

Abstract
Results have historically shown a broad plasticity in the origin of tumors and their functions, with significant heterogeneity observed in both morphologies and functional capabilities. Largely unknown, however, are the mechanisms by which these variations occur and how these events influence tumor formation and behavior. Contemporary views on the origin of tumors focuses mainly on the role of particular sets of driver transformations, mutational or epigenetic, with the occurrence of the observed heterogeneity as an accidental byproduct of oncogenesis. As such, we present a hypothesis that tumors form due to heterogeneous adaptive selection in response to environmental stress through intrinsic genomic sampling mechanisms. Specifically, we propose that eukaryotic cells intrinsically explore their available genomic information, the Greater Genomic Landscape, in response to stress under normal conditions, long before the formation of a cancerous lesion. Finally, considering the influence of chromatin heterogeneity on the Greater Genomic Landscape, we propose a new class of compounds, Chromatin Protective Therapies (CPTs), which target the physical variations in chromatin topology. In this approach, CPTs reduce the overall information space available to limit the formation of tumors or the development of drug-resistant phenotypes.
Discussion
Classically, evolution has been studied as the set of mechanisms that confer heritable traits from parents to their progeny. In this view evolutionary sampling confers traits that can be advantageous to the progeny under the appropriate circumstances. As such, under stress conditions that favor a given set of traits, the populations with those traits will clonally expand and predominate. In multicellular organisms the distinction between progeny and evolutionary fitness becomes blurred. Intuitively, clonal selection of cell populations within a tissue can be advantageous to the whole organism, but are not reproductively heritable to the multicellular progeny. For the cell population at the tissue level, the discovered adaptions are not classically selective but capacitive, i.e. the resulting heterogeneous population confers an advantage to a plurality of traits since a broader distribution can help in the face of new stresses. However, by definition, this increase in traits fundamentally changes the tissue over time.
The most studied model of this evolution-driven functional transformation in humans is cancer1–4. Largely unknown, however, are the mechanisms by which adaptive sampling occurs and how these events could result in the formation of tumors. Results have historically shown a plasticity in the origin of tumors, with heterogeneous mutational and epigenetic events occurring throughout a challenged organ preceding an eventual pathological expansion2,5,6. Furthermore, tissues under constant energetic and replicative pressures account for the demonstrable majority of tumors7. These observations, however, do not fully explain the broad distribution of molecular events that can precipitate tumor formation. Contemporary views on the origin of tumors derive from the monoclonal expansion of cells (tumor stem cells, clonal selection due to mutations or chromosome instability) into a lesion before the occurrence of the observed heterogeneous acceleration1. This view, however, does not explain the functional diversity in tissues under non-perturbed conditions even within cells of the same lineage8.
Here we present a hypothesis that tumors form due to heterogeneous adaptive selection in response to environmental stress through intrinsic genomic sampling mechanisms. Specifically, we propose that eukaryotic cells intrinsically explore their available genomic information in response to stress under normal conditions in real time, long before the formation of a cancerous lesion. This information, the Greater Genomic Landscape (GGL), is the available distribution of functional states: the current functions of the cell (proteomic/metabolic) and possible future states (genes that can be expressed/repressed or mutated). In essence, the GGL hypothesis merges critical traits of information theory and evolutionary biology to explain tumorigenesis as something other than an accidental byproduct, but a consequence of multicellular fitness. Specifically, the intrinsically encoded exploration of genomic information is a main adaptive advantage of multi-cellularity and occurs primarily at three levels and time scales: (1) post-translational proteomic (rapid – seconds/hours), (2) epigenomic (intermediate – minutes/days), and (3) mutational (days-weeks-years). For instance, let’s consider epigenomic sampling. The normal chromatin nanoenvironment helps restrict cells to a relatively small niche within the genomic information space formed by the estimated ~20,000 human protein-coding genes, however, deviations in chromatin structure, such as those observed in cancer cells, facilitate a greater genomic exploration. As such, the Greater Genomic Landscape should not be confused with the well-established cancer genome landscapes, which refers to the set of genes altered in carcinogenesis, while the GGL refers to the ability of a cell to sample its genome or, in other words, the cell’s potential trajectory in the ~20,000-dimensional space9.
Briefly, let’s consider some other intrinsically encoded mechanisms of information sampling for these three levels of genomic information. At the proteomic level, there are numerous non-transcriptional ways to alter cellular function. For instance, studies of yeast under stress demonstrate that eukaryotic cells employ a plurality of strategies to respond to conditions, including varying abundance and location of proteins (and mRNA), leading to a heterogeneity of initial conditions and variability of response to stress10,11. At the epigenomic level, there are both enzymatic and non-enzymatic ways to alter the information space. In tumorigenesis, there are numerous demonstrations of chromatin remodeling enzymes being critical drivers in chemoevasion and tumor formation. However, there is also an often overlooked level of epigenetic heterogeneity, which is to vary the initial configurations of chromatin structure to change accessibility and probability of expression for genes from cell to cell. Critically, both the proteomic and epigenetic mechanisms happen at time scales that are faster than the division of cells, allowing cells to discover new adaptions during exposure to stress. As demonstrated in Figure 1, the presence of rare subpopulations occurs at significant levels even while maintaining an “average” population. An increase in the heterogeneity of subpopulations does not necessarily transform the overall tissue function, but it can have a profound effect on the information space available to respond to stress conditions. Classically, this is considered at the time scale of cell division, with mutational alterations as the predominant mode of increasing the genomic information space by creating inherently new potential functions. In this way, mutational transformation is also the classical example of tumor heterogeneity, but occurs at time scales that are challenging to target pharmacologically.
Figure 1. Tumor formation models.
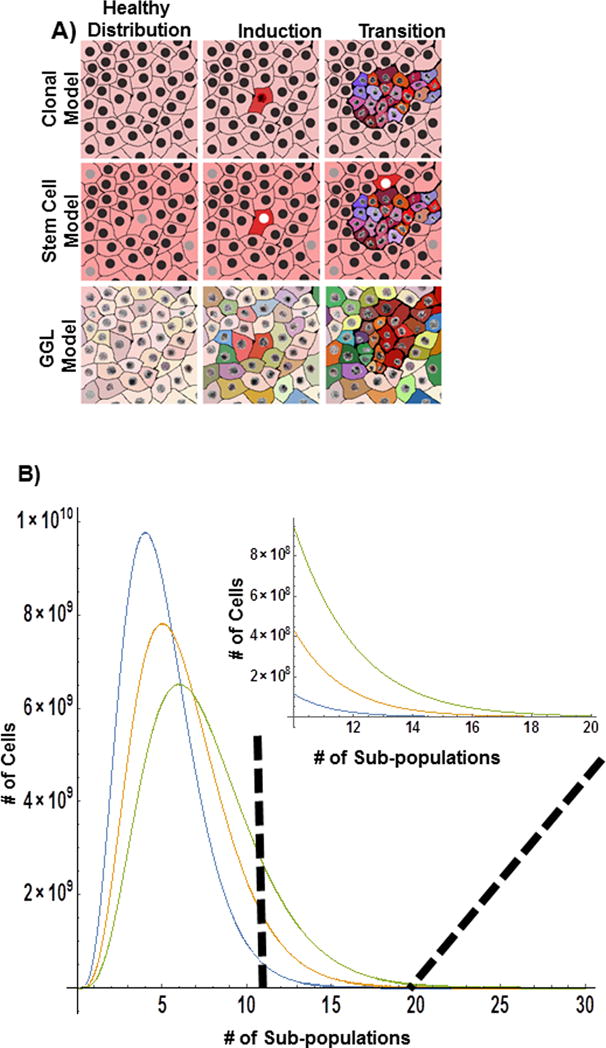
A) Clonal expansion secondary to perturbation is classically defined as the cause of tumorigenesis. Clonal expansion often well characterizes hematopoietic tumors and pediatric tumors, but often fails to explain the underlying heterogeneity observed in solid organ tumors. In the CSC model, tumors arise due to the formation of stem cells that give rise to new tumor with multiple subtypes, allowing for a partial heterogeneity in cell origin within a tumor. In contrast, the Greater Genomic Landscape focuses on the general feature of multicellular systems: their potential to change their function in the face of stress. In the GGL model, tumors arise due to the probability of a population arriving at a cancer state due to the selection of a large distribution of cell subpopulations (and functions) and from increased information sampling that it produces. B) Consider the case of 5 predominant subpopulations within the tissue for a given cell type. Assuming a population follows from a gamma distribution, small changes in the heterogeneity (scale parameter) result in large deviations in the number of subpopulations. As such, while the average population and tissue function does not change significantly, the total number of possible states (and functions) has increased.
Consequently, repeated and multidimensional stressors are will likely select for cells with traits that enhance the capacity to search the GGL, not just for a particular set of proteomic pathways or initial traits which in single cell systems is often termed “bet-hedging”. As a result, each perturbation increases the heterogeneity of the underlying tissues by favoring a broader distribution of semi-unique states and cells that have the greatest plasticity (capacity to search for new functions). Over time, this differential sampling of the genome produces an increasingly diverse population, commonly observed as the detection of overt tumors as they by definition have unique features. It is this tissue heterogeneity and intrinsic plasticity that acts as a conserved evolutionary mechanism that favors more exploratory cells in eukaryotic systems, resulting in tumor formation through the increased probability of proliferative configurations.
In this view, tumor formation is an evolution-driven information-sampling problem arising as stress induces the population of cells to sample the information coded within their genomes and proteomes to collectively maintain tissue function. The origins of these stresses are innumerable (alcohol, smoking, infections, etc) and as such, the tissue does not a priori know what mechanism of evasion will work for every perturbation. Instead, cells carry a limited repertoire of encoded proteins that include intrinsic samplers to rapidly and probabilistically search the GGL for solutions to maintain the underlying function of the tissue. This occurs not by just rapidly inducing all genes, but by combinatorially exploring the information space encoded across numerous subpopulations. Within an individual cell, these intrinsic samplers initiate a probabilistic search response at both the proteomic (post-translational modification) and genomic (chromatin remodeling, mutational transformation) levels. The cells that fail during this sampling under stress undergo apoptosis or mitotic arrest after a few hours.
For this mechanism to be a central evolutionary property of multicellular organisms, there must first be a distribution of time scales during which the levels of stress response occur. In particular, sampling must be relatively rapid in comparison to mechanisms of cellular clearance, i.e. apoptosis and immune-clearance. Interestingly, evidence of this separation of timescales has been observed previously, even indicating possible transition states between death and survival12. Irreversible commitment to apoptosis occurs over the course of several hours, while proteomic transformation and chromatin remodeling are very rapid (< a few minutes). This suggests that irreversible commitment to apoptosis is delayed in order to give cells time to find stress evasion mechanisms. Without this complementary intrinsic sampling mechanism, our tissues would fail under mild perturbation from unique stressors.
A second requirement of such a mechanism is the presence of central convergence points between exploration, apoptosis, and cellular arrest. As such, we consider that one potential regulator of intrinsic sampling of the GGL is mitochondrial membrane potential, Ψm. Mitochondria are ubiquitously implicated in diseases, specifically diseases of aging; e.g. tumors, neurodegeneration, and atherosclerosis13. Beyond this central association, disruption of Ψm has been shown to regulate the epigenetic structure of chromatin, molecular signaling cascades, and post-translational modification of cytoplasmic proteins13. Furthermore, processes directly linked to Ψm include apoptosis, proliferation, and senescence14. Consequently, Ψm could serve as the central barometer of cellular fitness, mediating sampling, apoptosis, and senescence concurrently. In this model, the disruption of Ψm would simultaneously induce proteomic and genomic exploration, initiate the apoptosome, and potentiate cell cycle arrest15,16. If the stressor is not resolved, either extrinsically or intrinsically, cells would commit to apoptosis to limit their use of resources required for the remaining cells.
The evolutionary selection of more robust samplers and an increasingly heterogeneous population of cells occurs primarily for two reasons. First, continuous maintenance of many traits is energetically unfavorable for an individual cell. Secondly, more robust samplers and a greater number of initial states will increase the likelihood of finding traits that prevent tissue failure during duress. With each perturbation event, selective pressures will transform tissues by increasingly favoring a broader distribution of cellular configurations and cells with increased plasticity. Over time, this accelerates the evasive fitness and increases the cellular heterogeneity present within the affected tissue1. Currently, this process is considered as an accidental byproduct of selective pressure favoring the initial configurations (tumor stem cells, clonal expansion) over the general feature (heterogeneous, elastic sampling in response to normal stress across entire cell populations)1,3–5,17. Unlike evolutionary fitness being derived from tumor stem cells or accumulated variations from clonal expansion, the GGL hypothesis indicates that differentiated cells are a major component in tumor formation because of their capacity to still explore their genomic potential in the face of repeated stressors at timescales preceding cell division.
