

Abstract
Taliglucerase alfa is the first available plant cell‐expressed human recombinant therapeutic protein. It is indicated for treatment of patients with type 1 Gaucher disease (GD) in adult and pediatric patients in several countries. Study PB‐06‐002 examined the safety and efficacy of taliglucerase alfa for 9 months in patients who previously received imiglucerase. The results of adult patients from Study PB‐06‐002 who continued receiving taliglucerase alfa in extension Study PB‐06‐003 for up to 36 months are reported here. Eighteen patients received at least one dose of taliglucerase alfa in Study PB‐06‐003; 10 patients completed 36 total months of therapy, and four patients who transitioned to commercial drug completed 30–33 months of treatment. In patients who completed 36 total months of treatment, mean percent (±standard error) changes from baseline/time of switch to taliglucerase alfa to 36 months were as follows: hemoglobin concentration, −1.0% (±1.9%; n = 10); platelet count, +9.3% (±9.8%; n = 10); spleen volume measured in multiples of normal (MN), −19.8% (±9.9%; n = 7); liver volume measured in MN, +0.9% (±5.4%; n = 8); chitotriosidase activity, −51.5% (±8.1%; n = 10); and CCL18 concentration, −36.5 (±8.0%; n = 10). Four patients developed antidrug antibodies, including one with evidence of neutralizing activity in vitro. All treatment‐related adverse events were mild or moderate and transient. The 36‐month results of switching from imiglucerase to taliglucerase alfa treatment in adults with GD provide further data on the clinical safety and efficacy of taliglucerase alfa beyond the initial 9 months of the original study. www.clinicaltrials.gov identifier NCT00705939. Am. J. Hematol. 91:661–665, 2016. © 2016 Wiley Periodicals, Inc.
Introduction
Gaucher disease (GD), caused by acid beta‐glucosidase deficiency, results in the accumulation of glucocerebroside in macrophages and other cell types leading to splenomegaly, hepatomegaly, anemia, and thrombocytopenia 1, 2. For patients with type 1 (nonneuronopathic) GD, enzyme replacement therapy (ERT) has been the standard of care for more than 20 years 3.
The necessity of additional ERT options for patients with GD became apparent during the shortage of imiglucerase in 2009 4, 5. Taliglucerase alfa is the first FDA‐approved plant cell‐expressed human recombinant therapeutic protein 6 and is indicated for treatment of adults with type 1 GD in the United States, Israel, Brazil, Australia, Canada, Chile, and other countries; it is also approved for treatment of children in the United States, Australia, Canada, Israel, Mexico, and other countries, and for hematologic manifestations in pediatric patients with type 3 GD in Canada. The taliglucerase alfa production system offers the ability for rapid scale‐up and is not susceptible to contamination with mammalian pathogens 7. The current report expands the database of the long‐term safety and efficacy of taliglucerase alfa in patients who have been previously treated with imiglucerase.
Study PB‐06‐002 (NCT00712348) 8 was a phase III, 9‐month, multicenter, open‐label, switchover trial of taliglucerase alfa in adult and pediatric patients with GD who had achieved disease stability with imiglucerase. Patients were switched from imiglucerase to the same dose of taliglucerase alfa for 9 months 9. Hemoglobin concentration, platelet count, spleen volume, and liver volume remained unchanged and treatment‐related adverse events (AEs) were mild or moderate in severity and transient in nature 9. Adult patients who completed Study PB‐06‐002 were eligible to participate in extension Study PB‐06‐003 (NCT00705939) 8, 10. The objective of the present analysis was to continue to follow the efficacy and safety of taliglucerase alfa through 36 total months of treatment.
Patients and Methods
Study design
This analysis evaluated the efficacy and safety of taliglucerase alfa treatment for adult patients with stable GD in the 9‐month, phase III, multicenter, open‐label Study PB‐06‐002 (pivotal “switchover study”) and its extension study PB‐06‐003, which included patients previously treated with imiglucerase. The design of “switchover study” PB‐06‐002 has been published 9; it incorporated a prospective 12‐week GD stability evaluation period for patients while on imiglucerase. Enrollment had been open during the period of imiglucerase shortage and, therefore, historical data were used to enable immediate treatment with taliglucerase alfa in eligible patients whose treatments were at risk for interruption. In the present report, data were analyzed from patients who completed Study PB‐06‐002 (9 months of treatment) and continued in Study PB‐06‐003 (27 months of treatment) for a total duration of up to 36 months of taliglucerase alfa treatment.
