Scholar]
- 5. Bos, J. L. ; Rehmann, H. ; Wittinghofer, A. Cell 2007, 129, 865–877. [DOI] [PubMed] [Google Scholar]
- 6. Vetter, I. R. ; Wittinghofer, A. Science 2001, 294, 1299–1304. [DOI] [PubMed] [Google Scholar]
- 7. Leipe, D. D. ; Wolf, Y. I. ; Koonin, E. V. ; Aravind, L. J Mol Biol 2002, 317, 41–72. [DOI] [PubMed] [Google Scholar]
- 8. Paulin, F. E. ; Campbell, L. E. ; O'Brien, K. ; Loughlin, J. ; Proud, C. G. Current Biol 2001, 11, 55–59. [DOI] [PubMed] [Google Scholar]
- 9. Unbehaun, A. ; Borukhov, S. I. ; Hellen, C. U. ; Pestova, T. V. Genes Dev 2004, 18, 3078–3093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Pestova, T. V. ; Lomakin, I. B. ; Lee, J. H. ; Choi, S. K. ; Dever, T. E. ; Hellen, C. U. Nature 2000, 403, 332–335. [DOI] [PubMed] [Google Scholar]
- 11. Atkinson, G. C. BMC Genomics 2015, 16, 78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Kjeldgaard, M. ; Nissen, P. ; Thirup, S. ; Nyborg, J. Structure (London, England: 1993) 1993, 1, 35–50. [DOI] [PubMed] [Google Scholar]
- 13. Polekhina, G. ; Thirup, S. ; Kjeldgaard, M. ; Nissen, P. ; Lippmann, C. ; Nyborg, J. Structure 1996, 4, 1141–1151. [DOI] [PubMed] [Google Scholar]
- 14. Berchtold, H. ; Reshetnikova, L. ; Reiser, C. O. A. ; Schirmer, N. K. ; Sprinzl, M. ; Hilgenfeld, R. Nature 1993, 365, 126–132. [DOI] [PubMed] [Google Scholar]
- 15. Davydov, I. I. ; Wohlgemuth, I. ; Artamonova, I. I. ; Urlaub, H. ; Tonevitsky, A. G. ; Rodnina, M. V. Nat Commun 2013, 4, 1387. [DOI] [PubMed] [Google Scholar]
- 16. Diaconu, M. ; Kothe, U. ; Schlunzen, F. ; Fischer, N. ; Harms, J. M. ; Tonevitsky, A. G. ; Stark, H. ; Rodnina, M. V. ; Wahl, M. C. Cell 2005, 121, 991–1004. [DOI] [PubMed] [Google Scholar]
- 17. Savelsbergh, A. ; Mohr, D. ; Wilden, B. ; Wintermeyer, W. ; Rodnina, M. V. J Biol Chem 2000, 275, 890–894. [DOI] [PubMed] [Google Scholar]
- 18. Mohr, D. ; Wintermeyer, W. ; Rodnina, M. V. Biochemistry 2002, 41, 12520–12528. [DOI] [PubMed] [Google Scholar]
- 19. Tourigny, D. S. ; Fernandez, I. S. ; Kelley, A. C. ; Ramakrishnan, V. Science 2013, 340, 1235490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Voorhees, R. M. ; Schmeing, T. M. ; Kelley, A. C. ; Ramakrishnan, V. Science 2010, 330, 835–838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Chen, Y. ; Feng, S. ; Kumar, V. ; Ero, R. ; Gao, Y. G. Nat Struct Mol Biol 2013, 20, 1077–1084. [DOI] [PubMed] [Google Scholar]
- 22. Fasano, O. ; Bruns, W. ; Crechet, J. B. ; Sander, G. ; Parmeggiani, A. Eur J Biochem/FEBS 1978, 89, 557–565. [DOI] [PubMed] [Google Scholar]
- 23. Gromadski, K. B. ; Wieden, H. J. ; Rodnina, M. V. Biochemistry 2002, 41, 162–169. [DOI] [PubMed] [Google Scholar]
- 24. Gromadski, K. B. ; Schummer, T. ; Stromgaard, A. ; Knudsen, C. R. ; Kinzy, T. G. ; Rodnina, M. V. J Biol Chem 2007, 282, 35629–35637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Louie, A. ; Ribeiro, N. S. ; Reid, B. R. ; Jurnak, F. J Biol Chem 1984, 259, 5010–5016. [PubMed] [Google Scholar]
