

Abstract
Background
Neuronal ceroid lipofuscinosis (NCL), a fatal neurodegenerative disease, has been diagnosed in young adult Australian Cattle Dogs.
Objective
Characterize the Australian Cattle Dog form of NCL and determine its molecular genetic cause.
Animals
Tissues from 4 Australian Cattle Dogs with NCL‐like signs and buccal swabs from both parents of a fifth affected breed member. Archived DNA samples from 712 individual dogs were genotyped.
Methods
Tissues were examined by fluorescence, electron, and immunohistochemical microscopy. A whole‐genome sequence was generated for 1 affected dog. A TaqMan allelic discrimination assay was used for genotyping.
Results
The accumulation of autofluorescent cytoplasmic storage material with characteristic ultrastructure in tissues from the 4 affected dogs supported a diagnosis of NCL. The whole‐genome sequence contained a homozygous nonsense mutation: CLN5:c.619C>T. All 4 DNA samples from clinically affected dogs tested homozygous for the variant allele. Both parents of the fifth affected dog were heterozygotes. Archived DNA samples from 346 Australian Cattle Dogs, 188 Border Collies, and 177 dogs of other breeds were homozygous for the reference allele. One archived Australian Cattle Dog sample was from a heterozygote.
Conclusions and Clinical Importance
The homozygous CLN5 nonsense is almost certainly causal because the same mutation previously had been reported to cause a similar form of NCL in Border Collies. Identification of the molecular genetic cause of Australian Cattle Dog NCL will allow the use of DNA tests to confirm the diagnosis of NCL in this breed.
Keywords: Autofluorescence, Lysosomal storage disease, Molecular genetics, Whole‐genome sequence
Abbreviations
- CSF
cerebrospinal fluid
- GFAP
glial fibrillary acid protein
- NCLs
neuronal ceroid lipofuscinoses
- VCF
variant call format
The neuronal ceroid lipofuscinoses (NCLs) are fatal progressive neurodegenerative diseases characterized by the accumulation of autofluorescent lysosomal storage material within the brain, the retina, and other tissues. Mutations in 13 different genes have been found to cause various forms of NCL in humans.1 Neuronal ceroid lipofuscinosis has been reported in a variety of wild and domestic animals including the dog.2, 3 At least 10 DNA sequence variants from 8 different genes have been identified as molecular genetic causes for the NCLs in dogs (Table 1).4, 5, 6, 7, 8, 9, 10, 11, 12, 13, 14, 15 Until recently, all of the previously identified NCL‐causing sequence variants in dogs have been reported to occur within members of single breeds, although there are 2 examples of different NCL‐causing alleles segregating independently in the same breed: mutations in PPT1 and TPP1 have been associated with different forms of NCL in Dachshunds4, 5 and mutations in CLN6 and CLN8 have been found in different NCLs of Australian Shepherds.8, 11 Faller et al15 were the first to report a previously identified NCL‐causing mutation in a second dog breed when they described NCL in Chihuahua siblings that were homozygous for a single‐base deletion and frameshift that had already been reported in a Chinese Crested Dog with NCL.9
Table 1.
Breed distributions of canine NCL‐associated mutations
| Disease | Gene | Mutation | Amino Acid Sequence | Affected Dog Breed |
|---|---|---|---|---|
| CLN1 | PPT1 | PPT1:c.736_737insC | p.F246Lfs*29 | Dachshund4 |
| CLN2 | TPP1 | TPP1:c.325delC | p.A108Pfs*6 | Dachshund5 |
| CLN5 | CLN5 | CLN5:c.619C>T | p.Q207X | Border Collie,6 Australian Cattle Dog (current report) |
| CLN5 | CLN5 | CLN5:c.934_935delAG | CLN5:p.E312Vfs*6 | Golden Retriever7 |
| CLN6 | CLN6 | CLN6:c.829T>C | p.W277R | Australian Shepherd8 |
| CLN7 | MFSD8 | MFSD8c.843delT | p.F282Lfs*13 | Chinese Crested Dog, Chihuahua9, 15 |
| CLN8 | CLN8 | CLN8:c.491T>C | p.L164P | English Setter10 |
| CLN8 | CLN8 | CLN8:c.585G>A | p.W195X | Australian Shepherd11 |
| CLN10 | CTSD | CTSD:c.597G>A | p.M199I | American Bulldog12 |
| CLN12 | ATP13A2 | ATP13A2:c.1623delG | p.P541 fs*56 | Tibetan Terrier13, 14 |
Still to be identified are the molecular genetic causes of the previously reported NCLs in other breeds, such as the Australian Cattle Dog (also known as Blue Heeler),16 Cocker Spaniel,17 Dalmatian,18 Labrador Retriever,19 Miniature Schnauzer,20 Polish Lowland Sheepdog,21 and Saluki.22 Mutations causing NCL are likely to be segregating in additional breeds for which the disease has not yet been reported. We had an opportunity to analyze biological samples from young adult Australian Cattle Dogs with NCL‐like clinical signs. Herein, we describe the disease phenotype and report that its molecular genetic cause is identical to a mutation previously identified as the cause of NCL in Border Collies.6 Thus, ours is the second report that a NCL‐causing mutation is present in a dog breed other than that of the original discovery.
Materials and Methods
All studies were conducted with approval from the University of Missouri Animal Care and Use Committee and with informed consent of the dogs' owners.
Subject Australian Cattle Dogs
Five dogs were evaluated for this study. Although pedigree information was not available for all dogs, all were believed by their owners to be purebred Australian Cattle Dogs, and according to their veterinarians, all had physical characteristics that were typical of purebred Australian Cattle Dogs (Fig 1). Australian Cattle Dog A, a 15‐month‐old spayed female (Fig 1A), was referred to the Veterinary Specialty Services Neurology Department near St. Louis for an acute onset of seizures and blindness in 2014. Approximately 2 weeks before presentation, the dog had experienced its first tonic‐clonic seizure. The dog was reportedly completely blind in the postictal period. Partial return of vision occurred within 24 hours, but the dog never regained full vision. Over the next few days, the dog developed persistent focal seizures and “fly biting.” After the initial seizure, the dog circled constantly when not sedated. The dog had been evaluated by the local veterinarian who initiated treatment with phenobarbital. There was no improvement with phenobarbital treatment and her care was transferred to the Neurology Department at Veterinary Specialty Services. The dog had been obtained from a breeder at a young age, but pedigree records and breeder contact information were lost, and it was not possible to locate closely related dogs. Although the owners originally described no visual deficits before the first seizure, they mentioned that the dog had once been quite athletic, but recently had not been successful playing Frisbee or fetch. The dog exhibited decreased coordination that included clumsiness and bumping into objects. Starting at approximately 1 year of age, the dog began to display signs of anxiety, particularly when encountering other dogs or unfamiliar people.
