

Abstract
Gain-of-function mutations of classic transient receptor potential channel 6 (TRPC6) were identified in familial FSGS, and increased expression of wild-type TRPC6 in glomeruli is observed in several human acquired proteinuric diseases. Synaptopodin, an actin binding protein that is important in maintaining podocyte function, is downregulated in various glomerular diseases. Here, we investigated whether synaptopodin maintains podocyte function by regulating podocyte surface expression and activity of TRPC6. We show indirect interaction and nonrandom association of synaptopodin and TRPC6 in podocytes. Knockdown of synaptopodin in cultured mouse podocytes increased the expression of TRPC6 at the plasma membrane, whereas overexpression of synaptopodin decreased it. Mechanistically, synaptopodin–dependent TRPC6 surface expression required functional actin and microtubule cytoskeletons. Overexpression of wild–type or FSGS–inducing mutant TRPC6 in synaptopodin-depleted podocytes enhanced TRPC6–mediated calcium influx and induced apoptosis. In vivo, knockdown of synaptopodin also caused increased podocyte surface expression of TRPC6. Administration of cyclosporin A, which stabilizes synaptopodin, reduced LPS-induced proteinuria significantly in wild-type mice but to a lesser extent in TRPC6 knockout mice. Furthermore, administration of cyclosporin A reversed the LPS-induced increase in podocyte surface expression of TRPC6 in wild-type mice. Our findings suggest that alteration in synaptopodin levels under disease conditions may modify intracellular TRPC6 channel localization and activity, which further contribute to podocyte dysfunction. Reducing TRPC6 surface levels may be a new approach to restoring podocyte function.
Keywords: podocyte, TRPC6, synaptopodin, surface expression, calcium, proteinuria
Transient receptor potential channel 6 (TRPC6) is a nonselective calcium–permeable cation channel that is expressed in a wide range of cells, including kidney podocytes. Gain-of-function mutations in TRPC6 were identified to cause autosomal dominant FSGS.1–3 It was also shown that glomerular expression of wild-type (wt) TRPC6 was elevated in acquired human glomerular diseases, including minimal change disease, FSGS, and membranous GN.4 Moreover, proteinuria caused by angiotensin II treatment was attenuated in transient receptor potential channel 6 knockout (TRPC6−/−) mice.5 These findings led to the hypothesis that hyperactive mutants of the channel or increased channel expression, possibly via increased TRPC6–mediated calcium signaling, cause podocyte dysfunction and glomerular damage. In support of this hypothesis, Krall et al.6 showed that transgenic mice overexpressing wt or mutated TRPC6 channels in kidney podocytes exhibited proteinuria and glomerular lesions that resembled human FSGS.
Many published studies concerning regulation and function of TRPC6 investigated overall expression of the channel. However, TRPC6 activity has been reported to be at least partially controlled by regulation of the channel’s expression at the cell surface.7 Disease-causing mutations of TRPC6 have been shown to have increased cell surface levels.1 Limiting surface levels of wtTRPC6 in cardiomyocytes by Klotho was shown to be protective against cardiac hypertrophy.8 Studies that focused on surface TRPC6 were mostly done in model cell lines.7,9–11 It was recently reported that TRPC6 surface expression in podocytes could be affected by insulin.12 However, mechanisms and functional consequences of modification of TRPC6 podocyte surface expression under physiologic and pathologic conditions have yet to be elucidated.
Synaptopodin is a podocyte–specific actin binding protein. By stabilizing stress fibers, synaptopodin plays a crucial role in regulating podocyte actin dynamics and sustaining glomerular filter function.13–15 Decreased synaptopodin expression in glomeruli has been observed in numerous kidney diseases, including FSGS and HIV-associated nephropathy16; idiopathic nephrotic syndrome of childhood, including minimal change disease; and diffuse mesangial hypercellularity.17 In addition, low synaptopodin levels were reported to be associated with poor response to steroid therapy.17,18 Reduction of synaptopodin was also found in several in vitro and in vivo kidney disease models, including LPS and angiotensin II mouse models.19–21 Restoring synaptopodin levels has been considered as a potential approach to improving glomerular functions. Cyclosporin A (CsA), a drug that has been used to treat FSGS, was shown to protect podocytes by preventing degradation of synaptopodin.19
In this study, we present evidence that TRPC6 surface expression in podocytes depends on expression levels of synaptopodin. We observed that downregulation of synaptopodin resulted in elevated TRPC6 at podocyte surface and increased apoptosis on TRPC6 activation. Conversely, overexpression of synaptopodin led to decreased surface TRPC6. The changes in TRPC6 surface levels were accompanied by correspondent changes in calcium influx mediated by the channel. We also show that in vivo knockdown of synaptopodin led to increased podocyte surface TRPC6. CsA was able to alleviate LPS-induced proteinuria in wt mice and to a lesser degree in TRPC6−/− mice. Finally, we show that, in wt mice, podocyte surface TRPC6 was elevated on LPS treatment and abrogated by administration of CsA.
Results
TRPC6 Interacts with Synaptopodin and Associates with Synaptopodin in Podocyte Foot Processes
To explore the association of TRPC6 with important regulators of podocyte function, we performed coimmunoprecipitation (Co-IP) in HEK293 cells transfected with TRPC6-GFP and flag-tagged synaptopodin (Synpo-flag) as well as slit diaphragm components podocin-flag, nephrin-flag, and CD2AP-flag (Figure 1A, Supplemental Figure 1A). We found that TRPC6 interacted with synaptopodin as well as podocin and nephrin. We confirmed the interaction between TRPC6 and synaptopodin in cultured mouse podocytes (Figure 1B). However, we did not detect direct interaction between the two proteins by far Western blotting (Figure 1C, Supplemental Figure 1, B and C). Nonrandom association of TRPC6 and synaptopodin in podocyte foot processes was found in mouse glomeruli by immunogold double labeling of synaptopodin and TRPC6 (Figure 1D, Supplemental Figure 1D).
Figure 1.

TRPC6 interacts and associates with synaptopodin in podocytes. (A) Co-IP performed in HEK293 cotransfected with TRPC6-GFP, Synpo-flag, podocin-flag, nephrin-flag, and CD2AP-flag. (B) Co-IP performed in cultured mouse podocytes. TRPC6 or synaptopodin was pulled down together with antibody against synaptopodin or TRPC6. (C) Far Western blotting shows no direct interaction between the indicated isoforms13 of synaptopodin and TRPC6. Podocin-flag was used as a positive control, and CD2AP-flag was used as a negative control. Notice that a major band appears between 66 kD and 146 kD and that another band is >480 kD, corresponding to the molecular masses of the TRPC6 monomer (approximately 110 kD) and tetramer (the functional unit of the TRPC6 channel; approximately 440 kD). (D) A representative image of immunogold double labeling of TRPC6 and synaptopodin shows TRPC6 and synaptopodin locating in close vicinity in podocytes foot processes. D, a and b are magnifications of areas highlighted in D. Quantification is shown in the table: numbers of foot processes that contained synaptopodin and/or TRPC6 gold particles or did not contain any gold particles were counted, and quantitative double–labeling analysis was performed according to the method described in the work by Mayhew et al.34 The Fisher exact test probability (P<0.001) together with the odds ratio (3.39; 95% confidence interval, 1.786 to 6.454) indicate that there is nonrandom association of TRPC6 and synaptopodin in foot processes. IP, immunoprecipitation; WB, Western blot. Scale bar, 500 nm in D; gold particle diameter for TRPC6, 15 nm; synaptopodin, 10 nm.