The study protocol and the informed consent form were reviewed and approved by the ethics committees/institutional review boards (IRBs) for each study site; protocol amendments were approved by the IRBs prior to implementation. This study was conducted in accordance with the International Conference on Harmonisation Good Clinical Practice guidelines and all local regulations. Written informed consent was obtained for all patients.
Patients
The main inclusion criteria were completion of Study PB‐06‐002 and written informed consent to continue in Study PB‐06‐003. Inclusion criteria for Study PB‐06‐002 included age ≥2 years, treatment with imiglucerase for ≥2 years, and being on a stable dose of imiglucerase for at least 6 months preceding study enrollment. Patients were also required to have stable GD determined during the 12‐week stability evaluation period for all patients except for those affected by the 2009 imiglucerase shortage, for whom historical data were used. Disease stability was based on criteria that implicated no clinically meaningful variation in laboratory values of hemoglobin and platelet counts or in liver and spleen volumes (see Supporting Information).
The main exclusion criteria from Study PB‐06‐002 affecting patients in this analysis were a previous infusion reaction to imiglucerase or alglucerase; presence of human immunodeficiency virus; hepatitis B surface antigen and/or hepatitis C infections; and unresolved anemia due to iron, folic acid, or vitamin B12 deficiency. Exclusion criteria for Study PB‐06‐003 included taking another experimental medication (for any disease), and presence of any condition that would interfere with compliance with study requirements as judged by the investigator.
Study treatment
Taliglucerase alfa was administered by intravenous infusion at 2‐week intervals. Patients started receiving taliglucerase alfa in Study PB‐06‐002 at the dosage equivalent to the patient's imiglucerase dose at screening or prior to the imiglucerase shortage. In Study PB‐06‐003, each patient continued treatment with the same dose of taliglucerase alfa the patient was receiving at the completion of Study PB‐06‐002. If a clinically relevant deterioration occurred in either Study PB‐06‐002 or Study PB‐06‐003, the dose for that patient could be increased up to 60 U/kg or treatment could be discontinued as previously described 9. Patients could exit the study upon marketing approval in their country and transition to a commercially available drug.
Assessments
Efficacy evaluations included hemoglobin concentration, platelet count, spleen volume, liver volume, and biomarkers (chitotriosidase activity and chemokine [C‐C motif] ligand 18 [CCL18] concentration). Organ volumes were determined via magnetic resonance imaging using a validated system as previously described with two readers blinded to treatment group, patient number, and sequence 11, 12. Two patients were unable to tolerate magnetic resonance imaging procedures and were excluded from the organ volume analyses; their organ volumes were monitored using ultrasonography. Another patient had a pre‐existing splenectomy and was excluded from the spleen volume analysis. Safety was assessed as previously described 9.
Statistical analysis
Descriptive statistics for continuous variables, sample size (n), mean and its standard error (SE), standard deviation, median, and range were calculated for hemoglobin concentration, platelet count, spleen volume, liver volume, and change in biomarker activity. Spleen and liver volumes were calculated as multiples of normal (MN) where normal spleen volume is 2 mL/kg of body weight and normal liver volume is 25 mL/kg of body weight. For categorical variables, count and percentages are presented. Time points were counted continuously from baseline at the start of Study PB‐06‐002 through Study PB‐06‐003. No inferential statistics were performed for testing the change from baseline and/or for comparing between or among treatment groups because of the small number of patients. For each parameter, efficacy analyses included those patients for whom data were available at each time point. The safety population included all patients originally enrolled in Study PB‐06‐002 who received the study drug in Study PB‐06‐003 (n = 18).
Results
Study patients
Patient disposition is provided in Supporting Information Fig. S1. A total of 19 adult patients from Study PB‐06‐002 were enrolled in extension Study PB‐06‐003. One patient was noncompliant after signing the informed consent form and was excluded from the study prior to receiving the study drug. A total of 18 patients received treatment in Study PB‐06‐003. Four patients discontinued the study for different reasons: (1) move to the compassionate use program for taliglucerase alfa after 28 infusions; (2) withdrew to pursue another clinical study after 25 infusions; (3) dissatisfaction with the results of treatment after 15 infusions despite lack of objective parameters of deterioration; and (4) protocol violation (refusal to return to all end‐of‐study visits) after 44 infusions. During this study, marketing approval for taliglucerase alfa occurred in the countries of four additional patients and, per protocol, these four patients exited the study and continued treatment with commercially available taliglucerase alfa. Treatment for these latter patients ranged from 30 to 33 total months across Studies PB‐06‐002 and PB‐06‐003.