Figure 1.

Photographs of four of the subject Australian Cattle Dogs. (A) Dog A from near St. Louis euthanized in 2014, (B) Dog B from Chicago euthanized in 2014. (C) Dog C from Seattle euthanized in 2007. (D) Dog E from Alabama euthanized in 2015.
The physical examination was limited because Dog A was extremely disoriented and hypersensitive to any auditory or tactile stimulation. With mild restraint, the dog would have severe myoclonic jerks. Cranial nerve examination identified a bilaterally absent menace reflex, but the remainder of the cranial nerve evaluation was normal. The dog was ambulatory with mild tetraparesis, proprioceptive ataxia, and delayed general proprioception in all 4 limbs. Dog A had intact segmental reflexes. A CBC and serum biochemistry analyses were unremarkable. Magnetic resonance imaging identified diffuse brain atrophy indicated by widening of the cerebral cortical sulci, diffuse dilatation of the ventricular system, subjectively decreased size of the interthalamic adhesion, and increased cerebrospinal fluid (CSF) surrounding the folia of the cerebellum (Fig 2). Because of the severity of the clinical signs, Dog A was euthanized the day after advanced diagnostic procedures were performed.
Figure 2.
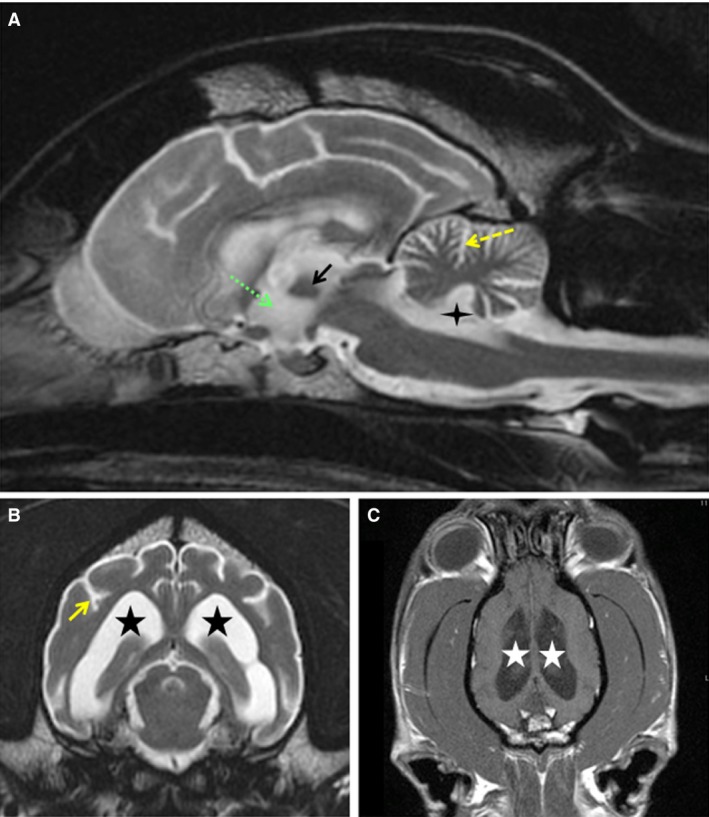
T2‐weighted MRI from Dog A in the sagittal (A) and transverse (B) planes. From Dog A, T1‐weighted postcontrast MRI in the dorsal plane (C). CSF is white and brain parenchyma gray in the T2‐weighted images. CSF is dark gray in the T1‐weighted image. Diffuse brain atrophy is indicated by atrophy of the interthalamic adhesion (black arrow in A), dilatation of the 3rd (green dashed arrow in A), 4th (4‐point star in A), and lateral ventricles (5‐point stars in B and C), widening of the sulci of the cerebral cortex (solid yellow arrow in B), and increased CSF surrounding the folia of the cerebellum (yellow dashed arrow in A).
Australian Cattle Dog B, a neutered male (Fig 1B), presented to the Chicago Veterinary Neurology and Neurosurgery Group in 2014 at approximately 18 months of age for evaluation of progressive neurologic changes and a recent severe seizure. The onset of signs occurred at approximately 12 months of age with periodic episodes of severe anxiety, particularly in response to loud noises. The severe anxiety persisted throughout the course of the disease. Over time, the dog's ability to recognize and respond to commands progressively decreased. As the disease progressed, other signs exhibited by Dog B included compulsive repetitive scanning of its surroundings, circling, loss of ability to navigate stairs, tremors, loss of coordination, visual impairment in both dim and bright light, periods of trance‐like behavior, aggression, ataxia, and clumsiness including increased bumping into obstacles. At the time of examination, the dog reacted aggressively to any attempts to control its head or body. Because of progressing neurologic signs that did not abate with zonisamide and diazepam treatment, Dog B was euthanized at approximately 20 months of age and a necropsy was performed to collect tissues as described below. No pedigree information was available for this dog.
Australian Cattle Dog C (Fig 1C) had been tentatively diagnosed with NCL in Pullman, Washington, in 2007. A DNA sample was received at the University of Missouri at that time and archived. The owners reported that during the 6 months before presentation, Dog C had begun showing abnormal behavior, including apparent incoordination, trance‐like episodes, increasing fear of other dogs, and aggression toward the owners. At examination, Dog C was disoriented, was overreactive to normal auditory and tactile stimuli, and had an intermittent left head tilt. Static knuckling was delayed in all 4 limbs, but no paresis was noted during observation of the gait. Tetra‐ataxia also was noted, especially during pivoting. The menace response was absent bilaterally, but pupillary light reflexes were normal. When the head was elevated, there was bilateral ventrolateral strabismus and vertical nystagmus. Segmental spinal reflexes were normal. Cerebrospinal fluid analysis disclosed a normal protein concentration (12 mg/dL; reference range, <25 mg/dL) and a normal nucleated cell count (1/μL; reference range,<5/μL) with normal cell cytology. A 2‐week course of prednisone was prescribed (1 mg/kg PO q12h for 1 week, then 0.5 mg/kg PO q12 h for 1 week) while awaiting additional testing (urine metabolic screen, CSF culture, serum titers for Toxoplasma and Neospora). Clinical signs were not altered by prednisone treatment. Results of the urine metabolic screen were normal, CSF culture was negative for bacteria at 3 days, and serum IgG and IgM titers for Toxoplasma and Neospora were negative. Because of the progression of its neurologic signs, the dog was euthanized at approximately 26 months of age.
