

Abstract
During recovery by regeneration after AKI, proximal tubule cells can fail to redifferentiate, undergo premature growth arrest, and become atrophic. The atrophic tubules display pathologically persistent signaling increases that trigger production of profibrotic peptides, proliferation of interstitial fibroblasts, and fibrosis. We studied proximal tubules after ischemia-reperfusion injury (IRI) to characterize possible mitochondrial pathologies and alterations of critical enzymes that govern energy metabolism. In rat kidneys, tubules undergoing atrophy late after IRI but not normally recovering tubules showed greatly reduced mitochondrial number, with rounded profiles, and large autophagolysosomes. Studies after IRI of kidneys in mice, done in parallel, showed large scale loss of the oxidant–sensitive mitochondrial protein Mpv17L. Renal expression of hypoxia markers also increased after IRI. During early and late reperfusion after IRI, kidneys exhibited increased lactate and pyruvate content and hexokinase activity, which are indicators of glycolysis. Furthermore, normally regenerating tubules as well as tubules undergoing atrophy exhibited increased glycolytic enzyme expression and inhibitory phosphorylation of pyruvate dehydrogenase. TGF-β antagonism prevented these effects. Our data show that the metabolic switch occurred early during regeneration after injury and was reversed during normal tubule recovery but persisted and became progressively more severe in tubule cells that failed to redifferentiate. In conclusion, irreversibility of the metabolic switch, taking place in the context of hypoxia, high TGF-β signaling and depletion of mitochondria characterizes the development of atrophy in proximal tubule cells and may contribute to the renal pathology after AKI.
Keywords: metabolism, pathology, proximal tubule, ischemia-reperfusion, mitochondria
After death of proximal tubule cells by AKI, surviving epithelial cells dedifferentiate, migrate, and proliferate. Recovery of normal structure and function occurs by redifferentiation of reconstituted epithelium.1 However, to varying degrees, proximal tubule cells proliferating after AKI fail to redifferentiate, undergo premature growth arrest, and become atrophic.2–6 Paradoxically, the atrophic epithelium continues to signal through multiple pathways activated early during proliferation. The pathologically persistent signaling increases progressively, triggering production of profibrotic peptides, proliferation of interstitial fibroblasts, and fibrosis.2–6 The phenotype of failed differentiation of tubules includes vicarious signaling through EGF receptor (EGFR), extracellular signal–regulated kinase–MAPK, c-Jun N–terminal kinase–MAPK, PI3K, and TGF-β pathways; decreased PTEN; increased expression of Akt and c-Jun, Smad2 and Smad3, TGF-β receptors 1 and 2, LPA receptors 1 and 2, lysophospholipase D, and β6-integrin; and enhanced production of TGF-β, PDGF-B, and CTGF.2–7
Why regenerating proximal tubules become atrophic after AKI is unknown. However, a metabolic switch could be involved. Transport functions in proximal tubules require high turnover of ATP derived almost exclusively through mitochondrial oxidative phosphorylation.8,9 After ischemic AKI, regenerating proximal tubule cells assume simplified structure as they dedifferentiate. These changes are pronounced in tubules that fail to redifferentiate and become atrophic.4 Conceivably, metabolic alterations are part of the dedifferentiation program that, if not reversed, leads to tubule atrophy. Alterations of energy metabolism occur in models of AKI, including diminished fatty acid oxidation during folic acid nephropathy,10 increased glycolysis after mercuric chloride AKI,11 increased lactate release into kidney interstitium after ischemic AKI,12 and elevated pyruvate kinase in kidney homogenates after ischemia-reperfusion injury (IRI).13 Although these changes could be components of physiologic metabolic alterations in regenerating epithelium, they could become pathologic if not reversed. On the basis of this hypothesis, we studied proximal tubules after ischemic AKI with a view to characterize possible mitochondrial pathologies and alterations of critical enzymes that govern energy metabolism. We found that proximal tubules undergoing atrophy but not tubules recovering normally late after IRI display striking mitochondrial alterations, increased glycolysis and glycolytic enzyme expression, and inhibitory pyruvate dehydrogenase (PDH) phosphorylation. These alterations were accompanied by expression of hypoxia markers and prevented by TGF-β antagonism. Hypoxia can trigger TGF-β, and both hypoxia and TGF-β are known to induce glycolysis and mitochondrial oxidant species in proximal tubule cells and other cell types.14–21 Structural, signaling, and metabolic alterations are not unexpected after AKI and may be required for early tubule regeneration and adaptation. Indeed, they may have protective roles during the early recovery phase. However, such modifications should be reversed later by redifferentiation. Our data provide support for a metabolic switch that takes place physiologically during early regeneration after AKI that progresses in tubules that fail to redifferentiate. The magnitude, persistence, and progression of mitochondrial and metabolic pathologies suggest that they are important components of proximal tubule atrophy with possible roles in its pathogenesis.
Results
Mitochondrial Pathology in Atrophic Tubules
As we have reported in detail previously, proximal tubules with failed differentiation and atrophy after IRI exhibited flat simplified cytoplasm.4 In such tubules, mitochondria were greatly reduced in number and of smaller size with round profiles, unlike tubules that recovered normally (Figure 1, A and B). Moreover, atrophic tubules showed large complex autophagolysosomes containing degenerate mitochondria, profiles of partially degraded endoplasmic reticulum, and myelin figures (Figure 1C, Supplemental Figure 1). TGF-β receptor antagonist SD208 promotes tubule differentiation during regeneration after IRI and prevents tubule atrophy,2,4 which is illustrated in this work by the morphologic phenotype of differentiation instead of atrophy in proximal tubules of SD208-treated rats (SD panels in Figures 1, 2, 4, 5, and 8). Figure 1D shows that promotion of differentiation by SD208 is accompanied by greater mitochondrial number and normal mitochondrial morphology. Immunohistochemistry (IHC) for mitochondrial markers Tom20 (Figure 1) or AIF (not shown) revealed drastic reduction of mitochondrial mass during tubule atrophy after IRI (Figure 1F) compared with normally recovering tubules (Figure 1E) and tubules of SD208-treated rats (Figure 1G). Immunofluorescence confirmed that atrophic tubules with less mitochondria expressed vimentin (Figure 1H), the marker that indicates early dedifferentiation as well as late atrophy of regenerating proximal tubules.4,22–25 Severe loss of mitochondrial mass during tubule atrophy was documented by low-power microscopy covering large areas of kidney parenchyma (Figure 1, I–K) and semiquantitative grading of mitochondrial granule density (Figure 1L).
Figure 1.

Mitochondrial pathology after IRI. (A–D) Electron micrographs showing (A) normal recovery, (B) atrophy with loss and simplification of mitochondria, (C) autophagolysosome associated with atrophy, and (D) SD208-induced recovery in proximal tubule epithelium 14 days after IRI. (E–L) IHC for mitochondrial marker Tom20 in (E) kidneys of nephrectomized control with sham left kidney ischemia, (F and H) IRI kidneys, and (G) SD208–protected IRI kidney 14 days after IRI. Tom20 is visualized brown by IHC in E–G and red by immunofluorescence in H. Vimentin, shown in green, indicates dedifferentiation/atrophy of epithelium and is also present in interstitial fibroblasts. Note the abundant red staining of Tom20 in the differentiated vimentin–negative tubule (upper left) but significantly less red staining in flat, undifferentiated vimentin–positive (green) tubules (upper right and lower right) in H. I–K show low-power micrographs of Tom20 staining by IHC in kidneys of rats (I) without IRI, (J) with IRI treated with vehicle only, and (K) with IRI treated with SD208 14 days after surgery. L shows results for semiquantitative grading of Tom20 staining by IHC (n=4 for each group). DAPI, 4′,6-diamidino-2-phenylindole; NCtrl, nephrectomized control; Veh, vehicle; Vim, vimentin. Scale bar, 1 μm in A–D; 50 μm in E–H; 100 μm in I–K. *P<0.01.
Figure 2.