Patients received taliglucerase alfa at the same dose as imiglucerase. The mean (standard error [±SE]) taliglucerase alfa dose was 32.4 (±3.9) U/kg (range, 12.0–59.0 U/kg) in the population of all patients who received taliglucerase alfa in the extension study (n = 18), with four patients receiving ≤15 U/kg, seven patients receiving >15 U/kg to ≤30 U/kg, and seven patients receiving >30 U/kg.
Baseline patient demographics and disease characteristics are shown in Supporting Information Tables SI and SII. All of the patients were Caucasian, half were male, and approximately two thirds were of Ashkenazi Jewish ethnicity. A similar demographic profile was observed in the population of 10 patients who had completed 36 months of treatment.
Efficacy results
Individual and mean efficacy parameters are shown in Figs. 1, 2, 3; detailed individual patient efficacy data can be found in Supporting Information Tables SIII and SIV.
Figure 1.
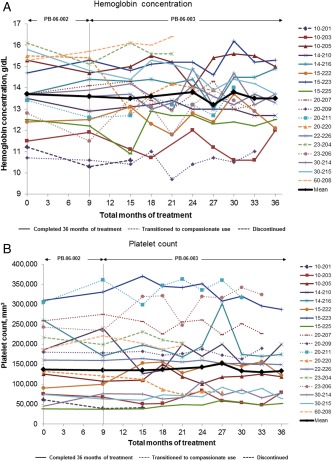
(A) Hemoglobin concentration and (B) platelet count during long‐term treatment with taliglucerase alfa. Values are shown for each patient from baseline to last observation or through 36 total months of treatment. Mean values represent the patients who completed 36 months of treatment (n = 10).
Figure 2.
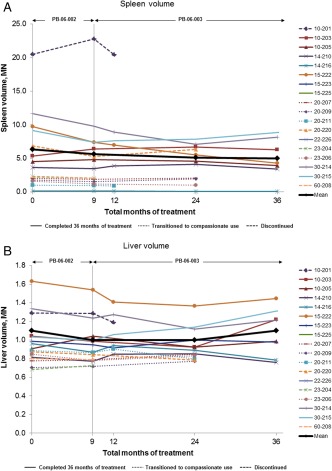
(A) Spleen volume, expressed as multiples of normal (MN), where normal spleen volume is 2 mL/kg of body weight (kg), and (B) liver volume, expressed as MN, where normal liver volume is 25 mL/kg of body weight (kg), during long‐term treatment with taliglucerase alfa. Values are shown for each patient from baseline to last observation or through 36 total months of treatment. One patient was splenectomized and not included in spleen volume analysis; two patients were unable to tolerate MRI procedures and were not included in spleen volume or liver volume analyses but were followed by ultrasound. Mean values represent the patients with available data who completed 36 months of treatment (spleen, n = 7; liver, n = 8). MRI, magnetic resonance imaging.
Figure 3.
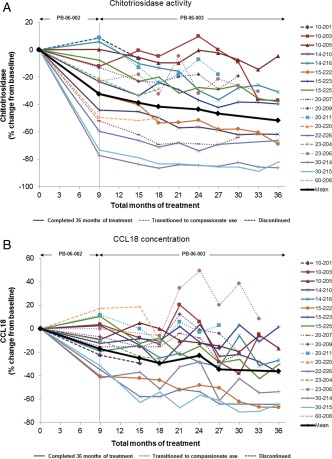
Percentage change in (A) chitotriosidase activity and (B) CCL18 concentration during long‐term treatment with taliglucerase alfa. Values are shown for each patient from baseline to last observation or through 36 total months of treatment. Mean values represent the patients who completed 36 months of treatment (n = 10).
Mean hemoglobin concentration was unchanged through 36 months of treatment with taliglucerase alfa (Fig. 1A). For the 10 patients who completed 36 months of treatment, mean absolute change (±SE) in hemoglobin concentration from baseline to 36 months was −0.2 (±0.3) g/dL, which represented a mean percentage change of −1.0% (±1.9%). Although some variations were observed within the 36 months of therapy for individual patients, hemoglobin concentration for each tended to be similar at month 36 compared with that at baseline (Fig. 1A).
