

Abstract
Loss of functional pancreatic β cells is a hallmark of both type 1 and 2 diabetes. Identifying the pathways that promote β cell proliferation and/or block β cell apoptosis is a potential strategy for diabetes therapy. The transcriptional coactivator Yes-associated protein (YAP), a major downstream effector of the Hippo signaling pathway, is a key regulator of organ size and tissue homeostasis by modulating cell proliferation and apoptosis. YAP is not expressed in mature primary human and mouse β cells. We aimed to identify whether reexpression of a constitutively active form of YAP promotes β cell proliferation/survival. Overexpression of YAP remarkably induced β cell proliferation in isolated human islets, while β cell function and functional identity genes were fully preserved. The transcription factor forkhead box M1 (FOXM1) was upregulated upon YAP overexpression and necessary for YAP-dependent β cell proliferation. YAP overexpression protected β cells from apoptosis triggered by multiple diabetic conditions. The small redox proteins thioredoxin-1 and thioredoxin-2 (Trx1/2) were upregulated by YAP; disruption of the Trx system revealed that Trx1/2 was required for the antiapoptotic action of YAP in insulin-producing β cells. Our data show the robust proproliferative and antiapoptotic function of YAP in pancreatic β cells. YAP reconstitution may represent a disease-modifying approach to restore a functional β cell mass in diabetes.
Introduction
β Cell failure (loss of β cell function and mass) is a hallmark of both type 1 and 2 diabetes (T1D/T2D). Over the past 20 years, little progress has been made in identifying efficient strategies to stop β cell failure. The further decline in β cell function during current therapies highlights the need for improved therapeutic approaches as well as better understanding of the molecular changes underlying functional β cell loss in diabetes. Apoptosis of insulin-producing β cells is the fundamental cause of T1D and a contributing factor to the reduced β cell mass in T2D (1–4). Given the varied and enigmatic nature of the causes of β cell failure, inhibition of apoptosis and/or enhancement of β cell regenerative capacity by augmenting β cell proliferation represent attractive therapeutic approaches to the treatment of diabetes (5).
Identification of signaling molecules that regulate both β cell apoptosis and proliferation, together with an in-depth knowledge of their mechanisms of action is a prerequisite for the discovery of new drugs for β cell–directed therapies in diabetes. Characterization of signaling cascades such as PI3K-AKT, MAPK, and Wnt has particularly contributed to the understanding of β cell pathogenesis in diabetes (6–12). A more recently discovered signal is the Hippo pathway that was proposed to regulate organ size. Initially defined in Drosophila, the conservation of this pathway in mammals has been firmly established, where it consists of the core kinase complexes mammalian sterile 20–like kinases (MST1/2) and large-tumor suppressors (LATS1/2), adaptor proteins (SAV1 for MST1/2 and MOB1 for LATS1/2), and downstream transcriptional coactivators (YAP and TAZ). MST1/2 kinases phosphorylate and activate LATS1/2 kinases. Active LATS1/2 phosphorylates YAP at serine 127 (S127) and provides the docking site for the 14-3-3 protein, which sequesters YAP in the cytoplasm and ultimately leads to ubiquitin-dependent degradation that thereby prevents YAP transcriptional activity (13–16). YAP acts mainly through TEA domain (TEAD) family transcription factors to promote the expression of target genes required for cell proliferation and survival. The loss of any component of the kinase core results in a YAP-dependent increase in proliferation and resistance to apoptosis in multiple tissues, suggesting Hippo as a powerful tool to regulate organ size (16–18).
Targeting the Hippo signaling pathway has recently emerged as an attractive therapeutic strategy for treatment of various pathological disorders (19–24). Although previously studied in pancreas development (25–27), little is known about the role of Hippo signaling components in the adult β cell in normal and disease states. We have recently identified MST1, the key core component of Hippo signaling, as a principal regulator of pancreatic β cell apoptosis and dysfunction in human and rodent β cells in vitro as well as in diabetic animal models in vivo (28, 29). At prenatal developmental stages, pancreas proliferation and cell-type specification is regulated by Hippo signaling. As major downstream effectors, TEAD and its coactivator YAP play a crucial role in the expansion of pancreatic progenitors by controlling key pancreatic signaling mediators and transcription factors in the embryonic phase of pancreas development (27). Consistently, YAP depletion is sufficient to block pancreatic progenitor cell proliferation (30). However, convincing studies have revealed that YAP is not expressed in terminally differentiated mature primary human and mouse β cells. YAP expression decreases as pancreas development proceeds and eventually switches off in the mature endocrine but not in the exocrine cells (25, 26); this correlates with the extremely low rate of β cell proliferation and β cell quiescence. Thus, while YAP signals are disconnected from the core Hippo kinases in mature islets, MST1/2 and LATS1/2 are still expressed and are able to activate Hippo signaling in the absence of YAP, suggesting the presence of alternative Hippo downstream effector(s). Notably, lack of MST1 alone (28) or both MST1 and MST2 (26) is not sufficient to drive pancreatic β cells out of quiescence and induce β cell proliferation. The absence of YAP as a critical signaling element of the Hippo pathway may explain the lack of endocrine cell proliferation in this context. Conversely, pancreas-specific MST1/2 knockout is sufficient to increase ductal and acinar proliferation, due to aberrant YAP activity in the exocrine compartments (26).
Since YAP has both antiapoptotic and proproliferative activity in other cell types, the reexpression of YAP may provide a strategy directed towards promoting β cell proliferation and protecting from stress-induced conditions relevant to diabetes. In this study, we aimed to identify whether reexpression of a constitutively active form of YAP promotes β cell proliferation in isolated human islets and whether such overexpression would also protect β cells from apoptosis under diabetic conditions.
Results
Overexpression of constitutively active YAP (YAPS127A) promotes humanβcell proliferation. Given the proproliferative function of YAP in other cell types, we asked whether reintroduction of the activated form of YAP is sufficient to drive β cell proliferation. Isolated human islets were infected with adenovirus expressing luciferase (Ad-Luc) as a control or Ad-YAP expressing activated YAP with a serine 127 to alanine substitution (YAPS127A) that abolishes MST/LATS-mediated YAP inactivation and provides a constitutively active form of YAP (31). YAP overexpression remarkably increased β cell proliferation in isolated primary human islets seen in multiple different organ donors by significantly increased Ki67-positive (Figure 1, A and B, and Figure 2, D and E) as well as BrdU-positive β cells (Figure 1, C and D, and Figure 2, F and G). Our triple-staining data show that most of the Ki67- and insulin-positive or BrdU- and insulin-positive cells are YAP positive (Supplemental Figure 1; supplemental material available online with this article; doi:10.1172/jci.insight.86326DS1), suggesting a cell-autonomous action of YAP on β cell proliferation.
Figure 1. YAP overexpression promotes human β cell proliferation.