A fourth Australian Cattle Dog (Dog D) presented at the Animal Medical Center in New York City in 2015 at the age of approximately 24 months after a 6‐month history of progressive behavior changes, including aggression toward people and other dogs, ataxia, anxiety, lethargy, and blindness. A neurologic examination identified the bilateral absence of the menace reflex and severely decreased nasal sensation. Pupillary light and palpebral reflexes were normal. The owners elected to have the dog euthanized immediately after the neurologic examination. Magnetic resonance imaging performed immediately after euthanasia showed diffuse symmetrical brain atrophy.
Australian Cattle Dog E (Fig 1D), a spayed female from Alabama, was euthanized in 2015 at approximately 27 months of age after having exhibited neurologic signs similar to the other 4 affected Australian Cattle Dogs. Starting at about 6 months of age, the dog began to occasionally bump into obstacles, but continued to engage in vision‐dependent activities, such as retrieving balls, without difficulty. Several months later, the dog began to exhibit signs of anxiety that became progressively more pronounced; it would become particularly agitated in response to loud noises. At approximately 2 years of age, the dog began to suffer from seizures and associated trance‐like behavior, lost interest in activities it previously had enthusiastically engaged in, would pace compulsively, and began to suffer from progressive vision loss and loss of coordination. Visual impairment was apparent in both dim and bright light. Upon neurologic examination at approximately 26 months of age, both direct and indirect pupillary responses could be elicited despite blindness. However, the dog did not exhibit a menace response on either side and physiologic nystagmus was observed in both eyes. Magnetic resonance imaging of the brain disclosed generalized brain atrophy with widened sulci and flattened gyri in the cortex, cerebellar atrophy, and enlarged ventricles. Cerebrospinal fluid analysis did not identify any abnormalities. Shortly after the neurologic examination, the dog's seizures became more frequent and severe and the dog was euthanized. No tissue or blood samples were saved, but buccal swab samples were collected from both parents.
Light and Electron Microscopic Procedures
Cerebral cortical (parietal lobe), cerebellar, and retinal samples were collected at necropsy from 4 of the affected Australian Cattle Dogs and examined by fluorescence and electron microscopy. The eyes from dogs A and B were enucleated and the corneas removed immediately. One eye from each dog was placed in a fixative consisting of 3.5% formaldehyde, 0.05% glutaraldehyde, 120 mM sodium cacodylate, 1 mM CaCl2, and pH 7.4 (immuno fix), and the other eye was placed in 2.5% glutaraldehyde, 100 mM sodium cacodylate, and pH 7.4 (EM fix). Slices of the cerebral cortex and cerebellum each were placed in the same fixatives. Before further processing, the eyecups were dissected to obtain regions from the posterior poles adjacent to the optic nerve heads, and these regions were used for examination. All samples were incubated in these fixatives at room temperature until being further processed for microscopic examination. The same tissues were collected postmortem from Dogs C and D in a similar manner except that they initially were placed in 10% buffered formalin for shipment. In addition, a sample of heart ventricle wall was collected from Dog C in the same fixative. Upon receipt at the University of Missouri, the tissue samples were washed briefly in 170 mM sodium cacodylate, pH 7.4, and then transferred to the same fixatives as used for the other dogs. These tissues were examined by previously described histopathologic, immunohistochemical, fluorescent microscopic, and electron microscopic procedures.7 Among these procedures was immunohistochemical staining of the cerebellar cortex for glial fibrillary acid protein (GFAP). Areas of the cerebellar cortex and closely associated medulla were dissected from the brains fixed in immuno fix (in some cases preceded by fixation in formalin). These tissues were embedded in paraffin and sectioned, and the sections were immunostained for GFAP.
Molecular Genetic Analysis
EDTA‐anticoagulated blood from affected Australian Cattle Dogs A, B, C, and D described above served as a source of DNA. Buccal swab samples from the sire and dam of affected Australian Cattle Dog E were collected on commercial cards.1 Previously described procedures were used to isolate DNA from the blood10 and cards.23 In addition, DNA samples from 347 other Australian Cattle Dogs, 188 Border Collies, and 177 dogs representing 88 other breeds also were used in the study. These samples were randomly selected from among the samples in the University of Missouri Animal DNA repository.
The DNA from Australian Cattle Dog A was submitted to the University of Missouri DNA Core Facility for the preparation of 2 PCR‐free libraries (fragment sizes of approximately 350 and 550 bp) with a commercial kit2 and for 2 × 100 paired‐end sequencing in 2 flow‐cell lanes in a massively parallel DNA sequencer.3 The adaptor sequences were trimmed with custom Perl scripts and the reads were error corrected using a previously described data analysis pipeline.9 These reads were deposited in the Sequence Read Archive (accession SRS834022) and aligned to the canine reference genome sequence (CanFam 3.1) using the default parameters of the bwa mem algorithm in BWA 0.7.12 software.24 Samtools 1.2 software was used to convert tab‐delimited text files that contained sequence alignment data (aligned.SAM file types) to binary versions (.BAM file types), merge the libraries, and sort and index the files by genomic coordinates.25 The Platypus 0.8.1 variant caller software26 was used to generate individual variant call format (VCF) files using the default parameters. Commercial software4 was used to annotate variants and to compare them with similarly generated and annotated VCF files from 43 other control dogs with different genetic diseases. The same software also was used to filter the variants to identify those with the following 2 characteristics: (1) they were located within canine orthologs of the 13 genes associated with NCL in humans1 and (2) they were predicted to alter the primary structure of the gene product.
The DNA samples from the 4 affected Australian Cattle Dogs were genotyped by direct automated Sanger sequencing of PCR amplicons produced with oligonucleotide primers 5′‐TTAACCAAATGGCAAAGTGGG‐3′ and 5′‐TTCTTGAACTCTGCTCCAAGT‐3′ which span a previously described causal CLN5:c.619C>T transition.6 A TaqMan allelic discrimination assay27 was used to genotype both parents of Australian Cattle Dog E and the archived DNA samples. The sequences of the PCR primers for the allelic discrimination assay were 5′‐GCGGGACAATGAAACAGGAATTTAT‐3′ and 5′‐TGTCTCAGCCCCCTTTGTTG‐3′. The competing probes were 5′‐CTGGCTTGAACAGTCC‐MGB‐3′ (reference allele) and 5′‐CTGGCTTAAACAGTCC‐MGB‐3′ (variant allele).
