

Abstract
Objective
Mutations in the spastic paraplegia gene 11 (SPG11), encoding spatacsin, cause the most frequent form of autosomal‐recessive complex hereditary spastic paraplegia (HSP) and juvenile‐onset amyotrophic lateral sclerosis (ALS5). When SPG11 is mutated, patients frequently present with spastic paraparesis, a thin corpus callosum, and cognitive impairment. We previously delineated a neurodegenerative phenotype in neurons of these patients. In the current study, we recapitulated early developmental phenotypes of SPG11 and outlined their cellular and molecular mechanisms in patient‐specific induced pluripotent stem cell (iPSC)‐derived cortical neural progenitor cells (NPCs).
Methods
We generated and characterized iPSC‐derived NPCs and neurons from 3 SPG11 patients and 2 age‐matched controls.
Results
Gene expression profiling of SPG11‐NPCs revealed widespread transcriptional alterations in neurodevelopmental pathways. These include changes in cell‐cycle, neurogenesis, cortical development pathways, in addition to autophagic deficits. More important, the GSK3ß‐signaling pathway was found to be dysregulated in SPG11‐NPCs. Impaired proliferation of SPG11‐NPCs resulted in a significant diminution in the number of neural cells. The decrease in mitotically active SPG11‐NPCs was rescued by GSK3 modulation.
Interpretation
This iPSC‐derived NPC model provides the first evidence for an early neurodevelopmental phenotype in SPG11, with GSK3ß as a potential novel target to reverse the disease phenotype. Ann Neurol 2016;79:826–840
Hereditary spastic paraplegias (HSPs) are a heterogeneous group of familial motor neuron diseases characterized by progressive spasticity and weakness of the lower limbs attributable to degeneration of axonal projections of corticospinal tracts and dorsal columns.1, 2 More than 75 different loci and 59 HSP genes, denoted as spastic paraplegia gene (SPG), have so far been identified.3 Mutations in SPG11 cause the most frequent form of autosomal‐recessive (AR)‐complex HSP,4, 5 and these patients, besides spastic paraparesis, present with cognitive impairment, cortical atrophy, a thin corpus callosum (TCC), and sensorimotor peripheral neuropathy,6, 7 indicative of a multisystem neurodegeneration. Interestingly, an additional clinical phenotype of mutations in this gene is AR juvenile‐onset amyotrophic lateral sclerosis, termed ALS5.8
SPG11 encodes the 2,443 amino acid protein spatacsin.9 Because of the lack of relevant disease models, the underlying molecular mechanisms and, in particular, the neuronal functions of spatacsin are still unclear. Previous studies employing non‐neuronal cellular models suggested stress‐related impairments within the lysosomal‐autophagy pathway attributed to loss of function of spatacsin in HeLa cells and patient‐derived fibroblasts.10, 11 We recently reported that SPG11‐iPSC‐derived patient neurites exhibited neurodegenerative changes on a functional and ultrastructural level.12
Indications of the combination of impaired cortical development and neurodegeneration were previously reported in induced pluripotent stem cell (iPSC)‐derived models for early‐onset diseases of the central nervous system (CNS), including models of Timothy syndrome and fragile‐X syndrome.13, 14 On account of early‐onset, cognitive deficits and a TCC,6, 7 SPG11, unlike other HSPs, has recently been grouped into the broad category of disorders with agenesis (hypoplasia) of the corpus callosum.15 The development of the human corpus callosum starts around E13, when cortical axons cross the midline.16 Axonal callosal outgrowth, neurite branching, dendritic arborization, and pruning continue throughout childhood and adolescence. Distinct structural changes, including callosal thickness, are temporally regulated and are closely linked to cortical progenitor development.17
Noting the presence of cortical atrophy and a TCC, we hypothesized a developmental defect in the cortical neural progenitor cells (NPCs) from SPG11 patients. We show that SPG11‐NPCs display widespread transcriptional dysregulation of genes associated with cortical development, including callosal developmental pathways and maintenance of neuronal homeostasis. The gene expression analysis was further substantiated by a significant decrease in proliferating SPG11‐NPCs, resulting in fewer neurons. Our data highlight specific defects in SPG11‐NPCs at the S phase and G2/M phase of the cell cycle. The developmental defects in SPG11‐NPCs were caused by dysregulation of GSK3ß signaling and, more important, could be rescued by GSK3 inhibitors.
Our data provide a novel perspective of a neurodevelopmental phenotype that precedes neurodegeneration in this motor neuron disease and suggest a novel GSK3ß‐mediated therapeutic approach for an early intervention in SPG11.
Patients and Methods
SPG11 Patients and CTRL Subjects
The patients (n = 3; hereafter referred to as SPG11‐1, SPG11‐2, and SPG11‐3) are Caucasians with clinically confirmed symptoms of AR‐HSP and previously described heterozygous mutations in SPG11.7, 12, 18 SPG11‐1 and SPG11‐2 are sisters (Fig 1A,B) with a heterozygous nonsense mutation at c.3036C>A/p.Tyr1012X in exon 16 and a c.5798 delC/p.Ala1933ValfsX18 mutation in exon 30.7, 18 SPG11‐3 has a heterozygous nonsense mutation at c.267G>A/p. Trp89X in exon 2 and a splice site mutation 1457‐2A > G in intron 6 (corresponding to the previously reported mutation c.1757‐2A > G).12 The CTRLs (n = 2; hereafter referred to as CTRL‐1 and CTRL‐2) are healthy Caucasian individuals with no history of movement disorder or neurological disease.12, 19 The detailed clinical and genetic characteristics are summarized in the Table 1.
Figure 1.
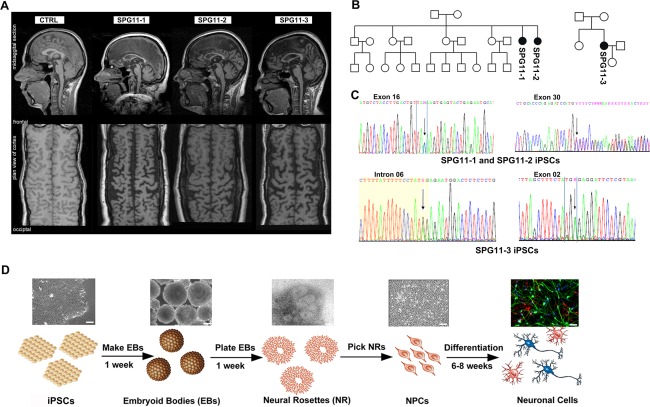
Generation of iPSCs from SPG11 patients and controls (CTRL). (A) MRI analysis of CTRL and SPG11 patients included in the study. (B) Pedigrees of SPG11 families. Female index patients are represented in black circles. (C) Mutations analysis in SPG11 patient fibroblasts. Patients 1 and 2 (SPG11‐1, SPG11‐2) have heterozygous nonsense mutations at c.3036C > A/p.Tyr1012X in exon 16 and c.5798 delC/p.Ala1933ValfsX18 in exon 30. Patient 3 (SPG11‐3) has a heterozygous nonsense mutation at c.267G > A/p. Trp89X in exon 2 and a splice site mutation 1457‐2A > G in intron 6. (D) Schematic representation of the neuronal differentiation paradigm. Scale bar = 50µm. iPSCs = induced pluripotent stem cells; MRI = magnetic resonance imaging; NPCs = cortical neural progenitor cells.
iPSC Derivation
Fibroblasts from the SPG11 patients and CTRLs were reprogrammed using retroviral transduction of the transcription factors Klf4, c‐Myc, Oct4, and Sox2 as previously described.12, 19, 20 We generated two iPSC lines from each SPG11 patient and CTRL, as described in Supplementary Table 1. The iPSC lines were characterized for pluripotency markers as described earlier.12, 19 The above‐mentioned SPG11 mutations were reconfirmed in the SPG11‐iPSC lines.
Table 1.
Clinic of SPG11 Patients and CTRL Subjects
| SPG11‐1 | SPG11‐2 | SPG11‐3 | CTRL‐1 | CTRL‐2 | |
|---|---|---|---|---|---|
| SPG11 mutations |
Exon 16: c.3036C > A heterozygote p.Tyr1012X Exon 30:c.5798 delC heterozygotp.Ala1933ValfsX18 |
Exon 16: c.3036C> A heterozygote p.Tyr1012X Exon 30:c.5798 delC heterozygotep.Ala1933ValfsX18 |
Exon 2: c.267G > A p. Trp89X Intron 6: 1457‐2 A > G splice mutation |
— | — |
| Sex | F | F | F | M | F |
| HSPRS (max.52) | 39 | 33 | 35 | 0 | 0 |
| MRI | TCC, WML, cortical atrophy | TCC, WML, cortical atrophy | TCC, WML, cortical atrophy | — | — |
| Age at onset/examination, (Y) | 25/40 | 25/35 | 31/44 | —/52 | —/45 |
| Barthel Index, % | 30 | 60 | 55 | 100 | 100 |
| Cognitive impairment | + | + | + | — | — |
| Landmark of disability (1–4) | 4 | 3 | 4 | — | — |
| Muscle wasting (upper/lower limbs) | + | + | + | — | — |
| Motor‐sensory neuropathy | + | + | + | — | — |
Patients: SPG11‐1, SPG11‐2, SPG11‐3; controls: CTRL‐1, CTRL‐2. F = female; HSPRS = Hereditary Spastic Paraplegia Rating Scale; MRI = magnetic resonance imaging; TCC = thin corpus callosum; WML = white matter lesion; Y = years, Barthel Index of Activity of Daily Living (maximum 100%).