- 26. Abrahamson, J. K. ; Laue, T. M. ; Miller, D. L. ; Johnson, A. E. Biochemistry 1985, 24, 692–700. [DOI] [PubMed] [Google Scholar]
- 27. Rosset, R. ; Julien, J. ; Monier, R. J Mol Biol 1966, 18, 308–320. [DOI] [PubMed] [Google Scholar]
- 28. Furano, A. V. Proc Natl Acad Sci USA 1975, 72, 4780–4784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Song, H. ; Parsons, M. R. ; Rowsell, S. ; Leonard, G. ; Phillips, S. E. J Mol Biol 1999, 285, 1245–1256. [DOI] [PubMed] [Google Scholar]
- 30. Leibundgut, M. ; Frick, C. ; Thanbichler, M. ; Bock, A. ; Ban, N. EMBO J 2005, 24, 11–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Hansson, S. ; Singh, R. ; Gudkov, A. T. ; Liljas, A. ; Logan, D. T. FEBS Lett 2005, 579, 4492–4497. [DOI] [PubMed] [Google Scholar]
- 32. al‐Karadaghi, S. ; Aevarsson, A. ; Garber, M. ; Zheltonosova, J. ; Liljas, A. Structure 1996, 4, 555–565. [DOI] [PubMed] [Google Scholar]
- 33. Salsi, E. ; Farah, E. ; Netter, Z. ; Dann, J. ; Ermolenko, D. N. J Mol Biol 2015, 427, 454–467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Lin, J. ; Gagnon, M. G. ; Bulkley, D. ; Steitz, T. A. Cell 2015, 160, 219–227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Yamamoto, H. ; Unbehaun, A. ; Loerke, J. ; Behrmann, E. ; Collier, M. ; Burger, J. ; Mielke, T. ; Spahn, C. M. Nat Struct Mol Biol 2014, 21, 721–727. [DOI] [PubMed] [Google Scholar]
- 36. Eiler, D. ; Lin, J. ; Simonetti, A. ; Klaholz, B. P. ; Steitz, T. A. Proc Natl Acad Sci USA 2013, 110, 15662–15667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Roll‐Mecak, A. ; Cao, C. ; Dever, T. E. ; Burley, S. K. Cell 2000, 103, 781–792. [DOI] [PubMed] [Google Scholar]
- 38. Andersen, G. R. ; Pedersen, L. ; Valente, L. ; Chatterjee, I. ; Kinzy, T. G. ; Kjeldgaard, M. ; Nyborg, J. Mol Cell 2000, 6, 1261–1266. [DOI] [PubMed] [Google Scholar]
- 39. Wang, Y. ; Jiang, Y. ; Meyering‐Voss, M. ; Sprinzl, M. ; Sigler, P. B. Nat Struct Biol 1997, 4, 650–656. [DOI] [PubMed] [Google Scholar]
- 40. Thirup, S. S. ; Van, L. B. ; Nielsen, T. K. ; Knudsen, C. R. J Struct Biol 2015, 191, 10–21. [DOI] [PubMed] [Google Scholar]
- 41. Peske, F. ; Kuhlenkoetter, S. ; Rodnina, M. V. ; Wintermeyer, W. Nucleic Acids Res 2014, 42, 1812–1820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Zavialov, A. V. ; Buckingham, R. H. ; Ehrenberg, M. Cell 2001, 107, 115–124. [DOI] [PubMed] [Google Scholar]
- 43. Koutmou, K. S. ; McDonald, M. E. ; Brunelle, J. L. ; Green, R. RNA 2014, 20, 609–620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Antoun, A. ; Pavlov, M. Y. ; Andersson, K. ; Tenson, T. ; Ehrenberg, M. EMBO J 2003, 22, 5593–5601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Wilden, B. ; Savelsbergh, A. ; Rodnina, M. V. ; Wintermeyer, W. Proc Natl Acad Sci USA 2006, 103, 13670–13675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Pulk, A. ; Cate, J. H. Science 2013, 340, 