Mean platelet count was unchanged through 36 months of treatment with taliglucerase alfa (Fig. 1B). For most of the 10 patients who completed 36 months of taliglucerase alfa treatment, platelet counts remained at similar levels at month 36 to those during imiglucerase treatment. The mean absolute change (±SE) in platelet count from baseline to 36 months was −3,800/mm3 (±10,072/mm3), which represented a nominal change.
Spleen and liver volumes were unchanged or decreased through 36 months of treatment with taliglucerase alfa (Fig. 2). For the patients with available data who completed 36 months of treatment (n = 7), mean absolute change (±SE) in spleen volume from baseline to 36 months was −1.3 (±0.9) MN, which represented a mean percentage change in volume (measured in MN) of −19.8% (±9.9%). There was no change in mean liver volume (measured in MN) across the patients with available data who completed 36 months of treatment. Most of the patients (n = 8) maintained a liver volume <1.5 MN, consistent with the therapeutic goal for hepatomegaly 13.
Chitotriosidase decreased by last observation for all patients assessed (Fig. 3A). Mean chitotriosidase activity (±SE) was 12,206 (±4,934) nmol/mL·hr at baseline and 6,551 (±3,018) nmol/mL·hr at 36 months (n = 10). This represented a mean (±SE) decrease in chitotriosidase activity of 51.5% (±8.1%) from baseline to 36 months. CCL18 decreased by last observation for most patients (Fig. 3B). One patient (23‐206) had a higher CCL18 level at the end of the study than at baseline but had lower chitotriosidase activity at study end. Mean CCL18 concentration (±SE) was 447.3 (±99.0) ng/mL at baseline; mean levels changed by −36.5% (±8.0%) from baseline to month 36 (n = 10).
Safety
Overall AE results for the safety population (n = 18) are listed in Table 1. Almost all AEs were mild to moderate in severity and transient. Serious AEs were reported in three patients, but none were considered to be treatment‐related. One patient (14‐216) had right gonarthrosis at baseline and developed left knee pain during the study; both knees were replaced with prostheses during the study. Another patient (20‐220) underwent renal stone removal and a third patient (15‐222) had traumatic rib fracture and pneumothorax.
Table 1.
Adverse Events in the Safety Population (n = 18)a
| Adverse events | No. of adverse events (n) | Percentage of total adverse events (percentage of patients) |
|---|---|---|
| Total | 136 (17) | 100 (94.4) |
| Mild or moderate in severity | 134 (17) | 98.5 (94.4) |
| Severe or very severe | 2 (2) | 1.5 (11.1) |
| Non–treatment‐related | 133 (17) | 97.8 (94.4) |
| Treatment‐related | 3 (1) | 2.2 (5.6) |
The most common adverse events were nasopharyngitis (n = 7), pyrexia (n = 4), arthralgia (n = 4), diarrhea (n = 3), vomiting (n = 3), upper respiratory tract infection (n = 3), cough (n = 3), and musculoskeletal pain (n = 3).
Antidrug antibodies
Four patients (14‐210, 20‐211, 20‐220, and 22‐226) tested positive for the presence of anti‐taliglucerase alfa IgG antibodies on at least one visit. Patient 14‐210 tested positive at months 3–30 with a maximal IgG titer at month 27, patient 20‐211 tested positive at months 18–27 with a maximal IgG titer at month 18, and patient 22‐226 tested positive at months 3, 6, and 18 with a maximal IgG titer at month 3. Patient 20‐220 tested positive at months 6–18 with a maximal IgG titer at month 18 and was found to have neutralizing antibodies between months 6 and 18 based on an in vitro enzymatic activity assay but was negative in a cell‐based assay. The other three patients tested negative for neutralizing activity. Disease parameters for these patients at baseline and last observation are available in Supporting Information Tables SIII and SIV. The significance of these findings is unclear at this time.
Discussion
In the present report of adult patients with type 1 GD receiving taliglucerase alfa following previous treatment with imiglucerase, hemoglobin concentration, platelet count, and liver volume remained unchanged while spleen volume remained unchanged or improved after a total of up to 36 months of treatment. Reductions were observed in the biomarkers chitotriosidase and CCL18. These findings indicate that the clinical stability of GD parameters previously reported in the first 9 months of treatment with taliglucerase alfa following previous treatment with imiglucerase 9 was sustained during longer‐term follow‐up in the present study. The AE findings did not present any new safety issues. Most AEs were mild or moderate and transient in nature.