Surface TRPC6 Expression Levels Are Affected by Synaptopodin Levels in Podocytes
To explore the biologic significance of the association of TRPC6 and synaptopodin, we first considered the possibility of synaptopodin regulating TRPC6 overall expression in podocytes. Effective knockdown and overexpression of synaptopodin in differentiated podocytes were achieved by lentiviral transduction (Supplemental Figure 2). We found that total TRPC6 mRNA and protein were not affected by downregulation or overexpression of synaptopodin (Supplemental Figure 2). We then considered if changes in synaptopodin expression would affect cell surface levels of endogenous TRPC6, overexpressed wtTRPC6, and FSGS–causing mutant TRPC6M131T.3 Surface TRPC6 expression was determined by surface biotinylation and compared between podocytes transduced with scrambled short hairpin RNA (SC) and synaptopodin short hairpin RNA (SynpoKD) lentiviruses (Figure 2A). Surface HA and TRPC6 were blotted in SC and SynpoKD podocytes that overexpressed wtTRPC6 (named SC-wtTRPC6 and SynpoKD-wtTRPC6, respectively) (Figure 2B) or TRPC6M131T (named SC-TRPC6M131T and SynpoKD-TRPC6M131T, respectively) (Figure 2C). Quantification showed an approximately 2.8-fold increase of surface TRPC6 in SynpoKD compared with SC podocytes (Figure 2, A and D), an approximately 2.5-fold increase in SynpoKD-wtTRPC6 compared with SC-wtTRPC6 podocytes (Figure 2, B and E), and an approximately 1.4-fold increase in SynpoKD-TRPC6M131T compared with SC-TRPC6M131T podocytes (Figure 2, C and F).
Figure 2.

Surface biotinylation assays show the effects of synaptopodin expression on cell surface TRPC6 levels. Representative Western blots of surface biotinylation assays and quantifications of TRPC6 levels in (A and D) SC and SynpoKD podocytes (n=5), (B and E) SC-wtTRPC6 and SynpoKD-wtTRPC6 podocytes (n=4), (C and F) SC-TRPC6M131T and SynpoKD-TRPC6M131T podocytes (n=4), (G and J) EV and SynpoOE podocytes (n=3), (H and K) EV-wtTRPC6 and SynpoOE-wtTRPC6 podocytes (n=4), and (I and L) EV-TRPC6M131T and SynpoOE-TRPC6M131T podocytes (n=4). Extra bands above the endogenous TRPC6 band in wtTRPC6-HA– and TRPC6M131T-HA–overexpressing podocyte lysates (Supplemental Figure 3B) were confirmed to be glycosylated forms of TRPC630,35 and used in quantification of TRPC6 intensity in addition to the endogenous TRPC6 band. Pan–Cadherin protein levels remained unchanged in SynpoKD– and SynpoOE– or wtTRPC6– and TRPC6M131T–overexpressing podocytes (Supplemental Figures 2, B and D and 3A); therefore, they were used as loading controls for total and surface fractions. ER protein calnexin served as an indicator of the purity of the cell surface fraction. Note that the glycosylated forms (upper bands) of TRPC6 appear more prominent in cell surface fraction, indicating that the glycosylated TRPC6 was enriched. GraphPad Software’s multiple t tests were used for statistical analysis. Graphs represent mean±SEM. *P<0.05; **P<0.01; ***P<0.001.
Alternatively, we tested whether overexpression of synaptopodin caused decreased surface TRPC6. Transduced podocytes were labeled as follows: empty vector (EV), overexpression of Synpo-long13 (SynpoOE), EV-wtTRPC6, SynpoOE-wtTRPC6, EV- TRPC6M131T, and SynpoOE-TRPC6M131T. Quantification showed a 28.9% decrease of surface TRPC6 in SynpoOE compared with EV podocytes (Figure 2, G and J), a 41.3% decrease in SynpoOE-wtTRPC6 compared with EV-wtTRPC6 podocytes (Figure 2, H and K), and a 23.2% decrease in SynpoOE-TRPC6M131T compared with EV-TRPC6M131T podocytes (Figure 2, I and L). Together, these results showed that downregulation of synaptopodin caused increased podocyte surface TRPC6 and that overexpression of synaptopodin led to reduced podocyte surface TRPC6.
Quantitative Immunocytochemistry Analyses of TRPC6 Plasma Membrane Expression
In addition to surface biotinylation assays, we used the Opera High Content Screening (HCS) System (PerkinElmer, Waltham, MA) to quantitatively measure the expression of TRPC6 on plasma membrane in response to up- or downregulated synaptopodin in podocytes. SC, SynpoKD, SC-wtTRPC6, SynpoKD-wtTRPC6, SC-TRPC6M131T, and SynpoKD-TRPC6M131T podocytes as well as EV, SynpoOE, EV-wtTRPC6, SynpoOE-wtTRPC6, EV-TRPC6M131T, and SynpoOE-TRPC6M131T podocytes were stained for TRPC6 and synaptopodin. Synaptopodin intensity was quantitated to confirm the knockdown and overexpression (Figure 3, B and D). Consistent with the results of surface biotinylation assays, TRPC6 membrane-to-total ratios were elevated in synaptopodin downregulated podocytes (Figure 3C) but decreased in synaptopodin-overexpressing podocytes (Figure 3E).
Figure 3.

TRPC6 expression in the cell membrane region is affected by synaptopodin levels in podocytes shown by quantitative immunocytochemistry with the Opera HCS System. (A) Membrane and cytoplasm regions of individual cells were designated by Columbus software as shown. Membrane region (defined as ±5% of total cell area along the edge of the cell) and cytoplasm regions (remaining cell area) of individual cells were designated, and the intensities of the two regions of each cell were recorded. For a particular well, the sum of mean fluorescence intensity (MFI) of TRPC6 at the membrane region and the MFI of TRPC6 at the cytoplasm region was calculated as total MFI of TRPC6: TRPC6 membrane-to-total ratio = MFI of TRPC6 at membrane/total MFI of TRPC6. In this manner, the fluorescence intensity of TRPC6 at plasma membrane was quantitated on the basis of all of the cells in a well (usually between 400 and 600 cells) by an unbiased methodology. (B) Synaptopodin intensity significantly decreased in SynpoKD podocytes. (C) TRPC6 membrane-to-total ratios were increased in SynpoKD, SynpoKD-wtTRPC6, and SynpoKD-TRPC6M131T podocytes compared with SC, SC-wtTRPC6, and SC- TRPC6M131T podocytes. (D) Synaptopodin intensity significantly increased in Synpo-flag virus–transduced podocytes. (E) TRPC6 membrane-to-total ratios in SynpoOE, SynpoOE-wtTRPC6, and SynpoOE-TRPC6M131T decreased compared with EV, EV-wtTRPC6, and EV-TRPC6M131T podocytes. Quantification was on the basis of four repeated wells for each condition. GraphPad Software’s multiple t tests were used for statistical analysis. Graphs represent mean±SEM. *P<0.05; **P<0.01; ****P<0.001.