Expression of hypoxia markers after IRI. (A) Western blotting of kidney lysates for HIF1α and CA9 in normal control, 14-day nephrectomy sham left kidney ischemia control, kidneys at serial time intervals up to 14 days of IRI, and kidney 14 days after IRI with SD208 treatment. In this blot, CA9 appears increased only after 14 days of IRI. A separate series of experiments (n=3) showed significant increases at 3, 7, and 14 days after IRI (not shown). (B–E) IHC for CA9 in nephrectomy control, kidneys 7 and 14 days after IRI, and SD208-treated kidney 14 days after IRI. (B) Compared with the time control 14 days after nephrectomy (NCtrl 14d), there is marked increase of staining for CA9 in (C) regenerating tubules (with flat epithelium) and fibroblasts proliferating in markedly widened interstitium of kidneys of rats treated with vehicle only 7 days after IRI (IRI 7d + Veh) and (D) flat atrophic epithelium of tubules in a fibrotic area of rat kidney treated with vehicle only 14 days after IRI (IRI 14d + Veh). The corresponding staining of kidney tubules from kidneys of rats treated with SD208 14 days after IRI (IRI 14d + SD) in (E) is reduced only modestly. These IHC findings are representative of a series of four separate experiments. ctrl, Control; GAPDH, glyceraldehyde-3-phosphate dehydrogenase; NCtrl, nephrectomized control; neph, nephrectomy; Veh, vehicle. Scale bar, 100 μm.
Figure 4.

Glycolytic enzyme protein expression in kidney tissue and tubules after IRI. (A) Western blotting of kidney lysates for hexokinase 2 (HK2), PFKP, PFKFB3, PKM2, and PKLR. Protocol and conditions are as outlined in Figure 2A. (B–D): IHC for (B) HK2, (C) PFKP, and (D) PFKFB3 in nephrectomy control, kidneys 7 and 14 days after IRI, and kidneys with SD208 treatment 14 days after IRI. Results for HK2 and PFKP are representative of four separate experiments. The accentuation of staining for PFKFB3 at the apical membranes of tubules seen after IRI in addition to increased cytoplasmic staining is not an artifact as shown by control IHC studies and the absence of such staining in the apical membranes of adjacent normally redifferentiating tubules as well as control tubules. ctrl, Control; GAPDH, glyceraldehyde-3-phosphate dehydrogenase; NCtrl, nephrectomized control; neph, nephrectomy; Veh, vehicle. Scale bar, 100 μm.
Figure 5.

Glycolytic enzyme protein expression in atrophic kidney tubules after IRI showing increase for PKM2 but decrease for PKLR. (A and B) IHC for (A) PKM2 and (B) PKLR in nephrectomy control, kidneys 7 and 14 days after IRI, and kidney with SD208 treatment 14 days after IRI. Results for PKM2 are representative of a series of four separate experiments. NCtrl, nephrectomized control; Veh, vehicle. Scale bar, 100 μm.
Figure 8.

Phosphorylation of pyruvate dehydrogenase in kidney tissue and tubules after IRI. (Upper panel) IHC for phosphopyruvate dehydrogenase E1α (pPDHE1α) in nephrectomy control, kidneys 7 and 14 days after IRI, and kidney with SD208 treatment 14 days after IRI. Arrows point to sharp transitions between pPDHE1α staining in atrophic epithelium and adjacent thicker differentiated epithelium with weak or no staining. Scale bar, 100 μm. (Lower panel) Western blotting of kidney lysates for pPDHE1α and PDHE1α in the time course and protocol outlined for Figure 2A. ctrl, Control; GAPDH, glyceraldehyde-3-phosphate dehydrogenase; NCtrl, nephrectomized control; neph, nephrectomy; Veh, vehicle.
Early dedifferentiation during regeneration of surviving proximal tubules after AKI is marked by massive physiologic loss of mitochondria,26–28 presumably by autophagy. Therefore, redifferentiation during tubule recovery axiomatically requires substantial mitochondrial biogenesis.29 However, some tubules failed to redifferentiate instead and became atrophic, with persistently low mitochondrial mass and abnormal autophagy. These findings raised the possibility that a mitochondrial pathology had developed that prevented epithelial recovery. Because TGF-β is involved in the pathogenesis of postischemic tubule atrophy,2,4 we assessed Mpv17L, a mitochondrial protein sensitive to TGF-β–induced oxidant stress16 that becomes decreased in tubules during TGF-β–induced CKD.16,30 Because mouse–directed Mpv17L antibody16 was unreactive with rat antigen, we performed IRI in C57BL/6 mice using a protocol similar to that of our rat model.2 Development of early fibrosis 14 days after IRI was accompanied by markedly reduced Mpv17L in kidney lysate (Supplemental Figure 2A) and in proximal tubule cells (Supplemental Figure 2B).
IRI Is Followed by Hypoxia Marker Expression That Persists in Tubules as They Become Atropic
IRI was followed by enhanced expression of hypoxia–inducible factor 1α (HIF1α) as reported before,31,32 remaining persistently increased in kidney lysates until 14 days; kidneys of SD208-treated rats showed modest decrease of HIF1α induction (Figure 2A). There also was increased expression of carbonic anhydrase 9 (CA9), the hypoxia marker that is directly transcribed by HIF133 (Figure 2A). CA9 showed convincing increase by Western blotting only at 14 days after IRI in the experiments depicted by Figure 2A. However, CA9 was found to be significantly increased by IHC in regenerating proximal tubules 7 days after IRI, which persisted in atrophic tubules at 14 days, with only modest decreases of the marker in SD208-treated kidneys (Figure 2B–E). Moreover, a separate series of experiments (n=3) showed significant increases at 3, 7, and 14 days after IRI (not shown). Thus, hypoxia, a major driver of glycolysis, was found to affect tubules regenerating after IRI and also, atrophic tubules late after IRI, when fibrosis develops.
Enhanced Kidney Glycolysis Associates with Increased Glycolytic Enzyme Expression in Regenerating Proximal Tubules That Increases Further in Atrophic Tubules
Glucose uptake and lactate production by kidneys were reported to be increased over several days after HgCl2-induced AKI in rats coinciding with tubule regeneration accompanied by enhanced glucose phosphorylating activity.11 Kidneys were reported to release more lactate and exhibit increased activities and mRNA content for pyruvate kinase M2 (PKM2; or M2-PK), with complementary decreases for pyruvate kinase liver/RBC isoform (PKLR; or L-PK) during early reperfusion after IRI.12,13 We have provided evidence for increased glycolysis in kidneys recovering from IRI. During reperfusion 3, 7, and 14 days after IRI, lactate and pyruvate concentrations in cortex and the outer stripe of outer medulla were increased relative to time controls, and at 7 and 14 days, this was accompanied by increased hexokinase activity (Figure 3). By Western blotting, there was early enhancement and pronounced and progressive late increases in protein expression of rate-limiting enzymes for glycolysis after IRI: hexokinase 2 (HK2), phosphofructokinase platelet isoform (PFKP), 6-phosphofructo-2-kinase/fructose-2,6-bisphosphatase 3 (PFKFB3), and PKM2 (Figure 4A). However, PKLR that is expressed by normal proximal tubules34,35 became depleted after an early increase 8 hours after reperfusion (Figure 4A). SD208 treatment largely reversed these changes observed by Western blotting with two exceptions—for PFKP and PKM2, which showed persistent increase, despite TGF-β antagonism (Figure 4A) (see below for results by IHC that do show decrease of both PFKP and PKM2 in SD208–treated IRI kidneys). By IHC, there was increased expression of hexokinase 2, PFKP, PFKFB3, and PKM2 in regenerating tubules 7 days after IRI, with striking further increases in atrophic tubules at 14 days (Figures 4–6). Increases were seen also at 3 and 5 days (shown for PKM2 in Supplemental Figure 3). Immunofluorescence confirmed the specificity of the strikingly increased glycolytic enzyme in atrophic proximal tubule epithelium. In mosaic tubules lined partially by well differentiated and atrophic epithelium,4 vimentin–positive atrophic cells but not well differentiated cells with intact PHAE-lectin staining brush borders expressed PKM2 (Figure 6). In a notable exception, proximal tubules that had recovered normal structure after SD208 treatment also expressed PKM2 focally, albeit to a lesser degree (Figure 5A), presumably accounting for persistently increased PKM2 seen by Western blotting in SD208-treated kidneys (Figure 4A). IHC for PKLR confirmed the decrease of this enzyme during reperfusion seen by Western blotting; atrophic tubules, but not well differentiated tubules, showed the decrease of PKLR protein expression (Figure 5B). Notably, fibroblasts that had proliferated in the interstitium around atrophic tubules also showed increased glycolytic enzyme expression, most pronounced for PKM2 (Figures 5A and 6). Treatment with SD208 largely prevented these alterations (Figures 4 and 5, Supplemental Figure 3). Strikingly, PKM2 colocalized with hypoxia marker CA9 in atrophic tubules (Supplemental Figure 4). In varying degrees, distal nephron segments but not proximal tubules in normal kidneys expressed glycolytic enzymes as might be expected from measurements of enzyme activities in microdissected nephrons and functional assays.8,9,36 Also in varying degrees, proximal tubules that recovered normally after IRI and proximal tubules from kidneys of sham ischemia controls that received contralateral nephrectomy showed modest increases of glycolytic enzyme expression, never approaching the dramatic increases seen in atrophic tubules (Figures 4 and 5).