Only one patient (20‐220) experienced AEs considered to be related to treatment, which were mild flushing, discomfort, and skin tightness, all occurring within 24 hr of the seventh infusion. The patient continued treatment without recurrence and without pre‐medication; the patient received an average dose of 31.8 U/kg and discontinued the study after the 44th infusion (due to protocol violation of refusing to return to all end‐of‐study visits), well after resolution of these events. This patient tested positive for neutralizing antibodies between months 6 and 18. This patient also underwent surgical renal stone removal, missed an infusion, but resumed therapy. Later, the patient discontinued therapy, refusing to return for all end‐of‐study visits because of time constraints. At baseline and last observation, respectively (see Supporting Information Table SIII), hemoglobin concentration was 15.5 and 13.9 g/dL, platelet count was 132,333/mm3 and 80,000/mm3, spleen volume was 6.9 MN and 6.3 MN, and liver volume was 0.9 MN and 0.8 MN; chitotriosidase activity was not available (not conducted at baseline).
Two other patients discontinued the study. One of these patients (23‐204; average dose, 28.8 U/kg) experienced one AE (wasp sting, not related to treatment). The patient decided to pursue another experimental medication after 20 total months of treatment; at baseline and last observation, respectively (see Supporting Information Table SIII), hemoglobin concentration was 16.1 and 15.6 g/dL, platelet count was 217,167/mm3 and 203,000/mm3, spleen volume was 2.3 MN and 2.0 MN, liver volume was 0.7 MN at both time points, and chitotriosidase activity was reduced by 32.6% from a baseline of 448 nmol/mL·hr.
The other patient (10‐201; average dose, 19.9 U/kg) discontinued after 16 total months of treatment with taliglucerase alfa because of dissatisfaction with treatment; at baseline and last observation for each parameter, respectively (see Supporting Information Table SIII), hemoglobin concentration was 11.2 and 10.6 g/dL, platelet count was 60,667/mm3 and 41,000/mm3, spleen volume was 20.5 MN at both time points, liver volume was 1.3 MN and 1.2 MN, with a 6.4% reduction in chitotriosidase activity from a baseline value of 15,209 nmol/mL·hr. None of the patients who completed 36 months of treatment experienced an AE considered to be related to taliglucerase alfa treatment.
It should be noted that the relatively broad range in the biomarkers reflects different baseline parameters of the patients entering the trial, such as different disease severities, different genotypes, and various ERT dosage regimens. The significance of the findings of anti‐taliglucerase alfa antibodies in four patients in this study is not clear. Comparison of the incidence of antibodies to taliglucerase alfa with the incidence of antibodies to other products may be misleading. This is because immunogenicity assay results can be highly dependent upon or influenced by extrinsic and intrinsic factors such as the sensitivity and specificity of the assay, assay methodology including sample handling as well as timing of sample collection, concomitant medication, and underlying disease 14. The limitations of this study lie primarily in its small size, which is common for studies of “orphan drugs” such as ERT for GD.
In conclusion, following 3‐year treatment with taliglucerase alfa in adult patients with type 1 GD who were previously treated with imiglucerase, mean organ volume and hematologic values were unchanged or improved. In addition, further reductions in both spleen volume and biomarkers were observed in several patients in this study. These results extend the clinical safety and efficacy profile of taliglucerase alfa. A major limitation of the study is the small number of patients assessed.
Supporting information
Supporting Information
Acknowledgments
Pfizer and Protalix entered into an agreement in November 2009 to develop and commercialize taliglucerase alfa. Statistical analyses were performed by Target Health, who provides clinical research services to Pfizer and Protalix BioTherapeutics. None of the authors received compensation for their contributions to this manuscript.