TRPC6–Mediated Calcium Influx Is Altered by Synaptopodin, and Apoptosis Is Induced in Synaptopodin Knockdown Podocytes
We further investigated whether changes in synaptopodin expression can affect the calcium influx mediated by TRPC6. We treated podocytes with Hyp9, a stabilized derivative of the TRPC6–specific activator hyperforin,22 for calcium imaging. There was no significant difference among baseline levels (Figure 4D). The integrated intracellular calcium ([Ca2+]i) (Figure 4E) as well as the normalized peaks of calcium influx (Figure 4F) were greater in SynpoKD-wtTRPC6 and SynpoKD-TRPC6M131T podocytes compared with their controls, with a larger difference between SynpoKD-TRPC6M131T and SC-TRPC6M131T than between SynpoKD-wtTRPC6 and SC-wtTRPC6 (Figure 4, E and F). However, in wt podocytes, the normalized peak and integrated [Ca2+]i were only slightly higher in SynpoKD than SC cells but not statistically significant, possibly because the changes to the very low levels of endogenous surface TRPC6 might not be enough to cause a significant difference in [Ca2+]i. In contrast, the integrated [Ca2+]i significantly decreased in SynpoOE-wtTRPC6 and SynpoOE-TRPC6M131T cells compared with their EV controls (Supplemental Figure 4E), except for wt podocytes. The normalized peaks showed decreased trends in all of the SynpoOE podocytes, but they were not statistically significant (Supplemental Figure 4F). This could be because of the fact that, in podocytes with adequate levels of synaptopodin, the effect of excessive synaptopodin is limited. These results were consistent with the changes in protein levels of podocyte surface TRPC6 shown above. Together, the effects of synaptopodin levels on TRPC6 cell surface levels resulted in modification of TRPC6 activity.
Figure 4.

TRPC6–mediated calcium influx is altered by synaptopodin, and apoptosis is induced in SynpoKD podocytes with TRPC6 overexpression. Calcium influx is shown by Fura-2AM calcium imaging and represented by 340-to-380–nm ratios in (A) SC and SynpoKD podocytes, (B) SC-wtTRPC6 and SynpoKD-wtTRPC6 podocytes, and (C) SC- TRPC6M131T and SynpoKD-TRPC6M131T podocytes. (D) Baselines of 340-to-380–nm ratios (average of the ratios before adding Hyp9). (E) Quantification of 340-to-380–nm ratio changes from 90 seconds (adding Hyp9) to 400 seconds; 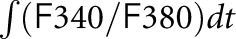 integrated the 340-to-380–nm ratio during 90 and 400 seconds. (F) Peak values of the 340-to-380–nm ratio normalized to the according baselines. (G) Representative Opera confocal images of Annexin V/PI–stained untreated podocytes showing negative Annexin V and PI staining and Hyp9 (4 μM for 2 hours) –treated SynpoKD-TRPC6M131T podocytes showing positive Annexin V and negative PI staining (cells were also stained with CellMask Blue to outline cytoplasm and nuclei). (H) Percentages of early apoptotic cells (Annexin V positive/PI negative) in SC, SynpoKD, SC-wtTRPC6, SynpoKD-wtTRPC6, SC-TRPC6M131T, and SynpoKD-TRPC6M131T podocytes treated with 4 μM Hyp9 for 2 hours. Quantification was on the basis of three repeated wells for each condition. GraphPad Software’s multiple t tests were used for statistical analysis. Graphs represent mean±SEM. *P<0.05; ***P<0.001.
integrated the 340-to-380–nm ratio during 90 and 400 seconds. (F) Peak values of the 340-to-380–nm ratio normalized to the according baselines. (G) Representative Opera confocal images of Annexin V/PI–stained untreated podocytes showing negative Annexin V and PI staining and Hyp9 (4 μM for 2 hours) –treated SynpoKD-TRPC6M131T podocytes showing positive Annexin V and negative PI staining (cells were also stained with CellMask Blue to outline cytoplasm and nuclei). (H) Percentages of early apoptotic cells (Annexin V positive/PI negative) in SC, SynpoKD, SC-wtTRPC6, SynpoKD-wtTRPC6, SC-TRPC6M131T, and SynpoKD-TRPC6M131T podocytes treated with 4 μM Hyp9 for 2 hours. Quantification was on the basis of three repeated wells for each condition. GraphPad Software’s multiple t tests were used for statistical analysis. Graphs represent mean±SEM. *P<0.05; ***P<0.001.
To investigate the functional consequence of the increased TRPC6 activity in synaptopodin knockdown podocytes, we performed the Annexin V/PI Apoptosis Assay (Life Technologies, Grand Island, NY) (Figure 4G) using the Opera HCS System. SynpoKD-TRPC6M131T podocytes had a significantly increased percentage of apoptotic cells (9.82%±1.92%) compared with SC-TRPC6M131T cells (3.89%±0.76%) (P<0.05) as well as all other conditions on Hyp9 stimulation (Figure 4H). Increased apoptosis was also observed in SynpoKD-wtTRPC6 podocytes (3.02%±0.18%) compared with SC-wtTRPC6 cells (1.32%±0.44%; P<0.05) (Figure 4H). These results suggest that the elevated [Ca2+]i in SynpoKD podocytes with TRPC6 overexpression causes damage to podocytes, which leads to apoptosis on channel activation, with TRPC6M131T causing more severe injury than wtTRPC6.
Actin and Microtubule Are Involved in Regulation of Surface TRPC6 by Synaptopodin
Both actin and microtubules have been shown to extensively participate in membrane protein trafficking to plasma membrane.23 We investigated how disruption of actin and microtubules would influence TRPC6 membrane-to-total ratios in SynpoKD podocytes using the Opera HCS System. We treated SynpoKD and SC podocytes with cytochalasin D (F–actin disruption agent)24 or nocodazole (microtubule disruption agent)25 overnight at the concentrations indicated in Figure 5, A and B and measured the TRPC6 membrane-to-total ratios with the Opera HCS System at the end of treatment. Dose-dependent disruption of F actin or microtubule cytoskeleton was confirmed with phalloidin or tubulin staining by confocal microscopy (Figure 5C, Supplemental Figures 6 and 7). TRPC6 membrane-to-total ratios decreased as the concentrations of cytochalasin D and nocodazole increased in both SC and SynpoKD podocytes, with a greater decrease in SynpoKD (Figure 5, A and B), and eventually, the differences diminished at 800 nM for cytochalasin D and 60 nM for nocodazole. These data suggest that synaptopodin–dependent TRPC6 membrane localization requires both functional actin and the microtubule system.
