

Abstract
The application of commonly used force spectroscopy in biological systems is often limited by the need for an invasive tether connecting the molecules of interest to a bead or cantilever tip. Here we present a DNA origami‐based prototype in a comparative binding assay. It has the advantage of in situ readout without any physical connection to the macroscopic world. The seesaw‐like structure has a lever that is able to move freely relative to its base. Binding partners on each side force the structure into discrete and distinguishable conformations. Model experiments with competing DNA hybridisation reactions yielded a drastic shift towards the conformation with the stronger binding interaction. With reference DNA duplexes of tuneable length on one side, this device can be used to measure ligand interactions in comparative assays.
Keywords: DNA hybridisation, DNA origami, DNA structures, self-assembly, sensors
In biological systems, the interaction of molecules often leads to conformational changes. The nature of these changes has been investigated with various single‐molecule force spectroscopy tools, most prominently magnetic and optical tweezers and atomic force microscopy.1 Although these techniques feature single‐nanometer resolution and piconewton force sensitivity, they suffer from two limitations: poor potential for parallelisation, and the need for an invasive connector between the biomolecule and the macroscopic device. In addressing the first of these, DNA has been used as a programmable reference bond to quantitatively measure biomolecular interactions in a highly parallel fashion.2 The second limitation could potentially be overcome by molecular tools such as DNA force sensors, as used to study DNA looping.3 Structural DNA nanotechnology, particularly DNA origami, opens up a promising route to construct such nanoscopic molecular tools.4 In DNA origami,5, 6 a long scaffold strand is folded into a designed shape by hundreds of short oligonucleotides with programmed sequences. The simple, computer‐aided design process7, 8, 9, 10 has enabled the self‐assembly of a variety of complex 2D5, 10 and 3D11, 12, 13, 14, 15 geometries with nanometer addressability. DNA origami structures have been frequently employed to arrange molecules, binding moieties and proteins in designed patterns.16, 17, 18 Furthermore, conformational changes of these structures have been demonstrated, for example by endonuclease activity,14 aptamer‐ligand interactions,19 toehold‐mediated branch migration20, 21 and with hydrophobic moieties such as cholesterol.21
In this work, we present a seesaw‐like DNA origami structure that can potentially serve in a differential molecular binding assay (Figure 1). A pair of binding partners on one side of the balance‐like structure is compared to a reference pair of binding partners on the other side. The binding pair with the stronger interaction locks the structure in a distinct conformation. This conformation can be easily identified by transmission electron microscopy (TEM). Moreover, there is no need for a physical tether to a macroscopic device, and thus, in principle, this approach allows efficient parallelisation.
Figure 1.
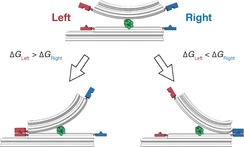
Seesaw‐like force balance. A pair of binding partners is conjugated to each side of the device. The stronger binding pair locks the structure in a geometrically distinguishable conformation.
The prototype DNA origami structure (Figure 2 A) consists of two 18‐helix bundles connected by a short six‐helix bundle hinge. The upper beam (lever) is 60 nm long and has a 60° bend along its length. The lower beam (base) is 60 nm long and straight, with a 25 nm extension of 12 helices on the left side to introduce asymmetry. This permits the detection of the orientation of the structure in TEM images. The 25 nm hinge (perpendicular between the base and the lever) serves to connect both beams by four scaffold double crossovers (two from base to hinge and two from hinge to lever). The scaffold crossovers are placed such that the lever and the base are free to pivot in one plane. Each crossover features a single‐stranded scaffold spacer of three nucleotides. Each side of the structure is equipped with three individually addressable positions on both base and lever. At these positions, the corresponding staple strands can be extended with single‐stranded DNA (ssDNA) either on the 5′‐end (right side of the base, left side of the lever) or on the 3′‐end (left side of the base, right side of the lever).
Figure 2.
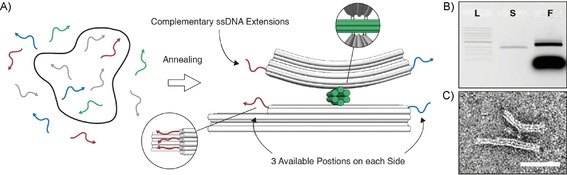
A) The circular scaffold molecule (black) and staple subsets for the hinge (green), base and lever (grey) with the three addressable positions on each side (red and blue) are thermally annealed to form the seesaw‐like balance. At the addressable positions, the corresponding staples are extended at either the 5′ or 3′‐end (3′‐ends depicted as arrow heads). B) Agarose gel electrophoresis confirms assembly after thermal annealing (L: 1kb DNA ladder; S: scaffold‐only control; F: folded structure). C) Representative TEM image of a gel‐purified structure shows correct assembly. Scale bar: 50 nm.