Conflict of interest: GMP is the recipient of research grants/support from Actelion, Amicus/GSK, BioMarin, Genzyme, Protalix BioTherapeutics, and Shire. SPS has been a site primary investigator in clinical trials; has received research support and educational grants sponsored by Actelion, Amicus, Genzyme, Protalix BioTherapeutics, and Shire; and has received honoraria and travel support as a speaker and for investigator meetings for Genzyme, Protalix BioTherapeutics, and Shire (these activities have been monitored and found to be in compliance with the conflict‐of‐interest policies at Emory University). MP, PG, HR, and JS are study investigators. DJA is a study investigator and has received honoraria and travel support from Actelion, Genzyme/Sanofi, Protalix/Pfizer, and Shire. RC and EB‐A are employees of Protalix BioTherapeutics. AZ is a consultant and member of the Scientific Advisory Board for Protalix BioTherapeutics; has stock options in Protalix BioTherapeutics; and has received honoraria from Genzyme/Sanofi, Pfizer, and Shire; in addition, the Gaucher Clinic receives research grants/support from Genzyme/Sanofi and Shire for participation in GOS and ICGG, respectively.
References
- 1. Grabowski GA, Petsko GA, Kolodny EH. Gaucher disease In: Valle D, Beaudet AL, Vogelstein B, et al., editors. The Online Metabolic and Molecular Basis of Inherited Disease. New York, NY: The McGraw Hill Companies, Inc; 2010. Chapter 146. [Google Scholar]
- 2. Grabowski GA, Kolodny EH, Weinreb NJ, et al. Gaucher disease: Phenotypic and genetic variation In: Valle D, Beaudet AL, Vogelstein B, et al., editors. The Online Metabolic and Molecular Bases of Inherited Disease. New York, NY: The McGraw Hill Companies, Inc; 2010. Chapter 146.1. [Google Scholar]
- 3. Barton NW, Brady RO, Dambrosia JM, et al. Replacement therapy for inherited enzyme deficiency―macrophage‐targeted glucocerebrosidase for Gaucher's disease. N Engl J Med 1991;324:1464–1470. [DOI] [PubMed] [Google Scholar]
- 4. Hollak CE, vom Dahl S, Aerts JM, et al. Force majeure: Therapeutic measures in response to restricted supply of imiglucerase (Cerezyme) for patients with Gaucher disease. Blood Cells Mol Dis 2010;44:41–47. [DOI] [PubMed] [Google Scholar]
- 5. Deegan PB, Cox TM. Imiglucerase in the treatment of Gaucher disease: A history and perspective. Drug Des Dev Ther 2012;6:81–106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Fox JL. First plant‐made biologic approved. Nat Biotechnol 2012;30:472. [Google Scholar]
- 7. Grabowski GA, Golembo M, Shaaltiel Y. Taliglucerase alfa: An enzyme replacement therapy using plant cell expression technology. Mol Genet Metab 2014;112:1–8. [DOI] [PubMed] [Google Scholar]
- 8.National Institutes of Health. Switchover trial from imiglucerase to plant cell expressed recombinant human glucocerebrosidase [NCT00712348]. June 10, 2013; Available at: http://clinicaltrials.gov/ct2/show/NCT00712348?term=nct00712348&rank=1. Accessed: January 27, 2015.
- 9. Pastores GM, Petakov M, Giraldo P, et al. A phase 3, multicenter, open‐label, switchover trial to assess the safety and efficacy of taliglucerase alfa, a plant cell expressed recombinant human glucocerebrosidase, in adult and pediatric patients with Gaucher disease previously treated with imiglucerase. Blood Cells Mol Dis 2014;53:253–260. [DOI] [PubMed] [Google Scholar]
- 10.National Institutes of Health. Plant cell expressed recombinant human glucocerebrosidase extension trial [NCT00705939]. August 13, 2013; Available at: http://clinicaltrials.gov/ct2/show/NCT00705939?term=nct00705939&rank=1. Accessed: October 8, 2015.
- 11. Zimran A, Brill‐Almon E, Chertkoff R, et al. Pivotal trial with plant cell‐expressed recombinant glucocerebrosidase, taliglucerase alfa, a novel enzyme replacement therapy for Gaucher disease. Blood 2011;118:5767–5773. [DOI] [PubMed] [Google Scholar]
- 12. Bracoud L, Ahmad H, Brill‐Almon E, Chertkoff R. Improving the accuracy of MRI spleen and liver volume measurements: A phase III Gaucher disease clinical trial setting as a model. Blood Cells Mol Dis 2011;46:47–52. [DOI] [PubMed] [Google Scholar]
- 13. Pastores GM, Weinreb NJ, Aerts H, et al. Therapeutic goals in the treatment of Gaucher disease. Semin Hematol 2004;41:4–14. [DOI] [PubMed] [Google Scholar]
- 14.Elelyso [package insert]. New York, NY: Pfizer Labs; 2015.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting Information