Results
Light and Electron Microscopy
All cerebellar, cerebral cortical, and retinal samples from the affected Australian Cattle Dogs exhibited abnormal accumulations of autofluorescent storage material with the fluorescence spectral characteristics typical of the NCLs (Fig 3A–D). The cardiac muscle of a heart ventricle also was found to harbor substantial amounts of autofluorescent inclusions within the muscle fibers in the 1 affected dog from which the heart was examined (Fig 3E).
Figure 3.
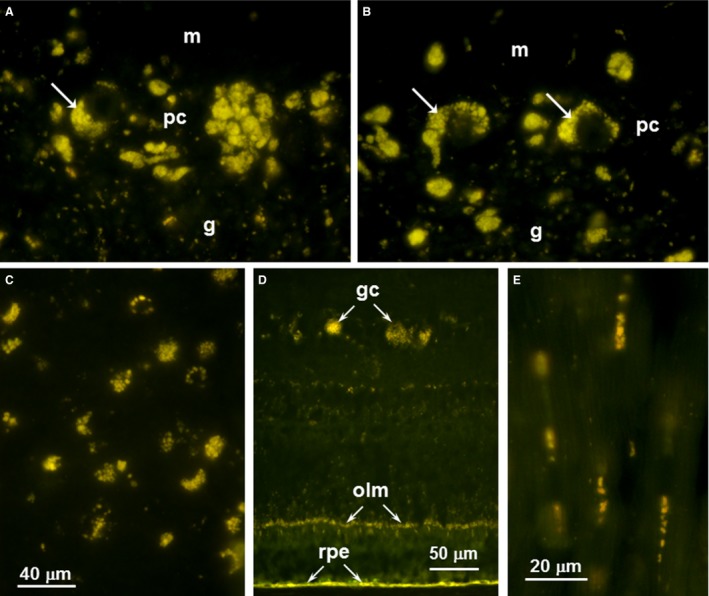
Fluorescence micrographs of cryostat sections the cerebellum (A and B), cerebral cortex (C), retina (D) from Dog A and heart ventricular wall (E) from Dog C. In the cerebellum, storage body accumulation was most abundant in the Purkinje cell layer (pc) with some but not all of the material within cells that could be identified as Purkinje cells (arrows in A and B). There were lesser accumulations of this material in the molecular (m) and granular (g) layers. In the cerebral cortex (B), the storage material was widely distributed throughout most of the tissue. In the retina (C), the most prominent accumulations of autofluorescent material were in the ganglion cells (gc) and along the outer limiting membrane (olm). The retinal pigment epithelium (rpe) also contained substantial amounts of material with similar fluorescence properties, but since the accumulation of such material occurs during normal aging, the presence of this material in the rpe is not diagnostic for NCL. Clusters of autofluorescent inclusions were present in the heart muscle fibers sections in longitudinal orientation (E).
The ultrastructural appearances of the storage material in brain and retina were essentially the same for Dogs A and B from which EM‐fixed samples were obtained. The contents of the storage bodies had a primarily membranous appearance in cerebellar Purkinje cells, cerebral cortical neurons, and retinal ganglion cells (Fig 4). In all 3 cell types, the inclusions were membrane bound and consisted of aggregated clusters of membrane‐like structures, but the arrangement of this membrane‐like material varied among cell types. The primary storage components in the Purkinje cells had short linear and vesicular appearances (Fig 4A). In the cerebral cortical neurons and retinal ganglion cells, storage bodies consisted almost exclusively of clusters of parallel stacks of relatively linear profiles (Fig 4B–D). The cerebral cortex contained storage bodies with scattered, extremely electron‐dense structures (Fig 4B). The degree of compaction of the membrane‐like structures in the retinal ganglion cell inclusions was variable (Fig 4C and D). The storage bodies along the outer limiting membrane of the retina were located primarily within the photoreceptor cells just interior to the junctions between these cells and the adjacent Mueller cells (Fig 4E and F). These storage bodies contained either aggregates of small vesicles (Fig 4E) or membrane‐like material in a variety of configurations (Fig 4F). The membrane‐like components were randomly distributed within primarily granular material, unlike the clustered distributions found in the other cell types. The only cardiac tissue received from an affected dog was fixed in formalin, which did not permit good ultrastructural preservation of the storage material.
Figure 4.
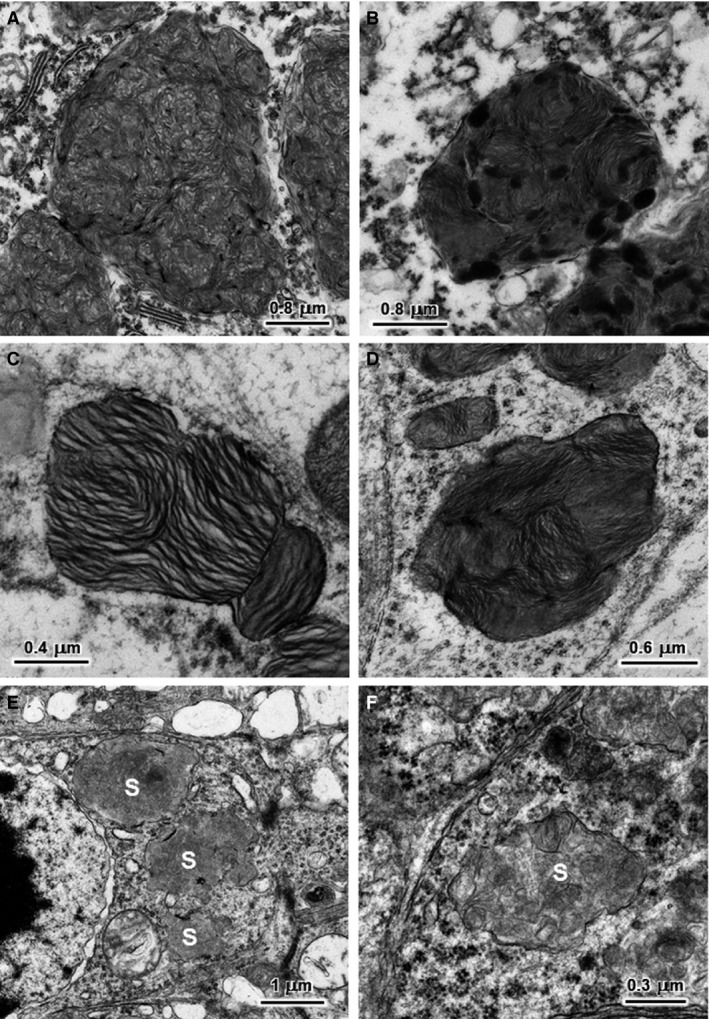
Electron micrographs of the disease‐related storage bodies from a cerebellar Purkinje cell (A), a cerebral cortical neuron (B), retinal ganglion cells (C and D), and retinal photoreceptor cells (E and F) of NCL‐affected Australian Cattle Dog D. The storage bodies (s) in the photoreceptor cells are primarily located just internal to the outer limiting membrane. The contents of storage bodies consisted of both aggregates of small vesicles (E) or of membrane‐like structures in a variety of configurations (F).