In order to verify the correct assembly of the structures, we analysed the folding by agarose gel electrophoresis directly after thermal annealing of the DNA origami structure. The gel demonstrated the successful, high‐yield folding of our prototype (Figure 2 B). Negative‐stain TEM imaging of gel‐purified structures (Figure 2 C) confirmed that the intended geometry had formed.
As a proof of principle, we developed a DNA hybridisation assay with competing complementary ssDNA extensions on each side. During annealing, the complementary extensions form a duplex in a zipper conformation and lock the structure in a geometrically distinct conformation. The probability of closing on one side depends on two variables: the number of hybridising extensions on each side (maximum of three per side) and the binding energies of all formed duplexes. We choose 20 nucleotides for each extension, and the same complementary sequences on both sides. The number of DNA extensions was varied and is denoted “vs” to represent the number of extensions per side.
Samples with different numbers of competing hybridisation partners on each side were prepared and thermally annealed. After folding, the structures were subjected to agarose gel electrophoresis (Figure S4 in the Supporting Information) and purification. TEM images were taken from several randomly selected areas on the grid (example zoom‐out images in Figure S5). The TEM images were analysed. Firstly, three distinct conformations of the structure were defined: left, open and right (Figure 3). Secondly, single seesaws immobilised on the TEM grid lying on the side were identified in the micrographs, and one of the three defined conformations was assigned to each particle (left or right when the lever was in contact with the base at the end; open otherwise). Finally, the number of particles in each of the three groups was counted. Figure 3 shows the results for different combinations of competing extensions. When none of the positions was addressed (0 vs 0), more than 60 % of the particles were in the open conformation. For equal numbers of extensions on each side (1 vs 1 and 3 vs 3), we observed an almost 50:50 distribution, with only a small fraction in the open conformation. Unequal numbers of extensions resulted in a dramatic shift of concentration of closed structures towards the side with the higher number of extensions (3 vs 2 and 2 vs 3). A greater difference in the number of extensions on each side resulted in a more pronounced concentration shift: 90 % correct closing was achieved by addressing all three positions on one side and none on the opposing side (0 vs 3 and 3 vs 0).
Figure 3.
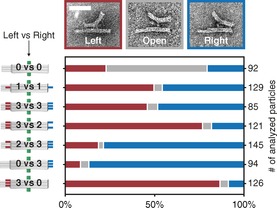
Analysis of TEM images of DNA origami structures with different numbers of competing DNA hybridisation partners. The TEM images show the three distinct conformations: left, open, and right. Scale bar: 50 nm.
In order to compare our results with a theoretical prediction, we calculated the probability of our structure being either in the left or right conformation. For this, we assigned a Boltzmann factor to each side based on the computed ΔG of the formed duplexes (calculated at www.nupack.org 22 for T=T m) and assumed that the probability of one conformation is proportional to the ratio of the Boltzmann factors. A 20 % increase in ΔG on one side should lead to virtually 100 % of the structures being closed on that side (see Figure S3 for details). For the 3 vs 2 and 2 vs 3 samples (i.e. 50 % increase in ΔG on one side), we observed only 80 % closed objects. Even 3 vs 0 and 0 vs 3 were not able to achieve the expected 100 % closing. We believe that two reasons explain the majority of incorrectly closed or open structures: firstly, the incorporation probability of each staple strand is not 100 % (previously reported values are 96–99 %, depending on the geometry of the origami design);23 and secondly, excess free extension oligomers in the solution (not incorporated as staples) can bind to complementary counterparts attached to the structure and thus saturate that position. Optimising the annealing process and implementing a hairpin‐based reaction hierarchy in the closing reaction24 could further improve the yields of structures in the correct conformation in the future.
In order to test our prototype in a comparative binding assay after folding instead of in a one‐pot reaction, we used a pre‐formed, locked structure to detect DNA–DNA binding events (Figure 4 A). We folded the structure with a 15‐nucleotide extension on the left and a 30‐nucleotide extension on the right (15 vs 30). This is equivalent to the 1 vs 1 sample (Figure 3), except that we expected a dramatic shift towards the conformation with the stronger binding (the 30‐nt extension on the right), similarly to the 2 vs 3 sample. Additionally, the long extension at the base on the right featured an eight‐nucleotide toehold. After folding, the sample was subjected to agarose gel purification as for the samples in Figure 3. TEM imaging revealed that about 80 % of the structures were closed on the right (Figure 4 B). This is close to the results for 2 vs 3, which has a similar relative binding mismatch. Next, we added an input strand complementary to the 30‐base toehold extension at 20× molar excess. TEM images were taken after 2 h incubation at RT. A dramatic shift in the concentration of closed structures towards the left was observed: about 75 % were now closed on the left (Figure 4 B).