Figure 5.

Actin and microtubule cytoskeletons are involved in the regulation of TRPC6 surface expression by synaptopodin in podocytes. (A) TRPC6 membrane-to-total ratios in cytochalasin D–treated podocytes decrease as the concentrations of cytochalasin D increase. (B) TRPC6 membrane-to-total ratios in nocodazole-treated podocytes decrease as the concentrations of nocodazole increase. GraphPad Software’s multiple t test was performed to compare the ratios between SynpoKD and SC podocytes at the same concentration of the same treatment. Graphs represent mean±SEM. **P<0.01; ***P<0.001. (C) Representative immunocytochemistry images by confocal microscopy show patterns of TRPC6, F-actin, and tubulin in cytochalasin D– or nocodzole-treated podocytes. Note that, in 800 nM cytochalasin D–treated podocytes, TRPC6 staining presented an aster–like aggregated pattern corresponding to actin pattern (middle panel). In nocodazole-treated podocytes, TRPC6 became unevenly distributed and appeared more concentrated in cytoplasm (bottom panel).
In Vivo Knockdown of Synaptopodin Leads to Increased Podocyte Surface TRPC6
To investigate if synaptopodin affects TRPC6 surface expression in podocytes in vivo, we knocked down synaptopodin in mice by intravenously injecting chemically stabilized Synpo siRNAs (Figure 6, A and B). To test if podocyte TRPC6 expression and localization were affected, we enriched podocytes from the glomeruli isolated from the Synpo siRNA– and nontargeting siRNA–injected mice (Supplemental Figure 8) and performed surface biotinylation. Synaptopodin protein levels in total cell lysates decreased by approximately 54% in podocytes from Synpo siRNA–injected mice (Figure 6, A and B). An approximately 1.6-fold increase of surface TRPC6 was detected in these cells, with no change in total expression (Figure 6, A and C). No proteinuria was detected in the SynpoKD mice (Supplemental Figure 9A), suggesting that 54% downregulation of synaptopodin is not sufficient to cause proteinuria.
Figure 6.

In vivo knockdown of synaptopodin leads to increased podocyte surface TRPC6, and CsA reduces podocyte surface TRPC6 of LPS-treated mice. (A) Representative Western blots showing TRPC6 and synaptopodin in total and surface fractions of enriched podocytes from nontargeting siRNA– and Synpo siRNA–injected mice. pan-Cadherin was used as loading control. HSP90 (heat shock protein 90) was used to indicate the purity of surface fraction. Synaptopodin was seen in the surface fraction, possibly because of its interaction with TRPC6 and other proteins on the plasma membrane, such as podocin and nephrin. (B) Synaptopodin expression was normalized to glyceraldehyde-3-phosphate dehydrogenase (GAPDH) in total cell lysates. (C) TRPC6 levels were normalized to pan-Cadherin, and fold changes were calculated by comparing TRPC6 levels of Synpo siRNAs mice with those of nontargeting mice in the same fraction. The in vivo knockdown experiment (four to five mice in each group) was performed three times for quantification of the protein levels; t tests and GraphPad Software’s multiple t tests were performed in the experiments shown in B and C. (D) Urinary albumin (milligrams)-to-creatinine (grams) ratios in wt and TRPC6−/− mice with LPS and/or CsA treatment. Albumin and creatinine levels were measured from urine samples taken 36 hours after LPS injection (Supplemental Figure 9B). Control mice were injected with saline and olive oil, LPS mice were injected with LPS and olive oil, and LPS+CsA mice were injected with LPS and CsA; six to nine mice were used in each group. Ordinary one–way ANOVA was performed to analyze proteinuria within wt or TRPC6 mice; multiple t tests were performed to compare proteinuria between wt and TRPC6−/− mice. (E) Representative Western blots showing TRPC6 and synaptopodin in total and surface fractions of isolated podocytes from LPS or LPS and CsA–treated mice. (F) Synaptopodin expression was normalized to GAPDH in total cell lysates. (G) TRPC6 levels were normalized to pan-Cadherin, and fold changes were calculated by comparing TRPC6 levels of LPS and LPS and CsA to that of the control in the same fraction. The LPS and CsA treatment (three mice in each group) for the surface biotinylation assays was performed three times for quantification of the protein levels. Ordinary one–way ANOVA was performed in F and G. Graphs represent mean±SEM. *P<0.05; **P<0.01; ***P<0.001.
CsA Reduces Podocyte Surface TRPC6 in LPS-Treated Mice
To further test our hypothesis in a disease model, we used an LPS mouse model, in which synaptopodin has been shown to be downregulated by LPS treatment and rescued by CsA.19 We hypothesized that CsA protects podocytes from LPS injury partially by reducing TRPC6 membrane expression through preservation of synaptopodin in podocytes. LPS induced proteinuria in wt C57BL/6 mice (81.6±5.0 mg/g versus 2108.7±219.2 mg/g; P<0.001) (Figure 6D). The LPS-induced proteinuria was significantly reduced by CsA cotreatment (2108.7±219.2 mg/g versus 668.5±253.2 mg/g; an approximately 68.3% decrease; P<0.01) (Figure 6D). In TRPC6−/− mice, LPS proteinuria was approximately 45% less than that in wt mice (1161.3±271.1 mg/g versus 2108.7±219.2 mg/g; P<0.05) (Figure 6D), suggesting that TRPC6 contributes to LPS-induced proteinuria. CsA treatment in TRPC6−/− mice was also able to reduce LPS-induced proteinuria but to a lesser extent (654.4±247.6 mg/g versus 1161.3±271.1 mg/g; an approximately 43.6% decrease; P>0.05) compared with its effect in wt mice (Figure 6D).
To further analyze the role of TRPC6 in CsA treatment, we enriched podocytes from control, LPS-treated, and LPS and CsA–treated wt mice and performed surface biotinylation assays. Synaptopodin protein levels in total fraction were decreased by LPS and rescued by CsA (Figure 6, E and F). Accordingly, surface TRPC6 was elevated on LPS treatment and lowered by CsA in wt mice with total TRPC6 unchanged (Figure 6, E and G). Together with the in vitro data, these observations suggested that CsA protects podocytes from LPS treatment in mice partially by lowering podocyte surface TRPC6 levels through stabilization of synaptopodin.
Discussion
Gain-of-function variants of TRPC6 have been identified as a cause of hereditary FSGS, and upregulation of TRPC6 was found in several forms of acquired proteinuric diseases.1–4 Synaptopodin, a key regulator o actin cytoskeleton homeostasis in podocytes, was reported to be downregulated in various human and rodent glomerular diseases.16,17,19–21 Our study provides evidence that expression of TRPC6 on plasma membrane and calcium entry via TRPC6 in podocytes are affected by synaptopodin. We first showed that TRPC6 and synaptopodin interact and significantly associate with each other in podocyte foot processes. In differentiated mouse podocytes, knocking down synaptopodin results in increased surface expression of TRPC6 accompanied by enhanced TRPC6–mediated calcium influx. Apoptosis is induced in synaptopodin-depleted podocytes with TRPC6 overexpression. On the contrary, overexpression of synaptopodin leads to decreased podocyte surface TRPC6 and calcium influx. These findings suggest that synaptopodin limits podocyte surface TRPC6 expression, and the interaction of these two proteins is likely to be involved in the mechanism.