Neural Differentiation of CTRL‐ and SPG11‐iPSCs
NPCs were generated and subsequently differentiated into neural cells from the iPSC lines as described earlier.12, 19 See also Supplementary Table 1.
Microarray Analysis
To compare the global differences in the gene expression profiles between SPG11‐NPCs and CTRL‐NPCs samples, total RNA was isolated from six SPG11‐NPC lines and four CTRL‐NPC lines using TRIzol reagent (Invitrogen, Carlsbad, CA). RNAs were DNase digested, followed by a cleanup with RNeasy MinElute (Qiagen, Hilden, Germany) and subjected to whole human genome expression array analysis, as described earlier.21 Gene Ontology term (GO‐term) biological process analysis was performed using the Gene Ontology enrichment analysis and visuaLizAtion tool (GOrilla) database (http://cbl-gorilla.cs.technion.ac.il) and the Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway representation (http://www.genome.jp/kegg/pathway.html).
Immunofluorescence Stainings and Analysis
Unless stated otherwise, SPG11‐ and CTRL‐NPCs (80,000 cells/cm2) were cultured on PORN/laminin precoated glass coverslips in neural proliferation medium (NPM). On day 3 of culture, NPCs were fixed either in ice‐cold methanol for 5 minutes (for phospho‐histone 3 [H3P] detection) or with freshly prepared 4% paraformaldehyde in phosphate‐buffered saline (PBS) at room temperature (RT) for 15 to 20 minutes. Immunofluorescence staining for proliferating cell nuclear antigen (PCNA), H3P, AuroraB, and 5‐bromo‐2’‐deoxyuridine (BrdU) were then performed as previously described.22 The primary and secondary antibodies used are listed in Supplementary Table 4. Cell viability assay was performed by immunostaining with cleaved Caspase‐3 antibody (1:500) and counting cleaved Caspase‐3‐positive cells out of Nestin/Sox2 double‐labeled cells.
Quantification of Cell Numbers
Images were acquired using a Zeiss inverted fluorescence microscope (Observer Z1; Carl Zeiss GmbH, Jena, Germany). Three random images per coverslip (triplicate coverslips per cell line) were acquired for analysis. N > 400 cells were counted per NPC line using ImageJ software (NIH, Bethesda, MD). For quantification of data, unless otherwise stated, Nestin/Sox2 double‐positive cells were used as a reference representing the total number of NPCs and the respective proliferation markers (PCNA, H3P, AuroraB, and BrdU) or dead cells (cleaved Caspase‐3) are expressed as percentage of the Nestin/Sox2 double‐positive cells. Data are represented as means ± SEM of each NPC line from at least three independent experiments performed in triplicates.
Flow cytometry/Propidium Iodide Analysis
Flow cytometry analysis was performed as described earlier23 on SPG11‐ and CTRL‐NPCs.
Transfections of Small Interfering RNA and Plasmid DNA
Spatacsin knockdown experiments and subsequent proliferation analysis in a human neuroblastoma cell line (SH‐SY5Y, Leibnitz Institute DSMZ–German Collection of Microorganisms and Cell Cultures) were performed as previously described.12, 24
TOP‐Flash/FOP‐Flash Luciferase Reporter Assay
To assess the transcriptional activity of β‐Catenin, SPG11‐ and CTRL‐NPCs (1 × 105 cells/well in a 24‐well plate) were cotransfected with β‐Catenin responsive firefly luciferase reporter plasmids containing either multimeric LEF/TCF cognate sequences (pTOP‐flash) or mutated binding sites (pFOP‐flash) and pUHD16‐1 (encoding the β‐galactosidase) using Lipofectamine LTX+ (Invitrogen) in NPM and subjected to luciferase activity analysis as previously described.25, 26 For Wnt activation and tideglusib treatment, Wnt3a (100ng/ml) or tideglusib (3µm) were respectively added in the culture medium of the transfected NPCs and incubated for 12 to 14 hours for subsequent analysis.
Electrophysiology
Whole‐cell patch clamp recordings were performed on 6‐week differentiated neural cells as previously described.19
Real‐Time Polymerase Chain Reaction Analysis
Quantitative real‐time PCR (qRT‐PCR) analysis was performed using the protocol described earlier12, 19 on RNA isolated from SPG11‐ and CTRL‐NPC lines. The primer pairs used are listed in Supplementary Table 3.
Western Blot
NPCs were washed with PBS and lysed in either modified radioimmunoprecipitation assay RIPA lysis buffer (150mM of NaCl, 50mM of Tris/HCl [pH 7.4], 1% NP‐40, 0.25% dexoycholic acid, 0.1% sodium dodecyl sulfate, 1mM of ethylenedimainetetraacetic acid [EDTA], 10mM of Na‐pyrophosphate, 2mM of Na‐orthovanadate, 1mM of Na‐fluoride, and protease inhibitor cocktail; Roche Diagnostics, Indianapolis, IN) or hypotonic buffer (25mM of Tris/HCl [pH 8.0], 1mM of EDTA, and protease inhibitor cocktail; Roche) at 4°C for 10 minutes. Lysates were cleared by centrifugation at 11,000g for 10 minutes at 4ºC, and immunoblot analysis was performed using the protocol described earlier.12, 19
Pharmacological Rescue
SPG11‐ and CTRL‐NPCs were plated at a cell density of 80,000 cells/cm2 on PORN/laminin‐coated glass coverslips in NPM. The next day, following results from the dose‐response curve (data not shown), cells were treated with 3µM of the GSK3 inhibitor, CHIR99021 (R&D Systems, Minneapolis, MN), and the clinically used GSK3 blocker, tideglusib (Selleckchem, Houston, TX). After 24 hours of exposure, cells were kept in culture for 1 additional day. Proliferation analyses were then performed on the treated NPCs using PCNA antibody as described above.
Statistical Analysis
Statistical analysis was performed using Prism software (version 5.0; GraphPad Software Inc., La Jolla, CA). The Student t test was applied when comparing the means between two groups representing means of each cell line. To compare three or more groups, one‐way analysis of variance (ANOVA), followed by Dunnett's post‐hoc test for multiple comparisons, was used. Probability values (p values) ≤ 0.05 were considered statistically significant (*p ≤ 0.05; ** p ≤ 0.01; *** p ≤ 0.001). All data are shown as mean ± SEM from ≥3 independent experiments performed in triplicates.
Results
Generation of Human iPSCs from SPG11 Patients and Controls
iPSC lines from 3 SPG11 patients (SPG11‐1, SPG11‐2, and SPG11‐3) and 2 controls (CTRL‐1 and CTRL‐2) were included in this study. Cerebral magnetic resonance imaging (MRI) revealed a TCC in all 3 SPG11 patients. The flat‐tire reconstruction (Fig 1A) of the cortex indicates severe frontal and parietal cortical atrophy. Pedigree analysis showed an AR mode of inheritance in the patients (Fig 1B). The spatacsin heterozygous mutations (c.3036C>A, c.5798 delC, c.267G > A, and c.1757‐2A > G), reported earlier,7, 18 were confirmed in the iPSC lines from all of the SPG11 patients (Fig 1C; Table 1 and Supplementary Table 1).12 The iPSC lines expressed pluripotency markers and maintained a normal karyotype, and undirected differentiation of these iPSC lines generated progeny of all three germ layers (data not shown). The iPSCs were differentiated into neurons, as previously described12, 19 (Fig 1D).
Global Transcriptome Analysis of SPG11‐NPCs and CTRL‐NPCs
The transcriptome of SPG11‐NPCs and CTRL‐NPCs was investigated by microarray analysis (Fig. 2A–G). We restricted the analysis to those transcripts with significantly altered expression in SPG11 compared to CTRL (fold change, ≥1.5; p ≤ 0.05). Hierarchical clustering visualized several gene clusters distinguishing the SPG11 patient group from CTRLs (Fig 2A). A total of 1,537 transcripts were differentially regulated (ANOVA; p ≤ 0.05) in SPG11 compared to CTRL (959 transcripts were upregulated and 578 downregulated; Fig 2B). Interestingly, GO analysis of differentially regulated transcripts revealed over‐represented biological processes mostly associated with distinct stages of neurodevelopmental pathways, including regulation of neurogenesis, nervous system development, and neuron differentiation (Fig 2C; Supplementary Table 2). The KEGG pathway representation of individual sets of genes outlined dysregulation of important components of Wnt/GSK3ß signaling in SPG11‐NPCs (data not shown), including components of the Wnt/ß‐Catenin destruction complex (Axin2, APC, and PKA) and negative regulators of Wnt signaling (FRZB, SMAD4, CBP, TJP2, and Slit1) that were differentially upregulated in SPG11‐NPCs. Conversely, the positive modulators of the Wnt/GSK3ß pathway (TCF7L2, FZD3, KIF3A, PLCD1, JAM, and FOSL2) and components of calcium signaling regulation (NFAT, CaN, PKC, and CaMK1D) were significantly downregulated, implicating defects in the downstream cell cycle and proliferation pathways (Fig 2D). Indeed, several positive modulators of cell cycle (CCNA1, PERP, DAB2, JAM2, USP53, MAPKAPK, and CDC42EP3) were significantly downregulated (Fig 2E). Negative modulators (CDH1, PPCDC, POGZ, ATMIN, MAPK8IP1, TP53RK, and RBBP6) were upregulated in SPG11‐NPCs (Fig 2E), indicative of early developmental defects in patients with mutations in SPG11.
Figure 2.
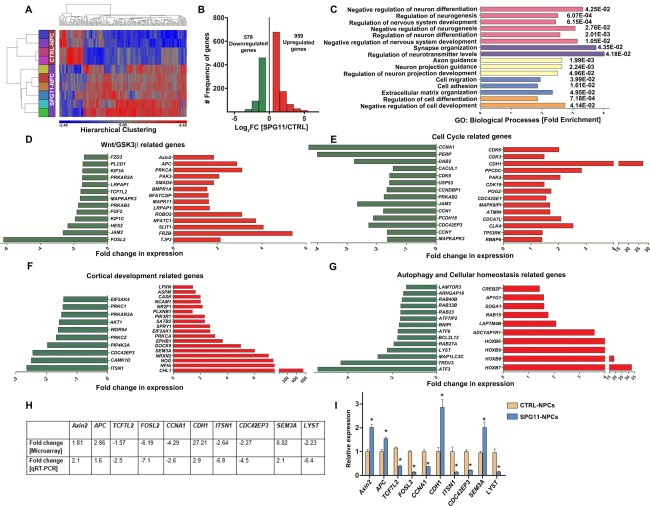
Global gene expression analysis of day 3 NPCs generated from SPG11‐ and CTRL‐iPSCs. n = 2 samples from each individual. (A) Heatmap showing hierarchical clustering of differentially expressed genes in SPG11‐NPCs compared to CTRL‐NPCs. (B) Histographs of total number of differentially expressed genes (upregulated genes in red, downregulated genes in green; p ≤ 0.05). (C) Gene Ontology term analysis of important biological processes enriched within differentially expressed genes (p values: Benjamini‐Hochberg corrected). (D) Wnt/GSK3ß pathway‐related genes differentially regulated in SPG11‐NPCs. (E) Cell‐cycle–related genes differentially regulated in SPG11‐NPCs. (F) Cortical development‐related genes differentially regulated in SPG11‐NPCs. (G) List of differentially regulated transcripts related to autophagy, endolysosomal, and ER stress pathways. Upregulated transcripts shown in red, downregulated transcripts shown in green (p ≤ 0.05), analyzed by one‐way ANOVA (D–G). (H) Validation of transcriptome data with qRT‐PCR in SPG11‐ and CTRL‐NPCs. Table showing fold‐change difference for 10 selected genes of Wnt/GSK3 signaling‐, cell‐cycle–, cortical development‐, and autophagy‐related pathways (p ≤ 0.05). (I) Bar graph showing relative gene expression of the 10 selected genes. Relative expression shows the average values of four CTRL‐NPCs (set at a value of 1) and six SPG11‐NPC lines. mRNA levels were normalized against GAPDH. Data are represented as mean ± SEM; *p ≤ 0.05 by two‐tailed Student t test. APC = adenomatous polyposis coli, TCF7L2 = transcription factor 7‐like 2, FOSL2 = Fos‐related antigen 2, CCNA1 = cyclin‐A1, CDH1 = cadherin‐1, ITSN1 = intersectin‐1, CDC42EP3 = Cdc42 effector protein 3, SEM3A = semaphorin‐3A, LYST = lysosomal trafficking regulator. ANOVA = analysis of variance; ER = endoplasmic reticulum; GAPDH = glyceraldehyde 3‐phosphate dehydrogenase; iPSCs = induced pluripotent stem cells; MRI = magnetic resonance imaging; mRNA = messenger RNA; NPCs = cortical neural progenitor cells; qRT‐PCR = quantitative real‐time polymerase chain reaction.
The proper development of the corpus callosum in humans is regulated by guidance cues and receptors that, when bound to their ligands, guide them along the midline to form layer‐specific neurons, modulating neurite development and regulating the temporal and spatial orientation of the callosal fibers.16, 17 Positive regulators of neuronal morphogenesis, including the attractive cytoskeleton modulators (ITSN1, CAMK1D, CDC42EP3, PIP4K2A, PRKCZ, WDR54, PRKCI, and EIF2AK4), were significantly downregulated, whereas various modulators for repulsive cytoskeleton modulators (SEMA3A, EPHB1, ASPM, LPXN, PLXNB1, NOG, NFIA, NR2F1, NCAM1, and PIK3R1), which tend to repel the callosal fibers away from the midline glia during formation of commissural fibers in brain, were upregulated in SPG11 patients (Fig 2F). Thus, the global transcriptome profiling suggested an early aberration of the neurodevelopmental pathways and detrimental changes in proliferation and neurogenesis in SPG11‐NPCs.
In addition, several transcripts associated with autophagy and the endolysosomal pathway were differentially regulated in SPG11‐NPCs (Fig 2G), confirming previously published findings in non‐neuronal cell systems.11 This includes components of the lysosomal biogenesis pathway such as LAMTOR3 and LYST (significantly downregulated), whereas LAPTM4B, regulating the endolysosomal pathway for autophagosome maturation, is upregulated in patients. Several members of the HOX gene family (HOXB6, HOXB7, HOXB8, and HOXB9), which have been shown to be strong repressors of developmental autophagy,27 were upregulated (10‐ to 30‐fold) in SPG11, suggesting a deleterious impact of defective autophagy on the developmental potential of SPG11‐NPCs during cortical neurogenesis and callosal morphogenesis (Fig 2G). Transcriptome data were validated by qRT‐PCR in a selected subset of 10 genes (Fig 2H,I). The Wnt/GSK3ß signaling positive regulator genes TCF7L2 and FOSL2 were more than 2‐fold downregulated whereas APC and Axin2 negative regulators of Wnt/GSK3ß signaling were significantly upregulated in SPG11‐NPCs, which are consistent with our transcriptome analysis (Fig 2D). Similarly, the cell‐cycle–related gene, CCNA1, was more than 2‐fold downregulated whereas CDH1 was almost 3‐fold upregulated. The cortical development‐related genes, ITSN1 and CDC42EP3, were more than 4‐fold downregulated, whereas SEM3A was more than 2‐fold upregulated in SPG11‐NPCs compared to CTRL‐NPCs. LYST (lysosomal trafficking regulator), a gene important for maintaining cellular homeostasis, was more than 6‐fold downregulated in SPG11‐NPCs compared to the CTRL‐NPCs.
Reduced Proliferation and Neurogenesis in SPG11‐NPCs
The SPG11‐NPCs, compared to CTRL‐NPCs, grew more slowly, resulting in a 36% to 50% decrease in the density of Nestin/Sox2 double‐positive NPCs in SPG11 (CTRL: 285 ± 14 cells/mm2 vs. SPG11: 159 ± 9 cells/mm2; p ≤ 0.0003; Fig 3A,B), whereas the proportion of the Nestin/Sox2 double‐positive cells was unchanged (CTRL: 83% vs. SPG11: 81%; Fig 3C). We differentiated the SPG11‐ and CTRL‐NPCs into functionally active forebrain glutamatergic neurons over a period of 6 weeks (Fig 3D). Both SPG11 and CTRL differentiated neurons, under voltage‐clamp recordings, displayed outward and inward currents, indicative of the presence of voltage‐gated potassium and sodium channels, respectively (data not shown). Significant reductions in total neural cell density and the percentage of Tuj1‐positive neurons were observed in SPG11 (CTRL: 82.90 ± 12.19 cells/mm2 vs. SPG11: 29.08 ± 5.052 cells/mm2; p = 0.0025; Tuj1: CTRL: 72.82% vs. SPG11: 59.11%; p < 0.0001) attributable to reduced cortical neurogenesis (Fig 3E,F).
Figure 3.
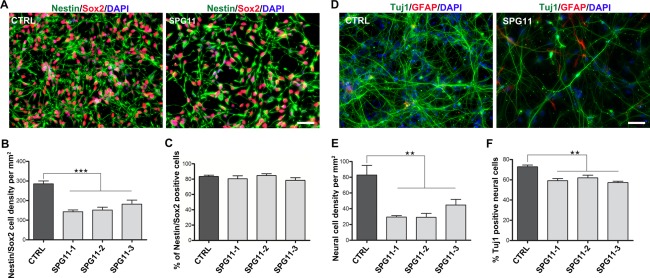
Reduced proliferation and neurogenesis in SPG11‐NPCs. (A) Representative images of Nestin/Sox2 double‐positive NPCs generated from SPG11- and CTRL-iPSCs. Nuclei were visualized with DAPI. Scale bar = 50µm. (B) SPG11‐NPCs exhibit a decreased Nestin/Sox2 cell density compared to CTRL-NPCs. (C) No difference in Nestin/Sox2 double‐positive cells (% over DAPI) between SPG11- and CTRL-NPC lines. (D) Differentiated neuronal cells expressing neuron‐specific (Tuj1) and glia‐specific (GFAP) markers. Nuclei were visualized with DAPI. Scale bar = 50µm. (E) SPG11‐NPCs exhibit a marked reduction in neuronal cell density compared to CTRL. (F) SPG11‐NPCs show reduced generation of Tuj1‐positive neurons compared to CTRL-NPCs, reflecting neurogenesis deficits in SPG11 patients. Data are represented as mean ± SEM. **p < 0.01; ***p < 0.001, by one‐way ANOVA followed by Dunnett's post‐hoc multiple comparison test (B, C, E, F). ANOVA = analysis of variance; DAPI = 4’,6‐diamidino‐2‐phenylindole; GFAP = glial fibrillary acidic protein; iPSCs = induced pluripotent stem cells; NPCs = cortical neural progenitor cells.
Altered Cell‐Cycle– and Stage‐Specific Anomalies in Checkpoint Progression in SPG11‐NPCs
Indications of a compromised proliferation of NPCs in SPG11 patients came from pulse labeling of BrdU experiments (Fig 4A), in which a significantly reduced number of BrdU‐labeled Nestin/Sox2 double‐positive cells was detected in SPG11‐NPCs (CTRL: 33.86 ± 1.5% vs. SPG11: 23.21 ± 1.1%; p < 0.0001; Fig 4A,B). The endogenous proliferation marker, PCNA (Fig 4C), confirmed these results in SPG11‐NPCs (CTRL: 40.84 ± 2.1% vs. SPG11: 21.59 ± 1.0%; p < 0.0001; Fig 4D). To identify the affected stages of the cell cycle, we analyzed the metaphase marker (Ser10) protein, H3P. We found a 2‐fold decrease in H3P‐labeled Nestin/Sox2 double‐positive cells in SPG11‐derived cells (CTRL: 10.05 ± 0.8% vs. SPG11: 4.87 ± 0.35%; p < 0.0001; Fig 4E).
Figure 4.
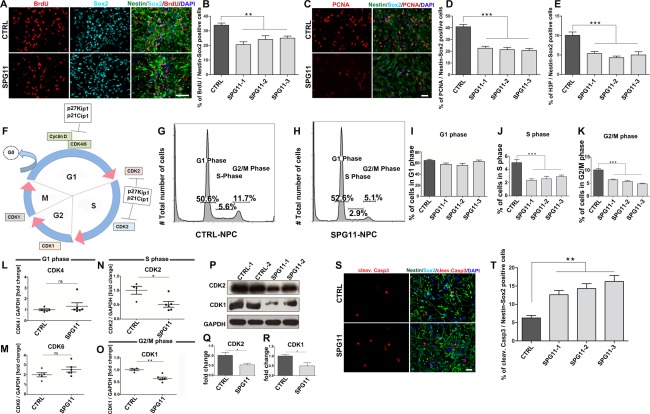
SPG11‐NPCs show altered cell‐cycle distribution and stage‐specific downregulation of important checkpoint genes. (A) Representative images of Nestin/Sox2‐positive cells colabeled with BrdU‐positive nuclei for SPG11‐ and CTRL‐NPCs. Nuclei were visualized with DAPI. Scale bar = 50µm. (B) SPG11‐derived Nestin/Sox2‐positive cells have significantly reduced numbers of BrdU‐labeled cells compared to CTRLs. (C) SPG11‐ and CTRL‐NPCs (Nestin/Sox2+) colabeled with the endogenous proliferation marker, PCNA. Nuclei were visualized with DAPI. Scale bar = 50µm. (D) SPG11‐derived Nestin/Sox2 double‐positive NPCs have significantly reduced numbers of PCNA colabeled cells. (E) Diminished number of Nestin/Sox2‐positive NPCs colabeled with mitotic marker H3P of SPG11‐NPCs, suggesting a compromised mitosis. (F) Schematic representation of important checkpoint and senescence genes of the cell cycle. CDK = cyclin‐dependent kinase; p21Cip1 = cyclin‐dependent kinase inhibitor 1; p27Kip1 = cyclin‐dependent kinase inhibitor 1B. (G–K) Flow cytometry/PI staining analysis shows the respective distribution of cells in G1, S, and G2/M phases of the cell cycle for SPG11‐ and CTRL‐NPC lines. (I) No significant difference in the percentage of viable cells in the G1 phase of cell cycle between SPG11‐ and CTRL‐NPCs. However, SPG11‐NPCs have a highly diminished percentage of viable cells in the S phase (J) and G2/M phase (K). Data represented as mean ± SEM: **p < 0.01; ***p < 0.001, by one‐way ANOVA followed by Dunnett's post‐hoc multiple comparison test (B, D, E, I–K). (L–O) mRNA expression analysis of SPG11- and CTRL-NPCs revealed no significant difference for G1 phase cell‐cycle checkpoint markers CDK4 (L) and CDK6 (M). ns = not significant. (N) Expression of cell‐cycle genes at the S phase CDK2 is significantly downregulated in SPG11‐NPCs. (O) Significant downregulation of G2/M phase cell‐cycle gene CDK1 highlighted a perturbed cell‐cycle activity in SPG11‐NPCs. mRNA levels were normalized against GAPDH. (P–R) Protein expression of CDK2 and CDK1 using Western blot analysis in SPG11‐NPCs compared to CTRL‐NPCs. Significant decrease in CDK2 protein levels (Q) and CDK1 (R) in SPG11‐NPCs compared to the CTRL. CDK2 and CDK1 expression was normalized against GAPDH. (S–T) Increased rate of apoptosis in SPG11‐NPCs. Immunofluorescent images of SPG11‐ and CTRL‐NPCs stained with apoptotic marker cleaved caspase 3. (S) SPG11‐NPCs have 2‐ to 3‐fold increase in number of apoptotic cells compared to CTRL-NPCs (T). Nuclei were visualized with DAPI. Scale bar = 20µm. Data represented as mean ± SEM: *p < 0.05; **p < 0.01, by two‐tailed Student t test (L–O, Q, R, T). ANOVA = analysis of variance; BrdU = 5‐bromo‐2’‐deoxyuridine; cleav. Casp3 = cleaved caspase‐3; DAPI = 4’,6‐diamidino‐2‐phenylindole; GAPDH = glyceraldehyde 3‐phosphate dehydrogenase; H3P = phospho‐histone 3; mRNA = messenger RNA; NPCs = cortical neural progenitor cells; PCNA = proliferating cell nuclear antigen.
We quantified the frequency of NPCs in distinct phases of the cell cycle using flow cytometry (Fig 4G–K). Whereas the percentage of cells in the G1 phase was unchanged in SPG11 (Fig 4I), a significantly reduced number of SPG11‐NPCs was present in the S phase (CTRL: 5.0% vs. SPG11: 2.6%; Fig 4J) and G2/M phase (CTRL: 10.0% vs. SPG11: 5.4%; Fig 4K). It is important to note that proliferative changes were not present in the matching fibroblasts and iPSCs of SPG11 (data not shown).
Dysregulation of Cell‐Cycle Checkpoint Genes in SPG11‐NPCs
Considering that the cell‐cycle distribution analysis revealed highly reduced numbers of SPG11‐NPCs in the S and G2/M phase, we subsequently analyzed the messenger RNA (mRNA) expression profiles of checkpoint genes regulating progression of NPCs from G0/G1, S, and G2/M stages of the cell cycle (Fig 4F,L–O). Whereas expression of cyclin‐dependent kinase genes regulating G0/G1 to S phase transit was not altered (CDK4 and CDK6; Fig 4L,M), genes indicative of the S phase were reduced in SPG11‐NPCs (CDK2: p = 0.0172; Fig 4N). We further analyzed CDK1, a gene regulating the transition to the G2/M phase, and observed an almost 2‐fold downregulation of CDK1 in SPG11‐NPCs (CDK1: p = 0.0013; Fig 4O), suggesting that altered expression of cell‐cycle genes might result in impaired proliferation of SPG11‐NPCs. We next performed Western blot analysis for the cell‐cycle markers in SPG11‐NPCs and CTRL‐NPCs. Corresponding to our mRNA expression data, we revealed a 2‐fold decreased expression of CDK2, an S phase marker (CDK2: p = 0.0445; Fig 4P,Q) and CDK1, a G2/M phase marker (CDK1: p = 0.0353; Fig 4P,R), thereby confirming the significant differences in cell‐cycle checkpoint controls in SPG11‐NPCs.
To test whether the impaired proliferation of the SPG11‐NPCs was associated with reduced numbers of newly formed daughter cells, we examined the NPCs using the cytokinesis marker, Aurora B, and detected a more than 2‐fold decrease in SPG11‐NPCs (CTRL: 3.0% vs. SPG11: 1.2%, data not shown). Thereupon, we compared cell viability and observed a 2‐ to 3‐fold increase in the number of cleaved Caspase‐3‐positive cells in SPG11‐NPCs (CTRL: 6.26% vs. SPG11: 16.84%; p < 0.0001; Fig 4S,T), suggesting that, in addition to the proliferation defect, mutations in SPG11 led to increased apoptosis.
Impaired GSK3ß/ß‐Catenin Signaling in SPG11‐NPCs
The GSK3ß/ß‐Catenin signaling pathway is a well‐known regulator of proliferation and differentiation during brain development.28, 29 Protein expression of the Ser9‐phosphorylated form of GSK3ß (p‐GSK3ß) was significantly decreased in SPG11‐NPCs (p < 0.0001; Fig 5A,B). Phosphorylation at Ser9 residue reduces the catalytic activity of GSK3ß,30, 31, thus indicating an increased GSK3ß activity in SPG11‐NPCs. We next assessed the expression of ß‐Catenin, an important downstream target of GSK3 signaling. Interestingly, ß‐Catenin exhibited a significant decrease in expression in SPG11‐NPCs compared to the CTRL‐NPCs (p < 0.0001; Fig 5C,D). To confirm the role of SPG11 mutations for regulation of ß‐Catenin levels, we made use of the TCF/LEF (T‐cell factor/lymphoid enhancer factor) ß‐Catenin reporter activity (performed using TOP/FOP luciferase assay).25 We transfected SPG11‐ and CTRL‐NPCs with the TOP/FOP construct and analyzed the relative TOP flash luciferase activity (Fig 5E–G). Interestingly, SPG11‐NPCs showed an almost 2‐fold reduced TOP luciferase activity (CTRL: 1.00 ± 0.12 vs. SPG11: 0.61 ± 0.03; p = 0.0214) compared to the CTRL‐NPCs (Fig 5F). We next investigated the effect of the Wnt pathway activation in SPG11‐ and CTRL‐NPCs. For this, we exogenously treated the NPCs with Wnt3a (100ng/ml) to activate the canonical Wnt/ß‐Catenin signaling pathway and examined the relative TOP flash luciferase activity (Fig 5G). SPG11‐NPCs exhibited an almost 2‐fold enhanced TOP luciferase activity (CTRL‐Wnt: 6.25 ± 0.89 vs. SPG11‐Wnt: 11.13 ± 1.26; p = 0.0200) compared to the CTRL‐NPCs, thereby rescuing the GSK3‐mediated aberrant ß‐Catenin reporter activity in SPG11‐NPCs. We then asked whether the impaired GSK3 signaling led to a subsequent increase in the senescence marker, p27Kip1 (Fig 5H). We found a 2‐ to 3‐fold increase in expression of p27Kip1 protein levels in SPG11‐NPCs compared to CTRL‐NPCs (p = 0.0007; Fig 5I), thereby highlighting a pronounced senescence activity of SPG11‐NPCs. Taken together, these findings suggested a dysregulation of GSK3ß/ß‐Catenin signaling modulating the proliferation of SPG11‐NPCs.
Figure 5.
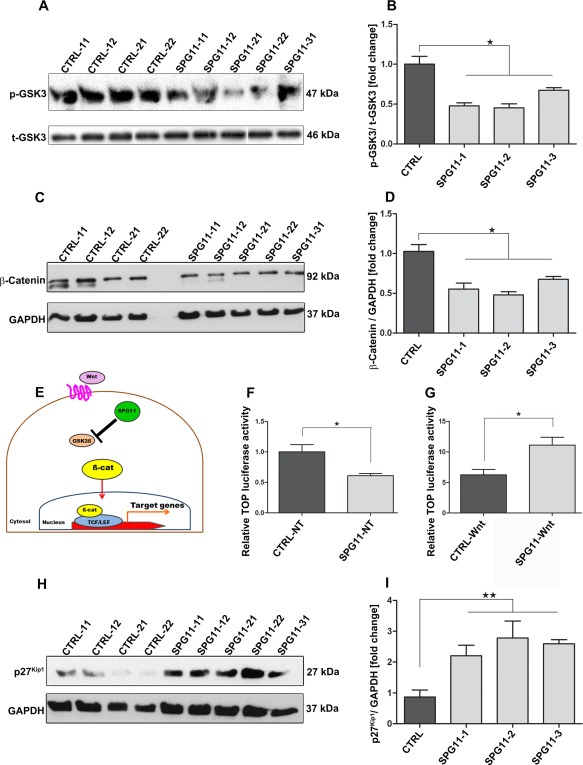
Increased GSK3ß activity leads to reduced ß‐Catenin levels in SPG11‐NPCs. (A) Phospho‐GSK3ß (Ser9) protein expression in SPG11‐ and CTRL‐NPC lines. (B) Protein expression of p‐GSK3ß (Ser9) was significantly reduced in SPG11‐NPCs compared to CTRL‐NPCs. p‐GSK3ß expression was normalized against total GSK3ß. (C) ß‐Catenin protein levels in SPG11‐ and CTRL‐NPC lines. (D) Significant decrease in the ß‐Catenin protein levels in SPG11‐NPCs compared to the CTRL. ß‐Catenin expression was normalized against GAPDH. (E) Schematic representation of ß‐Catenin (TCF/LEF) reporter activity assayed using TOP/FOP flash luciferase assay. (F) TOP flash luciferase activity is 2‐fold reduced in SPG11‐NPCs compared to the CTRL‐NPCs under normal conditions. (G) Wnt pathway activation by treatment with Wnt3a rescues ß‐Catenin signaling mediated TCF/LEF reporter activity in SPG11‐NPCs; n = 3. Data represented as mean ± SEM: * p < 0.05, by two‐tailed Student t test (F, G). (H) Representative Western blot for expression of senescence marker p27Kip1 in SPG11‐ and CTRL‐NPC lines. (I) Two‐ to three‐fold increase in p27Kip1 protein levels in SPG11‐NPCs compared to CTRL‐NPCs. Data represented as mean ± SEM: n = 3; * p < 0.05; ** p < 0.01, by one‐way ANOVA followed by Dunnett's post‐hoc multiple comparison test (B, D, I). ANOVA = analysis of variance; GAPDH = glyceraldehyde 3‐phosphate dehydrogenase; NPCs = cortical neural progenitor cells. See also Supplementary Table 1. [Color figure can be viewed in the online issue, which is available at www.annalsofneurology.org.]
Knockdown of Spatacsin Impairs Proliferation in SH‐SY5Y Cells
The interplay between loss of function of spatacsin and proliferation of neural cells was further investigated by knockdown of spatacsin in SH‐SY5Y cells using SPG11‐siRNA (siSPG1112). In line with previous experiments, we noted a significant reduction in the number of green fluorescent protein (GFP)‐positive cells colabeled with PCNA in siSPG11‐transfected SH‐SY5Y cells compared to CTRL siRNA (p = 0.0055), thereby confirming that loss of function of spatacsin was able to compromise proliferation of neural cells (data not shown).
GSK3 Inhibition Rescues the Proliferation and Neurogenesis Defect of SPG11‐NPCs
The global transcriptome analysis outlined several important transcripts of the canonical Wnt/GSK3β signaling to be differentially regulated in the modulation of the neurodevelopmental phenotype in SPG11. Inhibition of the GSK3 pathway was recently shown to enhance the proliferation of NPCs in rodent brain.32 We hypothesized that the inhibition of GSK3ß should enhance proliferation and neurogenesis in SPG11‐NPCs. We therefore treated SPG11‐NPCs with 3μM of the GSK3 inhibitor, CHIR99021, or with 3μM of the clinically used GSK3 blocker, tideglusib (Fig 6A). The GSK3 inhibitor significantly increased the percentage of PCNA‐labeled cells in the treated group (SPG11‐CHIR) compared to the untreated group (SPG11‐NT: 22.55 ± 1.6% vs. SPG11‐CHIR: 31.51 ± 1.1%; p = 0.0003; Fig 6B). Consistent with restored proliferation in SPG11‐1‐NPCs after GSK3 inhibition, we noted a similar restoration of proliferation in tideglusib‐treated SPG11‐NPCs (Fig 6C) compared to an untreated group (SPG11‐NT: 25.56 ± 1.38% vs. SPG11‐Tide: 35.06 ± 0.98%; p < 0.0001). To confirm the role of GSK3/ß‐Catenin signaling for the rescue of proliferation defects in SPG11‐NPCs, we next investigated the expression of ß‐Catenin levels by Western blot analysis in the tideglusib‐treated SPG11‐ and CTRL‐NPCs (Fig 6D,E). Interestingly, SPG11‐NPCs treated with tideglusib showed an almost 2‐fold enhanced expression of ß‐Catenin levels (SPG11‐NT: 0.46 ± 0.06 vs. SPG11‐Tide: 0.85 ± 0.07; p = 0.0101) compared to the nontreated SPG11‐NPCs (Fig 6E), suggesting a ß‐Catenin‐mediated increase in proliferation of SPG11‐NPCs. We did not observe any significant increase in the CTRL‐NPCs treated with tideglusib (Fig 6E). Furthermore, we analyzed the downstream ß‐Catenin‐mediated TCF/LEF reporter activity in SPG11‐ and CTRL‐NPCs treated with tideglusib using TOP/FOP luciferase assay. Consistent with the previous result of a Wnt3a‐mediated increase in TCF/LEF activity in SPG11‐NPCs (Fig 5G), we observed an almost 2‐fold increase in relative TOP flash luciferase activity (CTRL‐Tide: 2.71 ± 0.32 vs. SPG11‐Tide: 4.98 ± 0.38; p = 0.0028) in SPG11‐NPCs compared to CTRL‐NPCs (Fig 6F), thereby suggesting that modulation of Wnt/ß‐Catenin signaling by inhibition of the GSK3 pathway is sufficient to rescue the proliferation defects in SPG11‐NPCs. We further asked whether the restoration of Wnt/ß‐Catenin signaling is able to rescue the alteration in cell cycle of SPG11‐NPCs. We investigated the mRNA expression profile of CDK2 and CDK1 in SPG11‐ and CTRL‐NPCs treated with tideglusib (Fig 6G,H), Interestingly, whereas there was no significant increase in CTRL‐NPCs treated with tideglusib, an almost 2‐fold increase (SPG11‐NT: 0.48 ± 0.08 vs. SPG11‐Tide: 1.09 ± 0.06; p = 0.0308) in the expression of CDK2, an S phase marker (Fig 6G), and a more than 3‐fold increase (SPG11‐NT: 0.38 ± 0.05 vs. SPG11‐Tide: 1.23 ± 0.10; p = 0.0175) in CDK1, a G2/M phase marker, was evidenced in tideglusib‐treated SPG11‐NPCs compared with the untreated SPG11‐NPCs (Fig 6H). More important, treatment with tideglusib during neuronal differentiation of the progenitors substantially increased the number of neuronal cells in SPG11 patients (SPG11‐NT: 30.33 ± 2.22 vs. SPG11‐Tide: 61.71 ± 5.85 cells/mm2; p < 0.0001; Fig 6I).
Figure 6.
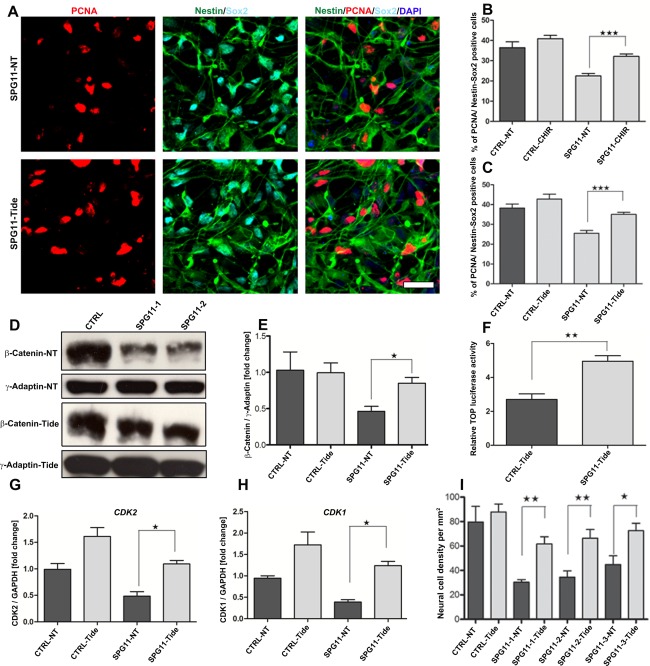
GSK3 antagonists (CHIR99021 and tideglusib) rescue neurodevelopmental defects of SPG11‐NPCs. (A) NPCs were treated with 3µM of the GSK3 inhibitor (tideglusib) for 24 hours. Representative images of untreated SPG11‐NPCs (SPG11‐NT) and tideglusib‐treated SPG11‐NPCs (SPG11‐Tide) on day 3. Cell proliferation was analyzed using colabeling of PCNA in Nestin/Sox2‐positive NPCs. Nuclei were visualized with DAPI. Scale bar = 50µm. (B) Increased numbers of Nestin/Sox2‐positive cells colabeled with PCNA in CHIR99021‐treated SPG11‐NPCs. (C) Tideglusib‐treated SPG11‐NPCs, compared to untreated NPCs, revealed restoration of cell proliferation similar to the CTRL‐NPCs. (D) ß‐Catenin protein levels in tideglusib‐treated group (CTRL‐Tide/SPG11‐Tide) and untreated group (CTRL‐NT/SPG11‐NT). (E) Almost 2‐fold increased expression of ß‐Catenin protein levels in Tide‐treated SPG11‐NPCs (SPG11‐Tide) compared to the untreated group (SPG11‐NT). ß‐Catenin expression was normalized against γ-Adaptin. (F) TOP flash luciferase activity is almost 2‐fold enhanced in SPG11‐NPCs treated with tideglusib, suggesting restoration of Wnt/ß‐Catenin signaling in SPG11‐NPCs. (G, H) Representative mRNA expression profile of important cell‐cycle checkpoint genes, CDK2 and CDK1 in SPG11‐ and CTRL‐NPCs treated with tideglusib. (H) Almost 2‐fold increase in mRNA expression of S phase marker (CDK2) and more than 3‐fold increase in expression of G2/M phase marker (CDK1) in SPG11‐NPCs treated with tideglusib (SPG11‐Tide). (I) Neural differentiation in presence of 3µM of the GSK3 inhibitor (tideglusib) increased the numbers of neural cells in tideglusib‐treated SPG11‐NPCs, compared to untreated NPCs. Data represented as mean ± SEM: *p ≤ 0.05; **p ≤ 0.01; ***p < 0.001, by two‐tailed Student t test; n = 3 (B, C, E–I). ß‐Catenin‐NT = ß‐Catenin expression in nontreated NPCs; ß‐Catenin‐Tide = ß‐Catenin expression in tideglusib‐treated NPCs, γ‐Adaptin‐NT = γ‐Adaptin expression in nontreated NPCs; γ‐Adaptin–Tide = γ‐Adaptin expression in tideglusib‐treated NPCs; ANOVA = analysis of variance; DAPI = 4’,6‐diamidino‐2‐phenylindole; mRNA = messenger RNA; NPCs = cortical neural progenitor cells; PCNA = proliferating cell nuclear antigen.
Discussion
The TCC and severe cortical atrophy in SPG11‐linked HSP, previously classified as a neurodegenerative disease, raised the question of whether cortical neurons were developmentally reduced in number and/or degenerated after maturity. The transcriptome analysis of cortical progenitors dissected a signature of early developmental dysregulated pathways in SPG11 patients. Roughly half of the differentially expressed genes in SPG11‐NPCs were related to neural development and included transcriptional changes in the regulation of neurogenesis, neuronal differentiation, nervous system development, and axonal projection. Cortical NPCs derived from SPG11 patients’ iPSCs had a reduced proliferative potential. This proliferation defect could be linked to a loss of function of spatacsin and cell‐cycle abnormalities. Fewer NPCs were present at the S phase and G2/M phase, paralleled by a decreased expression of these checkpoint genes in SPG11‐NPCs. Furthermore, our study revealed an impaired GSK3ß/ß‐Catenin signaling in SPG11‐NPCs that could be rescued by a GSK3 inhibitor (CHIR99021) and a clinically used GSK3 blocker (tideglusib). In addition to the previously known adult‐onset motor neuron disease phenotype, the present study provides a novel link between a GSK3‐dependent pathway and a cortical developmental phenotype in SPG11 patients.
Reduced Capacity of SPG11‐iPSCs to Generate NPCs and Neurons Attributed to Dysregulation of the Cell Cycle
In the current study, we observed a substantial decrease of SPG11‐NPCs attributable to a decrease in the S phase and G2/M phase of the cell cycle and an increase in cell death. We hypothesized that a loss‐of‐function mechanism of spatacsin is responsible for this proliferation deficit and confirmed this by knockdown of spatacsin in SH‐SY5Y cells. Whereas some HSP‐related genes, such as spastin (SPG4), spartin (SPG20), and spastizin (SPG15), are localized in the midbody and appear to facilitate cytokinesis during cell division,33, 34, 35 to our knowledge, proliferation of human cortical progenitors has only been studied in SPG4‐NPCs, where no proliferation deficit matching a pure spastic paraplegia phenotype was present.19 Our data point toward distinct temporal functions of spatacsin, causing axonal transport deficits in corticospinal projections in mature neurons in vitro, and most likely during adulthood, in patients.12 It is imperative to speculate that the cortical changes (TCC and cortical atrophy) in SPG11 patients, when they first present with spastic paraplegia, are rather the results of defects in proliferation during cortical development,5, 6, 7 thereby highlighting a novel role for spatacsin in early neural development. Our current model of SPG11 disease pathology starts with an early‐onset neurodevelopmental phenotype (first two decades), consisting of a proliferation deficit and cortical neurogenesis defects. This results in impairment of axonal outgrowth and guidance, consistent with a TCC and cortical atrophy. After transition around the second and third decades of life, an additional neurodegenerative phenotype is observed. The major adult‐onset phenotype is reflected by axonal degeneration, resulting in impaired axonal transport, with the clinical correlates of spastic paraparesis and peripheral neuropathy (Fig 7A).