Figure 4.
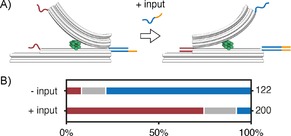
DNA–DNA binding detection on a pre‐formed, closed, seesaw structure. A) The structure is annealed with an extension that is twice as long on the right (30 bases) as on the left (15 bases). The extension on the right also carries a toehold. Upon the addition of an input strand that is complementary to the toehold strand on the right, the structure opens and subsequently closes on the left. B) Analysis of TEM images before and after the addition of the input strand (red: left; grey: open; blue: right).
In conclusion, we successfully assembled a DNA‐origami‐based prototype for a comparative binding assay. Discrete and geometrically distinguishable conformations were observed for DNA hybridisation reactions on both sides during one‐pot annealing, as a model system. DNA–DNA binding events were detected after the one‐pot annealing on a pre‐formed and locked structure, and this resulted in a switch of conformation. This device can potentially be used to study bio‐molecular interactions in a comparative fashion with DNA as a tuneable reference, for example to investigate molecule–aptamer interactions and DNA–protein interactions, such as DNA bending (possible implementations illustrated in Figure 5). The lack of coupling to a cantilever or micrometre bead together with the possibility of in situ FRET readout will allow applications in biological systems with a high degree of parallelism. In future experiments, similar DNA origami tools containing ssDNA sections acting as entropic spring elements9, 14 could greatly simplify the study of force dependence in dynamic DNA systems and for DNA–protein interactions.
Figure 5.
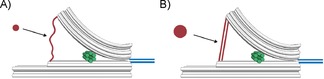
Potential binding assays to study A) molecule–aptamer interactions and B) DNA bending proteins in comparison to a reference DNA duplex of tuneable length.
Experimental Section
Sample preparation: The DNA origami structure was designed by using caDNAno software (version 0.2.3; http://cadnano.org/legacy;7 design schematics in Figure S1). Curvature along the length of the lever was introduced by a pattern of base deletions and insertions as described previously (deletion pattern in Figure S2).13 The 8634‐nucleotide single‐stranded scaffold DNA, derived from M13mp18, was prepared as previously described.12, 25 Scaffold DNA (10 nm) was mixed with each staple strand (100 nm; high purity salt free, MWG Eurofins Operon) in TE buffer (Tris (10 mm, pH 7.6), EDTA (1 mm)) containing MgCl2 (18 mm) and NaCl (5 mm). The mixture was subjected to a nonlinear thermal annealing ramp from 65 to 25 °C over 24 h (see the Supporting Information). Folded structures were electrophoresed on 0.7 % agarose gels containing 0.5×TBE buffer (Tris (45 mm), boric acid (45 mm0, EDTA (1 mm)) withMgCl2 (11 mm) and ethidium bromide (0.5 μg mL−1) at 5.5 V cm−1 for 2 h in an ice‐water bath. Bands were visualised with UV light and physically extracted. DNA was recovered by manually squeezing the excised gel slice and collecting the liquid.
TEM imaging: Gel‐purified origami solution (3 μL) was adsorbed onto glow‐discharged TEM grids (formvar/carbon, 300 mesh Cu; Ted Pella, Redding, CA) at 20 °C, and then stained with aqueous uranyl formate (2 %) containing sodium hydroxide (25 mm). Imaging was performed at 30 000× magnification (zoom‐out images in Figure S5 at 12 000× magnification) with a JEM1011 transmission electron microscope (JEOL) operated at 80 kV, equipped with a FastScan‐F114 camera (TVIPS, Gauting, Germany). Particles in TEM images were picked by the interactive boxing routine (e2boxer.py) of Eman2 software (http://blake.bcm.edu/emanwiki/EMAN; example set of particles in Figure S6).26
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary
Acknowledgements
We thank Ingo Stein, Robert Schreiber, and Tao Zhang for discussions and Susanne Kempter for technical assistance. We acknowledge funding from the Deutsche Forschungsgemeinschaft (DFG; LI 1743/2‐1, TI 329/6‐1, and SFB 1032 (TPA6)) the Nanosystems Initiative Munich (NIM), and the Center for NanoScience (CeNS) Munich.
