

Summary
Proteasome inhibitors (PIs) are highly active in multiple myeloma (MM) but resistance is commonly observed. All clinical stage PIs effectively inhibit chymotrypsin‐like (CT‐L) activity; one possible mechanism of resistance is compensatory hyperactivation of caspase‐like (C‐L) and trypsin‐like (T‐L) subunits, in response to CT‐L blockade. Marizomib (MRZ), an irreversible PI that potently inhibits all three 20S proteasome subunits with a specificity distinct from other PIs, is currently in development for treatment of MM and malignant glioma. The pan‐proteasome pharmacodynamic activity in packed whole blood and peripheral blood mononuclear cells was measured in two studies in patients with advanced solid tumours and haematological malignancies. Functional inhibition of all proteasome subunits was achieved with once‐ or twice‐weekly MRZ dosing; 100% inhibition of CT‐L was frequently achieved within one cycle at therapeutic doses. Concomitantly, C‐L and T‐L activities were either unaffected or increased, suggesting compensatory hyperactivation of these subunits. Importantly, this response was overcome by continued administration of MRZ, with robust inhibition of T‐L and C‐L (up to 80% and 50%, respectively) by the end of Cycle 2 and maintained thereafter. This enhanced proteasome inhibition was independent of tumour type and may underlie the clinical activity of MRZ in patients resistant to other PIs.
Keywords: marizomib, proteasome hyperactivation, pan‐proteasome inhibitor
The proteasome is a multi‐catalytic proteinase complex, responsible for the degradation of ubiquitinated proteins within normal and transformed cells. Malignant cells are more dependent on proteasome activity to remove mis‐folded or damaged proteins due to their genetic instability and rapid proliferation. Due to generally higher levels of proteasome activity in cancer cells, inhibition of the proteasome elicits pro‐apoptotic effects preferentially in malignant cells compared with normal cells, and is a well‐validated target in multiple myeloma (MM). Three proteasome inhibitors (PIs), bortezomib (BTZ: Kane et al, 2003; Richardson et al, 2003, 2005), carfilzomib (CFZ) and ixazomib (IXZ) are currently approved for the treatment of MM, with several others in development. Although important therapeutic advances, these PIs are associated with significant and dose‐limiting off‐target toxicities (Lonial et al, 2005; Richardson et al, 2006; Cai et al, 2014; Harvey, 2014; Atrash et al, 2015; Wanchoo et al, 2015) and the development of acquired resistance (Fall et al, 2014; Huber et al, 2015; Niewerth et al, 2015). Despite consistently high response rates with PI‐based combinations, almost all MM patients eventually relapse, with progressively lower rates and durations of response with each subsequent line of therapy and poor prognosis as resistance emerges (Kumar et al, 2012). Current therapeutic options for MM patients who have relapsed and refractory disease are limited, and effective new treatments that re‐establish tumour responsiveness are urgently needed to improve patient outcomes.
The proteasome consists of three types of subunits in the inner β rings of the 20S core particle, which carry the proteolytic chymotrypsin‐like (CT‐L, β5), trypsin‐like (T‐L, β2) and caspase‐like (C‐L, β1) activities, with CT‐L activity of the β5 subunit being the rate‐limiting step of proteolysis. BTZ, CFZ and IXZ are selective inhibitors of β5, as are several next‐generation clinical‐stage PIs, such as oprozomib, with little to no inhibitory activity on T‐L or C‐L (Teicher & Tomaszewski, 2015). In contrast, Marizomib (MRZ; salinosporamide A), a PI derived from a marine actinomycete, inhibits all 3 major catalytic activities of the 20S core particle (Chauhan et al, 2005; Fenical et al, 2009). MRZ rapidly enters cells and covalently binds to all three active enzyme sites (Groll et al, 2006). This irreversible binding elicits proteasome inhibition in vitro and in vivo (Chauhan et al, 2005, 2008; Singh et al, 2010), reversal of which requires cell replacement and/or proteasome re‐synthesis.
Resistance to PIs arises via multiple potential mechanisms (Niewerth et al, 2015). While BTZ‐adapted haematological cell line models frequently acquire mutations in β5 that confer resistance to BTZ, these mutations are absent in clinical specimens obtained from BTZ‐resistant patients (reviewed in Niewerth et al, 2015). Several studies indicate that overexpression of catalytic subunits is the primary cellular response mechanism to BTZ treatment and may precede acquisition of β5 mutations (Franke et al, 2012; Niewerth et al, 2013), as well as increased β2 and β1 activity (Ruckrich et al, 2009). Given that both pan‐proteasome inhibitory activity (Britton et al, 2009) and irreversible binding to the proteasome subunits (Orlowski, 2013; Dou & Zonder, 2014) have been postulated to overcome BTZ resistance and provide prolonged activity, the unique pharmacological profile of MRZ may confer therapeutic advantage by irreversibly inhibiting more than one proteasomal activity (reviewed in Kale & Moore, 2012).
MRZ began clinical development with a Phase 1 trial conducted in patients with solid tumours and refractory lymphoma (NPI‐0052‐100, NCT00396864), followed by two additional Phase 1/2 trials: a US trial exploring once‐ and twice‐weekly dosing schedules in relapsed and/or refractory MM (NPI‐0052‐101, NCT00461045), and a trial conducted in Australia and Estonia exploring once‐ and twice‐weekly dosing schedules in patients with solid and haematological tumours, including relapsed and/or refractory MM (NPI‐0052‐102, NCT00629473). The pharmacodynamic effects of MRZ on subunit‐specific proteasome activity observed in these two Phase 1/2 trials are reported here.
Patients and methods
NPI‐0052‐101P1. A total of 68 patients with relapsed or relapsed/refractory MM were enrolled in the trial (Richardson et al, 2016). Patients received MRZ by intravenous (IV) infusion by two different schedules: either once‐weekly infusions of MRZ on Days 1, 8 and 15 of 4‐week cycles, at dose ranges of 0·025 to 0·7 mg/m2, infused over 1 or 10 min; or twice‐weekly infusions of MRZ on Days 1, 4, 8 and 11 of 3‐week cycles, at dose ranges of 0·015 to 0·6 mg/m2, infused over 60 or 120 min. Of the 68 patients enrolled, 27 had blood samples collected for pharmacodynamic evaluation of proteasome inhibition – 18 treated on the weekly MRZ infusion schedule, from whom blood samples were collected immediately before and 1 h after MRZ infusion on Days 1 and 15 of Cycle 1 and 2, and 8 treated on the twice‐weekly MRZ infusion schedule, from whom blood samples were collected immediately before and 1 h after MRZ infusion on Days 1 and 11 of Cycle 1. In addition, blood samples from one patient on the twice‐weekly MRZ infusion schedule (0·075 mg/m2) were collected on Day 15 of Cycle 6.
NPI‐0052‐102. A total of 86 patients were enrolled on the trial (Harrison et al, 2016): 42 patients with advanced malignancies (Arm AM) including solid tumours (n = 24), lymphoma (n = 15) and leukaemia (n = 3), received MRZ administered IV once‐weekly, on Days 1, 8 and 15 in 4–week cycles, at doses ranging from 0·1 to 0·9 mg/m2 by infusion for 1–10 min; 44 patients with haematological malignancies (Arm MM) including MM (n = 35), non‐Hodgkin lymphoma (n = 6), Hodgkin lymphoma (n = 1) and chronic lymphocytic leukaemia (n = 2), received MRZ administered IV twice‐weekly, on Days 1, 4 8 and 11 in 3–week cycles, at doses ranging from 0·075 to 0·6 mg/m2 by infusion for 1, 10, or 120 min. Pharmacodynamics samples were to be obtained at baseline, Day 1 (before treatment and 1 h post‐infusion) and Day 15 (Arm AM)/Day 11 (Arm MM) (pre‐ and 1 h post‐infusion) of Cycles 1 and 2, and then every other cycle thereafter, so that (for both schedules) the second sampling in a cycle was always performed after the third dose. For patients that discontinued early in treatment, samples were obtained from Cycle 1 only.
Pharmacodynamic sample processing
Packed whole blood (PWB) pellets were prepared by centrifugation of ~10 ml anticoagulated (sodium heparin) human blood samples. After centrifugation, PWB pellets re‐suspended in 5 volumes of ice‐cold phosphate‐buffered saline (PBS) were aliquoted, re‐centrifuged and stored at −70°C. For peripheral blood mononuclear cell (PBMC) isolation, anticoagulated (sodium heparin) blood samples (2 ml) were diluted with an equal volume of PBS, layered over 3 ml Ficoll‐Paque™ PLUS and centrifuged. PBMC pellets were gently re‐suspended in 6 ml of PBS, re‐centrifuged twice more, and then stored at −70°C.
For pellet lysis, PWB and PBMC were thawed on ice for 1 h and then re‐suspended in ice‐cold 5 mmol/l EDTA (pH 8·0); PWB pellets were re‐suspended in three‐times their volume of EDTA, and PBMC pellets were re‐suspended with 100 μl EDTA. After thorough vortexing, the cells were lysed on ice for at least 1 h. Following centrifugation at 19 500 g at 4°C for 10 min, glycerol was added to the supernatants at a final concentration of 10% (v/v) and lysates were stored at −70°C in aliquots. Protein concentration was determined using a modified Lowry‐Assay (Pierce BCATM Protein Assay Kit, ThermoFisher Scientific, Carlsbad, CA, USA) per manufacturer's instructions.
Proteasome activity assays
Proteasome activity was measured as previously reported (Lightcap et al, 2000). Briefly, CT‐L, C‐L and T‐L activities were determined in 96‐well microtitre plates in 20 mmol/l HEPES/0·5 mmol/l EDTA, pH 8·0. Sodium dodecyl sulfate (0·05%) was added to the CT‐L and C‐L assays. The substrates Suc‐Leu‐Leu‐Val‐Tyr‐AMC, Z‐Leu‐Leu‐Glu‐AMC and Bz‐Val‐Gly‐Arg‐AMC were used for CT‐L, C‐L and T‐L activity, respectively. Lysates from PWB or PBMC were added to start the reaction. The plate was immediately placed in a pre‐warmed spectrofluorometer (37°C) and read every 5 min for 2 h (λex = 390 nm, λem = 460 nm with 435 nm cut‐off). Activity was reported as pmol AMC/mg/min (background subtracted). Two negative controls were included, one containing lysate diluted in assay buffer and one containing assay buffer and substrate. A positive control was included that consisted of rat PWB in the corresponding assay buffer to demonstrate maximal activity for the different enzymatic assays.
Data analysis
Proteasome inhibition in each post‐infusion sample is expressed as a percentage of the activity in the pre‐infusion sample from Day 1 of Cycle 1 (C1D1) of MRZ treatment, for each subunit. Data are presented as the observed inhibition on C1D1 and the peak effect, which was the largest inhibitory effect observed for each patient across all dosing cycles.
Results
The objective of these studies was to quantitatively assess the pharmacodynamic impact of MRZ using proteasome subunit‐specific assays to measure CT‐L, T‐L and C‐L activity in whole blood samples and mononuclear cells collected from patients with advanced solid tumours and haematological malignancies across clinical trials.
MRZ dose‐dependently inhibits CT‐L activity in packed whole blood (PWB) and peripheral blood mononuclear cells (PBMCs)
Dose‐dependent inhibition of CT‐L activity in PWB by MRZ was evident with the first dose (C1D1, Fig 1A). Maximal pharmacodynamic efficacy – 100% inhibition of CT‐L activity – was evident within the first dosing cycle, and observed in all patients at the MRZ dosages subsequently identified as the recommended phase 2 dose levels (0·7 mg/m2 for once‐weekly infusion and 0·5 mg/m2 for twice‐weekly infusion). Similarly, maximal inhibition of CT‐L activity by MRZ in PWB during the first dosing cycle within each patient (Peak Effect) was also dose‐dependent (Fig 1B), and apparently independent of the infusion regimen (once‐ vs. twice‐weekly). The inhibition of CT‐L activity in PWB samples, plotted as a function of cumulative dose, was described by a three‐parameter log dose versus response curve (Fig 1C). Increasing MRZ dose exposure resulted in increasing inhibition of CT‐L activity in PWB, with a 50% inhibitory dose of 0·6 mg/m2 [95% Confidence Intervals (CI) 0·18–2·0 mg/m2]. Complete inhibition of CT‐L activity in PWB samples was achieved at cumulative MRZ doses ≥1·6 mg/m2, occurring at the end of Cycle 1 for patients who received MRZ twice‐weekly at doses ≥0·4 mg/m2 or once‐weekly at the 0·7 mg/m2 dose. Peak inhibition of T‐L activity ranged from 25–78% after repeat dosing with moderate to high MRZ doses (≥0·4 mg/m2) and 14% to 26% inhibition of C‐L activity occurred at the end of the first cycle of repeat dosing with high MRZ doses (≥0·5 mg/m2, data not shown).
Figure 1.
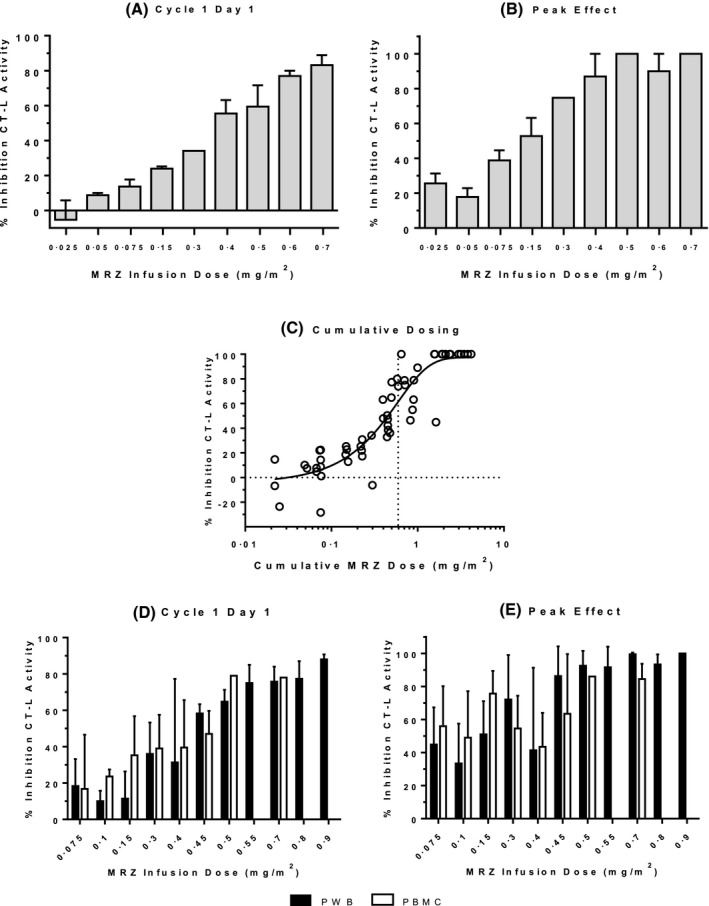
Inhibition of CT‐L activity by MRZ. (A & B) PWB samples from MM patients (Trial NPI‐0052‐101). All MRZ doses levels were infused once weekly, except 0·4, 0·5 and 0·6 mg/m2, which were infused twice‐weekly. Data are depicted as Mean + standard error (SE). (A) Effect of MRZ infusion by dose level on CT‐L activity on Day 1 of Cycle 1 (N = 3, 2, 5, 2, 1, 2, 3, 2 and 4, for doses of 0·025, 0·05, 0·075, 0·15, 0·3, 0·4, 0·5, 0·6 and 0·7 mg/m2, respectively). (B) Peak effect of MRZ infusion by dose level on CT‐L activity during the first 1‐2 cycles (one patient achieved peak effect on Day 15 of Cycle 6 in the 0·075 mg/m2 dose group; N = 3, 2, 6, 2, 1, 4, 3, 2 and 4, for doses of 0·025, 0·05, 0·075, 0·15, 0·3, 0·4, 0·5, 0·6 and 0·7 mg/m2, respectively). (C) Cumulative effect of MRZ infusion on CT‐L activity in PWB from MM patients after repeated infusions. Dotted vertical line denotes the dose level estimated to induce 50% inhibition of CT‐L activity (0·6 mg/m2). (D & E) PWB (solid bars) and PBMC (open bars) samples from patients with solid tumours (once‐weekly MRZ infusion regimen) and haematological malignancies (twice‐weekly MRZ infusion regimen)(Trial NPI‐0052‐102). Data are depicted as Mean + SE. For PWB: N = 6, 5, 7, 8, 4, 3, 4, 5, 4, 3, and 2 for doses of 0·075, 0·1, 0·15, 0·3, 0·4, 0·45, 0·5, 0·55, 0·7, 0·8 and 0·9 mg/m2, respectively. For PBMC: N = 4, 3, 3, 8, 2, 2, 1, 0, 2, 0, and 0 for doses of 0·075, 0·1, 0·15, 0·3, 0·4, 0·45, 0·5, 0·55, 0·7, 0·8 and 0·9 mg/m2, respectively. (D) Effect of MRZ infusion by dose level on CT‐L activity on Day 1 of Cycle 1. (E) Peak effect of MRZ infusion by dose level on CT‐L activity, which occurred for most patients during the first MRZ treatment cycle (for five patients the effect was observed on Day 1 or 15 of Cycle 2, for four patients on Day 15 of Cycle 4, for one patient on Day 15 of Cycle 6, and for one patient on Day 15 of Cycle 12). MRZ, marizomib; CT‐L, chymotrypsin‐like; PWB, packed whole blood; PBMC, peripheral blood mononuclear cells; MM, multiple myeloma
Inhibition of CT‐L proteasome activity on initial MRZ infusion and peak inhibition observed in PBMC after repeat MRZ dosing tended to be dose‐related (Table 1), as was observed in the PWB samples, despite small sample numbers. Inhibition of T‐L and C‐L activity by MRZ in PBMC also tended to be dose‐related and less robust than for inhibition of CT‐L activity (data not shown), similar to effects observed in the PWB samples.
Table 1.
Inhibition of CT‐L proteasome activity by MRZ in PBMC (Study NPI‐0052‐101)
| MRZ Dose (mg/m2) | Time point | % CT‐L Inhibition Mean (SD, N) |
|---|---|---|
| 0·025 |
Cycle 1 Day 1 Peak Effect |
−14·4 (29, 2) 8·89 (20, 2) |
| 0·05 |
Cycle 1 Day 1 Peak Effect |
−20·6 (0·0, 1) 29·8 (0·0, 1) |
| 0·075 |
Cycle 1 Day 1 Peak Effect |
−46·5 (103, 4) 49·8 (25, 5) |
| 0·15 |
Cycle 1 Day 1 Peak Effect |
ND |
| 0·3 |
Cycle 1 Day 1 Peak Effect |
ND |
| 0·4 |
Cycle 1 Day 1 Peak Effect |
−22·2 (0·0, 1) 36·1 (82, 2) |
| 0·5 |
Cycle 1 Day 1 Peak Effect |
66·7 (3·1, 3) 85·5 (14, 3) |
| 0·6 |
Cycle 1 Day 1 Peak Effect |
71·5 (14, 2) 88·1 (9·8, 2) |
| 0·7 |
Cycle 1 Day 1 Peak Effect |
92·4 (6·7, 3) 100·0 (0·0, 2) |
CT‐L, chymotrypsin‐like; MRZ, marizomib; PBMC, peripheral blood mononuclear cells; SD, standard deviation, N, number.
As in NPI‐0052‐101, dose‐dependent inhibition of CT‐L activity by MRZ was evident with the first dose (C1D1) in both PWB and PBMC (Fig 1D) in samples from NPI‐0052‐102. Similarly, maximal inhibition of CT‐L activity in PWB and in PBMC by MRZ during the first dosing cycle within each patient (Peak Effect) was also dose‐dependent (Fig 1E), and independent of the infusion regimen (once‐ vs twice‐weekly). At MRZ doses ≤0·15 mg/m2, inhibition of CT‐L activity in PWB ranged from 10–18% inhibition on C1D1, increasing to a peak effect of 33–51% CT‐L inhibition. Inhibition of CT‐L activity was more pronounced at intermediate dose levels (0·3–0·55 mg/m2), averaging 31–75% inhibition on C1D1, reaching a peak effect of 41–91% with repeat dosing. At the highest dose levels examined (0·7–0·9 mg/m2), an average of 75–88% inhibition of CT‐L was observed on C1D1 and a maximal 93–100% inhibition was observed during the first cycle at these dose levels. Maximum inhibition of CT‐L activity in PWB was observed in Cycle 1 for 40 of the 51 patients, with the remainder of the peak effects noted in Cycle 2 (four patients) or later (seven patients), demonstrating a rapid effect of MRZ on inhibition of CT‐L activity.
Importantly, proteasome inhibition in the nucleated cells (PBMCs) was comparable at all dose levels to that observed in PWB samples after both a single dose (C1D1, Fig 1D) or repeated doses (peak effect, Fig 1E), with near maximal inhibition at recommended Phase 2 doses of 0·5 mg/m2 (twice‐weekly) and 0·7 mg/m2 (once‐weekly), displaying an average of 78–79% inhibition on C1D1 and 84–86% inhibition at the peak effect. These data suggest that the irreversible binding mode of MRZ can overcome the re‐synthesis of proteasome subunits in nucleated cells, as expected for an irreversible PI.
When the inhibition of CT‐L activity in PWB samples was plotted as a function of cumulative dose, the resulting curve could again be described by a three‐parameter log dose versus response curve in both AM and MM patient cohorts (Fig 2A,B) from study NPI‐0052‐102. Increasing MRZ dose exposure resulted in increasing inhibition of CT‐L activity in PWB, with estimated 50% inhibitory dose levels of 0·3 and 0·8 mg/m2 in the AM and MM arms, respectively (95% CI: AM, 0·02–4·3; MM, 0·14–4·5), indicating equivalent proteasomal inhibitory activity of MRZ in PWB between tumour types or infusion regimens. Complete inhibition of CT‐L activity in PWB samples was achieved at cumulative MRZ doses ≥1·2 mg/m2, which were achieved by the end of Cycle 1 for patients who received MRZ twice‐weekly at doses ≥0·3 mg/m2 or once‐weekly doses ≥0·4 mg/m2.
Figure 2.
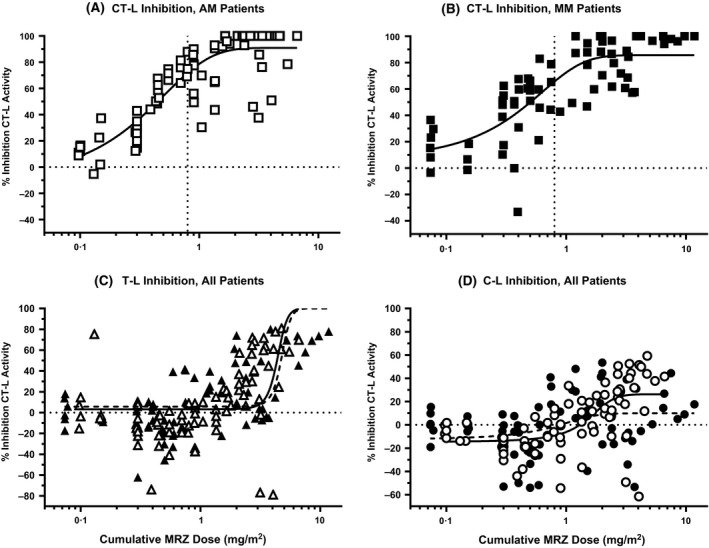
Cumulative effect of MRZ infusion on (A and B) CT‐L, (C) T‐L and (D) C‐L activity in PWB after repeated infusion. (A) CT‐L inhibition in Arm AM patients, (B) CT‐L inhibition in Arm MM patients; curves denote the nonlinear fit (log MRZ dose vs response, three parameters), dotted vertical lines denote the dose level estimated to induce 50% inhibition of CT‐L activity (0·3 mg/m2 for Arm AM, 0·8 mg/m2 for Arm MM). (C) T‐L inhibition, (D) C‐L inhibition; treatment effects are depicted in AM (open symbols) or MM (closed symbols) patients. Curves denote the nonlinear fit (log MRZ dose vs response, three parameters): solid line, AM patients; dashed line, MM patients. MRZ, marizomib; CT‐L, chymotrypsin‐like; T‐L, trypsin‐like; C‐L, caspase‐like; PWB, packed whole blood; AM, advanced malignancies; MM, multiple myeloma.
Repeated dosing with MRZ overcomes initial hyperactivation of T‐L and C‐L subunits
In contrast with the rapid and robust blockade of the β5 chymotrypsin‐like proteasome subunit by MRZ, initial effects on T‐L and C‐L subunits were modest, absent or, in many cases, apparently stimulatory. Upon initial dosing with MRZ, particularly at dose levels that produced ≥40% inhibition of CT‐L activity (≥0·3 mg/m2, see Fig 1D), an increase in T‐L and C‐L activity (Fig 3A, B) was routinely observed in PWB samples. This enhancement of T‐L and C‐L activity on C1D1 was as high as 41% to 50% at intermediate (0·3–0·55 mg/m2) and high dose ranges (0·7–0·9 mg/m2), and observed in patients in both the AM and MM arms of the study. This initial hyperactivation of T‐L and C‐L activity observed with the C1D1 MRZ dose was reversed with repeated dosing; average peak inhibitory effects in the range of 43–71% for T‐L activity and 16–51% for C‐L activity were observed at the recommended Phase 2 doses of 0·5 mg/m2 (twice‐weekly) and 0·7 mg/m2 (once‐weekly) with repeat dosing (Fig 3A, B), with peak T‐L and C‐L inhibition occurring after 1–2 cycles of dosing on the once‐weekly schedule, and after 1–6 cycles of dosing on the twice‐weekly schedule.
Figure 3.
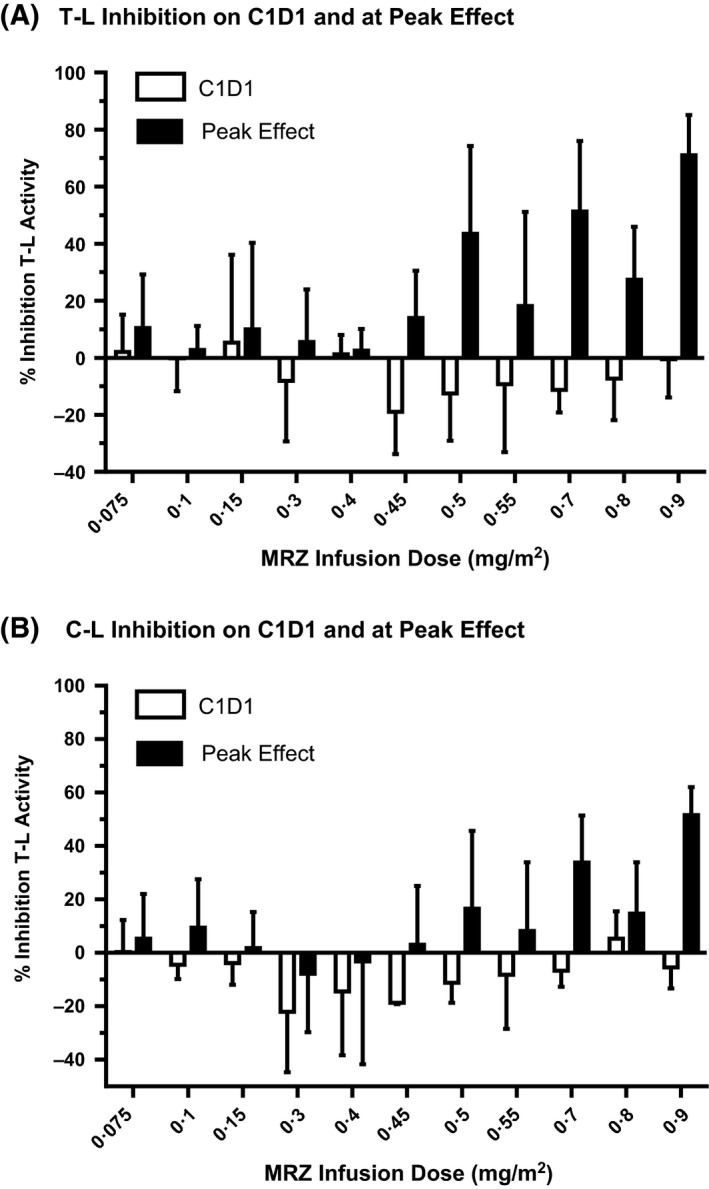
Inhibition of T‐L and C‐L activity by MRZ in PWB samples from patients with solid tumours (once‐weekly MRZ infusion regimen) and haematological malignancies (twice‐weekly MRZ infusion regimen). Data are from Day 1 of Cycle 1 (C1D1, open bars) and at peak effect (solid bars)(Trial NPI‐0052‐102). Data for the two regimens are combined, and depicted as Mean + standard error. N = 6, 5, 7, 8, 4, 3, 4, 5, 4, 3, and 2 for doses of 0·075, 0·1, 0·15, 0·3, 0·4, 0·45, 0·5, 0·55, 0·7, 0·8 and 0·9 mg/m2, respectively. (A) Effect of MRZ infusion by dose level on T‐L activity. (B) Effect of MRZ infusion by dose level on C‐L activity. MRZ, marizomib; T‐L, trypsin‐like; C‐L, caspase‐like; PWB, packed whole blood
The kinetics of cumulative inhibition of each of the three catalytically‐active subunits of the 20S proteasome by MRZ in PWB suggested that the effects of MRZ on the different subunits were functionally linked. For example, in patients dosed weekly (Arm AM), the recommended Phase 2 dose was determined to be 0·7 mg/m2, representing a dose intensity of 2·1 mg/m2 per cycle. As shown in Fig 2A, inhibition of CT‐L activity was maximal and ~100% within a single cycle of dosing at this dose level. Interestingly, this cumulative dose also represents the time at which significant inhibition of the T‐L and, to a lesser extent, C‐L subunits began to be observed (Fig 2C, D). With cumulative doses of 5–6 mg/m2 in Arm AM, the effects of MRZ on T‐L and C‐L activity increased to a maximum of ~80% (T‐L) and ~50% (C‐L), representing approximately three cycles at the once‐weekly recommended phase 2 dose. Similarly, in Arm MM, at the cycle cumulative dose of 2·0 mg/m2 per cycle (twice‐weekly recommended phase 2 dose of 0·5 mg/m2), the kinetics and magnitude of the inhibition of all three proteasomal subunits were similar to the effects observed in the solid tumour patients (Figs 2C and D vs. 2A and B). Estimated 50% inhibitory dose levels for T‐L activity were 4·4 and 4·8 mg/m2 in the AM and MM arms, respectively (95% CI: AM, 3·9–4·9; MM, 3·9–4·5), and for C‐L activity, 1·5 and 0·6 mg/m2 in the AM and MM arms, respectively (95% CI: AM, 0·8–2·2; MM, −1·2–2·4), indicating equivalent proteasomal inhibitory activity of MRZ in PWB between tumour types and infusion regimens. The initial hyperactivation of C‐L and T‐L subunits followed by progressively accumulating pan‐subunit inhibition by MRZ was also observed in the few PBMC samples that were of sufficient quality for assessment of C‐L and T‐L activities. Due to limited sample numbers it was not possible to determine the dose‐response of the drug against the C‐L and T‐L activities in PBMC in this study, however in those patients where data was analyzable, C‐L and T‐L activities were inhibited as much as 50% and 69%, respectively (data not shown).
Discussion
In these investigations, the pharmacodynamic effects of MRZ on subunit‐specific activity of the proteasome were measured in whole blood samples and mononuclear cells collected from patients with solid and haematological malignancies from two clinical trials. Partial or complete inhibition of all three proteasome subunits was achieved with both once‐ and twice‐weekly MRZ dosing, with the rank order of sensitivity (CT‐L > T‐L > C‐L) consistent with the biochemical potency of MRZ (Teicher & Tomaszewski, 2015). For CT‐L activity, both initial (C1D1) and peak proteasome inhibition was dose‐dependent, with complete (100%) inhibition of CT‐L activity in PWB and maximal (60–80%) inhibition of CT‐L activity in PBMC, within the first dosing cycle. In contrast, C‐L and T‐L activities were unchanged or increased in the first cycle of MRZ dosing, suggesting compensatory hyperactivation in response to effective blockade of CT–L activity. Importantly, this response was overcome by further treatment with MRZ, with inhibition of T‐L and C‐L activity noted across dose levels with repeated dosing. These data suggest that initial potent inhibition of CT‐L activity leads to a compensatory hyperactivation of the C‐L and T‐L subunits. As shown schematically in Fig 4, as CT‐L activity becomes fully inhibited by the irreversible activity of MRZ, progressive inhibition of the hyperactivated C‐L and T‐L subunits occurs, ultimately resulting in robust pan‐proteasome inhibition within two dosing cycles in the majority of patients.
Figure 4.
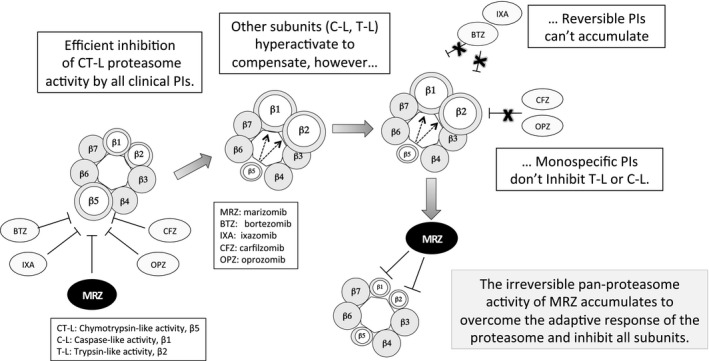
Hypothesis of compensatory activation and cumulative pan‐proteasome subunit inhibition by MRZ. Upon inhibition of CT‐L by all clinical proteasome inhibitors (PIs), the T‐L and C‐L subunits become hyperactive and/or increase in abundance, resulting in continued proteasome‐associated protein degradation, which cannot be inhibited by monospecific PIs. The irreversible binding of marizomib (MRZ) may afford a dual competitive advantage by (i) accomplishing more complete inhibition of CT‐L activity in the face of increased β5 subunit expression, while (ii) subsequently inhibiting the hyper‐activated T‐L and C‐L with repeat dosing.
Using proteasomes purified from rabbit muscle, Kisselev et al (1994) demonstrated dramatic activation of C‐L activity by substrates of the CT‐L sites, by an allosteric mechanism. Subsequently, compensatory upregulation of the activity of the T‐L and C‐L subunits following CT‐L inhibition was described in yeast model systems (Kisselev et al, 2003), MM cells (Altun et al, 2005; Chauhan et al, 2006) and MM xenograft‐bearing mice (Chauhan et al, 2006). Here, we have extended this observation to the clinical setting with MRZ; as MRZ treatment proceeded and the CT‐L subunit became maximally inhibited, increased activity of the T‐L and C‐L subunits was observed, followed by progressively increasing inhibition of these subunits. Of note, the adaptive hyperactivation of C‐L and T‐L subunits in the face of active CT‐L inhibition occurred in both PWB (predominantly anuclear red blood cells) and PBMC, supporting an allosteric interaction model between the different subunits of the 20S proteasome core, as described in a yeast model (Kisselev et al, 2003) rather than de novo resynthesis and overexpression of new proteasomes.
The three proteasomal enzymes exhibit unique substrate specificities – CT‐L cleaves downstream of hydrophobic residues, whereas C‐L and T‐L cleave downstream of acidic and basic residues, respectively – so it is not surprising that most proteins studied are not efficiently protected from degradation by monospecific proteasome inhibitors and that broad spectrum proteasome inhibition more profoundly alters target cell biology. In an elegant series of experiments employing subunit‐specific reagents, Kisselev and co‐workers (Kisselev et al, 2006; Fuchs et al, 2008; Britton et al, 2009; Mirabella et al, 2011) demonstrated that: (i) inhibition of CT‐L activity alone only rescued 11–50% of degradation, adding T‐L inhibition increased this to 40–68%, while blocking all three subunits prevented protein breakdown by 73–91%; (ii) approximately half of MM cell lines tolerated 80–95% specific inhibition of CT‐L activity for 48 h with no loss of viability; and (iii) the pro‐apoptotic activity of BTZ and CFZ in MM cells (but not in normal PBMCs) was enhanced by addition of specific inhibitors of C‐L or T‐L or, most effectively, both. Consistent with the latter point, MRZ has been shown to act synergistically with BTZ in vitro and in vivo (Chauhan et al, 2008).
Although all clinically‐active PIs display robust inhibition of the CT‐L activity, only MRZ has been observed to completely block all CT‐L activity in whole blood, as reported here and elsewhere (Millward et al, 2012; Spencer et al, 2015a, b). In comparison, maximal whole blood CT‐L inhibition in several clinical studies with BTZ was reported to be 65–69% by either the intravenous or subcutaneous routes of administration on a twice‐weekly schedule (Cortes et al, 2004; Dy et al, 2005; Moreau et al, 2008), although sporadic individuals with up to 84% CT‐L reduction have been described (Reece et al, 2011). CFZ is a highly potent, irreversible and specific inhibitor of the CT‐L subunit (Kuhn et al, 2007) and, accordingly, was able to block 75% of CT‐L activity after one dose (O'Connor et al, 2009) in whole blood or PBMCs and up to 80–90% after repeat dosing (Alsina et al, 2012), although the most impressive activity was seen on an unapproved schedule of five daily doses (O'Connor et al, 2009). In the studies reported here, the inhibition of CT‐L and also T‐L and C‐L activity by MRZ was dose‐dependent and comparable across the two dosing regimens (once‐ vs. twice‐weekly IV infusion), as might be expected for an irreversible mechanism of action.
Despite their notable success in MM and some other B‐lineage malignancies, PIs have to date proved relatively ineffective clinically in solid tumours (Dou & Zonder, 2014). MRZ displayed superior activity to BTZ in several solid tumour xenograft models and more potently impacted several hallmarks of cancer, including angiogenesis and invasion (reviewed in Potts et al, 2011), suggesting that a proteasome inhibitor with a broader spectrum of biochemical activity might be more active in solid tumours than those specific to the CT‐L subunit. Key proteasome target proteins in solid tumours are likely to be distinct from those in MM due to dependence on diverse oncogenic signalling pathways and the hypoxia‐driven accumulation of oncoproteins, so the pan‐subunit activity of marizomib may enhance the net ‘load/capacity’ stress (Shabaneh et al, 2013) in solid tumour cells, resulting in increased selective apoptosis of malignant cells. Uniquely among proteasome inhibitors described to date, MRZ crosses the blood‐brain barrier; this, combined with the observation of equivalent inhibition of CT‐L, T‐L and C‐L activity in circulating blood samples from solid tumour and MM patients summarized here, provides rationale for a Phase 1b trial with MRZ in combination with Avastin in malignant glioma which was initiated in 2015 (ClinicalTrials.gov Identifier: NCT02330562).
This is the first report of initial hyperactivation followed by robust inhibition of T‐L and C‐L activity by a PI in the clinic, an attribute that could have important implications for the development of MRZ in MM and other tumour types. All of the clinical‐stage PIs are active in MM, suggesting that inhibition of CT‐L activity alone is sufficient for clinical activity in this disease, however their efficacy is limited due to intrinsic and acquired resistance, the underlying mechanisms of which are poorly understood. While the trials presented here included MM patients previously exposed to PIs, there were no specific inclusion criteria to enroll patients refractory to specific PIs in their most recent regimen. Although data are limited, no differences were apparent in either the C1D1 or ‘peak effect’ of MRZ on proteasome inhibition across the dose range examined in patients refractory to PIs. Studies are in progress to assess the efficacy of MRZ in relapsed and relapsed refractory MM patients treated with MRZ in combination with pomalidomide and dexamethasone (ClinicalTrials.gov Identifier NCT02103335). The predictive power of the proteasomal inhibitory profile elicited by MRZ will be assessed in light of the substantial clinical response rate in this on‐going study (Spencer et al, 2015a, b).
The irreversible mode of action of MRZ, also seen with CFZ (O'Connor et al, 2009), resulted in similar efficacy in PBMCs and erythrocytes (i.e. whole blood), suggesting that the irreversible binding mode of these two newer drugs is able to overcome the re‐synthesis of proteasome subunits that occurs in nucleated cells. Taken together, the data suggest that MRZ may exert superior clinical activity in comparison with other clinical proteasome inhibitors due to their reversible binding mode of action (BTZ, ixazomib) or monospecificity for the CT‐L site (CFZ, oprozomib). In conclusion, we report for the first time the phenomenon of compensatory hyperactivation of the C‐L and T‐L proteasome subunits during the process of effective inhibition of the CT‐L activity in patients with MM and solid tumours. Detailed analyses of the clinical pharmacodynamics of MRZ indicate that this pan‐subunit, irreversible PI is able to overcome this physiological response and cumulatively block all three proteasome activities.
Author contributions
NL and FJB analysed data and wrote the manuscript; AS, DC, SDR, and MT interpreted data and provided critical review of the data and manuscript; AS, SJH, KCA, and PR provided clinical samples and critical review of the manuscript.
Disclosure of conflicts of interest
Levin: Employee of Triphase Accelerator Corp. Spencer: Celgene Corporation, Honoraria and Research Funding. Harrison: No disclosures. Chauhan: Consultant for Triphase Accelerator Corp. Burrows: Consultant for Triphase Accelerator Corp. Anderson: Bristol‐Myers Squibb Pharmaceuticals, Celgene Corporation, Gilead Pharmaceuticals, Millenium (The Takeda Oncology Company): Advisor Board. Acetylon Pharmaceutcials, OncoPep, Inc: Scientific Founder. Reich: Consultant for Triphase Accelerator Corp. Richardson: Celgene and Millenium (The Takeda Oncology Company); Service on Advisory Committees, Research Funding. Trikha: Employee of Triphase Accelerator Corp.
Acknowledgements
The diligent efforts of G. Kenneth Lloyd, Ph.D. and Natasha Reddinger in executing the pharmacodynamic sample assessments are gratefully acknowledged, as is critical review of the manuscript by Ann MacLaren, Ph.D. and review of the data by Karl Cremer, PharmD.
References
- Alsina, M. , Trudel, S. , Furman, R.R. , Rosen, P.J. , O'Connor, O.A. , Comenzo, R.L. , Wong, A. , Kunkel, L.A. , Molineaux, C.J. & Goy, A. (2012) A phase I single‐agent study of twice‐weekly consecutive‐day dosing of the proteasome inhibitor carfilzomib in patients with relapsed or refractory multiple myeloma or lymphoma. Clinical Cancer Research, 18, 4830–4840. [DOI] [PubMed] [Google Scholar]
- Altun, M. , Galardy, P.J. , Shringarpure, R. , Hideshima, T. , LeBlanc, R. , Anderson, K.C. , Ploegh, H.L. & Kessler, B.M. (2005) Effects of PS‐341 on the activity and composition of proteasomes in multiple myeloma cells. Cancer Research, 65, 7896–7901. [DOI] [PubMed] [Google Scholar]
- Atrash, S. , Tullos, A. , Panozzo, S. , Bhutani, M. , Van Rhee, F. , Barlogie, B. & Usmani, S.Z. (2015) Cardiac complications in relapsed and refractory multiple myeloma patients treated with carfilzomib. Blood Cancer Journal, 5, e272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Britton, M. , Lucas, M.M. , Downey, S.L. , Screen, M. , Pletnev, A.A. , Verdoes, M. , Tokhunts, R.A. , Amir, O. , Goddard, A.L. , Pelphrey, P.M. , Wright, D.L. , Overkleeft, H.S. & Kisselev, A.F. (2009) Selective inhibitor of proteasome's caspase‐like sites sensitizes cells to specific inhibition of chymotrypsin‐like sites. Cell Chemistry & Biology, 16, 1278–1289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cai, X. , Bhattacharyya, S. , Plitt, A. , Raibagkar, R. , LaBuzetta, J.N. , Schleicher, S.M. , Munshi, N.D. & Kleain, J.P. (2014) Management of posterior reversible encephalopathy syndrome induced by carfilzomib in a patient with multiple myeloma. Journal of Clinical Oncology, 34, e1–e5. [DOI] [PubMed] [Google Scholar]
- Chauhan, D. , Catley, L. , Li, G. , Podar, K. , Hideshima, T. , Velankar, M. , Mitsiades, C. , Mitsiades, N. , Yasui, H. , Letai, A. , Ovaa, H. , Berkers, C. , Nicholson, B. , Chao, T.H. , Neuteboom, S.T. , Richardson, P. , Palladino, M.A. & Anderson, K.C. (2005) A novel orally active proteasome inhibitor induces apoptosis in multiple myeloma cells with mechanisms distinct from Bortezomib. Cancer Cell, 8, 407–419. [DOI] [PubMed] [Google Scholar]
- Chauhan, D. , Hideshima, T. & Anderson, K.C. (2006) A novel proteasome inhibitor NPI‐0052 as an anticancer therapy. British Journal of Cancer, 95, 961–965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chauhan, D. , Singh, A. , Brahmandam, M. , Podar, K. , Hideshima, T. , Richardson, P. , Munchi, N. , Palladino, M.A. & Anderson, K.C. (2008) Combination of proteasome inhibitors bortezomib and NPI‐0052 trigger in vivo synergistic cytotoxicity in multiple myeloma. Blood, 111, 1654–1664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cortes, J. , Thomas, D. , Koller, C. , Giles, F. , Estey, E. , Faderl, S. , Garcia‐Manero, G. , McConkey, D. , Ruiz, S.L. , Guerciolini, R. , Wright, J. & Kantarjian, H. (2004) Phase I study of bortezomib in refractory or relapsed acute leukemias. Clinical Cancer Research, 10, 3371–3376. [DOI] [PubMed] [Google Scholar]
- Dou, Q.P. & Zonder, J.A. (2014) Overview of proteasome inhibitor‐based anti‐cancer therapies: perspective on bortezomib and second generation proteasome inhibitors versus future generation inhibitors of ubiquitin‐proteasome system. Current Cancer Drug Targets, 14, 517–536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dy, G.K. , Thomas, J.P. , Wilding, G. , Bruzek, L. , Mandrekar, S. , Erlichman, C. , Alberti, D. , Binger, K. , Pitot, H.C. , Alberts, S.R. , Hanson, L.J. , Marnocha, R. , Tutsch, K. , Kaufmann, S.H. & Adjei, A.A. (2005) A phase I and pharmacologic trial of two schedules of the proteasome inhibitor, PS‐341 (bortezomib, velcade), in patients with advanced cancer. Clinical Cancer Research, 11, 3410–3416. [DOI] [PubMed] [Google Scholar]
- Fall, D.J. , Stessman, H. , Patel, S.S. , Sachs, Z. , Van Ness, B.G. , Baughn, L.B. & Linden, M.A. (2014) Utilization of translational bioinformatics to identify novel biomarkers of bortezomib resistance in multiple myeloma. Journal of Cancer, 5, 720–727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fenical, W. , Jensen, P.R. , Palladino, M.A. , Lam, K.S. , Lloyd, G.K. & Potts, B.C. (2009) Discovery and development of the anticancer agent salinosporamide A (NPI‐0052). Bioorganic & Medicinal Chemistry, 17, 2175–2180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Franke, N.E. , Niewerth, D. , Assaraf, Y.G. , van Meerloo, J. , Vojtekova, K. , van Zantwijk, C.H. , Zweegman, S. , Chan, E.T. , Kirk, C.J. , Geerke, D.P. , Schimmer, A.D. , Kaspers, G.J. , Jansen, G. & Cloos, J. (2012) Impaired bortezomib binding to mutant β5 subunit of the proteasome is the underlying basis for bortezomib resistance in leukemia cells. Leukemia, 26, 757–768. [DOI] [PubMed] [Google Scholar]
- Fuchs, D. , Berges, C. , Opelz, G. , Daniel, V. & Naujokat, C. (2008) Increased expression and altered subunit composition of proteasomes induced by continuous proteasome inhibition establish apoptosis resistance and hyperproliferation of Burkitt lymphoma cells. Journal of Cellular Biochemistry, 103, 270–283. [DOI] [PubMed] [Google Scholar]
- Groll, M. , Huber, R. & Potts, B.C. (2006) Crystal structures of salinosporamide A (npi‐0052) and b (npi‐0047) in complex with the 20s proteasome reveal important consequences of beta‐lactone ring opening and a mechanism for irreversible binding. Journal of the American Chemical Society, 128, 5136–5141. [DOI] [PubMed] [Google Scholar]
- Harrison, S.J. , Mainwaring, P. , Price, T. , Millward, M.J. , Padrik, P. , Underhill, C.R. , Cannell, P.K. , Reich, S.D. , Trikha, M. & Spencer, A. (2016) Phase 1 clinical trial of marizomib (NPI‐0052) in patients with advanced malignancies including multiple myeloma: study NPI‐0052‐102 final results. Clinical Cancer Research, In press. [DOI] [PubMed] [Google Scholar]
- Harvey, R.D. (2014) Incidence and management of adverse events in patients with relapsed and/or refractory multiple myeloma receiving single‐agent carfilzomib. Clinical Pharmacology : Advances and Applications, 2014, 87–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huber, E.M. , Heinemeyer, W. & Groll, M. (2015) Bortezomib‐resistant mutant proteasomes: structural and biochemical evaluation with carfilzomib and ONX 0914. Structure, 23, 407–417. [DOI] [PubMed] [Google Scholar]
- Kale, A.J. & Moore, B.S. (2012) The molecular mechanisms of acquired proteasome inhibitor resistance. Journal of Medicinal Chemistry, 55, 10317–10327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kane, R.C. , Bross, P.F. , Farrell, A.T. & Pazdur, R. (2003) Velcade: U.S. FDA approval for the treatment of multiple myeloma progressing on prior therapy. Oncologist, 8, 508–513. [DOI] [PubMed] [Google Scholar]
- Kisselev, A.F. , Akopian, T.N. , Castillo, V. & Goldberg, A.L. (1994) Proteasome active sites allosterically regulate each other, suggesting a cyclical bite‐chew mechanism for protein breakdown. Molecular Cell, 4, 395–402. [DOI] [PubMed] [Google Scholar]
- Kisselev, A.F. , Garcia‐Calvo, M. , Overkleeft, H.S. , Peterson, E. , Pennington, M.W. , Ploegh, H.L. , Thornberry, N.A. & Goldberg, A.L. (2003) The caspase‐like sites of proteasomes, their substrate specificity, new inhibitors and substrates, and allosteric interactions with the trypsin‐like sites. Journal of Biological Chemistry, 278, 35869–35877. [DOI] [PubMed] [Google Scholar]
- Kisselev, A.F. , Callard, A. & Goldberg, A.L. (2006) Importance of the different proteolytic sites of the proteasome and the efficacy of inhibitors varies with the protein substrate. Journal of Biological Chemistry, 281, 8582–8590. [DOI] [PubMed] [Google Scholar]
- Kuhn, D.J. , Chen, Q. , Voorhees, P.M. , Strader, J.S. , Shenk, K.D. , Sun, C.M. , Demo, S.D. , Bennett, M.K. , van Leeuwen, F.W.B. , Chanan‐Khan, A.A. & Orlowski, R.Z. (2007) Potent activity of carfilzomib, a novel, irreversible inhibitor of the ubiquitin‐proteasome pathway, against preclinical models of multiple myeloma. Cancer Chemotherapy and Pharmacology, 110, 3281–3290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar, S.K. , Lee, J.H. , Lahuerta, J.J. , Morgan, G. , Richardson, P.G. , Crowley, J. , Haessler, J. , Feather, J. , Hoering, A. , Moreau, P. , LeLeu, X. , Hullin, C. , Kleain, S.K. , Sonneveld, P. , Siegel, D. , Blade, J. , Goldschmidt, H. , Jagannath, S. , San Miguel, J. , Orlowski, R. , Palumbo, A. , SEzer, O. & Durie, B.G.M. , on behalf of the International Myeloma Working Group . (2012) Risk of progression and survival in multiple myeloma relapsing after therapy with IMiDs and bortezomib: a multicenter international myeloma working group study. Leukemia, 26, 149–157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lightcap, E.S. , McCormack, T.A. , Pien, C.S. , Chau, V. , Adams, J. & Elliott, P.J. (2000) Proteasome inhibition measurements: clinical application. Clinical Chemistry, 46, 673–683. [PubMed] [Google Scholar]
- Lonial, S. , Waller, E.K. , Richardson, P.G. , Jagannath, S. , Orlowski, R.Z. , Giver, C.R. , Jaye, D.L. , Francis, D. , Giusti, S. , Torre, C. , Barlogie, B. , Berenson, J.R. , Singhal, S. , Schenkein, D.P. , Esseltine, D.L. , Anderson, J. , Xiao, H. , Heffner, L.T. & Anderson, K.C. (2005) SUMMIT/CREST Investigators. Risk factors and kinetics of thrombocytopenia associated with bortezomib for relapsed, refractory multiple myeloma. Blood, 106, 3777–3784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Millward, M. , Price, T. , Townsend, A. , Sweeney, C. , Spencer, A. , Sukumaran, S. , Longenecker, A. , Lee, L. , Lay, A. , Sharma, G. , Gemmill, R.M. , Drabkin, H.A. , Lloyd, G.K. , Neuteboom, S.T. , McConkey, D.J. , Palladino, M.A. & Spear, M.A. (2012) Phase 1 clinical trial of the novel proteasome inhibitor marizomib with the histone deacetylase inhibitor vorinostat in patients with melanoma, pancreatic and lung cancer based on in vitro assessments of the combination. Investigational New Drugs, 30, 2303–2317. [DOI] [PubMed] [Google Scholar]
- Mirabella, A.C. , Pletnev, A.A. , Downey, S.L. , Florea, B.I. , Shabaneh, T.B. , Britton, M. , Verdoes, M. , Filippov, D.V. , Overkleen, H.S. & Kisselev, A.F. (2011) Specific cell‐permeable inhibitor of proteasome trypsin‐like sites selectivity sensitizes myeloma cells to bortezomib and carfilzomib. Chemistry & Biology, 18, 608–618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moreau, P. , Coiteux, V. , Hulin, C. , Leleu, X. , van de Velde, H. , Acharya, M. & Harousseau, J.‐L. (2008) Prospective comparison of subcutaneous versus intravenous administration of bortezomib in patients with multiple myeloma. Haematologica, 93, 1908–1911. [DOI] [PubMed] [Google Scholar]
- Niewerth, C. , Franke, N.E. , Janesen, G. , Assaraf, Y.G. , vanMeerloo, J. , Kirk, C.J. , Degenhardt, J. , Anderl, J.L. , Schimmer, A.D. , deHaas, V. , Horton, T.M. , Zweegman, S. , Kaspers, G.J.L. & Cloos, J. (2013) Higher ratio immune vs. constitutive proteasome level as novel indicator of sensitivity of pediatric acute leukemia cells to proteasome inhibitors. Haematologica, 98, 1896–1904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niewerth, D. , Jansen, G. , Assaraf, Y.G. , Zweegman, S. , Kaspers, G.J. & Cloos, J. (2015) Molecular basis of resistance to proteasome inhibitors in hematological malignancies. Drug Resistance Updates : Reviews and Commentaries in Antimicrobial and Anticancer Chemotherapy, 18, 18–35. [DOI] [PubMed] [Google Scholar]
- O'Connor, O.A. , Stewart, A.K. , Vallone, M. , Molineaux, C.J. , Kunkel, L.A. , Gerecitano, J.F. & Orlowski, R.Z. (2009) A phase 1 dose escalation study of the safety and pharmacokinetics of the novel proteasome inhibitor carfilzomib (PR‐171) in patients with hematologic malignancies. Clinical Cancer Research, 15, 7085–7091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orlowski, R.Z. (2013) Novel agents for multiple myeloma to overcome resistance in phase III clinical trials. Seminars in Oncology, 40, 1–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Potts, B.C. , Albitar, M.X. , Anderson, K.C. , Baritaki, S. , Berkers, C. , Bonavida, B. , Chandra, J. , Chauhan, D. , Cusack, J.C. , Fenical, W. , Ghobrial, I.M. , Groll, M. , Jensen, P.R. , Lam, K.S. , Lloyd, G.K. , McBride, W. , McConkey, D.J. , Miller, C.P. , Neuteboom, S.T.C. , Oki, Y. , Ovaa, H. , Pajonk, F. , Richardson, P.G. , Roccaro, A.M. , Sloss, C.M. , Spear, M.A. , Valashi, E. , Younes, A. & Palladino, M.A. (2011) Marizomib, a proteasome inhibitor for all seasons: preclinical profile and a framework for clinical trials. Current Cancer Drug Targets, 11, 254–284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reece, D.E. , Sullivan, D. , Lonial, S. , Mohrbacher, A.F. , Chatta, G. , Shustik, C. , Burris, H. , Venkatakrishnan, K. , Neuwirth, R. , Riordan, W.J. , Karol, M. , van Moltke, L.L. , Acharya, M. , Zannikos, P. & Stewart, A.K. (2011) Pharmacokinetic and pharmacodynamics study of two doses of bortezomib in patients with relapsed multiple myeloma. Cancer Chemotherapy and Pharmacology, 67, 57–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richardson, P. , Barlogie, B. , Berenson, J. , Singhal, S. , Jagannath, S. , Irwin, D. , Rajkumar, S.V. , Srkalovic, G. , Alsina, M. , Alexanian, R. , Siegel, D. , Orlowski, R.Z. , Kuter, D. , Limentani, S.A. , Lee, S. , Hideshima, T. , Esseltine, D.‐L. , Kauffman, M. , Adams, J. , Schenkein, D.P. & Anderson, K.C. (2003) A multicenter phase II multicenter study of Bortezomib in patients with relapsed and refractory multiple myeloma. New England Journal of Medicine, 348, 2609–2617. [DOI] [PubMed] [Google Scholar]
- Richardson, P.G. , Sonneveld, P. , Schuster, M.W. , Irwin, D. , Stadtmauer, E.A. , Facon, T. , Harousseau, J.‐L. , Ben‐Yehuda, D. , Lonial, S. , Goldschmidt, H. , Reece, D. , San‐Miguel, J.F. , Blade, J. , Boccadoro, M. , Cavehnagh, J. , Dalton, W.S. , Boral, A.L. , Esseltine, D.L. , Porter, J.B. , Schenkein, D. & Anderson, K.C. (2005) Bortezomib or high‐dose dexamethasone for relapsed multiple myeloma. New England Journal of Medicine, 352, 2487–2498. [DOI] [PubMed] [Google Scholar]
- Richardson, P.G. , Briemberg, H. , Jagannath, S. , Wen, P.Y. , Barlogie, B. , Berenson, J. , Singhal, S. , Siegel, D.S. , Irwin, D. , Schuster, M. , Srkalovic, G. , Alexanian, R. , Rajkumar, S.V. , Limentani, S. , ALsina, M. , Orlowski, R.Z. , Najarian, K. , Esseltine, D. , Anderson, K.C. & Amato, A.A. (2006) Frequency, characteristics, and reversibility of peripheral neuropathy during treatment of advanced multiple myeloma with bortezomib. Journal of Clinical Oncology, 24, 3113–3120. [DOI] [PubMed] [Google Scholar]
- Richardson, P.G. , Zimmerman, T.M. , Hofmeister, C.C. , Talpaz, M. , Chanan‐Khan, A.A. , Kaufman, J.L. , Laubach, J.P. , Chauhan, D. , Jakubowiak, A.J. , Reich, S. , Trikha, M. & Anderson, K.C. (2016) Phase 1 study of marizomib in relapsed or relapsed and refractory multiple myeloma; NPI‐0052‐101 part 1. Blood, 2016 Mar 23. doi: 10.1182/blood‐2015‐12‐686378. [Epub ahead of print]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruckrich, T. , Kraus, M. , Gogel, J. , Beck, A. , Ovaa, H. , Verdoes, M. , Overkleeft, H.S. , Kalbacher, H. & Driessen, C. (2009) Characterization of the ubiquitin‐proteasome system in bortezomib‐adapted cells. Leukemia, 23, 1098–1105. [DOI] [PubMed] [Google Scholar]
- Shabaneh, T.B. , Downey, S.L. , Goddard, A.L. , Screen, M. , Lucas, M.M. , Eastman, A. & Kisselev, A.F. (2013) Molecular basis of differential sensitivity of myeloma cells to clinically relevant bolus treatment with bortezomib. PLoS ONE, 8, e56132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh, A.V. , Palladino, M.A. , Lloyd, G.K. , Potts, B. , Chauhan, D. & Anderson, K.C. (2010) Pharmacodynamic and efficacy studies of the novel proteasome inhibitor NPI‐0052 (marizomib) in a human plasmacytoma xenograft murine model. British Journal of Haematology, 149, 550–559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spencer, A. , Badros, A. , Laubach, J. , Harrison, S. , Zonder, J. , Khot, A. , Chauhan, D. , Anderson, K. , Reich, S. , Trikha, M. & Richardson, P. (2015a) Phase 1, multicenter, open‐label, dose‐escalation, combination study (NCT02103335) of pomalidomide (POM), marizomib (MRZ, NPI‐0052), and dexamethasone (DEX) in patients with relapsed and refractory multiple myeloma (MM); Study NPI‐0052‐107 Preliminary Results. Clinical Lymphoma, Myeloma & Leukemia, 15 (Suppl. 3), e43, abstract 0146. [Google Scholar]
- Spencer, A. , Laubach, J. , Zonder, J. , Badros, A. , Harrison, S. , Khot, A. , Chauhan, D. , Anderson, K. , Reich, S.D. , Trikha, M. & Richardson, P. (2015b) Phase 1, multicenter, open‐label, combination study (NPI‐0052‐107; NCT02103335) of pomalidomide (POM), marizomib (MRZ, NPI‐0052), and low‐dose dexamethasone (lo‐DEX) in patients with relapsed and refractory multiple myeloma. Blood (ASH Annual Meeting Abstracts), 126, 4220. [Google Scholar]
- Teicher, B.A. & Tomaszewski, J.E. (2015) Proteasome inhibitors. Biochemical Pharmacology., 96, 1–9. [DOI] [PubMed] [Google Scholar]
- Wanchoo, R. , Khan, S. , Kolitz, J.E. & Jhaveri, K.D. (2015) Carfilzomib‐related acute kidney injury may be prevented by N‐acetyl‐L‐cysteine. Journal of Oncology Pharmacy Practice: Official Publication of The International Society of Oncology Pharmacy Practitioners, 21, 313–316. [DOI] [PubMed] [Google Scholar]