Most of the TRPC6 mutations associated with FSGS exhibit detectable increases in channel activity compared with that in wt channels.3,26 The mechanism of how these mutants cause glomerular abnormalities is still unclear. In our in vitro study, we included the mouse mutant TRPC6M131T corresponding to the human childhood FSGS–causing mutant TRPC6M132T that gives rise to significantly higher currents and delayed inactivation compared with the wt channel.3 We observed by surface biotinylation assays that synaptopodin was able to modify surface expression of the wt and the mutant TRPC6 to different degrees (Figure 2). Specifically, in synaptopodin downregulated podocytes, surface wtTRPC6 was increased by 2.5-fold, whereas TRPC6M131T was increased by 1.4-fold. In synaptopodin-overexpressing podocytes, surface wtTRPC6 was reduced by 41.3%, whereas TRPC6M131T was reduced by 23.2%. These findings indicate that the same manipulation of synaptopodin expression results in less modification of surface expression of the mutant, suggesting that the mutation may alter the ability for synaptopodin to regulate the localization and function of the channel.
However, quantification of the calcium imaging data revealed that calcium influx invoked in SynpoKD-TRPC6M131T was significantly higher (both peak values and integrated [Ca2+]i) than that in all other podocytes, including SynpoKD-wtTRPC6 (Figure 4, E and F). In addition, our apoptosis assay showed that SynpoKD-TRPCM131T podocytes had the highest percentages of apoptotic cells on TRPC6 activation compared with those of all other conditions (Figure 4H). Although surface level of TRPC6M131T was less affected by synaptopodin than that of wtTRPC6 (Figure 2), the changes in [Ca2+]i and the damage caused to the cells were more significant with TRPC6M131T than with wtTRPC6. These observations suggest that, in podocytes expressing TRPC6M131T, when synaptopodin is downregulated (as often observed in glomerular damage), the mutant is able to cause severe cell damage because of abnormally high intracellular Ca2+. Physiologically or pathologically present TRPC6 activators, such as angiotensin II,27 may trigger podocyte injury or induce podocyte apoptosis through this mechanism. It is noteworthy that SynpoKD-wtTRPC6 podocytes also showed significantly higher calcium influx (Figure 4, B, E, and F) and more apoptosis (Figure 4H) than control SC-wtTRPC6 podocytes, suggesting that synaptopodin downregulation in wtTRPC6-overexpressing podocytes is still detrimental to the cells, although it is less significant than in TRPC6M131T.
Our study also provided evidence that regulation of podocyte surface TRPC6 levels depends on both microtubules and actin filaments, because disruption of both cytoskeleton structures resulted in abortion of the elevated surface TRPC6 levels in SynpoKD podocytes (Figure 5). It has been well accepted that microtubules and actin filaments are major components of protein trafficking in a cell.23 Considering the association shown in Figure 1, we postulate that TRPC6 and synaptopodin may form a protein complex (with possible mediators) in the cytoplasm under physiologic conditions. When synaptopodin is downregulated, TRPC6 dissociates and transports to the plasma membrane by a cytoskeleton-mediated mechanism. Thus, when this cytoskeleton structure is disrupted, less TRPC6 can be transported to the membrane, yielding a lower membrane-to-total ratio. As for scrambled podocytes, TRPC6 membrane-to-total ratio also generally followed a decreasing trend as the concentration of the drugs increased, but this was slower than in SynpoKD podocytes (Figure 5, A and B), possibly because of disruption of normal transport of TRPC6 to the membrane on cytochalasin D and nocodazole treatment, causing cytoplasmic retention of the channel (Figure 5C).
To verify our findings in vivo, we knocked down synaptopodin in mice by intravenously injecting Synpo siRNAs (Figure 6, A and B) and examined surface TRPC6 levels of the extracted podocytes. We found that podocyte surface TRPC6 increased by approximately 1.6-fold (P<0.05), whereas the overall TRPC6 remained unchanged (Figure 6, A and C). Therefore, we provided direct evidence that downregulation of synaptopodin in podocytes causes increased surface TRPC6. In another mouse model, we reproduced the previously published finding19 that rescue of synaptopodin by CsA partially protected wt mice from LPS-induced proteinuria (Figure 6D). It was suggested by Faul et al.19 that the protection was partially caused by preservation of podocyte actin cytoskeleton through stabilization of synaptopodin. We further showed that preserving synaptopodin by CsA is beneficial by maintaining low podocyte surface TRPC6 levels during glomerular damage. LPS caused a lower level of proteinuria in TRPC6−/− mice, and the protective effect of CsA was of lesser magnitude in TRPC6−/− mice compared with in wt mice (Figure 6D). These observations indicate that TRPC6 contributes to the induction of albuminuria, resulting from LPS–induced glomerular damage, and furthermore, that TRPC6 is involved in the protective effect of CsA. This was supported by our surface biotinylation results of the in vivo podocytes that showed reduced TRPC6 surface expression when cotreated with CsA (Figure 6, E and G).
In summary, our study provides evidence that podocyte surface expression of TRPC6 is limited by synaptopodin, showing a new role for this actin binding protein in protecting podocyte function. Synaptopodin requires functional actin and microtubule cytoskeletons for its regulation of cell surface TRPC6. Limiting TRPC6 surface levels and thereby, lowering its activity could provide a new approach for the promotion of glomerular health.
Concise Methods
Co-IP
Recombinant TRPC6-GFP was expressed in HEK293 cells with Synpo-flag, podocin-flag, nephrin-flag, or CD2AP-flag. Co-IP was performed using anti–flag-M2 beads (Sigma-Aldrich, St. Louis, MO) according to the manufacturer’s manual. Co-IP in cultured mouse podocytes was done as described previously.28 Protein A/G agarose (Santa Cruz Biotechnology, Santa Cruz, CA) was used for immunoprecipitation. Cell lysates and eluates were analyzed by Western blotting.
Far Western Blotting
Far Western blotting was performed on the basis of the protocol published by Wu et al.29 Briefly, bait proteins (Synpo-long-flag, Synpo-short-flag, SynpoT-flag, podocin-flag, and CD2AP-flag) were expressed in HEK293 cells and purified using anti–flag-M2 beads (Sigma-Aldrich). The prey protein TRPC6-HA and its negative control empty plasmid were transfected into HEK293 cells. Native Western blotting was then performed using the NativePAGE Novex Bis-Tris Gel System (Invitrogen, Carlsbad, CA) according to the manufacturer’s protocol; 50 μg HEK293 lysates containing TRPC6-HA or negative control were loaded per lane. PVDF membrane was incubated with purified bait proteins (total of 5 μg; 3.3 μg/ml) for 4 hours at room temperature. Binding of the bait proteins on the membrane was detected by probing the flag tag. Expression and location of TRPC6-HA were detected by probing the HA tag. The NativeMark standards (Invitrogen) were used as molecular weight markers.