P. C. Nickels, H. C. Høiberg, S. S. Simmel, P. Holzmeister, P. Tinnefeld, T. Liedl, ChemBioChem 2016, 17, 1093.
References
- 1. Neuman K. C., Nagy A., Nat. Methods 2008, 5, 491–505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Albrecht C., Blank K., Lalic-Mülthaler M., Hirler S., Mai T., Gilbert I., Schiffmann S., Bayer T., Clausen-Schaumann H., Gaub H. E., Science 2003, 301, 367–370. [DOI] [PubMed] [Google Scholar]
- 3. Shroff H., Reinhard B. M., Siu M., Agarwal H., Spakowitz A., Liphardt J., Nano Lett. 2005, 5, 1509–1514. [DOI] [PubMed] [Google Scholar]
- 4. Gu H., Yang W., Seeman N. C., J. Am. Chem. Soc. 2010, 132, 4352–4357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Rothemund P. W. K., Nature 2006, 440, 297–302. [DOI] [PubMed] [Google Scholar]
- 6. Seeman N. C., Annu. Rev. Biochem. 2010, 79, 65–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Douglas S. M., Marblestone A. H., Teerapittayanon S., Vazquez A., Church G. M., Shih W. M., Nucleic Acids Res. 2009, 37, 5001–5006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Ke Y., Douglas S. M., Liu M., Sharma J., Cheng A., Leung A., Liu Y., Shih W. M., Yan H., J. Am. Chem. Soc. 2009, 131, 15903–15908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Kim D.-N., Kilchherr F., Dietz H., Bathe M., Nucleic Acids Res. 2012, 40, 2862–2868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Andersen E. S., Dong M., Nielsen M. M., Jahn K., Lind-Thomsen A., Mamdouh W., Gothelf K. V., Besenbacher F., Kjems J., ACS Nano 2008, 2, 1213–1218. [DOI] [PubMed] [Google Scholar]
- 11. Andersen E. S., Dong M., Nielsen M. M., Jahn K., Subramani R., Mamdouh W., Golas M. M., Sander B., Stark H., Oliveira C. L. P., Pedersen J. S., Birkedal V., Besenbacher F., Gothelf K. V., Kjems J., Nature 2009, 459, 73–76. [DOI] [PubMed] [Google Scholar]
- 12. Douglas S. M., Dietz H., Liedl T., Högberg B., Graf F., Shih W. M., Nature 2009, 459, 414–418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Dietz H., Douglas S. M., Shih W. M., Science 2009, 325, 725–730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Liedl T., Högberg B., Tytell J., Ingber D. E., Shih W. M., Nat. Nanotechnol. 2010, 5, 520–524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Han D., Pal S., Nangreave J., Deng Z., Liu Y., Yan H., Science 2011, 332, 342–346. [DOI] [PubMed] [Google Scholar]
- 16. Voigt N. V., Tørring T., Rotaru A., Jacobsen M. F., Ravnsbæk J. B., Subramani R., Mamdouh W., Kjems J., Mokhir A., Besenbacher F., Gothelf K. V., Nat. Nanotechnol. 2010, 5, 200–203. [DOI] [PubMed] [Google Scholar]
- 17. Jahn K., Tørring T., Voigt N. V., Sørensen R. S., Kodal A. L. B., Andersen E. S., Gothelf K. V., Kjems J., Bioconjugate Chem. 2011, 22, 819–823. [DOI] [PubMed] [Google Scholar]
- 18. Shaw A., Lundin V., Petrova E., Fördős F., Benson E., Al-Amin A., Herland A., Blokzijl A., Högberg B., Teixeira A. I., Nat. Methods 2014, 11, 841–846. [DOI] [PubMed] [Google Scholar]
- 19. Douglas S. M., Bachelet I., Church G. M., Science 2012, 335, 831–834. [DOI] [PubMed] [Google Scholar]
- 20. Kuzyk A., Schreiber R., Zhang H., Govorov A. O., Liedl T., Liu N., Nat. Mater. 2014, 13, 862–866. [DOI] [PubMed] [Google Scholar]
- 21. List J., Weber M., Simmel F. C., Angew. Chem. Int. Ed. 2014, 53, 4236–4239; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2014, 126, 4321–4325. [Google Scholar]
- 22. Zadeh J. N., Steenberg C. D., Bois J. S., Wolfe B. R., Pierce M. B., Khan A. R., Dirks R. M., Pierce N. A., J. Comput. Chem. 2011, 32, 170–173. [DOI] [PubMed] [Google Scholar]
- 23. Wagenbauer K. F., Wachauf C. H., Dietz H., Nat. Commun. 2014, 5, 1–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Tomov T. E., Tsukanov R., Liber M., Masoud R., Plavner N., Nir E., J. Am. Chem. Soc. 2013, 135, 11935–11941. [DOI] [PubMed] [Google Scholar]
- 25. Douglas S. M., Chou J. J., Shih W. M., Proc. Natl. Acad. Sci. USA 2007, 104, 6644–6648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Tang G., Peng L., Baldwin P. R., Mann D. S., Jiang W., Rees I., Ludtke S. J., J. Struct. Biol. 2007, 157, 38–46. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary