

Abstract
Alcoholic liver disease (ALD) is mediated via activation of TLR-4 signaling; MyD88-dependent and -independent signals are important contributors to injury in mouse models. Adiponectin, an anti-inflammatory adipokine, suppresses TLR-4/MyD88-dependent responses via induction of heme oxygenase-1 (HO-1). Here we investigated the interactions between chronic ethanol, adiponectin and HO-1 in regulation of TLR4/MyD88-independent signaling in macrophages and an in vivo mouse model. After chronic ethanol feeding, LPS-stimulated expression of IFN-β and CXCL10 mRNA was increased in primary cultures of Kupffer cells compared to pair-fed controls. Treatment of Kupffer cells with globular adiponectin (gAcrp) normalized this response. LPS-stimulated IFN-β/CXCL10 mRNA and CXCL10 protein was also reduced in RAW 264.7 macrophages treated with gAcrp or full-length adiponectin. gAcrp and full-length adiponectin acted via adiponectin receptor-1 and receptor-2, respectively. gAcrp decreased TLR4 expression in both Kupffer cells and RAW 264.7 macrophages. siRNA knock-down of HO-1 or inhibition of HO-1 activity with zinc protoporphyrin blocked these effects of gAcrp. C57BL/6 mice were exposed to chronic ethanol feeding, with or without treatment with cobalt protoporphyrin (CoPP) to induce HO-1. After chronic ethanol feeding, mice were sensitized to in vivo challenge with LPS, expressing increased IFN-β/CXCL10 mRNA and CXCL10 protein in liver compared to controls. Pre-treatment with CoPP 24 h prior to LPS challenge normalized this effect of ethanol. Adiponectin and induction of HO-1 potently suppressed TLR4-dependent/MyD88-independent cytokine expression in primary Kupffer cells from rats and in mouse liver after chronic ethanol exposure. These data suggest that induction of HO-1 may be a useful therapeutic strategy in ALD.
Keywords: macrophage, alcoholic liver disease, inflammation, cobalt protoporphyrin, interferon
Introduction
The innate immune system is involved in many stages of an organism’s response to injury or stress. Components of the innate immune response, including natural killer (NK) and NKT cells (1), Kupffer cells (2) and the complement system (3–4), are involved in the hepatic response to chronic alcohol exposure. Kupffer cells, the resident macrophages in the liver, are particularly critical to the onset of ethanol-induced liver injury. Ablation of Kupffer cells prevents the development of fatty liver and inflammation in rats chronically exposed to ethanol via intragastric feeding. (5) Increased exposure of Kupffer cells to LPS during chronic ethanol consumption, associated with impaired barrier function of the intestine (6), results in activation of TLR4 and increased production of inflammatory mediators (7). Mice deficient in TLR4 or TNFα receptor 1 expression are protected from chronic ethanol-induced liver injury (7).
TLR4 signaling is mediated via MyD88-dependent and independent pathways (8). While the rapid LPS-stimulated expression of TNFα is characteristic of MyD88-dependent signaling, MyD88-independent signals are more slowly activated and result in increased expression of type I IFN and IFN-dependent genes (8). Chronic ethanol exposure sensitizes Kupffer cells to TLR4-MyD88-dependent responses, such as rapid activation of mitogen-activated protein kinases and NFκB, as well as TNFα expression (2). Treatment of Kupffer cells with adiponectin, an abundant 30-kDa adipokine with potent anti-inflammatory properties (9), normalizes these effects of chronic ethanol on LPS-induced MAPK and NFκB activation, as well as TNFα production in primary cultures of Kupffer cells (10). Treatment of mice with supra-physiological concentrations of adiponectin during chronic ethanol exposure prevents the development of liver injury, decreasing both steatosis and TNFα expression in the liver (11).
Despite the efficacy of adiponectin in decreasing LPS-stimulated responses in Kupffer cells and preventing liver injury in mice, the development of adiponectin for therapeutic interventions in patients with ALD is likely of limited utility, due to the high concentration of adiponectin in the circulation, as well as the complex oligomeric structure of adiponectin. Therefore, recent studies have focused on the down-stream molecular targets of adiponectin in order to identify anti-inflammatory targets that are more amenable to pharmacological intervention. We recently identified an IL-10 and heme-oxygenase-1 (HO-1)-dependent pathway in Kupffer cells that mediates the anti-inflammatory effects of gAcrp on LPS-stimulated TNF-α expression (12). Similarly, induction of HO-1 expression in mice by treatment with cobalt protoporphyrin (CoPP) normalizes TNFα expression in response to in vivo LPS challenge after chronic ethanol exposure (12).
A critical role for TLR4 has been identified in the progression of multiple forms of liver disease, including ALD, non-alcoholic liver disease, and viral hepatitis (13). More recently, data has suggested that the MyD88-independent/Toll-interleukin-1 receptor domain-containing adapter IFN-β (TRIF)-dependent pathway of TLR4 signaling is also a key contributor to chronic ethanol-induced liver injury (14–15) and acute hepatitis (16) in mice. Very little is known about the impact of chronic ethanol on the regulation of MyD88-independent signaling. Here we report that chronic ethanol exposure sensitizes Kupffer cells to LPS-stimulated expression of IFN-β and CXCL10 mRNA, two critical TLR4-MyD88-independent responses. This sensitization was also observed in an in vivo mouse model of LPS challenge after chronic ethanol exposure. Treatment of Kupffer cells with adiponectin decreased these MyD88-independent responses. Importantly, induction of HO-1 was sufficient to ameliorate ethanol-induced sensitization of MyD88-independent responses in both Kupffer cells and in mice.
Materials and Methods
Animals
Adult male Wistar rats weighing 140–150 g were purchased from Harlan Sprague Dawley (Indianapolis, IN). Female C57BL/6 mice were purchased from Jackson Laboratories (Bar Harbor, ME). Lieber-DeCarli ethanol diet (regular, no.710260) was purchased from Dyets (Bethlehem, PA).
Materials
Recombinant human gAcrp expressed in E. coli was purchased from Peprotech, Inc. (Rocky Hill, NJ). Recombinant human full-length Acrp expressed in HEK293 cells were purchased from Biovendor Research and Diagnostic Products (Candler, NC, USA). Full length TLR4-LUC (containing −2715 to +52 of the TLR4 promoter in a luciferase reporter vector) was previously described (17). HO-1 overexpression plasmid (18) was from Prof. MP Soares, Instituto Gulbenkian de Ciência, Oeiras, Portugal. Cell culture reagents were from GIBCO-BRL (Grand Island, NY). Antibodies were from the following sources: total ERK1/2 (Upstate Biotechnology; Lake Placid, NY) and phospho-STAT1 (Cell Signaling Technology, Inc., Danvers, MA), HO-1 monoclonal and HO-1 polyclonal antibodies (Assay Designs, Ann Arbor, MI), Hsc70 (Alpha Diagnostic, San Antonio, TX). Anti-rabbit and anti-mouse IgG-peroxidase antibodies were purchased from Boehringer Mannheim (Indianapolis, IN). LPS from Escherichia coli serotype 026:B6 (tissue culture-tested, L-2654) was purchased from Sigma; all experiments were carried out with a single lot of LPS (Lot number 064K4077). Adiponectin preparations contained less than 0.2 ng of LPS/μg of protein. Mouse RAW 264.7 cell nucleofection kit was purchased from Lonza (Cologne, Germany). PE-conjugated TLR4-MD2 antibody and rat anti-mouse IgG1 monoclonal antibodies and CD32/CD16 Fcγ were purchased from eBioscience (San Diego, CA). Co (III) protoporphyrin IX chloride (CoPP) was purchased from Frontier Scientific, Inc (Logan, Utah). Endotoxin-free plasmid preparation kits were from Qiagen (Valencia, CA).
Culture of RAW 264.7 Macrophages and Luciferase Assays
The murine RAW 264.7 macrophage-like cell line was routinely cultured in Dulbecco’s modified Eagle medium with 10% fetal bovine serum and penicillin-streptomycin at 37 °C and 5% CO2. For luciferase reporter assays, RAW 264.7 macrophages were co-transfected with a luciferase reporter plasmid containing the full-length TLR4 promoter (17) and pRL-TK (Promega, Madison, WI), an expression vector for Renilla luciferase under the control of the thymidine kinase promoter, as a control for transfection efficiency. After 24 h, medium was removed and cells stimulated or not with adiponectin and/or LPS, as described in the figure legends, in Dulbecco’s modified Eagle’s medium/fetal bovine serum. Cells were then lysed and luciferase activities measured using the Dual Luciferase assay system (Promega).