Immunogold Electron Microscopy
Mouse kidneys were harvested and fixed in 4% PFA with 0.001% glutaraldehyde overnight. Kidney tissues were dehydrated and embedded in LR White Resin (London Resin Company Limited, London, United Kingdom) according to the manufacturer’s instructions. Thin sections of the tissues were placed on nickel grids for immunostaining. Images were taken by a Gatan Erlangshen ES1000W Camera on a Philips CM10 Electron Microscope.
Lentiviral Transduction
For overexpression, C–terminal HA mouse TRPC6 cDNA and mouse TRPC6M131T cDNA were subcloned into the VVPW lentiviral expression vector.6 RNA interference was used to knock down synaptopodin expression in podocytes. Short hairpin RNAs (shRNAs) were designed to target SynpoT sequence (an isoform of full-length synaptopodin that is upregulated in Synpo−/− podocytes).13 Because SynpoT is the C-terminal fragment of Synpo-long (full-length synaptopodin),13 the shRNAs that target SynpoT should also target Synpo-long. shRNAs were delivered by a lentiviral system into cultured mouse podocytes. We used the pLKO-TRC2 vector (RNAi Consortium; supplied by Addgene). Lentiviral transduction of podocytes was performed as described previously.30
Cell Surface Biotinylation
These assays were done using the Pierce Cell Surface Protein Isolation Kit (Thermo Fisher Scientific, Vernon Hills, IL). Briefly, podocytes were washed twice with PBS and incubated with Sulfo-NHS-SS-Biotin for 40 minutes at 4°C with gentle rocking. Reaction was stopped by adding quenching solution. After washing twice with TBS, cells were scraped gently and collected by centrifugation. Cells were then lysed, and the biotin-labeled proteins were isolated following the manufacturer’s instructions. Labeled proteins were eluted with 1× LDS sample buffer with 1× reducing agent (Life Technologies) by incubating at 70°C for 15 minutes. Eluates were analyzed by Western blotting.
Quantitative Immunocytochemistry with the Opera HCS System
Podocytes were grown and differentiated in 96-well CellCarrier Plates (PerkinElmer). Cells were transduced with lentiviruses or treated with cytochalasin D or nocodazole. Cells were then fixed and stained with primary and fluorophore–conjugated secondary antibodies and labeled with HCS CellMask Blue Stain (Life Technologies) for high-throughput imaging and analysis. Images of all cells in the wells of interest were obtained by the Opera HCS System and analyzed as described in Results by the Columbus Image Data Storage and Analysis System (PerkinElmer).
Calcium Imaging
Podocytes were grown on 35-mm glass–bottomed, collagen–coated culture dishes (In Vitro Scientific, Sunnyvale, CA) and transduced with lentiviruses as described above. For calcium imaging, podocytes were incubated with 1 μg calcium–sensitive dye Fura-2AM in HBSS (with Ca2+ and Mg2+; Life Technologies) for 15 minutes at 37°C. Cells were rinsed, and fresh HBSS was added for baseline recording. TRPC6–specific activator Hyp9 (Sigma-Aldrich) was added 90 seconds after baseline recording at a final concentration of 4 μM. Fura-2AM absorbs light at 340 and 380 nM depending on the binding of free calcium and emits at 512 nm. As such, the 340-to-380–nm excitation ratio changes as a function of cytosolic free calcium. Ratiometric imaging was carried out in podocytes (12 to 20 cells per condition) using the Attofluor Ratio Vision Digital Fluorescence Microscopy System (Atto Instruments, Inc., Rockville, MD) equipped with a Zeiss Axiovert S100 Inverted Microscope (Carl Zeiss GmbH, Jena, Germany) and F-Fluor ×40 1.3–Numerical Aperture Oil Immersion Objective.
Apoptosis Assay
Differentiated mouse podocytes were cultured in 96-well CellCarrier Plates and transduced with lentiviruses. Before the apoptosis assay, cells were treated with 4 μM Hyp9 (Sigma-Aldrich) at 37°C for 2 hours (or 0 and 1 hour in Supplemental Figure 5, B and C, respectively). Apoptosis was examined with the Alexa Fluor 488 AnnexinV/Dead Cell Apoptosis Kit (Life Technologies). Briefly, cells were gently washed with PBS in the wells and then, incubated with Annexin V/PI solution for 15 minutes at room temperature according to the manufacturer’s instructions. Stained cells were then fixed with 4% PFA at room temperature for 15 minutes and stained with CellMask Blue (Invitrogen) to outline the cytoplasm and nuclei for the Opera HCS System. Cells that were Annexin V positive and PI negative were counted as early apoptotic cells and labeled as apoptotic cells in this study. Cells treated with 0, 0.2, 0.5, and 1 mM H2O2 (Sigma-Aldrich) for 1 hour at 37°C were used as positive controls for the assay,31 and the intensity of the Annexin V and PI staining was used as references to set up cutoffs for Annexin V– and PI-positive cells (Supplemental Figure 5A).
Animal Studies
All animal studies were approved by the Rush University Medical Center Animal Institute Committee; 12- to 15-week-old female C57BL/6J mice from The Jackson Laboratory (Bar Harbor, ME) were used as wt mice. TRPC6−/− mice32 (originally with 129Sv/J:C57BL/6J as 1:1 background) were backcrossed with wt C57BL/6J mice for more than eight generations to obtain a pure C57BL/6J background; 12- to 14-week-old female TRPC6−/− mice were used.
In Vivo Synaptopodin Knockdown Experiment
The siRNAs used in in vivo synaptopodin knockdown were on the basis of the two shRNA sequences used in in vitro studies. siSTABLE chemically modified siRNAs targeting synaptopodin mRNA and nontargeting siRNA (negative control) were manufactured by Dharmacon (Lafayette, CO); 120 μg per mouse Synpo siRNAs or the same amount of nontargeting siRNAs mixed with Kidney In Vivo Transfection Reagents (Altogen Biosystems, Las Vegas, NV) were injected into wt mice (four to five mice per group) through tail veins. A secondary injection was performed 12–14 hours after the first injection. Urine and kidneys were collected 24–26 hours after the secondary injection.