Figure 3.

Renal pyruvate and lactate content and hexokinase activity are increased after IRI. Concentrations of pyruvate and l-lactate and activity of hexokinase in kidney cortex and outer stripe of outer medulla 3, 7, and 14 days after IRI and corresponding time controls (n=3 for each group). Ctrl, control. *P<0.05; **P<0.01.
Figure 6.

Glycolytic enzyme PKM2 protein expression is increased in vimentin positive atrophic epithelium but not in well differentiated proximal tubule epithelium in whole tubules as well as in clusters of cells or single cells lining the same tubule. Fluorescence images for PHAE lectin brush border staining and immunofluorescence images for vimentin and PKM2 localization in an IRI kidney 14 days after IRI shown singly or merged. Small arrows point to sharp transitions between isolated cells or clusters of cells with vimentin–positive undifferentiated proximal tubule epithelium and adjacent well differentiated proximal tubule epithelium with PHAE staining brush border. Note that interstitial fibroblasts (arrowheads) as well as undifferentiated proximal tubule epithelium (large arrows) stain for both vimentin and PKM2. Distal tubules stain strongly for PKM2 but lack vimentin. DT, distal tubule; PT, proximal tubule; Vim, vimentin. Scale bar, 50 μm.
Hypoxic Induction of Glycolytic Enzymes in Cultured Proximal Tubule Cells Involves HIF1α
Proximal tubules regenerating after IRI are subjected to chronic hypoxia.37–39 Hypoxic tubule microenvironments were shown by pimonidazole adduct IHC early after reperfusion as well as late, when tubules become atrophic and fibrosis develops.37,39,40 Increased hypoxia marker CA9 in atrophic tubules (Figure 2B, Supplemental Figure 4) suggested that increased glycolytic enzymes in atrophic tubules (Figures 4–6) were induced by hypoxia, a possibility consistent with a role for HIF1α in controlling metabolism.41,42 As such, we examined the effects of shRNA HIF1α knockdown in cultured proximal tubule cells. Exposure to 0.5% O2 for 48 hours increased the expression of PFKP, PFKFB3, PKM2, and PKLR; HIF1α knockdown abrogated the hypoxic effect for PFKP, PFKFB3, and PKM2 but increased further the expression of PKLR (Figure 7A). These data showed that PKLR, the pyruvate kinase isoform normally expressed by proximal tubules, is regulated differently relative to PKM2.
Figure 7.
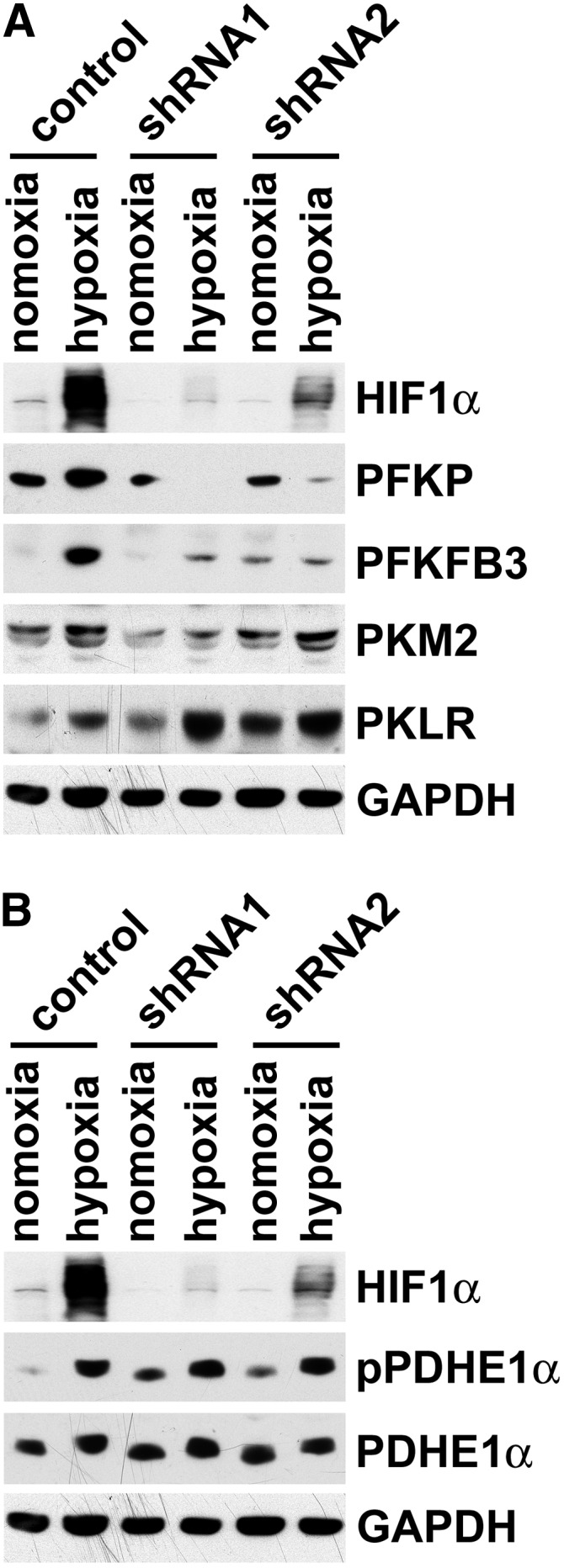
HIF1 dependent and independent effects on glycolytic enzyme protein expression and phosphorylation of pyruvate dehydrogenase. (A) Western blotting for HIF1α, PFKP, PFKFB3, PKM2, and PKLR in two groups of BUMPT cultured proximal tubule cells without and with HIF1α shRNA knockdown exposed to 0.5% O2 for 48 hours. (B) Western blotting for HIF1α, phosphopyruvate dehydrogenase E1α (pPDHE1α; S293), and PDHE1α in two groups of BUMPT cultured proximal tubule cells without and with HIF1α shRNA knockdown exposed to 0.5% O2 for 48 hours. GAPDH, glyceraldehyde-3-phosphate dehydrogenase.
