

TO THE EDITOR
Silver–Russell syndrome (SRS) is a representative imprinting disorder associated with growth failure and characteristic somatic features [Azzi et al., 2014]. Recent studies have identified epimutation (hypomethylation) of the paternally derived H19‐differentially methylated region (DMR) at the imprinting control region 1 (ICR1) on chromosome 11p15.5 in ∼45% of SRS patients and maternal uniparental disomy 7 (upd(7)mat) in 5–10% of SRS patients, in addition to rare underlying factors such as duplications of maternally derived chromosome 11p15.5 involving CDKN1C at the ICR2 just proximal to the ICR1, relatively mild gain‐of‐function mutations of CDKN1C, loss‐of‐function mutations of IGF2, epimutation (hypomethylation) of the IG‐DMR and/or the MEG3‐DMR on chromosome 14q32.2 imprinted region of paternal origin, upd(14)mat, upd(16)mat, and upd(20)mat [Eggermann, 2010; Binder et al., 2011; Brioude et al., 2013; Poole et al., 2013; Azzi et al., 2014; Begemann et al., 2015; Kagami et al., 2015; Nakashima et al., 2015]. Here, we report an SRS patient with somatic mosaicism for upd(11)mat that was identified by buccal cell analysis.
This Chinese boy was born to non‐consanguineous parents at 35 weeks gestation with a birth weight of 1.36 kg (−2.8 SD), after a prenatal history of intrauterine growth retardation since ∼24 weeks of gestation. At 1 month of age, he was referred to our clinical genetic service because of failure to thrive. Physical examination showed growth failure, relative macrocephaly, frontal bossing, left fifth finger clinodactyly, mild left hemihypoplasia, and glandular hypospadias (Fig. 1A). Routine laboratory tests were normal, and chromosome analysis revealed a 46,XY karyotype in all the 50 Lymphocytes examined. He also had feeding difficulty that required tube feeding for 1 year and subsequent gastrostomy. On the last examination at 2 years of age, he exhibited severe growth failure and mild speech delay, while his head circumference remained within the normal range (Fig. 1B). Thus, he was diagnosed as having typical SRS, on the basis of positive manifestations of all the six Netchine–Harbison clinical scoring system features [Azzi et al., 2014].
Figure 1.
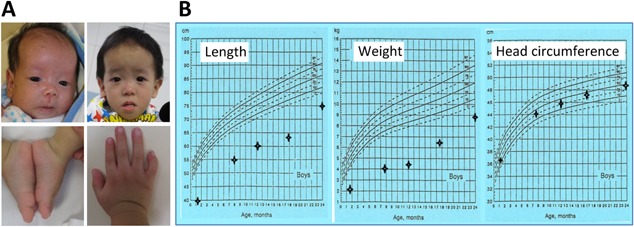
Clinical findings of this patient. (A) Photographs. Facial features at 1 month of age and 2 years of age, asymmetric feet, and left fifth finger clinodactyly. (B) Growth charts. [Color figure can be seen in the online version of this article, available at http://wileyonlinelibrary.com/journal/ajmga].
Thus, we attempted to clarify the underlying (epi)genetic cause. This study was approved by the Institutional Review Board Committees of the investigators, and performed after obtaining written informed consent. The primers used are summarized in Supplementary Table SI.
We first performed molecular studies using leukocyte genomic DNA. Methylation specific multiplex ligation dependent probe amplification (MS‐MLPA) was carried out with the probe mixture kit ME030‐C3 BWS/RSS for the ICR1/ICR2 (MRC‐Holland), revealing neither copy number change for untreated genomic DNA nor abnormal methylation pattern for HhaI‐digested DNA (Fig. 2A). Upd(7)mat was excluded by microsatellite analysis for nine loci widely dispersed on chromosome 7 (Supplementary Table SII). Furthermore, no copy number change was found by genomewide array comparative genomic hybridization (aCGH) using SurePrint G3 Human CGH Microarray Kit (1 × 1M, G4447A) (Agilent Technologies, Santa Clara, CA).
Figure 2.
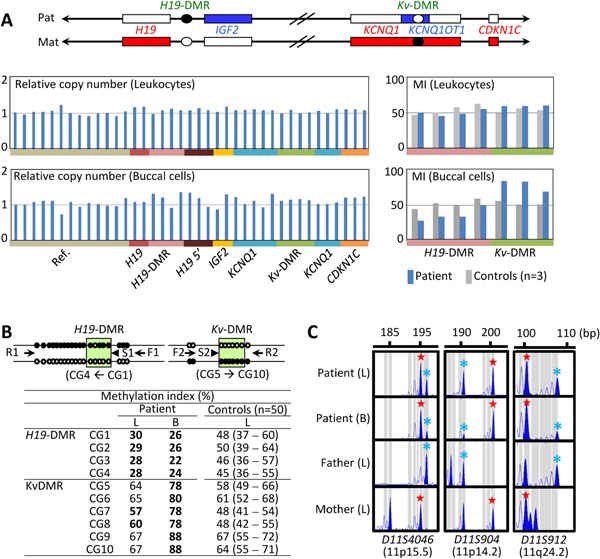
Molecular findings of this patient. (A) MS‐MLPA findings. In the upper diagram, paternally and maternally expressed genes are shown in blue and red, respectively; filled and open circles denote methylate and unmethylated DMRs, respectively. The mean values of three control subjects have been utilized for the assessment of relative copy number and methylation index (MI, the ratio of methylated clones). (B) Pyrosequencing analysis. For methylation analysis of the H19‐DMR, a segment encompassing 20 CpG dinucleotides was PCR‐amplified with F1 and R1 primers, and a sequence primer (S1) was hybridized to a single‐stranded PCR products. Subsequently, the methylation indices were obtained for four CpG dinucleotides (CG1–CG4) (indicated with a green rectangle). The Kv‐DMR was similarly examined using F2 and R2 primers and S2, and the MIs were obtained for CG5–CG10 (indicated with a green rectangle). The control data indicate the median (minimum–maximum). L, leukocytes; and B, buccal cells. (C) Microsatellite analysis. Maternally and paternally derived peaks are denoted with red stars and blue asterisks, respectively. [Color figure can be seen in the online version of this article, available at http://wileyonlinelibrary.com/journal/ajmga].
We next carried out similar studies using buccal cell genomic DNA, considering the possibility of somatic mosaicism. MS‐MLPA showed obvious hypomethylation of the H19‐DMR and hypermethylation of the Kv‐DMR, in the absence of a copy number alteration (Fig. 2A). Lack of copy number alteration was also confirmed by aCGH. To confirm the aberrant methylation pattern, we performed pyrosequencing with PyroMark Q24 (Qiagen, Hilden, Germany), using bisulfite‐treated genomic DNA. This revealed overt hypomethylation of the H19‐DMR and hypermethylation of the Kv‐DMR in the buccal cell DNA, and obviously hypomethylated H19‐DMR and equivocally hypermethylated Kv‐DMR in the leukocyte DNA (Fig. 2B). Furthermore, microsatellite analysis for 13 Loci on chromosome 11 showed not only the presence of biparentally inherited peaks, but also relatively large peaks of maternal origin for 12 informative loci on various parts of chromosome 11, especially in the buccal cell DNA (Fig. 2C, Supplementary Table SII). We further performed microsatellite analysis for multiple loci on various chromosomes using leukocyte and buccal cell genomic DNA samples of this patient and leukocyte genomic DNA samples of the parents, thereby excluding genome wide uniparental maternal isodisomy as an extremely rare condition for SRS [Yamazawa et al., 2010] (Supplementary Table SII and Fig. S1). These data indicate that this patient has a somatic mosaicism for full maternal isodisomy for chromosome 11. The ratio of upd(11)mat cells to normal cells was calculated as ∼20%:80% in the leukocytes and ∼74%:26% in the buccal cells, using the microsatellite data (Supplementary Note S1).
We identified mosaic 46,XY/46,XY,upd(11)mat in a patient with typical SRS. The mosaicism is most likely to be generated by mitotic non‐disjunction and subsequent trisomy rescue (Supplementary Fig. S2). To our knowledge, such mosaic upd(11)mat has been described only in a single case with SRS [Bullman et al., 2008], with no case with non‐mosaic upd(11)mat. Thus, it is likely that non‐mosaic upd(11)mat is a lethal condition, and that the frequency and distribution of 46,XY,upd(11)mat cells in this patient are sufficient to cause typical SRS phenotype and are below the threshold level for lethality.