With Ψm acting as one barometer of fitness, we expect evolutionary selection to produce cells with the following combinations of features. Cells that (1) more rapidly and thoroughly explore the genomic space; (2) have previously acquired a higher stress tolerance; (3) preferentially arrest to extend survival; and (4) have a broad distribution of initial states [Figure 1]. Most commonly, we study the mechanisms that increase damage tolerance, preferentially induce arrest, or more recently, increase the initial states1–5,17. Unfortunately, we are currently lacking thorough studies that focus on the primary feature of tumor formation: heterogeneous initial states and rapidly adaptive configurations that result in a larger exploration of the Greater Genomic Landscape in healthy tissues.
Previous characterizations of such an exploratory mechanism have too narrowly focused on global gene induction as a fitness mechanism. However, global gene induction should not be confused with differential exploration and tissue heterogeneity. In the GGL model, differential exploration selects for numerous populations of cells within a healthy (or unhealthy) tissue under the same stress. For example, at least two different mechanisms can favor cell survival in the presence of a toxin: (1) inactivating genes involved in the apoptotic cascade or (2) creating proteins that expel the stressor. As a result, repeated or multidimensional perturbations do not select for one trait, but instead broaden the distribution of initial cell states and favor more elastic samplers. Critically, this feature is likely conserved in normal tissues, and is not an adaption unique to carcinogenesis.
If evolutionary sampling of the GGL is a critical feature of tumorigenesis and normal tissue function, what are some potential mechanisms that would increase the exploration of the GGL and enhance the chance of cellular survival during stress conditions? One possible mechanism would be to delay the irreversible commitment to apoptosis, thereby extending the duration of exploration and allow the search of more possible evasive combinations18. A second mechanism would be the transformation of chromatin remodeling enzymes to increase the efficiency of combinatorial searches in response to stress19,20. A third mechanism could be to broaden the heterogeneity of chromatin structure of the cellular population, i.e. – vary the configurations to increase coverage across the entire population21. Conceptually, by increasing the distribution of chromatin organization across cells, each cell within the population has a different initial configuration state that produces a semi-unique exploration, enhancing the total information space [Figure 2]. As a result, 5 subpopulations would have ~3*1011 unique genomic configurations with only 1% variation in chromatin topology compared to 105 proteomic states with a similar level of proteomic variability.
Figure 2. Available information space for cellular subpopulations.
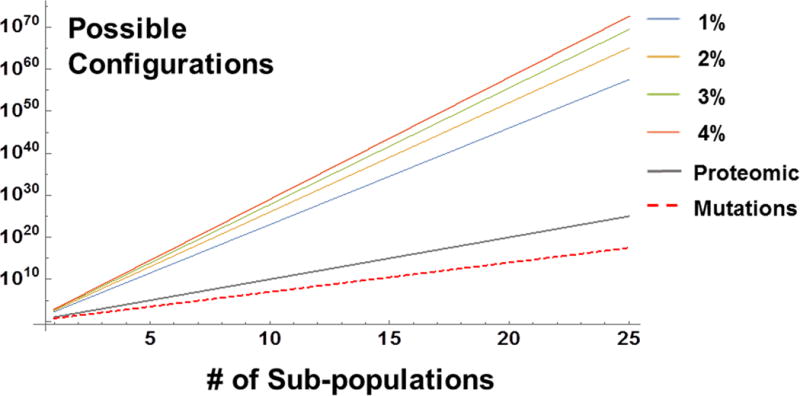
Conservative estimates in the heterogeneity of subpopulations for different types of cellular variance (assuming subpopulations are distinct, but largely share the same features). If cells each express 1000 proteins and only 10 are different between subpopulations (99% overlap in function), then 10n potential variations are possible. Likewise, if each subpopulation has 5 distinct mutations, ~3*1017 genetic states are possible for 25 distinct subpopulations. Often overlooked, however, is the effect of varying the physical configurations of chromatin. Even a 1% difference in the organizational topology would allow 3.3*1057 potential responses for 25 subpopulations. If physical heterogeneity increases to 4%, this increases to 3.7*1072, an ~1015 increase in possible responses! Given this asymmetry, drugs targeting variations in chromatin organization (Chromatin Protective Therapies) can greatly reduce the probability of either acquiring tumorigenic characteristics or chemoevasive expression.