Chronic ethanol feeding and Kupffer cell isolation
All procedures involving animals were approved by the Institutional Animal Care and Use Committee at the Cleveland Clinic. Chronic ethanol feeding to rats and mice, as well as the isolation and culture of Kupffer cells, were performed as previously described (19–20). Briefly, Rats were allowed free access to the Lieber-Decarli high-fat complete liquid diet for 2 days. Rats were then randomly assigned to pair-fed or ethanol-fed groups. Ethanol-fed rats were allowed free access to a liquid diet containing 17% of the calories (3.3% vol/vol) from ethanol for 2 days and then a liquid diet containing 35% of the calories (6.6% vol/vol) from ethanol for 4 weeks (19). Control rats were pair-fed a liquid diet in which maltose dextrins were substituted isocalorically for ethanol over the entire feeding period. Kupffer cells were isolated and cultured, as previously described (19, 21). Briefly, isolated Kupffer cells were suspended in CMRL media with 10% FBS, L-glutamine and antibiotic-antimycotic (CMRL-FBS) at a concentration of 2 × 106 cells/ml. Cell suspensions were immediately plated onto 96-well (0.2 × 106 cells/well for analysis of mRNA) or 24-well (1.5 × 106 cells/well for flow cytometry and Western blot analysis) culture plates. One hour after plating the isolated Kupffer cells, nonadherent cells were removed by aspiration and fresh media with or without gAcrp or CoPP was added. After 18 hours in culture, cells were treated with or without 100 ng/mL LPS, as indicated in the figure legends.
Mouse ethanol feeding
10–12 week old female C57BL/6 mice were randomly assigned to ethanol- or pair-fed groups. Ethanol-fed mice were allowed free access to a complete Lieber-DeCarli high fat diet with increasing concentrations of ethanol (5.5% of calories (1% vol/vol) for 2 days, 11% of calories (2% vol/vol) for 2 days, 22% of calories (4% vol/vol) for 7 days, 27% of calories (5% vol/vol)for 7 days and finally, 32% of calories (6% vol/vol) for 7 days) (22). Control mice were pair-fed diets that isocalorically substituted maltose dextrins for ethanol. HO-1 expression was induced using the following protocols. At the end of the feeding protocol, mice were treated with 5 mg/kg CoPP or vehicle (saline) via intraperitoneal injection. Twenty-four hours later, mice were injected intraperitoneally with 0.7 μg LPS/g body weight or an equivalent volume of sterile, endotoxin-free saline (0.09%). To terminate the experiment, mice were anesthetized, blood was collected into lithium tubes, and plasma was isolated and stored at −80°C. Livers were perfused and excised as previously described (22). Portions of each liver were either fixed in formalin or frozen in optimal cutting temperature compound (Sakura Finetek U.S.A., Inc., Torrance, CA) for histology, preserved in RNA later (Ambion, Austin, TX) and stored at −20°C for RNA isolation, or flash frozen in liquid nitrogen and stored at −80°C until further analysis.
Transfection of RAW 264.7 macrophages
For transfection with DNA plasmids, RAW 264.7 macrophages were grown in 6-well plates to 60–70% confluency and then transiently transfected with control and expression vectors using Superfect transfection reagent (for DNA-only transfections) according to the manufacturer’s instructions. Transfected cells were subcultured and seeded at 10.2 × 104/cm2 in 96-well plates.
Nucleofection in RAW 264.7 Macrophages
For siRNA knock-down experiments, RAW 264.7 cells were transfected using the Amaxa Nucleofector apparatus (Lonza, Cologne, Germany). Transfected cells were seeded at 10.2 × 104/cm2 in 96-well plates and cultured for 24 h prior to experimental manipulation. A nucleofection protocol was used to transfect RAW 264.7 macrophages with siRNA. Briefly, 2×106 cells were resuspended in 105 μl of Cell Line Nucleofector Solution V (Amaxa Biosystems) and were nucleofected with 100 nM of specific or scrambled siRNA (siRNA sequences are shown below) in the Nucleofector device using the D-032 program, according to the instructions of the manufacturer. After nucleofection, 500 μl of pre-warmed DMEM was added to the transfection mix and cells were transferred to 12-well plates containing 1.5 ml of pre-warmed DMEM per well. Following nucleofection, gene expression was analyzed at different times, as indicated in the figure legends. Validated Silencer Select siRNA pre-designed sequences were purchased from Ambion/Applied Biosystems. Mouse adipoR1 sequences: sense: 5′- GGCUGAAAGACA ACGA CUAtt-3′ and antisense: 5′-UAGUCGUUGUCUUUCAGCCag-3′. Mouse adipoR2 sequences: sense: 5′-GGCCCAUCAUGCUAUGGAAtt-3′ and antisense: 5′- UUCCAUAG CAUGAU GGGCCtg-3′. Nonspecific siRNA scrambled duplex (sense: 5′-GCG CGC UUU GUA GGA UUC G-3′, antisense: 5′-CGA AUC CUA CAA AGC GCG C-3′). Efficiency of knockdown was determined by Western blot analysis and qRT-PCR.
RNA isolation and quantitative real-time PCR (qRT-PCR)
Total RNA was isolated, reverse transcribed and qRT-PCR amplification was performed. The relative amount of target mRNA was determined using the comparative threshold (Ct) method by normalizing target mRNA Ct values to those of 18S or β-actin. RNA was isolated from Kupffer cells and RAW 264.7 macrophages using the RNeasy Micro Kit (Qiagen), with on-column DNA digestion using the RNase-free DNase set (Qiagen) according to the manufacturer’s instructions. Total RNA (200–300 ng) was reverse-transcribed using the RETROscript kit (Ambion/Applied Biosystems) with random decamers as primers. Real-time PCR amplification was performed in a Mx3000p (Stratagene, La Jolla, CA) using SYBR Green PCR Core Reagents (Applied Biosystems; Warrington, UK). All primers used for real-time PCR analysis were synthesized by Integrated DNA Technologies (Coralville, IA). Primer sequences are shown in Supplemental Table 1. Statistical analysis of real-time PCR data was performed using ΔCt values.
Immunohistochemistry of HO-1
To detect HO-1-protein in mouse liver, formalin-fixed paraffin embedded liver sections (4 μm) were de-paraffinized in Safeclear II ™ xylene substitute (Protocol, Kalamazoo, MI), (3 times 3 min each) and hydrated consecutively in 100% (2 times), 70% and 30% ethanol followed by two washes in PBS. Sections were then blocked with 2% bovine serum albumin (diluted in PBS) containing 0.1% Triton-X for 1 hour followed by overnight incubation with the primary antibody (1:300 dilution) at 4°C. All sections were then washed in PBS (3 times 5 min each), incubated with the fluorochrome-conjugated secondary antibody (Alexa fluor-488 labeled goat-anti-rabbit IgG, 1:300) for 2 hr in the dark at room temperature, washed again in PBS and mounted with VECTASHIELD containing DAPI and anti-fade reagent (Vector Laboratories, Inc., Burlingame, CA). Fluorescent images were acquired using a LEICA confocal microscope. No specific immunostaining was seen in sections incubated with PBS rather than the primary antibody.
Western blot analysis
After treatment of RAW 264.7 macrophages with gAcrp or CoPP for18h, cells were washed by cold PBS and lysed in RIPA lysis buffer containing 100 μM of sodium orthovanadate. Total cellular extracts (20 μg) were separated in 10% SDS polyacrylamide gel and transferred onto PVDF membranes. Membranes were first probed with HO-1 antibody, followed by secondary antibody conjugated with HRP and then visualized by ECL detection reagents (Amersham Biosciences). The membranes were then stripped and reprobed with either total ERK1/2 or Hsc70 antibody for loading control. Western blot analysis was performed using enhanced chemiluminescence for signal detection. Signal intensities were quantified by densitometry using Image J software (NIH, Bethesda, MD).
Flow cytometry Analysis
After 18h culture with or without gAcrp, RAW 264.7 cells were gently scraped and adjusted to 1 million per ml with culture media. Cells were greater than 90% viable as determined by Trypan blue exclusion. The cells were centrifuged at 100 × g for 10 minutes. The pellet was washed with PBS and resuspended in 100 μL PBS+ 0.1% sodium azide and then blocked with 1.0 μG anti-mouse CD32/CD16 Fcγ Block antibodies for 15 min at 4°C. Cells were then stained with 0.5 μg fluorochrome conjugated TLR4-MD2 (PE-conjugated TLR4-MD2) or isotype control (PE-conjugated IgG1) diluted in DPBS containing 0.1% sodium azide for 30 min. Cells were washed twice with PBS and resuspended in 0.5 ml wash buffer (final concentration ~106 cells in 0.5 ml) and kept on ice until flow cytometric measurements were performed on a FACScan flow cytometer (Becton Dickinson Immunocytometry systems, Mountain View, CA). Data were acquired and processed using FlowJo software (Becton Dickinson).
ELISA assays
Cell culture supernatants from RAW 264.7 macrophages were collected after treatment with adiponectin and LPS and CXCL10 concentration measured by ELISA according to the manufacturer’s instructions (R & D, Minneapolis, MN). The concentration of CXCL10 in mouse plasma and liver lysates was also assayed by ELISA.
Statistical analysis
Because of the limited number of Kupffer cells available from each animal, data from several feeding trials are presented in this study. All values are reported as means ± standard error of the mean (SEM). Data were analyzed by general linear models procedure (SAS; Carey, NC). Data were log transformed, if needed, to obtain a normal distribution. Follow-up comparisons were made by least square means testing.
Results
Adiponectin ameliorates the sensitization of Kupffer cells to TLR4-MyD88-independent cytokine expression after chronic ethanol
Chronic ethanol exposure sensitizes Kupffer cells to activation of TLR4/MyD88-dependent signaling by LPS, resulting in enhanced activation of MAPKs and NFκB and increased expression of TNFα. (2). Treatment of Kupffer cells with gAcrp normalizes these MyD88-dependent responses (10). Since recent studies have identified MyD88-independent/TRIF-dependent pathways as critical for the development of ethanol-induced liver injury in mice (14–15), we investigated the interactions between chronic ethanol and gAcrp on the sensitivity of MyD88-independent signals in Kupffer cells. Expression of IFN-β and CXCL10 mRNA, considered a molecular signature for TRIF signaling, was increased in response to a 4h challenge with LPS in Kupffer cells from both pair- and ethanol-fed rats. However, expression was 2-fold greater in Kupffer cells from ethanol-fed rats (Figure 1A/B). Treatment of Kupffer cells with gAcrp completely prevented LPS-stimulated IFN-β and CXCL10 mRNA expression (Figure 1A/B).
Figure 1. Effect of adiponectin on LPS-stimulated IFN-β and CXCL10 mRNA expression in macrophages.
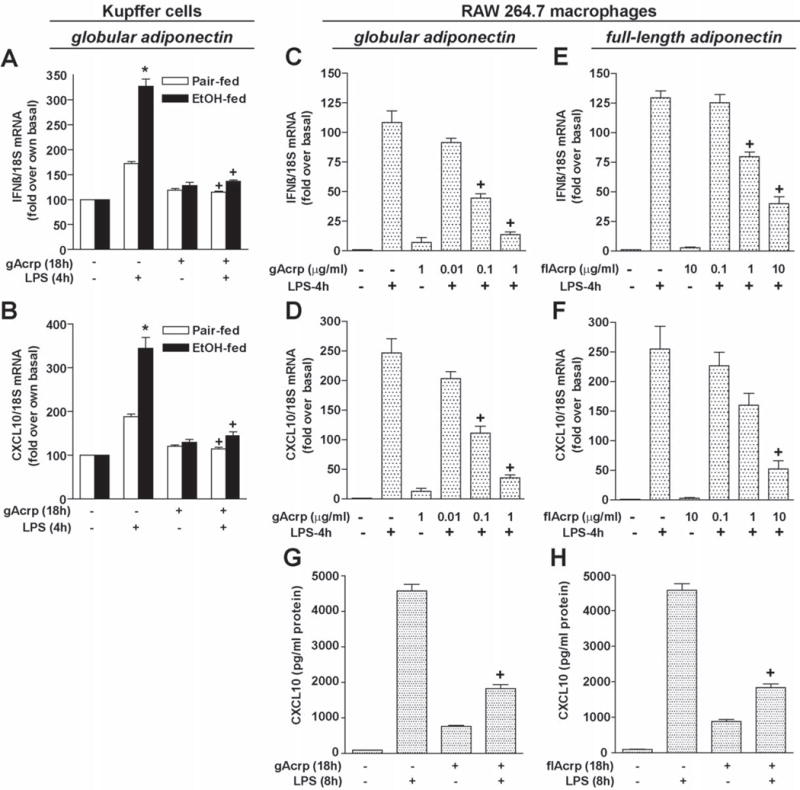
A and B) Kupffer cells isolated from pair- and ethanol-fed rats were cultured with 1μg/ml globular adiponectin (gAcrp) for 18h. Cells were then stimulated with 100 ng/ml LPS for 4h. The expression of IFN-β and CXCL10 mRNA was normalized to 18S mRNA. Values are expressed relative to Kupffer cells from pair-fed rats not treated with gAcrp. n = 4, * p < 0.05 ethanol-fed compared with pair-fed, +p < 0.05 compared with LPS-stimulated cells not treated with gAcrp. C-F) RAW 264.7 cells were cultured for 18h in the absence or presence of increasing doses of either gAcrp (0–1 μg/ml) or full-length adiponectin (flAcrp, 0–10 μg/ml). Cells were then stimulated with 100 ng/ml LPS for 4h and expression of IFN-β and CXCL10 mRNA was normalized to 18S mRNA. n = 4, + p < 0.05 compared with LPS-stimulated cells not treated with adiponectin. G and H) RAW 264.7 cells were cultured for 18h in the absence or presence of 10 μ/ml gAcrp or full-length adiponectin. Cells were then stimulated with 100 ng/ml LPS for 8h and the concentration of CXCL10 protein accumulated in the media was measured by ELISA. n = 6, + p < 0.05 compared with LPS-stimulated cells not treated with adiponectin.
Adiponectin receptors and suppression of MyD88-independent responses in RAW 264.7 macrophages
In RAW 264.7 macrophages, LPS increased expression of IFN-β and CXCL10 mRNA expression; both messages reached a maximal response at 4 h (Supplementary Figure 1). LPS-induced increases in IFN-β and CXCL10 were dose-dependently decreased by gAcrp at concentrations from 0.1–1 μg/ml (Figure 1 C/D). Treatment of RAW 264.7 macrophages with 1–10 μg/ml full-length adiponectin also decreased LPS-induced IFN-β and CXCL10 mRNA expression, but only at higher concentrations (Figure 1E/F). A similar suppression of CXCL10 expression was reported in human macrophages in response to 10 μg/ml full-length adiponectin (23). Pre-treatment of RAW 264.7 macrophages with either gAcrp and full-length adiponectin also suppressed LPS-stimulated accumulation of CXCL10 protein in the cell culture media after 8 h (Figure 1 G/H).
Adiponectin signals primarily via two receptors, adiponectin R1 (adipoR1) and adiponectin R2 (adipoR2) (24). To identify the adiponectin receptor utilized by gAcrp and full-length adiponectin in macrophages, RAW 264.7 macrophages were transfected or not with siRNA against adipoR1, adipoR2 or scrambled siRNA. Transfection with specific siRNAs effectively decreased expression of adipoR1 and adipoR2 (Supplementary Figure 2 A/B). In cells not transfected with siRNA or transfected with a scrambled siRNA control, both gAcrp and full-length adiponectin suppressed LPS-stimulated IFN-β and CXCL10 mRNA expression (Figure 2). siRNA knock-down of adipoR1 prevented the effects of gAcrp, but had no effect on the response to full-length adiponectin. In contrast, siRNA knock-down of adipoR2 prevented only the response to full-length adiponectin (Figure 2). Therefore, gAcrp and full-length adiponectin suppressed TLR-4 stimulated IFN-β and CXCL10 expression via adipoR1 and adipoR2, respectively, in line with the observation that gAcrp has a relatively higher affinity for adipoR1 and full-length adiponectin has a higher affinity for adipoR2 (24).