1235970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Zhou, J. ; Lancaster, L. ; Donohue, J. P. ; Noller, H. F. Science 2013, 340, 1236086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Zhou, J. ; Lancaster, L. ; Trakhanov, S. ; Noller, H. F. RNA 2012, 18, 230–240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Pape, T. ; Wintermeyer, W. ; Rodnina, M. V. EMBO J 1998, 17, 7490–7497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Johansson, M. ; Bouakaz, E. ; Lovmar, M. ; Ehrenberg, M. Mol Cell 2008, 30, 589–598. [DOI] [PubMed] [Google Scholar]
- 51. Rudorf, S. ; Thommen, M. ; Rodnina, M. V. ; Lipowsky, R. PLoS Comput Biol 2014, 10, e1003909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Savelsbergh, A. ; Katunin, V. I. ; Mohr, D. ; Peske, F. ; Rodnina, M. V. ; Wintermeyer, W. Mol Cell 2003, 11, 1517–1523. [DOI] [PubMed] [Google Scholar]
- 53. Adamczyk, A. J. ; Warshel, A. Proc Natl Acad Sci USA 2011, 108, 9827–9832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Prasad, B. R. ; Plotnikov, N. V. ; Lameira, J. ; Warshel, A. Proc Natl Acad Sci USA 2013, 110, 20509–20514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Liljas, A. ; Ehrenberg, M. Aqvist, J. Science 2011, 333, 37. author reply37. [DOI] [PubMed] [Google Scholar]
- 56. Wallin, G. ; Kamerlin, S. C. ; Aqvist, J. Nat Commun 2013, 4, 1733. [DOI] [PubMed] [Google Scholar]
- 57. Aleksandrov, A. ; Field, M. RNA 2013, 19, 1218–1225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Aqvist, J. ; Kamerlin, S. C. Biochemistry 2015, 54, 546–556. [DOI] [PubMed] [Google Scholar]
- 59. Grigorenko, B. L. ; Shadrina, M. S. ; Topol, I. A. ; Collins, J. R. ; Nemukhin, A. V. Biochim Biophys Acta 2008, 1784, 1908–1917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Schweins, T. ; Geyer, M. ; Scheffzek, K. ; Warshel, A. ; Kalbitzer, H. R. ; Wittinghofer, A. Nat Struct Mol Biol 1995, 2, 36–44. [DOI] [PubMed] [Google Scholar]
- 61. Maegley, K. A. ; Admiraal, S. J. ; Herschlag, D. Proc Natl Acad Sci USA 1996, 93, 8160–8166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Li, G. ; Zhang, X. C. J Mol Biol 2004, 340, 921–932. [DOI] [PubMed] [Google Scholar]
- 63. Schweins, T. ; Langen, R. ; Warshel, A. Nat Struct Mol Biol 1994, 1, 476–484. [DOI] [PubMed] [Google Scholar]
- 64. Carvalho, A. T. ; Szeler, K. ; Vavitsas, K. ; Aqvist, J. ; Kamerlin, S. C. Arch Biochem Biophys 2015, 582, 80–90. [DOI] [PubMed] [Google Scholar]
- 65. Lassila, J. K. ; Zalatan, J. G. ; Herschlag, D. Annual Rev Biochem 2011, 80, 669–702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Kamerlin, S. C. ; Sharma, P. K. ; Prasad, R. B. ; Warshel, A. Quarterly Rev Biophys 2013, 46, 1–132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Hengge, A. C. In Advances in Physical Organic Chemistry; Richard J. P., Ed.; Academic Press, 2005, pp 49–108. [Google Scholar]
- 68. Prakash, P. ; Gorfe, A. A. Mol Simulation 2014, 40, 839–847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Zeidler, W. ; Egle, C. ; Ribeiro, S. ; Wagner, A. ; Katunin, V. ; Kreutzer, R. ; Rodnina, M. ; Wintermeyer, W. ; Sprinzl, M. Eur J Biochem 1995, 229, 596–604. [DOI] [PubMed] [Google Scholar]
- 70. Scarano, G. ; Krab, I. M. ; Bocchini, V. ; Parmeggiani, A. FEBS Lett 1995, 365, 214–218. [DOI] [PubMed] [Google Scholar]
- 71. Daviter, T. ; Wieden, H. J. ; Rodnina, M. V. J Mol Biol 2003, 332, 689–699. [DOI] [PubMed] [Google Scholar]
- 72. Cool, R. H. ; Parmeggiani, A. Biochemistry 1991, 30, 362–366. [DOI] [PubMed] [Google Scholar]
- 73. Maracci, C. ; Peske, F. ; Dannies, E. ; Pohl, C. ; Rodnina, M. V. Proc Natl Acad Sci USA 2014, 111, 14418–14423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Cunha, C. E. ; Belardinelli, R. ; Peske, F. ; Holtkamp, W. ; Wintermeyer, W. ; Rodnina, M. V. Translation 2013, 1, e24315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Luchin, S. ; Putzer, H. ; Hershey, J. W. ; Cenatiempo, Y. ; Grunberg‐Manago, M. ; Laalami, S. J Biol Chem 1999, 274, 6074–6079. [DOI] [PubMed] [Google Scholar]
- 76. Laalami, S. ; Timofeev, A. V. ; Putzer, H. ; Leautey, J. ; Grunberg‐Manago, M. Mol Microbiol 1994, 11, 293–302. [DOI] [PubMed] [Google Scholar]
- 77. Dubiez, E. ; Aleksandrov, A. ; Lazennec‐Schurdevin, C. ; Mechulam, Y. ; Schmitt, E. Nucleic Acids Res 2015, 43, 2946–2957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Kuhle, B. ; Ficner, R. EMBO J 2014, 33, 2547–2563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Schmeing, T. M. ; Voorhees, R. M. ; Kelley, A. C. ; Gao, Y. G. ; Murphy, F. V. ; Weir, J. R. ; Ramakrishnan, V. Science 2009, 326, 688–694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Schuette, J. C. ; Murphy, F. V. ; Kelley, A. C. ; Weir, J. R. ; Giesebrecht, J. ; Connell, S. R. ; Loerke, J. ; Mielke, T. ; Zhang, W. ; Penczek, P. A. ; Ramakrishnan, V. ; Spahn, C. M. T. EMBO J 2009, 28, 755–765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Schweins, T. ; Warshel, A. Biochemistry 1996, 35, 14232–14243. [DOI] [PubMed] [Google Scholar]
- 82. Schweins, T. ; Geyer, M. ; Kalbitzer, H. R. ; Wittinghofer, A. ; Warshel, A. Biochemistry 1996, 35, 14225–14231. [DOI] [PubMed] [Google Scholar]
- 83. Nissen, P. ; Kjeldgaard, M. ; Thirup, S. ; Polekhina, G. ; Reshetnikova, L. ; Clark, B. F. C. ; Nyborg, J. Science 1995, 270, 1464–1472. [DOI] [PubMed] [Google Scholar]
- 84. Fasano, O. ; De Vendittis, E. ; Parmeggiani, A. J Biol Chem 1982, 257, 3145–3150. [PubMed] [Google Scholar]
- 85. Ivell, R. ; Sander, G. ; Parmeggiani, A. Biochemistry 1981, 20, 6852–6859. [DOI] [PubMed] [Google Scholar]
- 86. Ash, M. R. ; Maher, M. J. ; Mitchell Guss, J. ; Jormakka, M. FEBS Lett 2012, 586, 2218–2224. [DOI] [PubMed] [Google Scholar]
- 87. Scrima, A. ; Wittinghofer, A. EMBO J 2006, 25, 2940–2951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Allin, C. ; Gerwert, K. Biochemistry 2001, 40, 3037–3046. [DOI] [PubMed] [Google Scholar]