Hypoxia Induces Phosphorylation at the Inhibitory S293 Site of PDH E1α-Subunit in Cultured Proximal Tubule Cells
Entry of pyruvate into the Krebs cycle is controlled by PDH. Phosphorylation of PDH by pyruvate dehydrogenase kinases (PDKs) at inhibitory sites S232, S293, and S300 of pyruvate dehydrogenase E1α–subunit (PDHE1α) inhibits PDH activity and thereby, suppresses mitochondrial metabolism by decreasing the production of acetyl-CoA.43–45 Hypoxia induces PDKs.44,45 It was reported that hypoxic activation of HIF1 induces PDK1 in mouse embryo fibroblasts; phosphorylation of PDHE1α then inhibits PDH to slow the Krebs cycle and decrease mitochondrial oxidant stress.44 We found that exposure of cultured proximal tubule cells to 0.5% O2 for 48 hours induced the S293 phosphorylation of PDHE1α, which was, however, not decreased by HIF1α knockdown (Figure 7B). Regardless of whether hypoxic PDHE1α phosphorylation was mediated by HIF1α-independent mechanisms as our data suggest or HIF1α-dependent processes as proposed,44 these results highlight a fundamentally important effect of hypoxia in proximal tubule cells—the potential for suppressing the gatekeeper function of PDH in permitting pyruvate entry into the Krebs cycle for mitochondrial oxidative metabolism.
Fatty acid oxidation also supplies acetyl-CoA substrate for the Krebs cycle. We, therefore, asked if carnitine palmitoyl transferase 1, the rate-limiting enzyme for mitochondrial fatty acid import, is affected by hypoxia. Expression of CPT1A, the isoform significant for fatty acid metabolism in kidney, was strongly suppressed by hypoxia in an HIF1α-independent manner (data not shown), but we obtained conflicting findings in vivo after IRI (see below).
PDHE1α S293 Phosphorylation Is Increased during Late Regeneration and during the Development of Atrophy in Tubules Regenerating after IRI
By Western blotting, phospho-PDHE1α (S293) showed early decreases of overall signal in kidney lysates for several days after IRI followed by a slight increase after 14 days; SD208 treatment did not reduce the signal compared with that in IRI rats that received vehicle alone, whereas the corresponding 14-day nephrectomy control with sham ischemia also showed a modest increase. PDHE1α remained unaltered throughout (Figure 8). By IHC, the signal for phospho-PDHE1α was correspondingly reduced in regenerating tubules 3 days after IRI (not shown). Subsequently, the signal increased but still remained low (not shown). However, at day 7, strong staining was seen in regenerating tubules (Figure 8). After 14 days of IRI, when tubules were atrophic and surrounded by fibrosis, the atrophic tubule epithelium showed markedly increased signal for phospho-PDHE1α far exceeding the control; signals in tubules in SD208-treated rats were significantly lower than those in rats with vehicle alone (Figure 8). The specificity of increased phospho-PDHE1α signal in atrophic tubule epithelium was clearly shown by sharp transitions of high to low signal from atrophic to differentiated epithelium (arrows in Figure 8, upper panel), paralleling similarly sharp transitions for PKM2 (Figure 6).
Because hypoxia suppressed CPT1A in cultured proximal tubule cells, we assessed CPT1A protein after IRI. Western blotting of lysates showed no alterations of expression throughout 14 days after IRI; by IHC, staining was strong in distal nephron segments but weak in proximal tubules of normal controls before IRI. Counterintuitively, with progression of time during reperfusion, staining in regenerating proximal tubules showed increase rather than decrease (not shown). Similarly, we assessed PGC1α, a regulator of mitochondrial biogenesis, and PPARα; both of these transcription factors regulate fatty acid metabolism.29 Neither protein showed alterations post-IRI (not shown).
Discussion
Our findings provide pathologic and biochemical basis for a metabolic switch in proximal tubules regenerating after AKI. They also provide additional perspective on the phenotype of failed differentiation that often develops in such tubules and that, importantly, contributes to chronic progression. Cells of this unique phenotype exhibited greatly diminished numbers of mitochondria, altered mitochondrial shape and structure, and abnormally large autophagic bodies. Decrease of a mitochondrial protein that is sensitive to TGF-β–induced oxidant stress, Mpv17L, suggested that mitochondria suffer free radical damage during tubule atrophy. Furthermore, increased phosphorylation of PDH, the rate-limiting enzyme for pyruvate entry into the Krebs cycle, indicated that oxidative metabolism of pyruvate carbons derived from lactate, amino acids, and glucose must become inhibited in the fewer mitochondria that remained. In the other arm of the switch, kidney tissue glycolysis assessed as pyruvate and lactate content and hexokinase activity was enhanced, and the expression of key enzymes that are rate limiting for glycolysis was markedly increased in the same tubules that showed advanced mitochondrial pathology. The switch was remarkably cell specific. Mitochondrial pathology and increased glycolytic enzyme expression affected vimentin–positive whole tubules, clusters of cells, and even single cells sharply demarcated from normally recovering neighbors (shown for PKM2 in Figure 6). Together with older data showing increased renal glucose utilization for lactate production during tubule regeneration after toxic injury,11 our results suggest that energy metabolism in proximal tubules becomes profoundly altered as they dedifferentiate, migrate, and proliferate after AKI. This phase of early regeneration is marked by substantial loss of mitochondria that occurs as highly differentiated proximal tubule cells become simplified in structure.26–28 Thereafter, instead of the reversal expected for normal recovery, tubules that failed to redifferentiate continued to exhibit mitochondrial pathology and persistently showed marked and progressively increased expression of glycolytic enzymes.
Because fatty acids are mitochondrial substrates for oxidative metabolism in proximal tubules, we probed for key proteins that globally control fatty acid oxidation. CPT1A regulates fatty acid entry into mitochondria; PGC1α and PPARα are master regulators of mitochondrial biogenesis and transcription of proteins required for fatty acid metabolism.29 All three were downregulated during folic acid nephropathy,10 but we failed to detect altered expression early as well as late after IRI, when tubule atrophy developed. These results do not exclude a role for diminished fatty acid oxidation during tubule atrophy after IRI, because mitochondrial pathology after IRI and folic acid nephropathy could be caused differently. Importantly, extreme reduction of mitochondria in each atrophic cell implies overall decrease of mitochondrial metabolism per cell for all substrates, including fatty acids. It seems possible that actions of the most proximal upstream defect that causes failed differentiation—a defect that remains unknown—somehow induce uncontrolled autophagy, a process known to be involved in atrophy.
The other component of the metabolic switch—increased glycolysis and glycolytic enzyme expression—is more easily explained. Unlike the distal nephron, proximal tubules express very low levels of glycolytic enzymes,8,36 and although they do exhibit some glycolysis during hypoxia17 and hypoxia reoxygenation,18 their capacity for glycolysis is limited, unlike in the distal nephron.36,46 Zager et al.18 showed that lactate increased in kidneys during clamp ischemia but fell thereafter during reperfusion; conversely, pyruvate (an intermediary metabolite that can be a product of glycolysis, gluconeogenesis, or PDH inhibition, causing decreased pyruvate entry into the Krebs cycle) decreased during ischemia and remained decreased until 18 hours of reperfusion. Indeed, pyruvate infusions protected against ischemic AKI. Differences between these findings and the concurrent increases of pyruvate and lactate that we found during IRI are likely attributable to timing—our measurements were done at and after 3 days of IRI when tubules are dedifferentiated, regenerating and expressing more glycolytic enzymes, in contrast to in the study by Zager et al.,18 in which differentiated proximal tubules with predominantly oxidative metabolism were in the earliest stages of active injury and cell death (up to only 18 hours of reperfusion after ischemia). In other studies, basal levels of lactate released normally into the interstitium of renal cortex were found to be much lower than those in the medulla, but these levels became increased during ischemia and reperfusion.12 Such measurements cannot be ascribed to the production of lactate by proximal tubules alone, because vigorously glycolytic distal nephron segments,8,36,46 present in the cortex, will also contribute. The most persuasive evidence for increased glycolysis after AKI was provided by increased glucose utilization, lactate production, and glucose phosphorylation over many days as tubules regenerated after toxic AKI.11 However, interpretation of results from these studies is fraught with inability to discriminate between AKI events taking place in uninjured proximal tubules, distal tubules, and proximal tubules that recover normally after injury and AKI events taking place in those that become atrophic. IHC in conjunction with protein analysis of lysates now show that increased glycolysis during tubule regeneration and atrophy after AKI can be explained by enhanced glycolytic enzyme expression in tubules of the same undifferentiated phenotype, which show reduced mitochondrial mass and subsequently, display inhibitory PDH phosphorylation.