Figure 7.
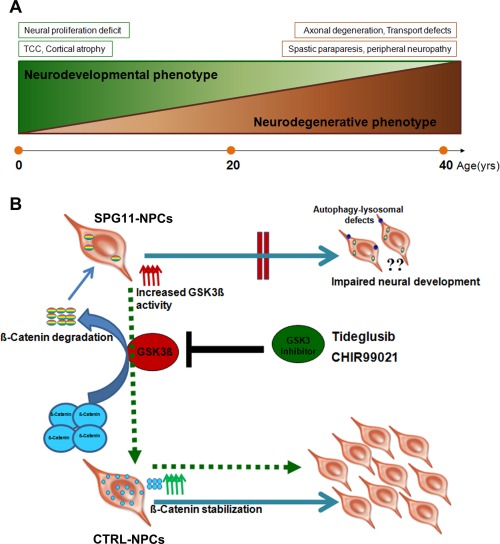
(A) Proposed two distinct stages of SPG11 pathogenesis. Neurodevelopmental phenotype represents early onset within the first two decades characterized by a proliferation deficit, impaired cortical development and consequently to cognitive impairment. The neurodegenerative phenotype, marked by progressive spasticity and paraparesis, leads to functional neuronal deficits, motor neuron degeneration, and peripheral sensorimotor neuropathy. (B) Schematic model of GSK3ß‐mediated neural development in SPG11‐ and CTRL‐NPCs. Increased GSK3 activity leads to reduced ß‐Catenin level in SPG11‐NPCs, thereby compromising proliferation and neurogenesis in SPG11 patients. Pharmacological treatment with the GSK3 inhibitors, tideglusib and CHIR99021, activates the canonical Wnt pathway by inhibiting GSK3 signaling and thereby restores proliferation and neurogenesis (shown by dashed green arrows). NPCs = cortical neural progenitor cells.
A neurodevelopmental phenotype consisting of defects in proliferation, combined with increased cell death, has been reported in other neurodegenerative diseases, including Down syndrome and myotonic dystrophy type1, partly accompanied with dysregulation of GSK3/mTOR signaling.36, 37 Similarly, reduced neuronal connectivity and altered neuronal gene expression profiles regulating neural migration, axonal outgrowth, guidance, and cell adhesion network were reported in a cohort of schizophrenia patient hiPSC‐derived neurons.38 An autism spectrum disease model of fragile‐X syndrome also revealed an aberrant neuronal differentiation, defective neurite extension, and altered calcium activity,39 reflecting early developmental phenotypes at the cellular level and likely contributing to disease phenotypes at the systems level in early‐onset disease of the CNS.
GSK3ß Inhibition Rescues Proliferation Phenotype of SPG11‐NPCs
The Wnt/β‐Catenin signaling pathway is an important regulator of the cell‐cycle machinery and proliferation of NPCs and regulates distinct stages of neural development, including cell proliferation, specification, differentiation, and migration.40 Impaired Wnt signaling, as observed in SPG11‐iPSC derived NPCs, suggested an adverse impact on cortical development in SPG11. Our model suggests that an increase in GSK3 activity leads to increased ß‐Catenin degradation. Reduced ß‐Catenin levels in SPG11‐NPCs result in a decreased NPC proliferation in SPG11 patients, resulting in impaired cortical development. Pharmacological treatment with the GSK3 inhibitors, tideglusib and CHIR99021, activates the canonical Wnt pathway by inhibiting GSK3 signaling and thereby restores proliferation and neurogenesis of SPG11‐NPCs (Fig 7B).
Dysregulation of GSK3 signaling has been associated with DISC1 and fragile‐X syndrome previously.29, 41 Increased GSK3ß activity results in proteolytic degradation of its downstream substrate, ß‐Catenin. Strikingly, we revealed a significant reduction in expression of ß‐Catenin in SPG11‐NPCs. The senescence marker, p27Kip1, has been shown to disrupt the cell‐cycle progression to S phase42; thus, the elevated senescence activity of SPG11‐NPCs (p27Kip1 expression levels) might cause a temporal dysregulation of progenitor proliferation and neurogenesis during cortical development.
Impairment of the GSK3 pathway has been shown to be associated with microcephaly, fragile‐X syndrome, and Alzheimer's disease.41, 43, 44 Given that inhibition of GSK3 is able to enhance the proliferation and neurogenesis of NPCs in rodent brain,32 we tested the pharmacological inhibition of the GSK3 pathway for the neurodevelopmental defects of SPG11‐NPCs. The GSK3 inhibitor, CHIR99021, and a clinically used GSK3 blocker, tideglusib, led to a significant increase of proliferating NPCs and neural cells selectively in SPG11 patients (Fig 7B). This finding provides confirmation that neurodevelopmental defects in the SPG11‐NPCs are linked to dysregulation of GSK3ß signaling. More important, it highlights the rescue of the impaired neurogenesis in SPG11 by inhibition of the GSK3 pathway and an important direction for the development of novel therapeutics for an early intervention of the disease.
In summary, our study highlights a neurodevelopmental phenotype of SPG11 and provides novel insights into GSK3/ß‐Catenin signaling‐mediated proliferation defects of SPG11‐NPCs, resulting in a compromised generation of cortical neurons in HSP patients. Our study further emphasizes the therapeutic potential of inhibiting the GSK3 pathway to restore the neurodevelopmental defects in HSP patients with SPG11 mutations. This pharmacological intervention presents a new direction for translating the molecular insights gained at the cellular level using human reprogramming technology to develop novel therapeutics for SPG11.
Author Contributions
H.K.M., B.W., and J.W. were responsible for concept and study design. H.K.M., I.P., S.H., G.S., Z.K., F.P.B., T.B., L.A., M.L., F.B.E., M.B., J.B., H.W., L.B., M.H., A.L., G.M., and J.K. were responsible for data acquisition and analysis. H.K.M., B.W., J.W., A.R., F.B.E., F.H.G., and A.L. were responsible for drafting the manuscript and the figures. I.P. and S.H. contributed equally.
Potential Conflicts of Interest
Nothing to report.
List of abbreviations
- APC
Adenomatous polyposis coli
- TCF7L2
Transcription factor 7‐like 2
- FOSL2
Fos‐related antigen 2
- CCNA1
Cyclin‐A1
- CDH1
Cadherin‐1
- E13
human gestation day 13
- ITSN1
Intersectin‐1
- CDC42EP3
Cdc42 effector protein 3
- SEM3A
Semaphorin‐3A
- LYST
Lysosomal trafficking regulator
- CTRLs
Controls
- HSP
Hereditary spastic paraplegia
- SPG11
Spastic paraplegia gene11
- MRI
Magnetic resonance imaging
- NPM
Neural proliferation medium
- PBST
Phosphate buffer saline with Tween20
- H3P
Phospho Histone3
- PCNA
Proliferating cell nuclear antigen
- cleav. Casp3
cleaved Caspase3
- CHIR
CHIR99021
- Tide
Tideglusib
- CDK
Cyclin‐dependent kinase
- p21Cip1
Cyclin‐dependent kinase inhibitor 1
- p27Kip1
Cyclin‐dependent kinase inhibitor 1B
Supporting information
Additional supporting information can be found in the online version of this article.
Acknowledgment
This work is dedicated to our patients, controls and in particular, to Dr. Tom Wahlig and his family. We thank Ben Campbell, Daniela Gräf, Dr. Arif Ekici, Dr. Ute Hehr, Dr. Johannes Schlachetzki, Johanna Käsbauer and Sonja Plötz for excellent support and critical input. Support for this study came from the Tom‐Wahlig Foundation Advanced Fellowship, the German Federal Ministry of Education and Research (BMBF; 01GQ113 and 01GM1520A), the Interdisciplinary Centre for Clinical Research (University Hospital of Erlangen; N3 and F3), the Emerging Fields Initiative CYDER (FAU Erlangen‐Nuernberg), the Bavarian Ministry of Education and Culture, Science and the Arts in the framework of the Bavarian Molecular Biosystems Research Network, and ForIPS.
References
- 1. Harding AE. Classification of the hereditary ataxias and paraplegias. Lancet 1983;1:1151–1155. [DOI] [PubMed] [Google Scholar]
- 2. McDermott CJ, Shaw PJ. Hereditary spastic paraplegia. Int Rev Neurobiol 2002;53:191–204. [DOI] [PubMed] [Google Scholar]
- 3. Klebe S, Stevanin G, Depienne C. Clinical and genetic heterogeneity in hereditary spastic paraplegias: from SPG1 to SPG72 and still counting. Rev Neurol (Paris) 2015;171:505–530. [DOI] [PubMed] [Google Scholar]
- 4. Blackstone C, O'Kane CJ, Reid E. Hereditary spastic paraplegias: membrane traffic and the motor pathway. Nat Rev Neurosci 2011;12:31–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Winner B, Uyanik G, Gross C, et al. Clinical progression and genetic analysis in hereditary spastic paraplegia with thin corpus callosum in spastic gait gene 11 (SPG11). Arch Neurol 2004;61:117–121. [DOI] [PubMed] [Google Scholar]
- 6. Stevanin G, Azzedine H, Denora P, et al. Mutations in SPG11 are frequent in autosomal recessive spastic paraplegia with thin corpus callosum, cognitive decline and lower motor neuron degeneration. Brain 2008;131:772–784. [DOI] [PubMed] [Google Scholar]
- 7. Hehr U, Bauer P, Winner B, et al. Long‐term course and mutational spectrum of spatacsin‐linked spastic paraplegia. Ann Neurol 2007;62:656–665. [DOI] [PubMed] [Google Scholar]
- 8. Orlacchio A, Babalini C, Borreca A, et al. SPATACSIN mutations cause autosomal recessive juvenile amyotrophic lateral sclerosis. Brain 2010;133:591–598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Stevanin G, Santorelli FM, Azzedine H, et al. Mutations in SPG11, encoding spatacsin, are a major cause of spastic paraplegia with thin corpus callosum. Nat Genet 2007;39:366–372. [DOI] [PubMed] [Google Scholar]
- 10. Renvoise B, Chang J, Singh R, et al. Lysosomal abnormalities in hereditary spastic paraplegia types SPG15 and SPG11. Ann Clin Transl Neurol 2014;1:379–389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Chang J, Lee S, Blackstone C. Spastic paraplegia proteins spastizin and spatacsin mediate autophagic lysosome reformation. J Clin Invest 2014;124:5249–5262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Perez‐Branguli F, Mishra HK, Prots I, et al. Dysfunction of spatacsin leads to axonal pathology in SPG11‐linked hereditary spastic paraplegia. Hum Mol Genet 2014;23:4859–4874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Pasca SP, Portmann T, Voineagu I, et al. Using iPSC‐derived neurons to uncover cellular phenotypes associated with Timothy syndrome. Nat Med 2011;17:16571662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Sheridan SD, Theriault KM, Reis SA, et al. Epigenetic characterization of the FMR1 gene and aberrant neurodevelopment in human induced pluripotent stem cell models of fragile X syndrome. PLoS One 2011;6:e26203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Paul LK, Brown WS, Adolphs R, et al. Agenesis of the corpus callosum: genetic, developmental and functional aspects of connectivity. Nat Rev Neurosci 2007;8:287–299. [DOI] [PubMed] [Google Scholar]
- 16. Edwards TJ, Sherr EH, Barkovich AJ, Richards LJ. Clinical, genetic and imaging findings identify new causes for corpus callosum development syndromes. Brain 2014;137:1579–1613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Luders E, Thompson PM, Toga AW. The development of the corpus callosum in the healthy human brain. J Neurosci 2010;30:10985–10990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Bauer P, Winner B, Schule R, et al. Identification of a heterozygous genomic deletion in the spatacsin gene in SPG11 patients using high‐resolution comparative genomic hybridization. Neurogenetics 2009;10:43–48. [DOI] [PubMed] [Google Scholar]
- 19. Havlicek S, Kohl Z, Mishra HK, et al. Gene dosage‐dependent rescue of HSP neurite defects in SPG4 patients' neurons. Hum Mol Genet 2014;23:2527–2541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Takahashi K, Tanabe K, Ohnuki M, et al. Induction of pluripotent stem cells from adult human fibroblasts by defined factors. Cell 2007;131:861–872. [DOI] [PubMed] [Google Scholar]
- 21. Prots I, Skapenko A, Lipsky PE, Schulze‐Koops H. Analysis of the transcriptional program of developing induced regulatory T cells. PLoS One 2011;6:e16913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Engel FB, Hauck L, Cardoso MC, et al. A mammalian myocardial cell‐free system to study cell cycle reentry in terminally differentiated cardiomyocytes. Circ Res 1999;85:294–301. [DOI] [PubMed] [Google Scholar]
- 23. Ahmed A, Smoot D, Littleton G, et al. Helicobacter pylori inhibits gastric cell cycle progression. Microbes Infect 2000;2:1159–1169. [DOI] [PubMed] [Google Scholar]
- 24. Engel FB, Hauck L, Boehm M, et al. p21(CIP1) Controls proliferating cell nuclear antigen level in adult cardiomyocytes. Mol Cell Biol 2003;23:555–565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Veeman MT, Slusarski DC, Kaykas A, et al. Zebrafish prickle, a modulator of noncanonical Wnt/Fz signaling, regulates gastrulation movements. Curr Biol 2003;13:680–685. [DOI] [PubMed] [Google Scholar]
- 26. Dehner M, Hadjihannas M, Weiske J, et al. Wnt signaling inhibits Forkhead box O3a‐induced transcription and apoptosis through up‐regulation of serum‐ and glucocorticoid‐inducible kinase 1. J Biol Chem 2008;283:19201–19210. [DOI] [PubMed] [Google Scholar]
- 27. Banreti A, Hudry B, Sass M, et al. Hox proteins mediate developmental and environmental control of autophagy. Dev Cell 2014;28:56–69. [DOI] [PubMed] [Google Scholar]
- 28. Kim WY, Wang X, Wu Y, et al. GSK‐3 is a master regulator of neural progenitor homeostasis. Nat Neurosci 2009;12:1390–1397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Mao Y, Ge X, Frank CL, et al. Disrupted in schizophrenia 1 regulates neuronal progenitor proliferation via modulation of GSK3beta/beta‐catenin signaling. Cell 2009;136:1017–1031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Cohen P, Frame S. The renaissance of GSK3. Nat Rev Mol Cell Biol 2001;2:769–776. [DOI] [PubMed] [Google Scholar]
- 31. Engelman JA, Luo J, Cantley LC. The evolution of phosphatidylinositol 3‐kinases as regulators of growth and metabolism. Nat Rev Genet 2006;7:606–619. [DOI] [PubMed] [Google Scholar]
- 32. Nedachi T, Kawai T, Matsuwaki T, et al. Progranulin enhances neural progenitor cell proliferation through glycogen synthase kinase 3beta phosphorylation. Neuroscience 2011;185:106–115. [DOI] [PubMed] [Google Scholar]
- 33. Connell JW, Lindon C, Luzio JP, Reid E. Spastin couples microtubule severing to membrane traffic in completion of cytokinesis and secretion. Traffic 2009;10:42–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Renvoise B, Stadler J, Singh R, et al. Spg20‐/‐ mice reveal multimodal functions for Troyer syndrome protein spartin in lipid droplet maintenance, cytokinesis and BMP signaling. Hum Mol Genet 2012;21:3604–3618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Sagona AP, Nezis IP, Pedersen NM, et al. PtdIns(3)P controls cytokinesis through KIF13A‐mediated recruitment of FYVE‐CENT to the midbody. Nat Cell Biol 2010;12:362–371. [DOI] [PubMed] [Google Scholar]
- 36. Denis JA, Gauthier M, Rachdi L, et al. mTOR‐dependent proliferation defect in human ES‐derived neural stem cells affected by myotonic dystrophy type 1. J Cell Sci 2013;126:1763–1772. [DOI] [PubMed] [Google Scholar]
- 37. Hibaoui Y, Grad I, Letourneau A, et al. Modelling and rescuing neurodevelopmental defect of Down syndrome using induced pluripotent stem cells from monozygotic twins discordant for trisomy 21. EMBO Mol Med 2014;6:259–277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Brennand KJ, Simone A, Jou J, et al. Modelling schizophrenia using human induced pluripotent stem cells. Nature 2011;473:221–225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Liu J, Koscielska KA, Cao Z, et al. Signaling defects in iPSC‐derived fragile X premutation neurons. Hum Mol Genet 2012;21:3795–3805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Clevers H. Wnt/beta‐catenin signaling in development and disease. Cell 2006;127:469–480. [DOI] [PubMed] [Google Scholar]
- 41. Portis S, Giunta B, Obregon D, Tan J. The role of glycogen synthase kinase‐3 signaling in neurodevelopment and fragile X syndrome. Int J Physiol Pathophysiol Pharmacol 2012;4:140–148. [PMC free article] [PubMed] [Google Scholar]
- 42. Kaproth‐Joslin KA, Li X, Reks SE, Kelley GG. Phospholipase C delta 1 regulates cell proliferation and cell‐cycle progression from G1‐ to S‐phase by control of cyclin E‐CDK2 activity. Biochem J 2008;415:439–448. [DOI] [PubMed] [Google Scholar]
- 43. Novorol C, Burkhardt J, Wood KJ, et al. Microcephaly models in the developing zebrafish retinal neuroepithelium point to an underlying defect in metaphase progression. Open Biol 2013;3:130065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Hooper C, Killick R, Lovestone S. The GSK3 hypothesis of Alzheimer's disease. J Neurochem 2008;104:1433–1439. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Additional supporting information can be found in the online version of this article.