- 89. Kraal, B. ; de Graaf, J. M. ; Mesters, J. R. ; van Hoof, P. J. ; Jacquet, E. ; Parmeggiani, A. Eur J Biochem/FEBS 1990, 192, 305–309. [DOI] [PubMed] [Google Scholar]
- 90. Mesters, J. R. ; Martien de Graaf, J. ; Kraal, B. FEBS Lett 1993, 321, 149–152. [DOI] [PubMed] [Google Scholar]
- 91. Hazlett, T. L. ; Higashijima, T. ; Jameson, D. M. FEBS Lett 1991, 278, 225–228. [DOI] [PubMed] [Google Scholar]
- 92. Scheffzek, K. ; Ahmadian, M. R. ; Kabsch, W. ; Wiesmuller, L. ; Lautwein, A. ; Schmitz, F. ; Wittinghofer, A. Science 1997, 277, 333–338. [DOI] [PubMed] [Google Scholar]
- 93. Chung, H. ; Benson, D. ; Schultz, P. Science 1993, 259, 806–809. [DOI] [PubMed] [Google Scholar]
- 94. Fischer, N. ; Neumann, P. ; Konevega, A. L. ; Bock, L. V. ; Ficner, R. ; Rodnina, M. V. ; Stark, H. Nature 2015, 520, 567–570. [DOI] [PubMed] [Google Scholar]
- 95. Koch, M. ; Flur, S. ; Kreutz, C. ; Ennifar, E. ; Micura, R. ; Polacek, N. Proc Natl Acad Sci USA 2015, 112, E2561–E2568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Aqvist, J. ; Kamerlin, S. C. Sci Rep 2015, 5, 15817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. Sharma, P. K. ; Xiang, Y. ; Kato, M. ; Warshel, A. Biochemistry 2005, 44, 11307–11314. [DOI] [PubMed] [Google Scholar]
- 98. Rodnina, M. V. ; Pape, T. ; Fricke, R. ; Kuhn, L. ; Wintermeyer, W. J Biol Chem 1996, 271, 646–652. [DOI] [PubMed] [Google Scholar]
- 99. Blanchard, S. C. ; Gonzalez, R. L. ; Kim, H. D. ; Chu, S. ; Puglisi, J. D. Nat Struct Mol Biol 2004, 11, 1008–1014. [DOI] [PubMed] [Google Scholar]
- 100. Geggier, P. ; Dave, R. ; Feldman, M. B. ; Terry, D. S. ; Altman, R. B. ; Munro, J. B. ; Blanchard, S. C. J Mol Biol 2010, 399, 576–595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101. Ogle, J. M. ; Brodersen, D. E. ; Clemons, W. M. ; ; Tarry, M. J. ; Carter, A. P. ; Ramakrishnan, V. Science 2001, 292, 897–902. [DOI] [PubMed] [Google Scholar]
- 102. Pape, T. ; Wintermeyer, W. ; Rodnina, M. EMBO J 1999, 18, 3800–3807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103. Voorhees, R. M. ; Ramakrishnan, V. Annual Rev Biochem 2013, 82, 203–236. [DOI] [PubMed] [Google Scholar]
- 104. Rodnina, M. V.,F. R. ; Kuhn, L. ; Wintermeyer, W. EMBO J 1995, 14, 2613–2619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105. Kothe, U. ; Rodnina, M. V. Biochemistry 2006, 45, 12767–12774. [DOI] [PubMed] [Google Scholar]
- 106. Gromadski, K. B. ; Rodnina, M. V. Mol Cell 2004, 13, 191–200. [DOI] [PubMed] [Google Scholar]
- 107. Gromadski, K. B. ; Daviter, T. ; Rodnina, M. V. Mol Cell 2006, 21, 369–377. [DOI] [PubMed] [Google Scholar]