The NCLs, like many other lysosomal storage diseases, are characterized by astrogliosis, as indicated by increased amounts of glial fibrillary acid protein (GFAP) in astrocytes.7, 9, 11, 28 Examination of the cerebellar cortex from Australian Cattle Dog A by immunohistochemistry disclosed very high numbers of cells in both the cerebellar medulla and the granule cell layer that stained strongly with an anti‐GFAP antibody (Fig 5A and C). By comparison, little GFAP immunostaining was observed in either of these cerebellar areas in a normal healthy dog of similar age (Fig 5B and D).
Figure 5.
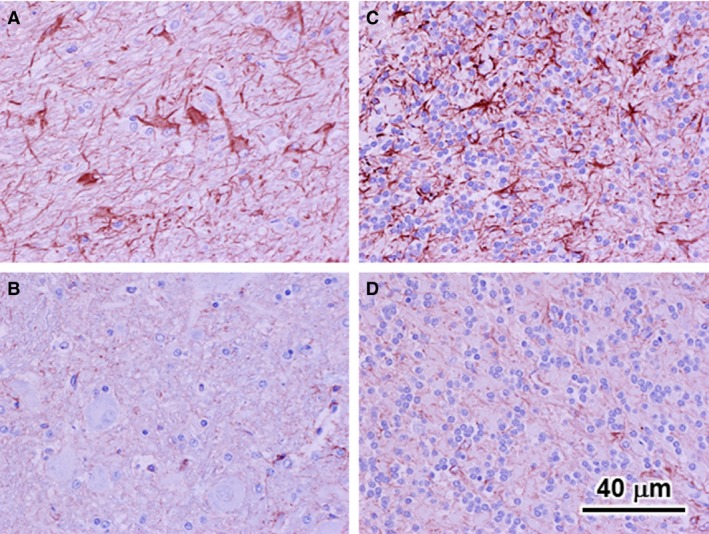
Light micrographs of sections of the cerebellar medulla (A and B) and granule cell layer (B and D). All sections were immunostained for GFAP. GFAP staining is a reddish brown color. All photomicrographs are from the cerebellum. Micrographs (A) and (C) are from the affected Australian Cattle Dog A; micrographs (B) and (D) are from a normal healthy Beagle of similar age. Bar in (D) indicates magnification of all 4 micrographs. GFAP‐positive cells with the typical profiles of reactive astrocytes were abundant in both the granule cell layer and medulla of affected dog, but were not present in either of these areas of the cerebellar cortex from the normal dog.
Molecular Genetics
We used massively parallel sequencing technology to generate a whole‐genome sequence from NCL‐affected Australian Cattle Dog A and aligned the sequence reads to the canine genome reference sequence (CanFam3.1). The average depth of coverage of the aligned sequence was 28‐fold. The 6.2 million potential sequence variants detected in this alignment were filtered to retain only those that were predicted to alter the amino acid sequences of any of the canine orthologs of the 13 genes known to have harbored mutations in human NCL patients.1 As shown in Table 2, only 5 sequence variants survived this filtration process. Four were missense mutations that occurred in the heterozygous state. These 4 variants also were present in from 5 to 13 of the 43 control whole‐genome sequences from dogs with no signs of NCL and, thus, were unlikely to have been responsible for Australian Cattle Dog A's NCL. In contrast, a C to T transition at position 30,574,637 bp on CFA22 was homozygous in Australian Cattle Dog A and produced a premature stop codon that was predicted to severely truncate the encoded CLN5. This variant was almost certainly causal because it previously had been reported to be responsible for NCL in Border Collies.6 The homozygous CLN5:c.619C>T transition, detected in the whole‐genome sequence from Australian Cattle Dog A, was confirmed by automated Sanger sequencing. In addition, automated Sanger sequencing showed that Australian Cattle Dogs B, C, and D also were homozygous for the CLN5:c.619T allele.
Table 2.
Variants in NCL‐associated genes in WGS from affected Australian Cattle Dog A
| Chromosomal Coordinate | Gene | Nucleotide Change | Predicted Amino Acid Change | Zygosity | Comments |
|---|---|---|---|---|---|
| 6:18,257,856 | CLN3 | c.209A>G | p.Glu70Gly | Heterozygous | Variant in 10 other WGSs |
| 18:46,010,727 | CTSD | c.1168T>C | p.Cys390Arg | Heterozygous | Variant in 10 other WGSs |
| 18:46,011,691 | CTSD | c.761A>C | p.Lys254Thr | Heterozygous | Variant in 13 other WGSs |
| 21:29,922,715 | TPP1 | c.1280C>T | p.Ala427Val | Heterozygous | Variant in 5 other WGSs |
| 22:30,574,637 | CLN5 | c.619C>T | p.Gln207* | Homozygous | Unique to affected WGS |
A TaqMan allelic discrimination assay27 was used to determine the CLN5:c.619 genotypes for another 714 dogs, including both parents of affected Australian Cattle Dog E. These 2 parents and 1 other Australian Cattle Dog, represented in our DNA collection since 2014, were CLN5:c.619C/T heterozygotes. The variant c.619T allele was not detected in archived DNA samples from 346 other Australian Cattle Dogs previously obtained for a variety of reasons unrelated to NCL. In addition, all 188 randomly selected Border Collie DNA samples and all 177 randomly selected samples from representatives of 88 other breeds were homozygous for the reference c.619C allele.
Discussion
We used fluorescence and electron microscopy to confirm the diagnosis of NCL in 4 apparently unrelated Australian Cattle Dogs that had exhibited neurologic signs consistent with this disorder. Previously, NCL had been diagnosed in 2 pairs of Australian Cattle Dog littermates over 35 years ago.16, 29, 30 Both of the previously reported litters were from the United States, but there was no known familial relationship between them.30 Both sexes were affected. One litter included an affected pair and 7 clinically normal littermates.16 The clinical status of the littermates to the other affected pair was not reported, but it was reported that their parents were normal.30 These observations are consistent with an autosomal recessive mode of inheritance. The earlier‐reported NCL‐affected Australian Cattle Dogs and those that we studied had brains with similar distributions of autofluorescent storage bodies with similar ultrastructural appearance. In addition, the ages at onset and euthanasia of these earlier‐reported Australian Cattle dogs with NCL are similar to those of the NCL‐affected Australian Cattle dogs reported here (Table 3). Thus, it is likely that a single founding mutation was responsible both for the earlier‐reported Australian Cattle Dog NCL and for the affected Australian Cattle Dogs reported here. Nonetheless, biological samples from the earlier‐reported dogs were not available for testing and the existence of NCL phenocopies within the Australian Cattle Dog breed cannot be excluded.