Normal proximal tubules use pyruvate derived from lactate (also from amino acids, oxaloacetate, and other metabolites) to make glucose in a predominantly gluconeogenic pathway. Inferring from overall increase of glycolysis and the markedly increased glycolytic enzyme expression in regenerating tubules, and the enhanced glycolysis reported for kidneys recovering from toxic AKI,11 we surmise that lactate to pyruvate conversion for gluconeogenesis in regenerating proximal tubules would be suppressed. Instead, lactate dehydrogenase would work in reverse to generate lactate from pyruvate, accommodating the needs of glycolysis. Later, when some tubules recover normal structure and biochemical function, others do not; the metabolic anomaly in tubules with failed differentiation is likely made worse by progressive mitochondrial pathology and autophagy as well as inhibitory PDH phosphorylation. Pyruvate made by glycolysis would be denied entry into the Krebs cycle and converted largely to lactate. Furthermore, in view of reduced mitochondrial mass, oxaloacetate substrate for gluconeogenesis would also fall, because oxaloacetate is made in mitochondria, and the Krebs cycle would be slowed by PDH inhibition. More evidence for the metabolic switch was provided by observing that PKLR, the pyruvate kinase isoform expressed by normal proximal tubules, decreased during tubule regeneration and atrophy in opposite direction from PKM2, the isoform responsible for glycolysis in the distal nephron. Notably, modulation of PKLR activity is required to regulate gluconeogenic flux in the normal state—to convert excess phosphoenol pyruvate back to pyruvate and avoid futile cycles.35,47 Decreased PKLR is, therefore, not unexpected after IRI when PKM2, a major engine for glycolysis, was overexpressed. Also in support of the metabolic switch after IRI is increased expression in atrophic tubules of PFKFB3, the hypoxia–induced PFK2 isoform48 also known as inducible phosphofructokinase 2 (iPFK2).49 PFKFB3 has the highest 6-phosphofructo-2-kinase/fructose-2-to-6-bisphosphatase-3 activity ratio (700:1) among PFK2 isozymes, greatly favoring glycolytic PFK1 activity and decreasing gluconeogenic flux in the opposite direction.50 Not surprisingly, PFKFB3/iPFK2 is highly expressed in contexts associated with markedly increased glycolysis, including cancer.49
Stimulated glycolytic enzyme expression in vivo after IRI becomes easier to understand in the context of chronic hypoxia and enhanced TGF-β signaling, both of which come into play during tubule regeneration after IRI.2,3,37–39,51 Both HIF1–mediated hypoxic actions and TGF-β signaling have pronounced effects to enhance glycolytic gene expression and glycolysis in proximal tubule cells.14,15,17,18 The paramount role of HIF1 in giving rise to increased enzyme expression required to sustain enhanced glycolysis is well established for diverse cell types.41,42 Particularly interesting, given the dramatic increase of PKM2 expression, enhanced expression of this isoform was shown to form part of a positive hypoxic feedback loop to potentiate HIF1 signaling.42 In this study, we show specific effects of HIF1 to regulate glycolytic gene expression using shRNA knockdown of HIF1α in cultured proximal tubule cells. HIF1 also regulates glycolytic genes of kidney tubules in vivo. Specific and stable induction of HIF1α in kidney tubules (by Pax8-rtTA–based VHL deletion) led to increased expression of glycolytic enzymes and protected proximal tubules at risk for injury and cell death during AKI.52 Increased EGF signaling could also contribute importantly to glycolytic shift in proximal tubules during AKI. Recovery from AKI is facilitated by increased signaling through EGFR during early AKI.53–55 EGF signaling promotes proximal tubule cell dedifferentiation, migration, and proliferation after injury.55–58 EGF stimulates glycolysis14,59 and furthermore, gives rise to downstream TGF-β signaling,54,55 which also stimulates glycolysis in kidney tubule cells.14 The situation is complicated by the fact that EGFR signaling that increases glycolysis and protects kidneys early during AKI can also promote tubule atrophy and fibrosis.54,55,60 Moreover, interactions between the several signaling pathways physiologically required for dedifferentiation and redifferentiation of kidney tubules early after injury but pathologically causing tubule atrophy and fibrosis late after injury are likely to be complex as well. For example, EGF gives rise to downstream TGF-β signaling, but the reverse (i.e., transactivation of EGFR by TGF-β) has also been reported.55 IRI does not heal completely and transitions to CKD (the AKI-CKD transition), which is accompanied by sustained EGFR activation.7 Relationships between the several factors that promote glycolysis (EGF, TGF-β, HIF1, and others), the metabolic switch that we now show, and kidney fate (normal recovery of tubule or tubule atrophy) are, therefore, likely to be complex, variable during the course of disease, and difficult to unravel. Additional detailed investigation of these relationships is clearly required.
The information presented here provides a basis for additional research on why tubules often fail to enter a differentiation program during recovery from AKI. Because a metabolic switch occurs as an early component of the physiologic program of dedifferentiation in regenerating epithelium, it needs to be determined why such a program does not reverse in tubules that become atrophic. Given that increased TGF-β signaling may contribute to altered metabolism through mitochondrial damage and stimulated glycolysis, the question must be asked: what are the upstream events that drive TGF-β signaling in regenerating cells that, if not reversed, lead to atrophy? Does chronic hypoxia have HIF1-independent effects that damage tubules to the extent that it produces irreversible dedifferentiation? Additional studies are needed to answer these questions using both in vivo models of tubule injury and cultured proximal tubule cells. A suitable proximal tubule culture model, in which dedifferentiation and redifferentiation can be modulated at will under normoxic and hypoxic conditions, will be particularly informative to unravel the interplay of TGF-β with HIF1-dependent and -independent processes in the pathogenesis of tubule atrophy.
Concise Methods
Antibodies and Reagents
Antibody sources were HIF1α (Cayman Chemicals, Ann Arbor, MI); CA9 and phospho-PDHE1α (S293; Novus Biologicals, Littleton, CO); hexokinase 2, C64G5, and PKM2 (D78A4; Cell Signaling Technology, Danvers, MA); PFKP (H44), iPFK2 br/pl (C-11), PKLR (E-2), and Tom20 (FL-145; Santa Cruz Biotechnology, Santa Cruz, CA); PFKFB3 (iPFK-2 and uPFK-2; Sigma-Aldrich, St. Louis, MO); pyruvate kinase L, AD12, BH3, and PKM2 DF4 (ScheBo Biotech, Giessen, Germany); PDHE1α (GeneTex, Irvine, CA); CPT1A and PGC1α (Abcam, Inc., Cambridge, MA); PPARα (Rockland Immunochemicals Inc., Gilbertsville, PA); vimentin clone V9 (Thermo Fisher Scientific, Vernon Hills, IL); glyceraldehyde-3-phosphate dehydrogenase (Research Diagnostics, Flanders, NJ); ImmPRESS HRP polymer–conjugated secondary antibodies (Vector Laboratories, Burlingame, CA); and HRP–labeled secondary antibodies (Jackson ImmunoResearch Laboratories, West Grove, PA). Sources of other reagents were MISSION shRNA Lentiviral Transduction Particles (Sigma-Aldrich), FITC–conjugated Phaseolus vulgaris Lectin PHA-E (EY Laboratories, Inc., San Mateo, CA), SD208 (2-[5-chloro-2-fluorophenyl]pteridin-4-yl)pyridin-4-yl amine; SCIOS, Inc., Johnson and Johnson, New Brunswick, NJ), and ImmPACT DAB reagent (Vector Laboratories).