- 108. Cochella, L. ; Green, R. Science 2005, 308, 1178–1180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109. Shepotinovskaya, I. ; Uhlenbeck, O. C. RNA 2013, 19, 510–516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110. McClory, S. P. ; Leisring, J. M. ; Qin, D. ; Fredrick, K. RNA 2010, 16, 1925–1934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111. Liu, W. ; Chen, C. ; Kavaliauskas, D. ; Knudsen, C. R. ; Goldman, Y. E. ; Cooperman, B. S. Nucleic Acids Res 2015, 43, 9519–9528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112. Wohlgemuth, I. ; Pohl, C. ; Mittelstaet, J. ; Konevega, A. L. ; Rodnina, M. V. Phil Trans Roy Soc Lond Series B, Biol Sci 2011, 366, 2979–2986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113. Wohlgemuth, I. ; Pohl, C. ; Rodnina, M. V. EMBO J 2010, 29, 3701–3709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114. Zhang, J. ; Ieong, K. W. ; Johansson, M. ; Ehrenberg, M. Proc Natl Acad Sci USA 2015, 112, 9602–9607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115. Valle, M. ; Sengupta, J. ; Swami, N. K. ; Grassucci, R. A. ; Burkhardt, N. ; Nierhaus, K. H. ; Agrawal, R. K. ; Frank, J. EMBO J 2002, 21, 3557–3567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116. Agirrezabala, X. ; Valle, M. Int J Mol Sci 2015, 16, 9866–9895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117. Agirrezabala, X. ; Schreiner, E. ; Trabuco, L. G. ; Lei, J. ; Ortiz‐Meoz, R. F. ; Schulten, K. ; Green, R. ; Frank, J. EMBO J 2011, 30, 1497–1507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118. Mittelstaet, J. ; Konevega, A. L. ; Rodnina, M. V. J Biol Chem 2011, 286, 8158–8164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119. Piepenburg, O. ; Pape, T. ; Pleiss, J. A. ; Wintermeyer, W. ; Uhlenbeck, O. C. ; Rodnina, M. V. Biochemistry 2000, 39, 1734–1738. [DOI] [PubMed] [Google Scholar]
- 120. Kapp, L. D. ; Lorsch, J. R. J Mol Biol 2004, 335, 923–936. [DOI] [PubMed] [Google Scholar]
- 121. Rodnina, M. V. ; Savelsbergh, A. ; Katunin, V. I. ; Wintermeyer, W. Nature 1997, 385, 37–41. [DOI] [PubMed] [Google Scholar]
- 122. Pan, D. ; Kirillov, S. V. ; Cooperman, B. S. Mol Cell 2007, 25, 519–529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123. Holtkamp, W. ; Cunha, C. E. ; Peske, F. ; Konevega, A. L. ; Wintermeyer, W. ; Rodnina, M. V. EMBO J 2014, 33, 1073–1085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124. Adio, S. ; Senyushkina, T. ; Peske, F. ; Fischer, N. ; Wintermeyer, W. ; Rodnina, M. V. Nature Commun 2015, 6, 7442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125. Kaziro, Y. Biochimica et biophysica acta 1978, 505, 95–127. [DOI] [PubMed] [Google Scholar]
- 126. Tomsic, J. ; Vitali, L. A. ; Daviter, T. ; Savelsbergh, A. ; Spurio, R. ; Striebeck, P. ; Wintermeyer, W. ; Rodnina, M. V. ; Gualerzi, C. O. EMBO J 2000, 19, 2127–2136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127. Grigoriadou, C. ; Marzi, S. ; Kirillov, S. ; Gualerzi, C. O. ; Cooperman, B. S. J Mol Biol 2007, 373, 562–572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128. Goyal, A. ; Belardinelli, R. ; Maracci, C. ; Milon, P. ; Rodnina, M. V. Nucleic Acids Res 2015, 43, 10700–10712. [DOI] [PMC free article] [PubMed] [Google Scholar]