Table 3.
Comparison of ages at onset and death or euthanasia for Australian Cattle Dogs, Border Collies, and Golden Retrievers with NCL
| Breed | Number of Dogs | Causal Mutation | Location | Age at Onset, Months | Age at Death or Euthanasia, Months | Reference |
|---|---|---|---|---|---|---|
| Australian Cattle Dog | 4 | CLN5:c.619T | United States | 12–19 | 15–26 | Current report |
| Australian Cattle Dog | 4 | Unknown | United States | 12–14 | 18–26 | 16, 30 |
| Border Collie | 23 | Unknown | Australia | 16–23 | 18–29 | 33 |
| Border Collie | 1 | Unknown | United States | 27 | 29 | 34 |
| Border Collie | 27 | CLN5:c.619T | Japan | 15–20 | 23–32 | 36 |
| Golden Retriever | 4 | CLN5:c.934_935delAG | United States | 13–17 | 30–34 | 7 |
Whole‐genome sequencing has proven to be an efficient strategy for the identification of the mutations responsible for NCL in dogs.7, 9, 11, 15 Thus, in search of the molecular genetic cause of NCL in Australian Cattle Dogs, we generated a whole‐genome sequence with DNA from affected Australian Cattle Dog A and identified a homozygous nonsense mutation, CLN5:c.619C>T, that had already been reported to cause NCL in Border Collies.6 The NCL‐affected Australian Cattle Dogs B, C, and D also were homozygous for the variant c.619T allele, whereas there were no c.619T homozygotes among 349 other Australian Cattle Dogs not known to have exhibited clinical signs of NCL. Three of these 349 Australian Cattle Dogs were c.619C/T heterozygotes, including both parents of Australian Cattle Dog E. The similarities between the clinical history of Dog E and the clinical histories of the other 4 NCL‐affected Australian Cattle Dogs, and the fact that both of Dog E's parents carried the rare c.619T allele strongly imply that Dog E was a c.619T homozygote. The association of NCL in 2 dog breeds with the same rare truncating mutation in a candidate gene strongly indicates that the mutation is causal for NCL in both Australian Cattle Dogs and Border Collies.
Although there are anecdotal accounts of “dog show” Border Collies with NCL‐like signs in Australia and New Zealand in the 1970s,31 the first published report of NCL in Border Collies appeared in 1988.32 This report described the disease in 3 male and 2 female Border Collies. Three years later, the clinical signs and laboratory findings for these 5 dogs and 18 other Border Collies were summarized.33 As shown in Table 3, the age at disease onset and the disease duration in these Border Collies from Australia closely resembled that of the affected Australian Cattle Dogs, including those shown here to be CLN5:c.619C>T homozygotes. Subsequently, a Border Collie from the United States was reported to have NCL with similar clinical and laboratory findings but a later age at onset.34
In 2005, linkage analysis with DNA markers from candidate regions placed the Border Collie NCL locus on a CLN5‐containing segment of canine chromosome 22 and the CLN5:c.619C>T transition was identified as the causal mutation.6 Since then, it has been possible to use DNA tests to identify c.619T homozygotes and to investigate the Border Collie NCL with genetically defined cohorts of NCL‐affected dogs. Two reports described CLN5:c.619T homozygous Border Collies from Japan.35, 36 The clinical and laboratory findings for 27 Border Collies36 with genetically defined NCL were similar to those from earlier reports of affected Border Collies and Australian Cattle Dogs and to those of the 5 Australian Cattle Dogs in our report. The ages at onset and death also were similar in each group of affected dogs (Table 3). The Japanese investigators found a CLN5:c.619T allele frequency >0.3 in dogs from certain kennels that unknowingly included asymptomatic heterozygotes in their breeding stock36 and estimated an overall CLN5:c.619T allele frequency of 0.04, based on a random survey of over 400 Japanese Border Collies.35 The mutant T allele appears to be less common in North America because 188 Border Collie samples from our DNA collection all were CLN5:c.619C allele homozygotes. Although possible, it is very unlikely that the CLN5:c.619C>T transition arose independently in Border Collies and Australian Cattle Dogs. Both breeds are of similar size and both breeds are used for herding. Some kennels produce puppies from both dog breeds. Thus, there is the potential for occasional planned or inadvertent exchange of genetic material between breeds. Published reports indicate that the disease has been present in both breeds for many generations,16, 32 but it is not possible from the available evidence to identify the breed of origin. Regardless of the origin, a DNA test for the mutation can be used to aid in the diagnosis of NCL.
A different CLN5 mutation, CLN5:c.934_935delAG, was found in the homozygous state in a Golden Retriever with NCL.7 This mutation produces a reading‐frame shift and is predicted to encode a truncated gene product: CLN5:p.E312Vfs*6. As shown in Table 3, the disease characteristics of the affected Golden Retrievers are similar to those of the affected Australian Cattle Dogs and Border Collies.
Mutations in human CLN5 were first reported to cause NCL in 1998.37 Since then, at least 36 potentially causal CLN5 sequence variants have been found in human NCL patients (http://www.ucl.ac.uk/ncl/cln5.shtml). For most CLN5 patients, the onset of clinical signs occurred between 4 and 7 years of age,38, 39 but some presumably hypomorphic mutations have produced much later disease onsets.40 Although CLN5 mutations have been recognized as causes of NCL for over 16 years, the biological functions of the gene product and the mechanisms leading to neurodegeneration remain unknown.41 Ovine and murine Cln5‐deficient animal models share disease features with human CLN5 patients42, 43 and have been used to investigate the underlying disease pathways.44, 45, 46 The ovine model is involved with ongoing and planned experiments to evaluate potential therapies for human CLN5 patients.47 Several canine models have been used to develop therapies for lysosomal storage diseases,48 and dogs have obvious advantages over sheep in urban research settings. A TPP1‐deficient canine NCL model5 has proven useful in the evaluation of therapies for the human CLN2 form of NCL including an enzyme replacement therapy that is currently in clinical trials in human patients (https://clinicaltrials.gov/ct2/show/NCT01907087).49, 50, 51 Dogs with CLN5 deficiency have the potential for similar use.