This suggests that the underlying heterogeneity of chromatin organization (and the ability to modulate the structure) has a disproportionate influence on tissue function, cellular diversity, and fitness. Even without taking into consideration additional influences such as cell communication, distinct cellular populations, and the time evolution of chromatin structure, this suggests an overwhelming influence of physical organization of chromatin on the probability of tumor formation. While not every potential configuration would be attempted in every stress, it is the distribution (the total number of possibilities) that assist the tissue over long periods of time, as it allows tissues to function across many different exposures. The obvious tradeoff is that increased variation increases the probability of acquiring negative traits. Interestingly, the observation of physical heterogeneity of chromatin (variations in fractal dimension) as a prognostic marker in cancer is well conserved in solid tumors and may be a proxy for the underlying information space within a tissue (higher fractal dimension produces greater variability in structure)22.
Finally, the exploration of the GGL may have critical implications for early carcinogenesis and chemotherapy. In this context, expansion of the population heterogeneity can stabilize otherwise deleterious gene mutations, and eventually potentiate tumor formation by increasing the likelihood of finding stable negative states. Furthermore, increased exploration of the GGL would be expected to aid in the development of new traits unique to tumors, such as angiogenic induction or stabilization of abnormal metabolism. Likewise, this has important ramifications for chemotherapy. The current strategy behind most existing anti-cancer chemotherapies is to kill as many cancer cells as possible while preserving non-cancer cells. Consider a highly potent drug that kills 99.9% of cancer cells. After therapy, ~105 cancer cells will still survive per each gram of the original tumor23. However, clonal expansion alone does not characterize the distribution of evasive mechanisms found within the surviving cells. We can gain some insight into why cancer cells can evade chemotherapies by going back to our hypothesis that the heterogeneity of the chromatin nanoenvironment may help cells to explore a larger genomic information space. Coupled with a strong selective pressure, e.g. a chemotherapeutic agent, this leads to the emergence of new drug-resistant clones due to cells finding new evasive mechanisms during treatment. This is somewhat reminiscent of antibiotic treatment of bacterial infections: bacteria evolve at the timescale of treatment, which eventually leads to the emergence of drug-resistant organisms. Following this analogy, instead of–or in addition to–developing new targeted anti-cancer compounds, is it feasible to change the cancer cells’ ability to evolve and develop drug resistance, thus improving the efficacy of the existing therapies? Furthermore, can such a strategy be adaptable to the prevention of tumor formation, a long sought-after but largely elusive strategy24?
This approach would focus beyond targeting only drivers for each tumor, to limiting genomic exploration by targeting variations in the physical structure of chromatin using low-dose Chromatin Protective Therapies (CPTs). Current epigenetic chemotherapy mirrors other driver-based strategies: epigenetic regulators, such as HDAC inhibitors used to modulate focal gene expression. Instead, a CPT approach limits the degrees of freedom present within chromatin by regulating the overall physical structure, i.e. targeting topological variations. As described above, variations in chromatin structure from cell-to-cell allows cells to search for new mechanisms that aid in survival at low energetic cost. Furthermore, work from our lab suggests a correlation between heterogeneity of chromatin organization (fractal dimension) and the heterogeneity of gene expression for critical processes, including proliferation and apoptosis. We have consistently found an increased chromatin heterogeneity preceding the development of tumors in both human and animal models of carcinogenesis25–28. Likewise, theoretical modeling and experimental results have shown that changes in the physical environment can independently modulate transcription29,30. As such, the physical transformation of chromatin could have a significant role in tumor formation and chemoresistance independent of effects mediated by epigenetic chemical modifications. Therefore, ideal CPTs would focus on physiochemical regulators that can control the overall heterogeneity of chromatin structure, possibly by targeting metal-ion homeostasis or Ψm.
Through this approach, CPTs would complement existing strategies by decreasing the cumulative adaptive potential of tumor cells. Specifically, an adjuvant CPT would work by decreasing the probability of emergence of secondary proliferative and evasive mechanisms through restriction of the possible configurations of chromatin. By acting on the overall physical structure, CPTs restrict the global sampling capacities of cells to reduce the combinatorial dimensions of evasion [Figure 2]. Likewise, CPTs could be a long-sought-after prophylactic approach for patients with high-risk mutations by preventing accumulated sampling in addition to the known drivers of tumor formation. These prophylactic CPTs could additionally be employed therapeutically to restrict the accumulation of adaptions between courses of conventional treatments. In this approach, CPTs would prevent the possible sampling of different states during stress – considerably reducing the population of surviving cancer cells to those that previously acquired a favorable initial evasive state.
In summary, we propose an evolutionarily conserved mechanism derived from information sampling that drives the observed heterogeneity in tumor formation at the origin of healthy tissues. Specifically, we propose that tumor formation results from repeated stressors driving normal tissues to explore their Greater Genomic Landscape, i.e. the collective information space available to cells, to prevent organismal death in the face of stress. The exploration of the Greater Genomic Landscape is potentially mediated by mitochondrial membrane potential in conjunction with the potentiation of apoptosis and induction of cell cycle arrest. We further propose that this could lead to the development of Chromatin Protective Therapies, which would target global genomic exploration by controlling the physical topology of chromatin and the ability of cells generally to access genomic information. These Chromatin Protective Therapies would be a new class of prophylactics and neoadjuvants that lower the probability of premalignant transformation and the development of chemoevasion mechanisms by restricting the cellular capacity to explore their greater genomic landscape.
Acknowledgments
This material is based upon work supported by National Science Foundation Graduate Research Fellowship under Grant DGE-0824162. This work is also supported by the MSTP NIH T32 training grant, T32GM008152. Additional support was provided by the Lefkofsky Foundation, the National Science Foundation Grants No. CBET-1249311, and the National Institute of Health Grants No. U54CA193419, R01CA155284, R01CA165309.
Footnotes
Conflict of interest: Drs. Backman and Roy are shareholders of Nanocytomics, LLC
References
- 1.Magee JA, Piskounova E, Morrison SJ. Cancer stem cells: impact, heterogeneity, and uncertainty. Cancer Cell. 2012;21:283–296. doi: 10.1016/j.ccr.2012.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Gerlinger M, et al. Intratumor heterogeneity and branched evolution revealed by multiregion sequencing. N Engl J Med. 2012;366:883–892. doi: 10.1056/NEJMoa1113205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Swanton C. Intratumor heterogeneity: evolution through space and time. Cancer Res. 2012;72:4875–4882. doi: 10.1158/0008-5472.CAN-12-2217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Burrell RA, McGranahan N, Bartek J, Swanton C. The causes and consequences of genetic heterogeneity in cancer evolution. Nature. 2013;501:338–345. doi: 10.1038/nature12625. [DOI] [PubMed] [Google Scholar]
- 5.Ling S, et al. Extremely high genetic diversity in a single tumor points to prevalence of non-Darwinian cell evolution. Proc Natl Acad Sci U S A. 2015;112:E6496–6505. doi: 10.1073/pnas.1519556112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Easwaran H, Tsai HC, Baylin SB. Cancer epigenetics: tumor heterogeneity, plasticity of stem-like states, and drug resistance. Mol Cell. 2014;54:716–727. doi: 10.1016/j.molcel.2014.05.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Tomasetti C, Vogelstein B. Cancer etiology. Variation in cancer risk among tissues can be explained by the number of stem cell divisions. Science. 2015;347:78–81. doi: 10.1126/science.1260825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Sun J, et al. Clonal dynamics of native haematopoiesis. Nature. 2014;514:322–327. doi: 10.1038/nature13824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Vogelstein B, et al. Cancer genome landscapes. Science. 