Animals
Animals were cared for in accordance with National Institutes of Health and institutional guidelines, and studies were approved by the Institutional Animal Care and Use Committee at University of texas Health Science Center, San Antonio, TX. Studies were performed on tissue obtained from rats subjected to IRI without or with SD208 treatment during reperfusion using methods identical to those we reported before.2 SD208 (SCIOS, Inc.) is a highly specific Alk5 TGF-β receptor antagonist that suppresses TGF-β signaling61,62 and TGF-β–dependent organ fibrosis,61,62 which we found to promote tubule differentiation and protect against tubulointerstitial fibrosis after IRI.2 Briefly, male Sprague–Dawley rats under isoflurane anesthesia received 45 minutes of left kidney ischemia and right nephrectomy followed by reperfusion for periods up to 14 days. The procedures used here were identical to those we have reported in previous studies.2 After 4 hours of reperfusion, SD208 in 1% methylcellulose at 60 mg/kg body wt or vehicle alone was provided by gavage two times daily for 4 days. SD208 is a specific inhibitor of Alk5 kinase used for in vivo studies to inhibit TGF signaling given according to this dose regimen.61,62 Controls were rats with right nephrectomy and sham surgery to the left kidney. IRI was monitored by serum creatinine and renal histology. In a separate series of studies, rats were studied after 60 minutes of ischemia using a similar IRI protocol. Studies were also done on 11- to 12-week-old male C57BL/6 mice after a surgery protocol for IRI exactly the same as that used for rats. Mice were euthanized after 3, 7, or 14 days.
Cell Culture and shRNA Knockdown
Boston University mouse proximal tubule cells (BUMPT-Clone 306; from W. Lieberthal and J. Schwartz) were grown at 37°C in DMEM (Gibco, Carlsbad, CA) with 10% FBS. BUMPT cells were derived from primary cultures of kidney proximal tubules of F1 hybrid mice with single copies of the H-2Kb-tsA58 transgene.63 Expression of large T antigen by the transgene at 39.5°C without γ-IFN is inhibited by >95% relative to cells at 33°C with the cytokine.63 Confluent BUMPT cells display proximal tubule characteristics and develop transepithelial resistance of approximately 300 Ω/cm2 when grown at 37°C.64 For HIF1α knockdown, BUMPT cells were infected with lentiviral particles with scrambled or HIF1α-specific shRNA and selected with puromycin. Two cultures, one with the lowest expression, and another with very low but perceptibly higher expression of HIF1α, and one culture with scrambled control were used for studies.
Morphology, IHC, and Immunofluorescence
Rat kidneys were perfusion fixed with periodic acid–lysine-paraformaldehyde using methods identical to those described in our previous studies.65 After overnight fixation at 4°C, kidney slices were washed twice in PBS and once in PBS and 100 mM glycine, and they were transferred to 70% ethanol for dehydration in increasing concentrations of ethanol and embedding in paraffin. After antigen retrieval in 99°C 1 mM Tris-EDTA for 20–30 minutes, deparaffinized sections were blocked with 2.5% horse serum before IHC with primary antibodies followed by ImmPRESS HRP polymer–conjugated secondary antibodies. For immunofluorescence, deparaffinized sections with antigen retrieval were used. Sections were examined by confocal fluorescence microscopy. Periodic acid–lysine-paraformaldehyde–fixed tissue was postfixed in 2% glutaraldehyde in 0.1 M Na Cacodylate buffer (pH 7.2) and 1% OsO4 and processed for electron microscopy.2
Semiquantitative Grading for Mitochondrial Mass
Mitochondrial mass was assessed semiquantitatively in four separate experiments. After IHC for mitochondrial marker Tom20, five different fields in kidney cross-sections from each kidney of each group were graded (by M.A.V.) on a zero to five scale using a system identical to that we described before in a previous study.25 The five fields evaluated covered the entire inner cortex and outer stripe of the outer medulla (where most of the injury took place after IRI). They were examined at low power (×100) and then, high power (×200 and as needed, ×400) to grade the density of brown–staining mitochondrial granules in entire tubule profiles. The grades represented an index of approximate mitochondrial number per tubule profile (or per tubule cell). Averaged grades from each of four kidneys from each group were used for quantitation and statistical analysis.
Western Blotting
Inner cortex and outer stripe of outer medulla were dissected, frozen, ground in liquid nitrogen, and extracted with 4× SDS Laemmli buffer. Protein loading was assessed by densitometry of Coomassie Blue–stained gels and loading controls. Cultured cells were extracted with 1× SDS buffer. SDS-PAGE and Western blotting were done as described.2
Lactate and Pyruvate Content and Hexokinase Activity
Measurements were made using kits purchased from BioVision, Inc. (Milipitas, CA). Kidney cortex and outer stripe of outer medulla were dissected on a cold plate, cut into small pieces, frozen in liquid nitrogen, and ground under liquid nitrogen with pestle and mortar. Aliquots of ground tissue were stored at −80°C. For analysis, ground tissue was rapidly homogenized in specific assay buffers provided by the manufacturer. Supernatants after centrifugation at 15,000×g were then processed according to the manufacturer’s instructions for assay with minor modifications. Lactate and pyruvate were assayed by fluorimetry, and hexokinase activity was assayed by colorimetry.
Disclosures
None.
Supplementary Material
Acknowledgments
E.P.B. received National Institutes of Health (NIH) grant 5R01DK060043, and M.A.V. is funded by NIH grant 1 R01 DK104128. J.M.W was was supported by Merit Review I01 BX002367 from the US Department of Veterans Affairs. The contents do not represent the views of the US Department of Veterans Affairs or the US Government.
Footnotes
Published online ahead of print. Publication date available at www.jasn.org.
This article contains supplemental material online at http://jasn.asnjournals.org/lookup/suppl/doi:10.1681/ASN.2015020177/-/DCSupplemental.