Acknowledgments
The authors thank the dog owners and veterinarians who provided the biological samples and clinical histories.
This work has not been presented at any meeting.
Grant Support: This work was supported by the University of Missouri Research Incentive Fund (MLK).
Conflict of Interest Declaration: Authors declare no conflict of interest.
Off‐label Antimicrobial Declaration: Authors declare no off‐label use of antimicrobials.
The work was performed at the University of Missouri and at collaborating private veterinary clinics.
Footnotes
Whatman FTA Elute card, catalog number WB120411, Fisher Scientific, Chicago IL
TruSeq DNA PCR‐Free Sample Preparation Kit, Illumina, San Diego, CA
HiSeq 2500 System, Illumina, San Diego, CA
SNP & Variation Suite v8.x, Golden Helix, Inc., Bozeman, MT
References
- 1. Mole SE, Cotman SL. Genetics of the neuronal ceroid lipofuscinoses (Batten disease). Biochim Biophys Acta 2015;1852:2237–2241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Jolly RD, Palmer DN. The neuronal ceroid‐lipofuscinoses (Batten disease): Comparative aspects. Neuropathol Appl Neurobiol 1995;21:50–60. [DOI] [PubMed] [Google Scholar]
- 3. Anderson GW, Goebel HH, Simonati A. Human pathology in NCL. Biochim Biophys Acta 2013;1832:1807–1826. [DOI] [PubMed] [Google Scholar]
- 4. Sanders DN, Farias FH, Johnson GS, et al. A mutation in canine PPT1 causes early onset neuronal ceroid lipofuscinosis in a Dachshund. Mol Genet Metab 2010;100:349–356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Awano T, Katz ML, O'Brien DP, et al. A frame shift mutation in canine TPP1 (the ortholog of human CLN2) in a juvenile Dachshund with neuronal ceroid lipofuscinosis. Mol Genet Metab 2006;89:254–260. [DOI] [PubMed] [Google Scholar]
- 6. Melville SA, Wilson CL, Chiang CS, et al. A mutation in canine CLN5 causes neuronal ceroid lipofuscinosis in Border collie dogs. Genomics 2005;86:287–294. [DOI] [PubMed] [Google Scholar]
- 7. Gilliam D, Kolicheski A, Johnson GS, et al. Golden Retriever dogs with neuronal ceroid lipofuscinosis have a two‐base‐pair deletion and frameshift in CLN5. Mol Genet Metab 2015;115:101–109. [DOI] [PubMed] [Google Scholar]
- 8. Katz ML, Farias FH, Sanders DN, et al. A missense mutation in canine CLN6 in an Australian shepherd with neuronal ceroid lipofuscinosis. J Biomed Biotechnol 2011;2011:198042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Guo J, O'Brien DP, Mhlanga‐Mutangadura T, et al. A rare homozygous MFSD8 single‐base‐pair deletion and frameshift in the whole genome sequence of a Chinese Crested dog with neuronal ceroid lipofuscinosis. BMC Vet Res 2014;10:960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Katz ML, Khan S, Awano T, et al. A mutation in the CLN8 gene in English Setter dogs with neuronal ceroid‐lipofuscinosis. Biochem Biophys Res Commun 2005;327:541–547. [DOI] [PubMed] [Google Scholar]
- 11. Guo J, Johnson GS, Brown HA, et al. A CLN8 nonsense mutation in the whole genome sequence of a mixed breed dog with neuronal ceroid lipofuscinosis and Australian Shepherd ancestry. Mol Genet Metab 2014;112:302–309. [DOI] [PubMed] [Google Scholar]
- 12. Awano T, Katz ML, O'Brien DP, et al. A mutation in the cathepsin D gene (CTSD) in American Bulldogs with neuronal ceroid lipofuscinosis. Mol Genet Metab 2006;87:341–348. [DOI] [PubMed] [Google Scholar]
- 13. Farias FH, Zeng R, Johnson GS, et al. A truncating mutation in ATP13A2 is responsible for adult‐onset neuronal ceroid lipofuscinosis in Tibetan terriers. Neurobiol Dis 2011;42:468–474. [DOI] [PubMed] [Google Scholar]
- 14. Wohlke A, Philipp U, Bock P, et al. A one base pair deletion in the canine ATP13A2 gene causes exon skipping and late‐onset neuronal ceroid lipofuscinosis in the Tibetan terrier. PLoS Genet 2011;7:e1002304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Faller KM, Bras J, Sharpe SJ, et al. The Chihuahua dog: A new animal model for neuronal ceroid lipofuscinosis CLN7 disease? J Neurosci Res 2016;94:339–347. [DOI] [PubMed] [Google Scholar]
- 16. Cho DY, Leipold HW, Rudolph R. Neuronal ceroidosis (ceroid‐lipofuscinosis) in a Blue Heeler dog. Acta Neuropathol 1986;69:161–164. [DOI] [PubMed] [Google Scholar]
- 17. Nimmo Wilkie JS, Hudson EB. Neuronal and generalized ceroid‐lipofuscinosis in a cocker spaniel. Vet Pathol 1982;19:623–628. [DOI] [PubMed] [Google Scholar]
- 18. Goebel HH, Bilzer T, Dahme E, et al. Morphological studies in canine (Dalmatian) neuronal ceroid‐lipofuscinosis. Am J Med Genet Suppl 1988;5:127–139. [DOI] [PubMed] [Google Scholar]
- 19. Rossmeisl JH Jr, Duncan R, Fox J, et al. Neuronal ceroid‐lipofuscinosis in a Labrador Retriever. J Vet Diagn Invest 2003;15:457–460. [DOI] [PubMed] [Google Scholar]
- 20. Jolly RD, Sutton RH, Smith RI, et al. Ceroid‐lipofuscinosis in miniature Schnauzer dogs. Aust Vet J 1997;75:67. [DOI] [PubMed] [Google Scholar]
- 21. Narfstrom K, Wrigstad A. Clinical, electrophysiological, and morphological findings in a case of neuronal ceroid lipofuscinosis in The Polish Owczarek Nizinny (PON) dog. Vet Q 1995;17:46. [PubMed] [Google Scholar]
- 22. Appleby EC, Longstaffe JA, Bell FR. Ceroid‐lipofuscinosis in two Saluki dogs. J Comp Pathol 1982;92:375–380. [DOI] [PubMed] [Google Scholar]