It is notable that mosaic upd(11)mat was recognized after the MS‐MLPA analysis of buccal cells, and that abnormal methylation patterns in leukocytes were undetected by MS‐MLPA but were detected by pyrosequencing. This indicates the importance of the examinations of different tissues and the usefulness of the analyses with different methods, when no abnormality was identified by investigating one tissue (usually leukocytes) with one method. For the methylation analysis, the MS‐MLPA and pyrosequencing methods were different in the examined CpG sites, PCR cycle numbers (35 vs. 45), DNA treatment (HhaI vs. bisulfite), and the number of control subjects (3 vs. 50). Such differences may underlie the apparent discrepancy of the methylation data obtained by the two methods.
In summary, we identified mosaic upd(11)mat in a patient with typical SRS. Further careful studies will reveal such apparently hidden mosaicism.
Supporting information
Additional supporting information may be found in the online version of this article at the publisher's web‐site.
Supplementary Fig. S1
Supplementary Fig. S2
Supplementary Table S1
Supplementary Table S2
Luk H‐M, Ivan Lo F‐M, Sano S, Matsubara K, Nakamura A, Ogata T, Kagami M. 2016. Silver–Russell syndrome in a patient with somatic mosaicism for upd(11)mat identified by buccal cell analysis. Am J Med Genet Part A 170A: 1938–1941.
Conflict of interest: The authors declare no conflict of interest.
REFERENCES
- Azzi S, Salem J, Thibaud N, Chantot‐Bastaraud S, Lieber E, Netchine I. 2014. A prospective study validating a clinical scoring system and demonstrating phenotypical‐genotypical correlations in Silver‐Russell syndrome. J Med Genet 52:446–453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Begemann M, Zirn B, Santen G, Wirthgen E, Soellner L, Büttel HM, Schweizer R, van Workum W, Binder G, Eggermann T. 2015. Paternally inherited IGF2 mutation and growth restriction. N Engl J Med 373:349–356. [DOI] [PubMed] [Google Scholar]
- Binder G, Begemann M, Eggermann T, Kannenberg K. 2011. Silver‐Russell syndrome. Best Pract Res Clin Endocrinol Metab 25:153–160. [DOI] [PubMed] [Google Scholar]
- Brioude F, Oliver‐Petit I, Blaise A, Praz F, Rossignol S, Le Jule M, Thibaud N, Faussat AM, Tauber M, Le Bouc Y, Netchine I. 2013. CDKN1C mutation affecting the PCNA‐binding domain as a cause of familial Russell Silver syndrome. J Med Genet 50:823–530. [DOI] [PubMed] [Google Scholar]
- Bullman H, Lever M, Robinson D, Mackay D, Holder S, Wakeling L. 2008. Mosaic maternal uniparental disomy of chromosome 11 in a patient with Silver‐Russell syndrome. J Med Genet 45:396–399. [DOI] [PubMed] [Google Scholar]
- Eggermann T. 2010. Russell‐Silver syndrome. Am J Med Genet C Semin Med Genet 154C:355–364. [DOI] [PubMed] [Google Scholar]
- Kagami M, Mizuno S, Matsubara K, Nakabayashi K, Sano S, Fuke T, Fukami M, Ogata T. 2015. Epimutations of the IG‐DMR and the MEG3‐DMR at the 14q32.2 imprinted region in two patients with Silver‐Russell syndrome‐compatible phenotype. Eur J Hum Genet 23:1062–1067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakashima S, Kato F, Kosho T, Nagasaki K, Kikuchi T, Kagami M, Fukami M, Ogata T. 2015. Silver‐Russell syndrome without body asymmetry in three patients with duplications of maternally derived chromosome 11p15 involving CDKN1C. J Hum Genet 60:91–95. [DOI] [PubMed] [Google Scholar]
- Poole RL, Docherty LE, Al Sayegh A, Caliebe A, Turner C, Baple E, Wakeling E, Harrison L, Lehmann A, Temple IK; International Clinical Imprinting Consortium. 2013. Targeted methylation testing of a patient cohort broadens the epigenetic and clinical description of imprinting disorders. Am J Med Genet A 161A:2174–2182. [DOI] [PubMed] [Google Scholar]
- Yamazawa K, Nakabayashi K, Kagami M, Sato T, Saitoh S, Horikawa R, Hizuka N, Ogata T. 2010. Parthenogenetic chimaerism/mosaicism with a Silver‐Russell syndrome‐like phenotype. J Med Genet 47:782–785. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Additional supporting information may be found in the online version of this article at the publisher's web‐site.
Supplementary Fig. S1
Supplementary Fig. S2
Supplementary Table S1
Supplementary Table S2