References
- 1.Bonventre JV, Weinberg JM: Recent advances in the pathophysiology of ischemic acute renal failure. J Am Soc Nephrol 14: 2199–2210, 2003 [DOI] [PubMed] [Google Scholar]
- 2.Geng H, Lan R, Wang G, Siddiqi AR, Naski MC, Brooks AI, Barnes JL, Saikumar P, Weinberg JM, Venkatachalam MA: Inhibition of autoregulated TGFbeta signaling simultaneously enhances proliferation and differentiation of kidney epithelium and promotes repair following renal ischemia. Am J Pathol 174: 1291–1308, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Geng H, Lan R, Singha PK, Gilchrist A, Weinreb PH, Violette SM, Weinberg JM, Saikumar P, Venkatachalam MA: Lysophosphatidic acid increases proximal tubule cell secretion of profibrotic cytokines PDGF-B and CTGF through LPA2- and Gαq-mediated Rho and αvβ6 integrin-dependent activation of TGF-β. Am J Pathol 181: 1236–1249, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lan R, Geng H, Polichnowski AJ, Singha PK, Saikumar P, McEwen DG, Griffin KA, Koesters R, Weinberg JM, Bidani AK, Kriz W, Venkatachalam MA: PTEN loss defines a TGF-β-induced tubule phenotype of failed differentiation and JNK signaling during renal fibrosis. Am J Physiol Renal Physiol 302: F1210–F1223, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Yang L, Besschetnova TY, Brooks CR, Shah JV, Bonventre JV: Epithelial cell cycle arrest in G2/M mediates kidney fibrosis after injury. Nat Med 16: 535–543, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Yang L, Humphreys BD, Bonventre JV: Pathophysiology of acute kidney injury to chronic kidney disease: Maladaptive repair. Contrib Nephrol 174: 149–155, 2011 [DOI] [PubMed] [Google Scholar]
- 7.Geng H, Lan R, Singha PK, Saikumar P, Weinberg JM, Venkatachalam MA: Lysophosphatidic acid (LPA) transactivates epidermal growth factor receptors (EGFR) via LPAR1/Gαi/o signaling to potentiate LPAR2/Gαq/αvβ6 integrin dependent TGFβ signaling and increase the production of PDGF-B and CTGF by proximal tubule (PT) cells [abstract]. J Am Soc Nephrol 24: 115A, 2013
- 8.Guder WG, Ross BD: Enzyme distribution along the nephron. Kidney Int 26: 101–111, 1984 [DOI] [PubMed] [Google Scholar]
- 9.Weinberg JM, Molitoris BA: Illuminating mitochondrial function and dysfunction using multiphoton technology. J Am Soc Nephrol 20: 1164–1166, 2009 [DOI] [PubMed] [Google Scholar]
- 10.Kang HM, Ahn SH, Choi P, Ko YA, Han SH, Chinga F, Park AS, Tao J, Sharma K, Pullman J, Bottinger EP, Goldberg IJ, Susztak K: Defective fatty acid oxidation in renal tubular epithelial cells has a key role in kidney fibrosis development. Nat Med 21: 37–46, 2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ash SR, Cuppage FE: Shift toward anaerobic glycolysis in the regenerating rat kidney. Am J Pathol 60: 385–402, 1970 [PMC free article] [PubMed] [Google Scholar]
- 12.Eklund T, Wahlberg J, Ungerstedt U, Hillered L: Interstitial lactate, inosine and hypoxanthine in rat kidney during normothermic ischaemia and recirculation. Acta Physiol Scand 143: 279–286, 1991 [DOI] [PubMed] [Google Scholar]
- 13.Fukuhara Y, Yamamoto S, Yano F, Orita Y, Fujiwara Y, Ueda N, Kamada T, Noguchi T, Tanaka T: Changes in activities and mRNA levels of glycolytic enzymes of ischemia-reperfused rat kidney. Contrib Nephrol 95: 222–228, 1991 [DOI] [PubMed] [Google Scholar]
- 14.Nowak G, Schnellmann RG: Autocrine production and TGF-beta 1-mediated effects on metabolism and viability in renal cells. Am J Physiol 271: F689–F697, 1996 [DOI] [PubMed] [Google Scholar]
- 15.de Laplanche E, Gouget K, Cléris G, Dragounoff F, Demont J, Morales A, Bezin L, Godinot C, Perrière G, Mouchiroud D, Simonnet H: Physiological oxygenation status is required for fully differentiated phenotype in kidney cortex proximal tubules. Am J Physiol Renal Physiol 291: F750–F760, 2006 [DOI] [PubMed] [Google Scholar]
- 16.Krick S, Shi S, Ju W, Faul C, Tsai SY, Mundel P, Böttinger EP: Mpv17l protects against mitochondrial oxidative stress and apoptosis by activation of Omi/HtrA2 protease. Proc Natl Acad Sci U S A 105: 14106–14111, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Dickman KG, Mandel LJ: Differential effects of respiratory inhibitors on glycolysis in proximal tubules. Am J Physiol 258: F1608–F1615, 1990 [DOI] [PubMed] [Google Scholar]
- 18.Zager RA, Johnson AC, Becker K: Renal cortical pyruvate depletion during AKI. J Am Soc Nephrol 25: 998–1012, 2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Casalena G, Daehn I, Bottinger E: Transforming growth factor-β, bioenergetics, and mitochondria in renal disease. Semin Nephrol 32: 295–303, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Jain M, Rivera S, Monclus EA, Synenki L, Zirk A, Eisenbart J, Feghali-Bostwick C, Mutlu GM, Budinger GR, Chandel NS: Mitochondrial reactive oxygen species regulate transforming growth factor-β signaling. J Biol Chem 288: 770–777, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Finkel T: Signal transduction by mitochondrial oxidants. J Biol Chem 287: 4434–4440, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Suzuki T, Kimura M, Asano M, Fujigaki Y, Hishida A: Role of atrophic tubules in development of interstitial fibrosis in microembolism-induced renal failure in rat. Am J Pathol 158: 75–85, 2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Gröne HJ, Weber K, Gröne E, Helmchen U, Osborn M: Coexpression of keratin and vimentin in damaged and regenerating tubular epithelia of the kidney. Am J Pathol 129: 1–8, 1987 [PMC free article] [PubMed] [Google Scholar]
- 24.Ward JM, Stevens JL, Konishi N, Kurata Y, Uno H, Diwan BA, Ohmori T: Vimentin metaplasia in renal cortical tubules of preneoplastic, neoplastic, aging, and regenerative lesions of rats and humans. Am J Pathol 141: 955–964, 1992 [PMC free article] [PubMed] [Google Scholar]
- 25.Polichnowski AJ, Lan R, Geng H, Griffin KA, Venkatachalam MA, Bidani AK: Severe renal mass reduction impairs recovery and promotes fibrosis after AKI. J Am Soc Nephrol 25: 1496–1507, 2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ormos J, Elemér G, Csapó Z: Ultrastructure of the proximal convoluted tubules during repair following hormonally induced necrosis in rat kidney. Virchows Arch B Cell Pathol Incl Mol Pathol 13: 1–13, 1973 [DOI] [PubMed] [Google Scholar]
- 27.Kempczinski RF, Caulfield JB: A light and electron microscopic study of renal tubular regeneration. Nephron 5: 249–264, 1968 [DOI] [PubMed] [Google Scholar]
- 28.Venkatachalam MA, Bernard DB, Donohoe JF, Levinsky NG: Ischemic damage and repair in the rat proximal tubule: Differences among the S1, S2, and S3 segments. Kidney Int 14: 31–49, 1978 [DOI] [PubMed] [Google Scholar]
- 29.Weinberg JM: Mitochondrial biogenesis in kidney disease. J Am Soc Nephrol 22: 431–436, 2011 [DOI] [PubMed] [Google Scholar]
- 30.Ju W, Eichinger F, Bitzer M, Oh J, McWeeney S, Berthier CC, Shedden K, Cohen CD, Henger A, Krick S, Kopp JB, Stoeckert CJ Jr., Dikman S, Schröppel B, Thomas DB, Schlondorff D, Kretzler M, Böttinger EP: Renal gene and protein expression signatures for prediction of kidney disease progression. Am J Pathol 174: 2073–2085, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Conde E, Alegre L, Blanco-Sánchez I, Sáenz-Morales D, Aguado-Fraile E, Ponte B, Ramos E, Sáiz A, Jiménez C, Ordoñez A, López-Cabrera M, del Peso L, de Landázuri MO, Liaño F, Selgas R, Sanchez-Tomero JA, García-Bermejo ML: Hypoxia inducible factor 1-alpha (HIF-1 alpha) is induced during reperfusion after renal ischemia and is critical for proximal tubule cell survival. PLoS One 7: e33258, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Heyman SN, Rosenberger C, Rosen S: Experimental ischemia-reperfusion: Biases and myths-the proximal vs. distal hypoxic tubular injury debate revisited. Kidney Int 77: 9–16, 2010 [DOI] [PubMed] [Google Scholar]