LPS and CsA Injection Experiment
The wt and TRPC6−/− mice were divided into three groups for each genotype (seven to nine mice per group for wt and six to seven mice per group for TRPC6−/−). Group 1 (control group) mice were subcutaneously injected with olive oil (Sigma-Aldrich) at 100 μl/d for 4 consecutive days as control for CsA. On day 3, mice were intraperitoneally injected with 300 μl saline (G-Biosciences, St. Louis, MO) as control for LPS. Group 2 (experimental group with LPS) mice were subcutaneously injected with olive oil at 100 μl/d for 4 consecutive days. On day 3, mice were intraperitoneally injected with ultrapure LPS (Invivogen, San Diego, CA) at 7 mg/kg in 300 μl saline. Group 3 (experimental group with LPS and CsA) mice were subcutaneously injected with CsA dissolved in 100 μl olive oil at 15 mg/kg per day for 4 consecutive days. On day 3, mice were intraperitoneally injected with one dose of ultrapure LPS at 7 mg/kg in 300 μl saline; 16 hours after LPS injection, 500 μl saline was injected into all mice to prevent hypovolemia, and 36 hours after LPS injection, urine samples were collected. Urinary albumin levels were determined by the Mouse Albumin ELISA Kit (Bethyl Laboratories, Montgomery, TX). Urinary creatinine levels were measured by the Creatinine Assay Kit (Cayman Chemicals, Ann Arbor, MI).
Glomerular Isolation and Podocyte Enrichment
The method used in this study was adapted from the glomerular and podocyte preparation method described by Boerries et al.33 Briefly, kidneys were harvested, minced into 1-mm3 pieces, and incubated in digestion buffer (DMEM; Life Technologies) containing collagenase (300 U/ml), collagenase type 4 (Worthington, Lakewood, NJ), and DNase I (50 U/ml; New England Biolabs, Ipswich, MA) at 37°C for 20 minutes with gentle shaking. The digested kidneys were sieved sequentially through two 100-μm cell strainers. Rinsed flow through was added onto a 40-μm cell strainer (glomeruli remained on the mesh). After extensive washes, the glomeruli were then suspended with DMEM and removed from the mesh. Biotinylation of the isolated glomeruli was carried out as described above. The biotinylated glomeruli were further digested into single cells with digestion buffer for 45 minutes at 37°C with vigorous shaking. Single-cell suspension was rinsed with DMEM and then, added to antibody-coated dishes (details are in Supplemental Figure 8). Cells were allowed to attach at 4°C for 4 hours. The unattached cells were gently washed off, and the cells remaining on the dish were processed for isolation of the biotinylated proteins.
Statistical Analyses
Statistical comparison was made between control and experimental groups with experiments conducted during the same time period. Each experiment was repeated at least three times. Fisher exact tests, Student's t-test, multiple t tests, and one-way ANOVAs followed by Tukey multiple comparisons were performed as indicated in the figures using Prism software (GraphPad Software, La Jolla, CA).
Disclosures
J.R. is a cofounder of TRISAQ, Inc, a biotechnology company in which he has financial interest, including stock. J.R. also has issued and pending patents on strategies for kidney therapeutics and stands to gain royalties from their commercialization.
Supplementary Material
Acknowledgments
We thank Dr. Dony Maiguel for extensive technical assistance with the Opera High Content Screening System and valuable advice on the manuscript.
This work was supported by National Institutes of Health grant 5R01DK089394, with resources from the Rush University Medical Center.
Footnotes
Published online ahead of print. Publication date available at www.jasn.org.
This article contains supplemental material online at http://jasn.asnjournals.org/lookup/suppl/doi:10.1681/ASN.2015080896/-/DCSupplemental.
References
- 1.Winn MP, Conlon PJ, Lynn KL, Farrington MK, Creazzo T, Hawkins AF, Daskalakis N, Kwan SY, Ebersviller S, Burchette JL, Pericak-Vance MA, Howell DN, Vance JM, Rosenberg PB: A mutation in the TRPC6 cation channel causes familial focal segmental glomerulosclerosis. Science 308: 1801–1804, 2005 [DOI] [PubMed] [Google Scholar]
- 2.Reiser J, Polu KR, Möller CC, Kenlan P, Altintas MM, Wei C, Faul C, Herbert S, Villegas I, Avila-Casado C, McGee M, Sugimoto H, Brown D, Kalluri R, Mundel P, Smith PL, Clapham DE, Pollak MR: TRPC6 is a glomerular slit diaphragm-associated channel required for normal renal function. Nat Genet 37: 739–744, 2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Heeringa SF, Möller CC, Du J, Yue L, Hinkes B, Chernin G, Vlangos CN, Hoyer PF, Reiser J, Hildebrandt F: A novel TRPC6 mutation that causes childhood FSGS. PLoS One 4: e7771, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Möller CC, Wei C, Altintas MM, Li J, Greka A, Ohse T, Pippin JW, Rastaldi MP, Wawersik S, Schiavi S, Henger A, Kretzler M, Shankland SJ, Reiser J: Induction of TRPC6 channel in acquired forms of proteinuric kidney disease. J Am Soc Nephrol 18: 29–36, 2007 [DOI] [PubMed] [Google Scholar]
- 5.Eckel J, Lavin PJ, Finch EA, Mukerji N, Burch J, Gbadegesin R, Wu G, Bowling B, Byrd A, Hall G, Sparks M, Zhang ZS, Homstad A, Barisoni L, Birbaumer L, Rosenberg P, Winn MP: TRPC6 enhances angiotensin II-induced albuminuria. J Am Soc Nephrol 22: 526–535, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Krall P, Canales CP, Kairath P, Carmona-Mora P, Molina J, Carpio JD, Ruiz P, Mezzano SA, Li J, Wei C, Reiser J, Young JI, Walz K: Podocyte-specific overexpression of wild type or mutant trpc6 in mice is sufficient to cause glomerular disease. PLoS One 5: e12859, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Cayouette S, Lussier MP, Mathieu EL, Bousquet SM, Boulay G: Exocytotic insertion of TRPC6 channel into the plasma membrane upon Gq protein-coupled receptor activation. J Biol Chem 279: 7241–7246, 2004 [DOI] [PubMed] [Google Scholar]
- 8.Xie J, Cha S-K, An S-W, Kuro-O M, Birnbaumer L, Huang C-L: Cardioprotection by Klotho through downregulation of TRPC6 channels in the mouse heart. Nat Commun 3: 1238, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lussier MP, Lepage PK, Bousquet SM, Boulay G: RNF24, a new TRPC interacting protein, causes the intracellular retention of TRPC. Cell Calcium 43: 432–443, 2008 [DOI] [PubMed] [Google Scholar]
- 10.Cayouette S, Bousquet SM, Francoeur N, Dupré E, Monet M, Gagnon H, Guedri YB, Lavoie C, Boulay G: Involvement of Rab9 and Rab11 in the intracellular trafficking of TRPC6. Biochim Biophys Acta 1803: 805–812, 2010 [DOI] [PubMed] [Google Scholar]