2013;339:1546–1558. doi: 10.1126/science.1235122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Levy SF, Ziv N, Siegal ML. Bet hedging in yeast by heterogeneous, age-correlated expression of a stress protectant. PLoS Biol. 2012;10:e1001325. doi: 10.1371/journal.pbio.1001325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Breker M, Gymrek M, Schuldiner M. A novel single-cell screening platform reveals proteome plasticity during yeast stress responses. J Cell Biol. 2013;200:839–850. doi: 10.1083/jcb.201301120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Yadav N, Chandra D. Mitochondrial and postmitochondrial survival signaling in cancer. Mitochondrion. 2014;16:18–25. doi: 10.1016/j.mito.2013.11.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lane RK, Hilsabeck T, Rea SL. The role of mitochondrial dysfunction in age-related diseases. Biochim Biophys Acta. 2015;1847:1387–1400. doi: 10.1016/j.bbabio.2015.05.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hay N. Interplay between FOXO, TOR, Akt. Biochim Biophys Acta. 2011;1813:1965–1970. doi: 10.1016/j.bbamcr.2011.03.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Antico Arciuch VG, Elguero ME, Poderoso JJ, Carreras MC. Mitochondrial regulation of cell cycle and proliferation. Antioxid Redox Signal. 2012;16:1150–1180. doi: 10.1089/ars.2011.4085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Czabotar PE, Lessene G, Strasser A, Adams JM. Control of apoptosis by the BCL-2 protein family: implications for physiology and therapy. Nat Rev Mol Cell Biol. 2014;15:49–63. doi: 10.1038/nrm3722. [DOI] [PubMed] [Google Scholar]
- 17.Welch DR. Tumor Heterogeneity-A ‘Contemporary Concept’ Founded on Historical Insights and Predictions. Cancer Res. 2016;76:4–6. doi: 10.1158/0008-5472.CAN-15-3024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144:646–674. doi: 10.1016/j.cell.2011.02.013. [DOI] [PubMed] [Google Scholar]
- 19.Morgan MA, Shilatifard A. Chromatin signatures of cancer. Genes Dev. 2015;29:238–249. doi: 10.1101/gad.255182.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Cancer Genome Atlas, N. Comprehensive molecular characterization of human colon and rectal cancer. Nature. 2012;487:330–337. doi: 10.1038/nature11252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Feinberg AP, Ohlsson R, Henikoff S. The epigenetic progenitor origin of human cancer. Nat Rev Genet. 2006;7:21–33. doi: 10.1038/nrg1748. [DOI] [PubMed] [Google Scholar]
- 22.Bedin V, Adam RL, de Sa BC, Landman G, Metze K. Fractal dimension of chromatin is an independent prognostic factor for survival in melanoma. BMC Cancer. 2010;10:260. doi: 10.1186/1471-2407-10-260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Rasnick D. Aneuploidy theory explains tumor formation, the absence of immune surveillance, and the failure of chemotherapy. Cancer Genet Cytogenet. 2002;136:66–72. doi: 10.1016/s0165-4608(01)00665-3. [DOI] [PubMed] [Google Scholar]
- 24.Brown K, Rufini A. New concepts and challenges in the clinical translation of cancer preventive therapies: the role of pharmacodynamic biomarkers. Ecancermedicalscience. 2015;9:601. doi: 10.3332/ecancer.2015.601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Cherkezyan L, et al. Nanoscale changes in chromatin organization represent the initial steps of tumorigenesis: a transmission electron microscopy study. BMC Cancer. 2014;14:189. doi: 10.1186/1471-2407-14-189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Stypula-Cyrus Y, et al. HDAC up-regulation in early colon field carcinogenesis is involved in cell tumorigenicity through regulation of chromatin structure. PLoS One. 2013;8:e64600. doi: 10.1371/journal.pone.0064600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Subramanian H, et al. Nanoscale cellular changes in field carcinogenesis detected by partial wave spectroscopy. Cancer Res. 2009;69:5357–5363. doi: 10.1158/0008-5472.can-08-3895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Subramanian H, et al. Optical methodology for detecting histologically unapparent nanoscale consequences of genetic alterations in biological cells. Proc Natl Acad Sci U S A. 2008;105:20118–20123. doi: 10.1073/pnas.0804723105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Matsuda H, Putzel GG, Backman V, Szleifer I. Macromolecular crowding as a regulator of gene transcription. Biophys J. 2014;106:1801–1810. doi: 10.1016/j.bpj.2014.02.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Tan C, Saurabh S, Bruchez MP, Schwartz R, Leduc P. Molecular crowding shapes gene expression in synthetic cellular nanosystems. Nat Nanotechnol. 2013;8:602–608. doi: 10.1038/nnano.2013.132. [DOI] [PMC free article] [PubMed] [Google Scholar]