- 33.Potter C, Harris AL: Hypoxia inducible carbonic anhydrase IX, marker of tumour hypoxia, survival pathway and therapy target. Cell Cycle 3: 164–167, 2004 [PubMed] [Google Scholar]
- 34.Domingo M, Einig C, Eigenbrodt E, Reinacher M: Immunohistological demonstration of pyruvate kinase isoenzyme type L in rat with monoclonal antibodies. J Histochem Cytochem 40: 665–673, 1992 [DOI] [PubMed] [Google Scholar]
- 35.Schering B, Reinacher M, Schoner W: Localization and role of pyruvate kinase isoenzymes in the regulation of carbohydrate metabolism and pyruvate recycling in rat kidney cortex. Biochim Biophys Acta 881: 62–71, 1986 [DOI] [PubMed] [Google Scholar]
- 36.Wirthensohn G, Guder WG: Renal substrate metabolism. Physiol Rev 66: 469–497, 1986 [DOI] [PubMed] [Google Scholar]
- 37.Basile DP, Donohoe DL, Roethe K, Mattson DL: Chronic renal hypoxia after acute ischemic injury: Effects of L-arginine on hypoxia and secondary damage. Am J Physiol Renal Physiol 284: F338–F348, 2003 [DOI] [PubMed] [Google Scholar]
- 38.Basile DP: Rarefaction of peritubular capillaries following ischemic acute renal failure: A potential factor predisposing to progressive nephropathy. Curr Opin Nephrol Hypertens 13: 1–7, 2004 [DOI] [PubMed] [Google Scholar]
- 39.Goldfarb M, Rosenberger C, Abassi Z, Shina A, Zilbersat F, Eckardt KU, Rosen S, Heyman SN: Acute-on-chronic renal failure in the rat: Functional compensation and hypoxia tolerance. Am J Nephrol 26: 22–33, 2006 [DOI] [PubMed] [Google Scholar]
- 40.Abdelkader A, Ho J, Ow CP, Eppel GA, Rajapakse NW, Schlaich MP, Evans RG: Renal oxygenation in acute renal ischemia-reperfusion injury. Am J Physiol Renal Physiol 306: F1026–F1038, 2014 [DOI] [PubMed] [Google Scholar]
- 41.Semenza GL, Roth PH, Fang HM, Wang GL: Transcriptional regulation of genes encoding glycolytic enzymes by hypoxia-inducible factor 1. J Biol Chem 269: 23757–23763, 1994 [PubMed] [Google Scholar]
- 42.Semenza GL: Regulation of metabolism by hypoxia-inducible factor 1. Cold Spring Harb Symp Quant Biol 76: 347–353, 2011 [DOI] [PubMed] [Google Scholar]
- 43.Rardin MJ, Wiley SE, Naviaux RK, Murphy AN, Dixon JE: Monitoring phosphorylation of the pyruvate dehydrogenase complex. Anal Biochem 389: 157–164, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kim JW, Tchernyshyov I, Semenza GL, Dang CV: HIF-1-mediated expression of pyruvate dehydrogenase kinase: A metabolic switch required for cellular adaptation to hypoxia. Cell Metab 3: 177–185, 2006 [DOI] [PubMed] [Google Scholar]
- 45.Lu CW, Lin SC, Chen KF, Lai YY, Tsai SJ: Induction of pyruvate dehydrogenase kinase-3 by hypoxia-inducible factor-1 promotes metabolic switch and drug resistance. J Biol Chem 283: 28106–28114, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Bagnasco S, Good D, Balaban R, Burg M: Lactate production in isolated segments of the rat nephron. Am J Physiol 248: F522–F526, 1985 [DOI] [PubMed] [Google Scholar]
- 47.Kraus-Friedmann N: Hormonal regulation of hepatic gluconeogenesis. Physiol Rev 64: 170–259, 1984 [DOI] [PubMed] [Google Scholar]
- 48.Minchenko A, Leshchinsky I, Opentanova I, Sang N, Srinivas V, Armstead V, Caro J: Hypoxia-inducible factor-1-mediated expression of the 6-phosphofructo-2-kinase/fructose-2,6-bisphosphatase-3 (PFKFB3) gene. Its possible role in the Warburg effect. J Biol Chem 277: 6183–6187, 2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Chesney J, Mitchell R, Benigni F, Bacher M, Spiegel L, Al-Abed Y, Han JH, Metz C, Bucala R: An inducible gene product for 6-phosphofructo-2-kinase with an AU-rich instability element: Role in tumor cell glycolysis and the Warburg effect. Proc Natl Acad Sci U S A 96: 3047–3052, 1999 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Sakakibara R, Kato M, Okamura N, Nakagawa T, Komada Y, Tominaga N, Shimojo M, Fukasawa M: Characterization of a human placental fructose-6-phosphate, 2-kinase/fructose-2,6-bisphosphatase. J Biochem 122: 122–128, 1997 [DOI] [PubMed] [Google Scholar]
- 51.Spurgeon KR, Donohoe DL, Basile DP: Transforming growth factor-beta in acute renal failure: Receptor expression, effects on proliferation, cellularity, and vascularization after recovery from injury. Am J Physiol Renal Physiol 288: F568–F577, 2005 [DOI] [PubMed] [Google Scholar]
- 52.Fähling M, Mathia S, Paliege A, Koesters R, Mrowka R, Peters H, Persson PB, Neumayer HH, Bachmann S, Rosenberger C: Tubular von Hippel-Lindau knockout protects against rhabdomyolysis-induced AKI. J Am Soc Nephrol 24: 1806–1819, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Chen J, Chen JK, Harris RC: Deletion of the epidermal growth factor receptor in renal proximal tubule epithelial cells delays recovery from acute kidney injury. Kidney Int 82: 45–52, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Zeng F, Singh AB, Harris RC: The role of the EGF family of ligands and receptors in renal development, physiology and pathophysiology. Exp Cell Res 315: 602–610, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Tang J, Liu N, Zhuang S: Role of epidermal growth factor receptor in acute and chronic kidney injury. Kidney Int 83: 804–810, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Zhuang S, Dang Y, Schnellmann RG: Requirement of the epidermal growth factor receptor in renal epithelial cell proliferation and migration. Am J Physiol Renal Physiol 287: F365–F372, 2004 [DOI] [PubMed] [Google Scholar]
- 57.Zhuang S, Yan Y, Han J, Schnellmann RG: p38 kinase-mediated transactivation of the epidermal growth factor receptor is required for dedifferentiation of renal epithelial cells after oxidant injury. J Biol Chem 280: 21036–21042, 2005 [DOI] [PubMed] [Google Scholar]
- 58.Hallman MA, Zhuang S, Schnellmann RG: Regulation of dedifferentiation and redifferentiation in renal proximal tubular cells by the epidermal growth factor receptor. J Pharmacol Exp Ther 325: 520–528, 2008 [DOI] [PubMed] [Google Scholar]
- 59.Nowak G, Schnellmann RG: Integrative effects of EGF on metabolism and proliferation in renal proximal tubular cells. Am J Physiol 269: C1317–C1325, 1995 [DOI] [PubMed] [Google Scholar]
- 60.Chen J, Chen JK, Nagai K, Plieth D, Tan M, Lee TC, Threadgill DW, Neilson EG, Harris RC: EGFR signaling promotes TGFβ-dependent renal fibrosis. J Am Soc Nephrol 23: 215–224, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Bonniaud P, Margetts PJ, Kolb M, Schroeder JA, Kapoun AM, Damm D, Murphy A, Chakravarty S, Dugar S, Higgins L, Protter AA, Gauldie J: Progressive transforming growth factor beta1-induced lung fibrosis is blocked by an orally active ALK5 kinase inhibitor. Am J Respir Crit Care Med 171: 889–898, 2005 [DOI] [PubMed] [Google Scholar]
- 62.Kapoun AM, Gaspar NJ, Wang Y, Damm D, Liu YW, O’young G, Quon D, Lam A, Munson K, Tran TT, Ma JY, Murphy A, Dugar S, Chakravarty S, Protter AA, Wen FQ, Liu X, Rennard SI, Higgins LS: Transforming growth factor-beta receptor type 1 (TGFbetaRI) kinase activity but not p38 activation is required for TGFbetaRI-induced myofibroblast differentiation and profibrotic gene expression. Mol Pharmacol 70: 518–531, 2006 [DOI] [PubMed] [Google Scholar]
- 63.Jat PS, Noble MD, Ataliotis P, Tanaka Y, Yannoutsos N, Larsen L, Kioussis D: Direct derivation of conditionally immortal cell lines from an H-2Kb-tsA58 transgenic mouse. Proc Natl Acad Sci U S A 88: 5096–5100, 1991 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Sinha D, Wang Z, Price VR, Schwartz JH, Lieberthal W: Chemical anoxia of tubular cells induces activation of c-Src and its translocation to the zonula adherens. Am J Physiol Renal Physiol 284: F488–F497, 2003 [DOI] [PubMed] [Google Scholar]
- 65.McLean IW, Nakane PK: Periodate-lysine-paraformaldehyde fixative. A new fixation for immunoelectron microscopy. J Histochem Cytochem 22: 1077–1083, 1974 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.