- 11.Kanda S, Harita Y, Shibagaki Y, Sekine T, Igarashi T, Inoue T, Hattori S: Tyrosine phosphorylation-dependent activation of TRPC6 regulated by PLC-γ1 and nephrin: Effect of mutations associated with focal segmental glomerulosclerosis. Mol Biol Cell 22: 1824–1835, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kim EY, Anderson M, Dryer SE: Insulin increases surface expression of TRPC6 channels in podocytes: Role of NADPH oxidases and reactive oxygen species. Am J Physiol Renal Physiol 302: F298–F307, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Asanuma K, Kim K, Oh J, Giardino L, Chabanis S, Faul C, Reiser J, Mundel P: Synaptopodin regulates the actin-bundling activity of alpha-actinin in an isoform-specific manner. J Clin Invest 115: 1188–1198, 2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Asanuma K, Yanagida-Asanuma E, Faul C, Tomino Y, Kim K, Mundel P: Synaptopodin orchestrates actin organization and cell motility via regulation of RhoA signalling. Nat Cell Biol 8: 485–491, 2006 [DOI] [PubMed] [Google Scholar]
- 15.Yanagida-Asanuma E, Asanuma K, Kim K, Donnelly M, Young Choi H, Hyung Chang J, Suetsugu S, Tomino Y, Takenawa T, Faul C, Mundel P: Synaptopodin protects against proteinuria by disrupting Cdc42:IRSp53:Mena signaling complexes in kidney podocytes. Am J Pathol 171: 415–427, 2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Barisoni L, Kriz W, Mundel P, D’Agati V: The dysregulated podocyte phenotype: A novel concept in the pathogenesis of collapsing idiopathic focal segmental glomerulosclerosis and HIV-associated nephropathy. J Am Soc Nephrol 10: 51–61, 1999 [DOI] [PubMed] [Google Scholar]
- 17.Srivastava T, Garola RE, Whiting JM, Alon US: Synaptopodin expression in idiopathic nephrotic syndrome of childhood. Kidney Int 59: 118–125, 2001 [DOI] [PubMed] [Google Scholar]
- 18.Hirakawa M, Tsuruya K, Yotsueda H, Tokumoto M, Ikeda H, Katafuchi R, Fujimi S, Hirakata H, Iida M: Expression of synaptopodin and GLEPP1 as markers of steroid responsiveness in primary focal segmental glomerulosclerosis. Life Sci 79: 757–763, 2006 [DOI] [PubMed] [Google Scholar]
- 19.Faul C, Donnelly M, Merscher-Gomez S, Chang YH, Franz S, Delfgaauw J, Chang JM, Choi HY, Campbell KN, Kim K, Reiser J, Mundel P: The actin cytoskeleton of kidney podocytes is a direct target of the antiproteinuric effect of cyclosporine A. Nat Med 14: 931–938, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Tian D, Jacobo SM, Billing D, Rozkalne A, Gage SD, Anagnostou T, Pavenstädt H, Hsu HH, Schlondorff J, Ramos A, Greka A: Antagonistic regulation of actin dynamics and cell motility by TRPC5 and TRPC6 channels. Sci Signal 3: ra77, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Markó L, Kvakan H, Park JK, Qadri F, Spallek B, Binger KJ, Bowman EP, Kleinewietfeld M, Fokuhl V, Dechend R, Müller DN: Interferon-γ signaling inhibition ameliorates angiotensin II-induced cardiac damage. Hypertension 60: 1430–1436, 2012 [DOI] [PubMed] [Google Scholar]
- 22.Müller M, Essin K, Hill K, Beschmann H, Rubant S, Schempp CM, Gollasch M, Boehncke WH, Harteneck C, Müller WE, Leuner K: Specific TRPC6 channel activation, a novel approach to stimulate keratinocyte differentiation. J Biol Chem 283: 33942–33954, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Alfonso A, Payne GS, Donaldson J, Segev N: Trafficking Inside Cells: Pathways, Mechanisms and Regulation, Berlin, Springer Science & Business Media, 2010 [Google Scholar]
- 24.Casella JF, Flanagan MD, Lin S: Cytochalasin D inhibits actin polymerization and induces depolymerization of actin filaments formed during platelet shape change. Nature 293: 302–305, 1981 [DOI] [PubMed] [Google Scholar]
- 25.Vasquez RJ, Howell B, Yvon AM, Wadsworth P, Cassimeris L: Nanomolar concentrations of nocodazole alter microtubule dynamic instability in vivo and in vitro. Mol Biol Cell 8: 973–985, 1997 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Mottl AK, Lu M, Fine CA, Weck KE: A novel TRPC6 mutation in a family with podocytopathy and clinical variability. BMC Nephrol 14: 104, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ilatovskaya DV, Palygin O, Chubinskiy-Nadezhdin V, Negulyaev YA, Ma R, Birnbaumer L, Staruschenko A: Angiotensin II has acute effects on TRPC6 channels in podocytes of freshly isolated glomeruli. Kidney Int 86: 506–514, 2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kim EY, Alvarez-Baron CP, Dryer SE: Canonical transient receptor potential channel (TRPC)3 and TRPC6 associate with large-conductance Ca2+-activated K+ (BKCa) channels: Role in BKCa trafficking to the surface of cultured podocytes. Mol Pharmacol 75: 466–477, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wu Y, Li Q, Chen XZ: Detecting protein-protein interactions by Far western blotting. Nat Protoc 2: 3278–3284, 2007 [DOI] [PubMed] [Google Scholar]
- 30.Kistler AD, Singh G, Altintas MM, Yu H, Fernandez IC, Gu C, Wilson C, Srivastava SK, Dietrich A, Walz K, Kerjaschki D, Ruiz P, Dryer S, Sever S, Dinda AK, Faul C, Reiser J: Transient receptor potential channel 6 (TRPC6) protects podocytes during complement-mediated glomerular disease. J Biol Chem 288: 36598–36609, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Liu BC, Song X, Lu XY, Li DT, Eaton DC, Shen BZ, Li XQ, Ma HP: High glucose induces podocyte apoptosis by stimulating TRPC6 via elevation of reactive oxygen species. Biochim Biophys Acta 1833: 1434–1442, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Dietrich A, Mederos Y Schnitzler M, Gollasch M, Gross V, Storch U, Dubrovska G, Obst M, Yildirim E, Salanova B, Kalwa H, Essin K, Pinkenburg O, Luft FC, Gudermann T, Birnbaumer L: Increased vascular smooth muscle contractility in TRPC6-/- mice. Mol Cell Biol 25: 6980–6989, 2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Boerries M, Grahammer F, Eiselein S, Buck M, Meyer C, Goedel M, Bechtel W, Zschiedrich S, Pfeifer D, Laloë D, Arrondel C, Gonçalves S, Krüger M, Harvey SJ, Busch H, Dengjel J, Huber TB: Molecular fingerprinting of the podocyte reveals novel gene and protein regulatory networks. Kidney Int 83: 1052–1064, 2013 [DOI] [PubMed] [Google Scholar]
- 34.Mayhew TM, Lucocq JM: Multiple-labelling immunoEM using different sizes of colloidal gold: Alternative approaches to test for differential distribution and colocalization in subcellular structures. Histochem Cell Biol 135: 317–326, 2011 [DOI] [PubMed] [Google Scholar]
- 35.Schlöndorff J, Del Camino D, Carrasquillo R, Lacey V, Pollak MR: TRPC6 mutations associated with focal segmental glomerulosclerosis cause constitutive activation of NFAT-dependent transcription. Am J Physiol Cell Physiol 296: C558–C569, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.