- 23. Zeng R, Coates JR, Johnson GC, et al. Breed distribution of SOD1 alleles previously associated with canine degenerative myelopathy. J Vet Intern Med 2014;28:515–521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Li H, Durbin R. Fast and accurate short read alignment with Burrows‐Wheeler transform. Bioinformatics 2009;25:1754–1760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Li H, Handsaker B, Wysoker A, et al. The sequence alignment/map format and SAMtools. Bioinformatics 2009;25:2078–2079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Rimmer A, Phan H, Mathieson I, et al. Integrating mapping‐, assembly‐ and haplotype‐based approaches for calling variants in clinical sequencing applications. Nat Genet 2014;46:912–918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Livak KJ. Allelic discrimination using fluorogenic probes and the 5' nuclease assay. Genet Anal 1999;14:143–149. [DOI] [PubMed] [Google Scholar]
- 28. Rama Rao KV, Kielian T. Astrocytes and lysosomal storage diseases. Neuroscience 2016;26:195–206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Wood PA, Sisk DB, Styer E, et al. Animal model: Ceroidosis (ceroid‐lipofuscinosis) in Australian cattle dogs. Am J Med Genet 1987;26:891–898. [DOI] [PubMed] [Google Scholar]
- 30. Sisk DB, Levesque DC, Wood PA, et al. Clinical and pathologic features of ceroid lipofuscinosis in two Australian cattle dogs. J Am Vet Med Assoc 1990;197:361–364. [PubMed] [Google Scholar]
- 31. Taylor RM, Farrow BR. Ceroid lipofuscinosis in the Border Collie dog: Retinal lesions in an animal model of juvenile Batten disease. Am J Med Genet 1992;42:622–627. [DOI] [PubMed] [Google Scholar]
- 32. Taylor RM, Farrow BR. Ceroid‐lipofuscinosis in Border Collie dogs. Acta Neuropathol 1988;75:627–631. [DOI] [PubMed] [Google Scholar]
- 33. Studdert VP, Mitten RW. Clinical features of ceroid lipofuscinosis in Border Collie dogs. Aust Vet J 1991;68:137–140. [DOI] [PubMed] [Google Scholar]
- 34. Franks JN, Dewey CW, Walker MA, et al. Computed tomographic findings of ceroid lipofuscinosis in a dog. J Am Anim Hosp Assoc 1999;35:430–435. [DOI] [PubMed] [Google Scholar]
- 35. Mizukami K, Chang HS, Yabuki A, et al. Novel rapid genotyping assays for neuronal ceroid lipofuscinosis in Border Collie dogs and high frequency of the mutant allele in Japan. J Vet Diagn Invest 2011;23:1131–1139. [DOI] [PubMed] [Google Scholar]
- 36. Mizukami K, Kawamichi T, Koie H, et al. Neuronal ceroid lipofuscinosis in Border Collie dogs in Japan: Clinical and molecular epidemiological study (2000‐2011). Scientific World Journal 2012;2012:383174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Savukoski M, Klockars T, Holmberg V, et al. CLN5, a novel gene encoding a putative transmembrane protein mutated in Finnish variant late infantile neuronal ceroid lipofuscinosis. Nat Genet 1998;19:286–288. [DOI] [PubMed] [Google Scholar]
- 38. Xin W, Mullen TE, Kiely R, et al. CLN5 mutations are frequent in juvenile and late‐onset non‐Finnish patients with NCL. Neurology 2010;74:565–571. [DOI] [PubMed] [Google Scholar]
- 39. Kousi M, Lehesjoki AE, Mole SE. Update of the mutation spectrum and clinical correlations of over 360 mutations in eight genes that underlie the neuronal ceroid lipofuscinoses. Hum Mutat 2012;33:42–63. [DOI] [PubMed] [Google Scholar]
- 40. Mancini C, Nassani S, Guo Y, et al. Adult‐onset autosomal recessive ataxia associated with neuronal ceroid lipofuscinosis type 5 gene (CLN5) mutations. J Neurol 2015;262:173–178. [DOI] [PubMed] [Google Scholar]
- 41. Carcel‐Trullols J, Kovacs AD, Pearce DA. Cell biology of the NCL proteins: What they do and don't do. Biochim Biophys Acta 2015;1852:2242–2255. [DOI] [PubMed] [Google Scholar]
- 42. Kopra O, Vesa J, von Schantz C, et al. A mouse model for Finnish variant late infantile neuronal ceroid lipofuscinosis, CLN5, reveals neuropathology associated with early aging. Hum Mol Genet 2004;13:2893–2906. [DOI] [PubMed] [Google Scholar]
- 43. Frugier T, Mitchell NL, Tammen I, et al. A new large animal model of CLN5 neuronal ceroid lipofuscinosis in Borderdale sheep is caused by a nucleotide substitution at a consensus splice site (c.571 + 1G>A) leading to excision of exon 3. Neurobiol Dis 2008;29:306–315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Amorim IS, Mitchell NL, Palmer DN, et al. Molecular neuropathology of the synapse in sheep with CLN5 Batten disease. Brain Behav 2015;5:e00401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Hughes SM, Hope KM, Xu JB, et al. Inhibition of storage pathology in prenatal CLN5‐deficient sheep neural cultures by lentiviral gene therapy. Neurobiol Dis 2014;62:543–550. [DOI] [PubMed] [Google Scholar]
- 46. Perentos N, Martins AQ, Watson TC, et al. Translational neurophysiology in sheep: Measuring sleep and neurological dysfunction in CLN5 Batten disease affected sheep. Brain 2015;138:862–874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Palmer DN, Neverman NJ, Chen JZ, et al. Recent studies of ovine neuronal ceroid lipofuscinoses from BARN, the Batten Animal Research Network. Biochim Biophys Acta 2015;1852:2279–2286. [DOI] [PubMed] [Google Scholar]
- 48. Bradbury AM, Gurda BL, Casal ML, et al. A review of gene therapy in canine and feline models of lysosomal storage disorders. Hum Gene Ther Clin Dev 2015;26:27–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Katz ML, Coates JR, Sibigtroth CM, et al. Enzyme replacement therapy attenuates disease progression in a canine model of late‐infantile neuronal ceroid lipofuscinosis (CLN2 disease). J Neurosci Res 2014;92:1591–1598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Vuillemenot BR, Kennedy D, Cooper JD, et al. Nonclinical evaluation of CNS‐administered TPP1 enzyme replacement in canine CLN2 neuronal ceroid lipofuscinosis. Mol Genet Metab 2015;114:281–293. [DOI] [PubMed] [Google Scholar]
- 51. Katz ML, Tecedor L, Chen Y, et al. AAV gene transfer delays disease onset in a TPP1‐deficient canine model of the late infantile form of Batten disease. Sci Transl Med 2015;7:313ra180. [DOI] [PMC free article] [PubMed] [Google Scholar]