Figure 2. AdipoR1 and AdipoR2 mediated the inhibitory effects of gAcrp and flAcrp, respectively, on LPS-induced IFN-β and CXCL10 mRNA expression in RAW 264.7 macrophages.
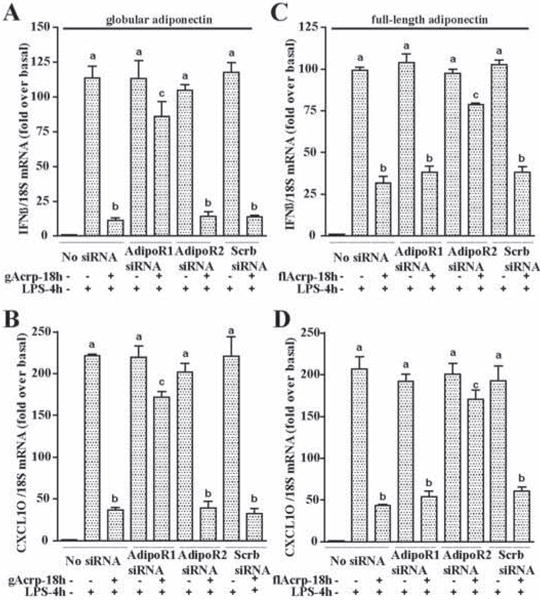
RAW 264.7 cells were transfected or not with 100 nM of AdipoR1 siRNA, AdipoR2 siRNA or scrambled siRNA and then cultured with or without 1 μg/ml gAcrp or 10 μg/ml flAcrp for 18h. Cells were then stimulated with LPS for 4h. IFN-β and CXCL10 mRNA was normalized to 18S mRNA. n=4, Values with different superscripts are significantly different from each other, p < 0.05.
gAcrp decreases expression of TLR4 in Kupffer cells and RAW 264.7 macrophages
Since adiponectin suppressed both TLR4-MyD88-dependent(10) and MyD88-independent/TRIF-dependent responses, we hypothesized that the anti-inflammatory effects of gAcrp may be mediated via decreased expression of TLR4 or CD14, components of the LPS-receptor complex required for LPS sensing by innate immune cells. TLR4 and CD14 mRNA expression was increased in Kupffer cells after ethanol feeding (Figure 3 A/B) (25). Treatment of Kupffer cells with increasing concentrations of gAcrp dose-dependently decreased TLR4 mRNA (Figure 3A), with no effect on CD14 mRNA (Figure 3B). Kupffer cells from ethanol-fed rats were more sensitive to the effects of gAcrp on TLR4 mRNA (Figure 3A).
Figure 3. Adiponectin suppressed TLR4 expression in Kupffer cells and RAW 264.7 macrophages.
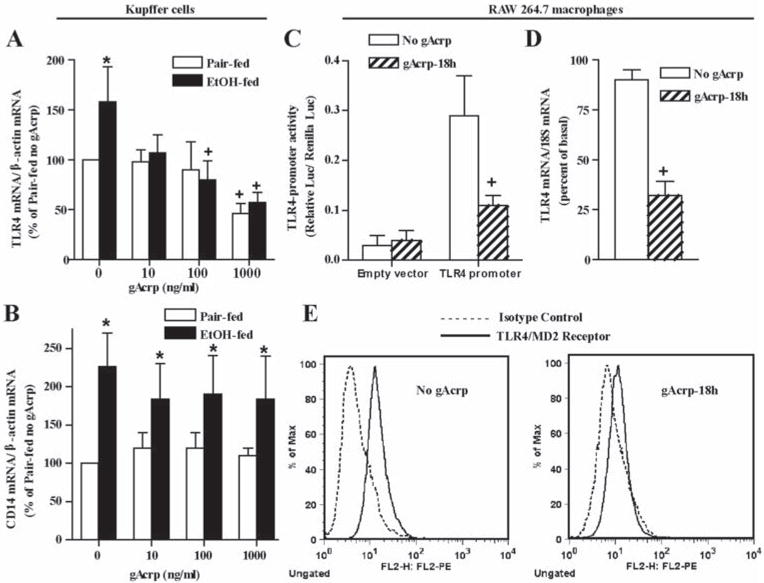
A and B) Kupffer cells from pair- and ethanol-fed rats were isolated and cultured overnight with 0–1000 ng/ml gAcrp for 18 h. Expression of TLR4 and CD14 mRNA was normalized to 18S mRNA. Values are expressed as fold increases relative to Kupffer cells from pair-fed rats not treated with gAcrp. n = 4, * p < 0.05 ethanol-fed compared to pair-fed, +p < 0.05 compared with cells not treated with gAcrp. C) TLR4 promoter activity was measured in RAW 264.7 macrophages transiently transfected with a full-length TLR4 promoter-luciferase reporter construct and a Renilla luciferase control plasmid. After 24 h, cells were treated with or without 1 μg/ml gAcrp for 18 h. Values represent relative luciferase activity. n = 4; + p < 0.05. D) RAW 264.7 macrophages were treated without or with 1 μg/ml gAcrp for 18h and expression of TLR4 mRNA measured and normalized to 18S mRNA. n=4, + p <.05 compared with cells not treated with adiponectin. E) RAW 264.7 macrophages were cultured with or without 1 μg/ml gAcrp for 18h and cell surface expression of TLR4-MD2 (solid line), relative to isotype controls (dotted line), was measured by flow cytometry. Data are representative of four experiments.
Using a luciferase reporter construct driven by the full-length TLR4-promoter, we assessed the effect of gAcrp on the transcription of TLR4 in RAW 264.7 macrophages. Treatment of RAW 264.7 macrophages with gAcrp decreased TLR4 promoter activity by ~45–55% (Figure 3C). gAcrp decreased the expression of TLR4 mRNA by 60% (Figure 3D) and had no effect on the mRNA stability of TLR4 (Supplementary Figure 3). This decrease in TLR4 mRNA was associated with decreased surface expression of TLR4/MD2 complex, measured by flow cytometry (66 ± 14%, n=4, p <0.03) (Figure 3E).
Heme oxygenase-1 is necessary and sufficient to decrease TLR4 expression and MyD88-independent responses in RAW 264.7 macrophages
In primary Kupffer cells from rats, the anti-inflammatory effects of gAcrp on LPS-stimulated TNFα expression are mediated via a HO-1-dependent pathway (12). Therefore, we investigated whether HO-1 was involved in gAcrp-mediated decrease in TLR4 expression and reduction in LPS-stimulated IFN-β and CXCL10. gAcrp increased the expression of HO-1 mRNA and protein in RAW 264.7 macrophages (Figure 4A/B). When HO-1 was overexpressed in RAW 264.7 macrophages, at expression levels comparable to cells treated with gAcrp (Supplementary Figure 4), TLR4 mRNA expression was reduced by approximately 50% (Figure 4C). Transfection with the empty vector had no effect on TLR4 mRNA expression (Figure 4C). Further, overexpression of HO-1 ameliorated LPS-stimulated IFN- β and CXCL10 mRNA to a similar extent as treatment with gAcrp (Figure 4D/E).
Figure 4. HO-1 expression and suppression of TLR4-mediated MyD88-independent signaling in RAW 264.7 macrophages.
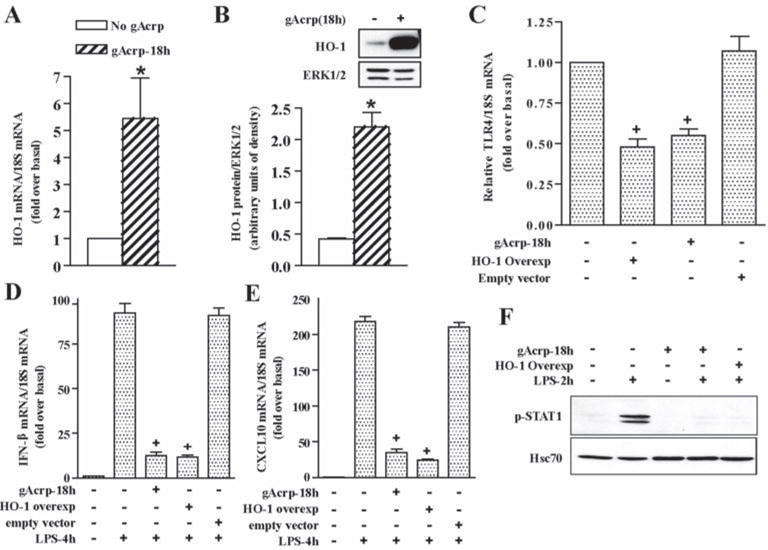
A and B) RAW 264.7 macrophages were treated with or without 1 μg/ml gAcrp for 18h. HO-1 mRNA expression was normalized to 18S mRNA. B) HO-1 protein expression was measured by Western blot and normalized to total ERK1/2. n=4, * p < 0.05 compared with cells not treated with adiponectin C-F) RAW 264.7 macrophages were transfected with an empty vector or an HO-1 overexpression plasmid. Twenty four hour after transfection, cells were treated or not with 1 μg/ml gAcrp for 18h. C) TLR4 mRNA was measured and normalized to 18S mRNA. n=4, * p < 0.05 compared to non-treated cells. D and E) Transfected cells were then stimulated with LPS for 4h. IFN-β (D) and CXCL10 (E) mRNA were measured normalized to 18S mRNA. n=4, * p < 0.05 compared with LPS-stimulated cells not treated with gAcrp or HO-1 overexpression. F) Transfected cells were then stimulated with LPS for 2h and Western blot analysis was performed using antibodies to phosphorylated STAT1 and Hsc70 (loading control). Images are representative of four experiments.
In macrophages, TLR4/MyD88-independent signals lead to increased IFN-β expression; IFN-β then activates IFN-inducible gene transcription, primarily via activation of the transcription factor STAT1 (26). LPS increased STAT-1 phosphorylation at 1–2 h in RAW 264.7 macrophages (data not shown), consistent with the time courses for IFN-β and CXCL10 mRNA expression (Supplementary Figure 1). LPS-activated STAT1 phosphorylation was completely blocked by treatment with gAcrp or overexpression of HO-1 (Figure 4F).
HO-1 mediates the inhibitory effects of gAcrp on LPS-stimulated MyD88-independent pathway
RAW 264.7 macrophages were transfected with siRNA against HO-1 or scrambled siRNA (Supplemental Figure 4C). When HO-1 expression was knocked-down, the anti-inflammatory effects of gAcrp on LPS-stimulated IFN-β and CXCL10 were attenuated (Figure 5A/B). Similarly, if the activity of HO-1 was inhibited by treating RAW 264.7 macrophages with zinc protoporphyrin, inhibition of LPS-stimulated IFN-β and CXCL10 mRNA expression was reduced (Figure 5C/D). Taken together, these data demonstrate that increased expression of HO-1 is necessary and sufficient to reduce TLR4 expression, as well as TRIF-dependent signaling, in response to gAcrp.
Figure 5. HO-1 mediates the inhibitory effects of gAcrp on LPS-stimulated MyD88-independent pathway.
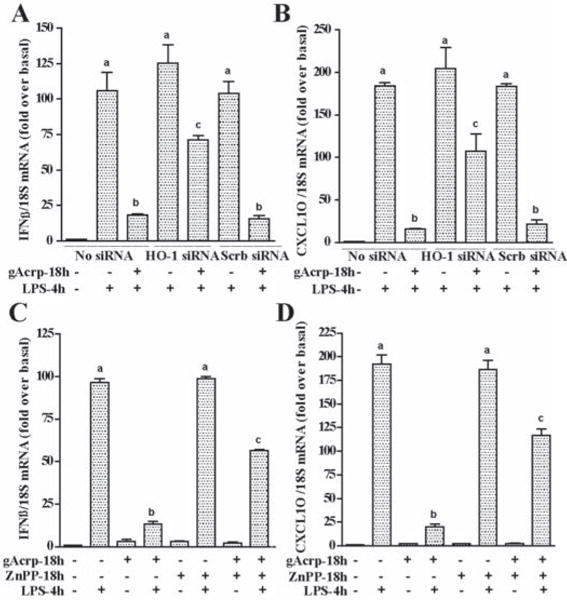
A) RAW 264.7 cells were transfected or not with 100 nM of HO-1 siRNA or scrambled siRNA and then cultured with or without 1 μg/ml gAcrp for 18h. Cells were then stimulated with LPS for 4h. IFN-β and CXCL10 mRNA was normalized to 18S mRNA. n=4, Values with different superscripts are significantly different from each other, p < 0.05. B) RAW264.7 cells were cultured with or without 0.5 μM ZnPP in the presence and absence of 1 μg/ml gAcrp for 18h. Cells were then stimulated with LPS for 4h. IFN-β and CXCL10 mRNA was normalized to 18S mRNA. n=4, Values with different superscripts are significantly different from each other, p < 0.05.
Induction of HO-1 decreases LPS-stimulated IFN-β and CXCL10 expression in macrophages
We next tested the ability of two different small molecules to mimic the anti-inflammatory effects of gAcrp or overexpression of HO-1 in macrophages. Treatment of cells with the heme analogue, CoPP, induced the expression of HO-1 in RAW 264.7 macrophages (Supplementary Figure 4B). When RAW 264.7 macrophages were pre-treated with CoPP for 18 h, LPS-stimulated expression of IFN-β and CXCL10 mRNA was suppressed to a similar extent as in cells pre-treated with gAcrp (Figure 6A/B). The anti-inflammatory effects of HO-1 are primarily mediated by its enzymatic products, carbon monoxide (CO) and/or biliverdin/bilirubin (27). Pretreatment of RAW 264.7 macrophages with the CO releasing molecule-2 (CORM-2) for 4 h prior to challenge with LPS was as effective as CoPP or gAcrp in ameliorating the effects of LPS on IFN-β and CXCL10 expression (Figure 6A/B). Pre-treatment of Kupffer cells with CoPP was equally effective at suppressing TLR4-stimulated IFN-β and CXCL10 mRNA expression (Figure 6C/D).
Figure 6. Induction of HO-1 decreased LPS-stimulated IFN-β and CXCL10 expression in RAW 264.7 macrophages and Kupffer cells.
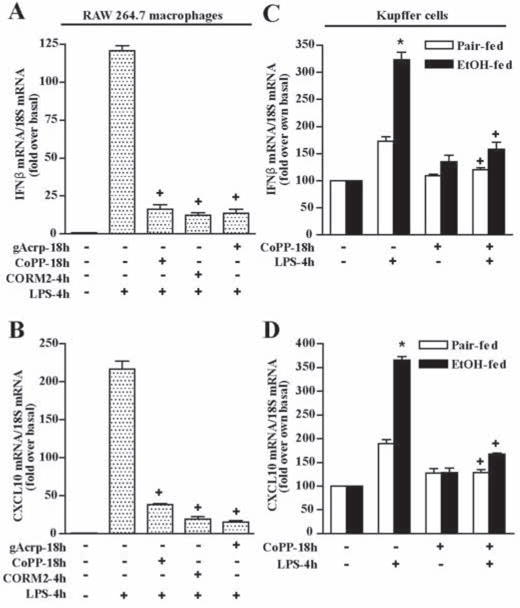
A and B) RAW 264.7 cells were cultured for 18h in the absence or presence of either gAcrp (1 μg/ml), CoPP (10μM) or CORM2 (100 μM). Cells were then stimulated without or with 100 ng/ml LPS for 4h and expression of IFN-β and CXCL10 mRNA normalized to 18S mRNA. n = 4, +p < 0.05 compared with LPS-stimulated cells not treated with inducers of HO-1. C and D) Kupffer cells isolated from pair- and ethanol-fed rats were cultured with 10μM CoPP for 18h. Cells were then stimulated with 100 ng/ml LPS for 4h. Expression of IFN-β and CXCL10 mRNA was normalized to 18S mRNA. Values are expressed relative to Kupffer cells from pair-fed rats not treated with gAcrp. n = 4, * p < 0.05 ethanol-fed compared with pair-fed, +p < 0.05 compared with LPS-stimulated cells not treated with CoPP.
Treatment of mice with CoPP reduced chronic ethanol-induced sensitization to TLR4-MyD88-independent responses in mouse liver
Since CoPP effectively prevented TLR4-MyD88-independent responses in isolated Kupffer cells, we next asked whether chronic ethanol feeding would sensitize mice to LPS-stimulated IFN-β and CXCL10 expression and whether treatment of mice with CoPP could ameliorate this effect of chronic ethanol. C57BL/6 mice were allowed free access to an ethanol-containing liquid diet for 28 days, at a final concentration of 6% ethanol (vol/vol), equivalent to 32% of total calories, or pair-fed control diets. Mice were then treated with a single injection of CoPP or vehicle. Twenty four hours after the CoPP treatment, mice were challenged with LPS or saline.
Body weights and food intake for all experimental groups are shown in Supplemental Table 2. Expression of HO-1 in liver was low in both pair- and ethanol-fed mice at baseline (Figure 7A). While challenge with LPS modestly increased expression of HO-1 in both treatment groups, treatment with CoPP robustly increased expression of HO-1 in livers from both pair-and ethanol-fed mice, in a predominantly sinusoidal distribution (Figure 7A). LPS challenge increased expression of IFN-β and CXCL10 mRNA in both pair- and ethanol-fed mice at 4 h; this response was greater after chronic ethanol feeding (Figure 7B and 7C). Similarly, the LPS-stimulated increase in CXCL10 protein in plasma (Figure 7D) and liver lysates (Figure 7E) was increased after chronic ethanol. Importantly, pre-treatment with CoPP reduced the sensitivity of both pair-and ethanol-fed mice to LPS-stimulated responses (Figure 7 B–D).
Figure 7. Chronic ethanol feeding sensitizes mice to an in vivo challenge with LPS: Induction of HO-1 rapidly normalizes this effect of ethanol.
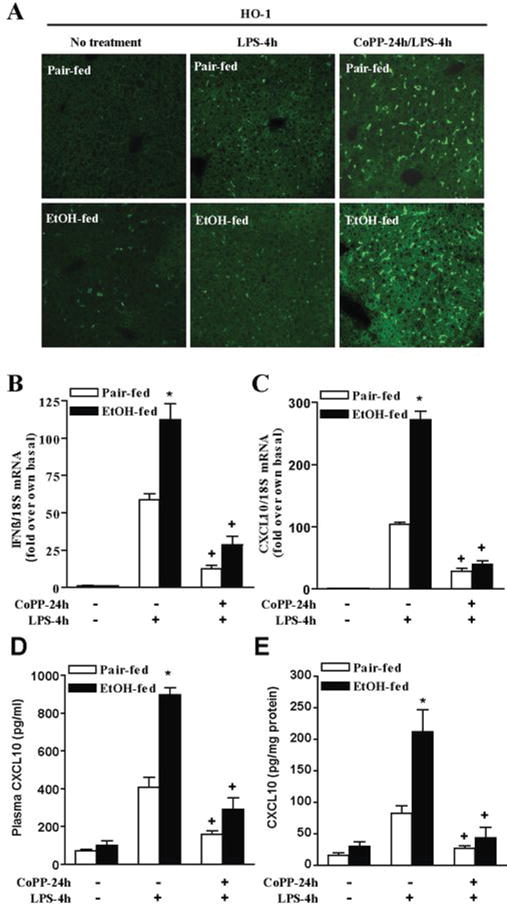
Female C57BL/6 mice were allowed free access to increasing concentrations of ethanol as part of a complete liquid diet to a maximum concentration of 6% vol/vol (32% of kcal) over 25 days or pair-fed control diets. Mice were then injected with 5 mg/kg cobalt protoporphyrin (CoPP) or vehicle (saline). After 24h, mice were challenged with 0.7 μg/kg LPS or saline. A) HO-1 positive cells were detected by immunohistochemistry. Data are representative of 4 pair-fed mice and 6 ethanol-fed mice. B and C) Expression of IFN-β and CXCL10 mRNA was measured after 4h and normalized to 18S mRNA. D and E) CXCL10 protein in plasma and liver lysates was measured by ELISA. n=3–4, *p < 0.05 compared to pair-fed, +p <.05 compared to LPS-treated mice without CoPP treatment.
Discussion
The development of ALD is a complex process involving both the parenchymal and non-parenchymal cells in the liver. Enhanced inflammation in the liver during ethanol exposure is associated with liver injury; activation of TLR4 by LPS and signaling via MyD88-dependent and independent pathways contribute to chronic ethanol-induced liver injury (7, 14–15). Increased activation of TLR4 by LPS during chronic ethanol exposure is due to increased exposure to gut-derived endotoxins, as well as a sensitization of macrophages to stimulation by LPS (2). Here we report that, in addition to the sensitization of MyD88-dependent signaling reported in the past (2), chronic ethanol also enhances TLR4 signaling via MyD88-independent pathways, both in isolated Kupffer cells and in an in vivo mouse model of chronic ethanol exposure. Chronic ethanol exposure increased LPS-stimulated IFN-β mRNA expression, as well as the IFN-inducible gene, CXCL10; expression of these cytokines is a signature for TLR4-TRIF-dependent signaling. Moreover, here we have identified an adiponectin/HO-1 pathway that attenuates TLR4 signaling via both MyD88-dependent and -independent pathways (summarized in Figure 8). Induction of HO-1 after chronic ethanol feeding to mice countered this sensitization, suppressing LPS-stimulated IFN-β/CXCL10 expression in liver within 24 h.
Figure 8. Model illustrating the role of HO-1 in the anti-inflammatory effect of gAcrp.
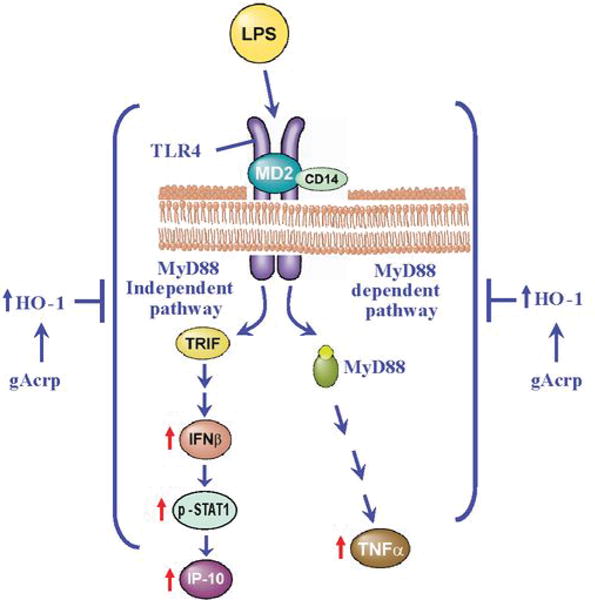
After chronic ethanol feeding, TLR4 signaling via MyD88-dependent (10) and –independent pathways is enhanced. Induction of HO-1, either by treatment with gAcrp or small molecule therapeutics, suppresses TLR4-mediated signaling via both MyD88 (10) and TRIF pathways.
Recent studies have explored the potential utility of adiponectin as an anti-inflammatory agent to treat ALD (11). Importantly, we find that adiponectin treatment normalized both LPS-induced TNFα production, (10, 12) as well as IFN-β and CXCL10 mRNA expression (Figure 1), in primary cultures of Kupffer cells after chronic ethanol exposure. Interestingly, we find that both globular and full-length adiponectin suppressed LPS-stimulated expression of IFN-β and CXCL10 mRNA. The response to gAcrp was mediated by AdipoR1, while the response to full-length adiponectin was dependent on AdipoR2 (Figure 2). These data are consistent with the differential affinities of gAcrp and full-length adiponectin for AdipoR1 and AdipoR2, respectively (24). Studies investigating the specific down-stream signaling events activated by AdipoR1/R2 in macrophages are currently under investigation.
Chronic ethanol feeding increased expression of TLR4 mRNA in Kupffer cells isolated from rats (Figure 3A) and in mouse liver after chronic ethanol exposure (28). In cellular models, changes in the expression of TLR4 mRNA influence TLR4-dependent responses (29). Therefore, increased expression of TLR4 likely contributes to ethanol-induced increases in TLR4-MyD88-dependent and –independent signaling in the liver. Treatment of macrophages with gAcrp or CoPP to induce HO-1 reduced expression of TLR4 mRNA and protein, likely via a decrease in TLR4 transcription.
We show that induction of HO-1 was sufficient to suppress TLR4-TRIF-dependent responses in macrophages and in mice after chronic ethanol exposure. Induction of HO-1 is also effective at the prevention of steatohepatitis induced by feeding a methyl-choline deficient (MCD) diet, a model of non-alcoholic steatohepatitis (NASH) (30) and protects against liver ischemia-reperfusion injury (31). However, it is not yet clear which cell(s) in the liver contribute to these cytoprotective effects of HO-1. In our studies, HO-1 expression is primarily sinusoidal, consistent with previous reports that HO-1 is typically expressed in Kupffer cells in the liver (32). However, in MCD-fed mice, HO-1 was observed in fatty hepatocytes (30). Similarly, while induction of HO-1 dampens TLR4-TRIF-dependent responses in Kupffer cells and RAW 264.7 macrophages (Figure 1) (31), as well as in mice after chronic ethanol exposure (Figure 7), other studies find that HO-1 up-regulates IFN-β production in some disease models (33–34). Taken together, these studies suggest that there are complex cell-cell interactions involving HO-1 within the liver that contribute to the protective effect of HO-1 and its enzymatic products in the liver in response to different insults.
Chronic ethanol-induced sensitization to the TLR4-TRIF-dependent pathway, associated with increased expression of TLR4 mRNA (28), likely contributes to ethanol-induced liver injury. TRIF-dependent signaling is critical to mediating the innate immune response to LPS, as the expression of TLR4 target genes is mediated primarily through MyD88-independent, rather than MyD88-dependent pathways (35). Moreover, recent studies have identified MyD88-independent/TRIF-dependent signaling as an essential element in chronic ethanol-induced liver injury in mice (14–15). Here we find that chronic ethanol exposure sensitized mice to LPS-stimulated activation of typical MyD88-independent/TRIF-dependent responses, including expression of IFN-β mRNA and the IFN-inducible CXCL10 mRNA in liver, as well as CXCL10 protein in plasma and liver lysates. However, the specific contributions of type I interferons to the pathophysiological progression of ALD have not been well characterized. The protective effects of CoPP treatment in supressing signature cytokines in both the MyD88 (12) and TRIF pathways (Figure 8) in Kupffer cells and in mice after chronic ethanol exposure suggests that CoPP would likely be effective in preventing ethanol-induced liver injury, independent of the relative contributions of MyD88-dependent and independent pathways in the progression of ethanol-induced liver injury.
The controlled and appropriate resolution of inflammation is an essential feature of the innate immune response; failure to terminate an inflammatory response likely contributes to a number of chronic inflammatory diseases, including ALD (36). Importantly, the resolution of inflammation is an active, highly coordinated response, with inflammatory signals initiating the induction of negative regulators (37). During ethanol exposure, despite high expression of inflammatory mediators, there appears to be a generalized failure in the resolution of inflammation. Here we have identified an adiponectin/HO-1 mediated anti-inflammatory pathway that, when activated, can effectively dampen the effects of chronic ethanol on TLR4-mediated signaling; resulting in decreased activation of MyD88-mediated signaling (10, 12), as well as MyD88-independent signaling. Induction of HO-1, either in Kupffer cells isolated from rats after chronic ethanol feeding or in an in vivo mouse model of chronic ethanol exposure, suppressed LPS-stimulated expression of IFN-β and CXCL10 mRNA expression. In summary, these data suggest that strategies, such as the induction of HO-1 identified here, aimed at decreasing TLR4 expression and/or TLR4-mediated signaling via both the MyD88-dependent and -independent pathways will likely be useful in treatment and/or prevention of ethanol-induced liver injury, as well as other types of liver injury, such as NASH and ischemia-reperfusion injury (30–31).
Supplementary Material
Acknowledgments
We thank MP Soares for his kind gift of the hemeoxygenase-1 overexpression plasmid. We would also like to thank Drs. A. Stavitsky and M.T. Pritchard for useful discussions during the course of this study, as well as their critical reading of the manuscript.
Financial support: This work was supported in part by NIH grant RO1AA011975, R56AA011975, P20AA17069 to LEN and the Swiss National Science Foundation (3100-118266.07 and 310000-114073) to TR.
Abbreviations
- AdipoR
Adiponectin receptor
- ALD
Alcoholic liver disease
- CO
carbon monoxide
- CoPP
cobalt protoporphyrin
- CORM2
carbon monoxide releasing molecule
- Ct
comparative threshold
- CXCL10
Chemokine (C-X-C motif) ligand 10
- EtOH
ethanol
- flAcrp
full length adiponectin
- gAcrp
globular adiponectin
- HO-1
heme oxygenase-1
- MD-2
Lymphocyte antigen 96
- Scrb siRNA
scrambled small interfering RNA
- TRIF
TIR-domain-containing adapter-inducing interferon-β
- ZnPP
zinc protoporphyrin
References
- 1.Minagawa M, Deng Q, Liu ZX, Tsukamoto H, Dennert G. Activated natural killer T cells induce liver injury by Fas and tumor necrosis factor-alpha during alcohol consumption. Gastroenterology. 2004;126:1387–1399. doi: 10.1053/j.gastro.2004.01.022. [DOI] [PubMed] [Google Scholar]
- 2.Nagy LE. Recent insights into the role of the innate immune system in the development of alcoholic liver disease. Exp Biol Med (Maywood) 2003;228:882–890. doi: 10.1177/153537020322800803. [DOI] [PubMed] [Google Scholar]
- 3.Bykov I, Junnikkala S, Pekna M, Lindros KO, Meri S. Complement C3 contributes to ethanol-induced liver steatosis in mice. Ann Med. 2006;38:280–286. doi: 10.1080/07853890600664608. [DOI] [PubMed] [Google Scholar]
- 4.Pritchard MT, McMullen MR, Stavitsky AB, Cohen JI, Lin F, Medof ME, Nagy LE. Differential contributions of C3, C5, and decay-accelerating factor to ethanol-induced fatty liver in mice. Gastroenterology. 2007;132:1117–1126. doi: 10.1053/j.gastro.2007.01.053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Adachi Y, Bradford BU, Gao W, Bojes HK, Thurman RG. Inactivation of Kupffer cells prevents early alcohol-induced liver injury. Hepatology. 1994;20:453–460. [PubMed] [Google Scholar]
- 6.Rao R. Endotoxemia and gut barrier dysfunction in alcoholic liver disease. Hepatology. 2009;50:638–644. doi: 10.1002/hep.23009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Thurman RG. II. Alcoholic liver injury involves activation of Kupffer cells by endotoxin. Am J Physiol. 1998;275:G605–611. doi: 10.1152/ajpgi.1998.275.4.G605. [DOI] [PubMed] [Google Scholar]
- 8.Takeuchi O, Akira S. Pattern Recognition Receptors and Inflammation. Cell. 2010;140:805–820. doi: 10.1016/j.cell.2010.01.022. [DOI] [PubMed] [Google Scholar]
- 9.Berg AH, Combs TP, Scherer PE. ACRP30/adiponectin: an adipokine regulating glucose and lipid metabolism. Trends Endocrinol Metab. 2002;13:84–89. doi: 10.1016/s1043-2760(01)00524-0. [DOI] [PubMed] [Google Scholar]
- 10.Thakur V, Pritchard MT, McMullen MR, Nagy LE. Adiponectin normalizes LPS-stimulated TNF-alpha production by rat Kupffer cells after chronic ethanol feeding. Am J Physiol Gastrointest Liver Physiol. 2006;290:G998–1007. doi: 10.1152/ajpgi.00553.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Xu A, Wang Y, Keshaw H, Xu LY, Lam KS, Cooper GJ. The fat-derived hormone adiponectin alleviates alcoholic and nonalcoholic fatty liver diseases in mice. J Clin Invest. 2003;112:91–100. doi: 10.1172/JCI17797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Mandal P, Park PH, McMullen MR, Pratt BT, Nagy LE. The anti-inflammatory effects of adiponectin are mediated via a heme oxygenase-1-dependent pathway in rat Kupffer cells. Hepatology. 2009 doi: 10.1002/hep.23427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Testro AG, Visvanathan K. Toll-like receptors and their role in gastrointestinal disease. J Gastroenterol Hepatol. 2009;24:943–954. doi: 10.1111/j.1440-1746.2009.05854.x. [DOI] [PubMed] [Google Scholar]
- 14.Hritz I, Mandrekar P, Velayudham A, Catalano D, Dolganiuc A, Kodys K, Kurt-Jones E, Szabo G. The critical role of toll-like receptor (TLR) 4 in alcoholic liver disease is independent of the common TLR adapter MyD88. Hepatology. 2008;48:1224–1231. doi: 10.1002/hep.22470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zhao XJ, Dong Q, Bindas J, Piganelli JD, Magill A, Reiser J, Kolls JK. TRIF and IRF-3 binding to the TNF promoter results in macrophage TNF dysregulation and steatosis induced by chronic ethanol. J Immunol. 2008;181:3049–3056. doi: 10.4049/jimmunol.181.5.3049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Imamura M, Tsutsui H, Yasuda K, Uchiyama R, Yumikura-Futatsugi S, Mitani K, Hayashi S, Akira S, Taniguchi S, Rooijen NVan, Tschopp J, Yamamoto T, Fujimoto J, Nakanishi K. Contribution of TIR domain-containing adapter inducing IFN-beta-mediated IL-18 release to LPS-induced liver injury in mice. J Hepatol. 2009;51:333–341. doi: 10.1016/j.jhep.2009.03.027. [DOI] [PubMed] [Google Scholar]
- 17.Roger T, Miconnet I, Schiesser AL, Kai H, Miyake K, Calandra T. Critical role for Ets, AP-1 and GATA-like transcription factors in regulating mouse Toll-like receptor 4 (Tlr4) gene expression. Biochem J. 2005;387:355–365. doi: 10.1042/BJ20041243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Brouard S, Otterbein LE, Anrather J, Tobiasch E, Bach FH, Choi AM, Soares MP. Carbon monoxide generated by heme oxygenase 1 suppresses endothelial cell apoptosis. J Exp Med. 2000;192:1015–1026. doi: 10.1084/jem.192.7.1015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Aldred A, Nagy LE. Ethanol dissociates hormone-stimulated cAMP production from inhibition of TNF-alpha production in rat Kupffer cells. Am J Physiol. 1999;276:G98–G106. doi: 10.1152/ajpgi.1999.276.1.G98. [DOI] [PubMed] [Google Scholar]
- 20.Thakur V, McMullen MR, Pritchard MT, Nagy LE. Regulation of macrophage activation in alcoholic liver disease. J Gastroenterol Hepatol. 2007;22(Suppl 1):S53–56. doi: 10.1111/j.1440-1746.2006.04650.x. [DOI] [PubMed] [Google Scholar]
- 21.Kishore R, Hill JR, McMullen MR, Frenkel J, Nagy LE. ERK1/2 and Egr-1 contribute to increased TNF-alpha production in rat Kupffer cells after chronic ethanol feeding. Am J Physiol Gastrointest Liver Physiol. 2002;282:G6–15. doi: 10.1152/ajpgi.00328.2001. [DOI] [PubMed] [Google Scholar]
- 22.Roychowdhury S, McMullen MR, Pritchard MT, Hise AG, Rooijen Nvan, Medof ME, Stavitsky AB, Nagy LE. An early complement-dependent and TLR-4-independent phase in the pathogenesis of ethanol-induced liver injury in mice. Hepatology. 2009;49:1326–1334. doi: 10.1002/hep.22776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Folco EJ, Rocha VZ, Lopez-Ilasaca M, Libby P. Adiponectin inhibits pro-inflammatory signaling in human macrophages independent of interleukin-10. J Biol Chem. 2009;284:25569–25575. doi: 10.1074/jbc.M109.019786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Yamauchi T, Kamon J, Ito Y, Tsuchida A, Yokomizo T, Kita S, Sugiyama T, Miyagishi M, Hara K, Tsunoda M, Murakami K, Ohteki T, Uchida S, Takekawa S, Waki H, Tsuno NH, Shibata Y, Terauchi Y, Froguel P, Tobe K, Koyasu S, Taira K, Kitamura T, Shimizu T, Nagai R, Kadowaki T. Cloning of adiponectin receptors that mediate antidiabetic metabolic effects. Nature. 2003;423:762–769. doi: 10.1038/nature01705. [DOI] [PubMed] [Google Scholar]
- 25.Jarvelainen HA, Oinonen T, Lindros KO. Alcohol-induced expression of the CD14 endotoxin receptor protein in rat Kupffer cells. Alcohol Clin Exp Res. 1997;21:1547–1551. [PubMed] [Google Scholar]
- 26.Toshchakov V, Jones BW, Perera PY, Thomas K, Cody MJ, Zhang S, Williams BR, Major J, Hamilton TA, Fenton MJ, Vogel SN. TLR4, but not TLR2, mediates IFN-beta-induced STAT1alpha/beta-dependent gene expression in macrophages. Nat Immunol. 2002;3:392–398. doi: 10.1038/ni774. [DOI] [PubMed] [Google Scholar]
- 27.Otterbein LE, Soares MP, Yamashita K, Bach FH. Heme oxygenase-1: unleashing the protective properties of heme. Trends Immunol. 2003;24:449–455. doi: 10.1016/s1471-4906(03)00181-9. [DOI] [PubMed] [Google Scholar]
- 28.Gustot T, Lemmers A, Moreno C, Nagy N, Quertinmont E, Nicaise C, Franchimont D, Louis H, Deviere J, Moine OLe. Differential liver sensitization to toll-like receptor pathways in mice with alcoholic fatty liver. Hepatology. 2006;43:989–1000. doi: 10.1002/hep.21138. [DOI] [PubMed] [Google Scholar]
- 29.Babu S, Blauvelt CP, Kumaraswami V, Nutman TB. Cutting edge: diminished T cell TLR expression and function modulates the immune response in human filarial infection. J Immunol. 2006;176:3885–3889. doi: 10.4049/jimmunol.176.7.3885. [DOI] [PubMed] [Google Scholar]
- 30.Yu J, Chu ES, Wang R, Wang S, Wu CW, Wong VW, Chan HL, Farrell GC, Sung JJ. Heme oxygenase-1 protects against steatohepatitis in both cultured hepatocytes and mice. Gastroenterology. 2010;138:694–704. 704 e691. doi: 10.1053/j.gastro.2009.09.058. [DOI] [PubMed] [Google Scholar]
- 31.Tsuchihashi S, Zhai Y, Bo Q, Busuttil RW, Kupiec-Weglinski JW. Heme oxygenase-1 mediated cytoprotection against liver ischemia and reperfusion injury: inhibition of type-1 interferon signaling. Transplantation. 2007;83:1628–1634. doi: 10.1097/01.tp.0000266917.39958.47. [DOI] [PubMed] [Google Scholar]
- 32.Goda N, Suzuki K, Naito M, Takeoka S, Tsuchida E, Ishimura Y, Tamatani T, Suematsu M. Distribution of heme oxygenase isoforms in rat liver. Topographic basis for carbon monoxide-mediated microvascular relaxation. J Clin Invest. 1998;101:604–612. doi: 10.1172/JCI1324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lehmann E, El-Tantawy WH, Ocker M, Bartenschlager R, Lohmann V, Hashemolhosseini S, Tiegs G, Sass G. The heme oxygenase 1 product biliverdin interferes with hepatitis C virus replication by increasing antiviral interferon response. Hepatology. 2010;51:398–404. doi: 10.1002/hep.23339. [DOI] [PubMed] [Google Scholar]
- 34.Tzima S, Victoratos P, Kranidioti K, Alexiou M, Kollias G. Myeloid heme oxygenase-1 regulates innate immunity and autoimmunity by modulating IFN-beta production. J Exp Med. 2009;206:1167–1179. doi: 10.1084/jem.20081582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Bjorkbacka H, Fitzgerald KA, Huet F, Li X, Gregory JA, Lee MA, Ordija CM, Dowley NE, Golenbock DT, Freeman MW. The induction of macrophage gene expression by LPS predominantly utilizes Myd88-independent signaling cascades. Physiol Genomics. 2004;19:319–330. doi: 10.1152/physiolgenomics.00128.2004. [DOI] [PubMed] [Google Scholar]
- 36.Vidali M, Stewart SF, Albano E. Interplay between oxidative stress and immunity in the progression of alcohol-mediated liver injury. Trends Mol Med. 2008;14:63–71. doi: 10.1016/j.molmed.2007.12.005. [DOI] [PubMed] [Google Scholar]
- 37.Serhan CN, Savill J. Resolution of inflammation: the beginning programs the end. Nat Immunol. 2005;6:1191–1197. doi: 10.1038/ni1276. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.