(A–D) Human islets were infected with either control adenovirus expressing luciferase (Ad-Luc) or Ad-YAP for 2 days. β Cell proliferation was analyzed by double staining for Ki67 (A and B) or BrdU (C and D) (red) and insulin (green). Pooled data from 7 (A, n = 7) or 5 (C, n = 5) different human islet donors. Scale bars: 100 μm. Data are the mean ± SEM. *P < 0.001, **P < 0.01 for Ad-YAP– compared with Ad-Luc–infected islets by 2-tailed Student’s t test.
Figure 2. YAP-induced human β cell proliferation is FOXM1 dependent.

(A) FOXM1 gene expression was analyzed by RT-PCR and levels normalized to cyclophilin and shown as change from control human islets infected with adenovirus expressing luciferase (Ad-Luc). Pooled data from 3 independent experiments from 3 different human islet donors (n = 3). (B and C) Western blots (B) and quantitative densitometry analysis (C) of FOXM1 protein level is shown. Western blots show results from 4 independent experiments from 4 different human islet donors (n = 4). (D–G) Human islets were infected with either control Ad-Luc or Ad-YAP and exposed to 10 μM FOXM1 inhibitor (thiostrepton; FOXM1i) for 2 days. β Cell proliferation was analyzed by double staining for Ki67 (D and E) or BrdU (F and G) (red) and insulin (green). (D and F) Pooled data from 4 independent experiments from 4 different human islet donors (n = 4). These 4 individual human islet data sets for Ad-Luc and Ad-YAP were also used for the quantification in Figure 1, A and C. Scale bar: 100 μm (applies to both E and G). Data are the mean ± SEM. *P < 0.05 for Ad-YAP compared with Ad-Luc–infected islets. **P < 0.05 for FOXM1i-treated Ad-YAP–infected compared with untreated Ad-YAP–infected islets. All 2-tailed Student’s t test except A (1-tailed).
Gene expression profiling showed that YAP potently and coordinately initiates gene programs that promote cell-cycle activity and proliferation in different cell types (32, 33). As many of these YAP-target genes are able to promote β cell proliferation, e.g., the transcription factor forkhead box M1 (FOXM1) (34), their identification as regulators of YAP might provide a molecular explanation of how YAP induces β cell proliferation. To identify the downstream YAP effector and mediator of YAP-induced proliferation in β cells, we investigated expression levels of FOXM1. YAP overexpression significantly induced a 2-fold elevated FOXM1 mRNA expression in human islets (Figure 2A) together with an increase in FOXM1 protein (Figure 2, B and C) levels. To investigate the potential of FOXM1 as a mediator of YAP-induced β cell proliferation, we used a pharmacologic inhibitor of FOXM1 expression, thiostrepton (35, 36). Thiostrepton fully blocked β cell proliferation in the Ad-YAP–infected human islets (Figure 2, D–G), suggesting that increased FOXM1 is a crucial contributor of β cell proliferation in YAP-reconstituted β cells. Together, our data show that YAP robustly induced β cell proliferation in a FOXM1-dependent way.
Overexpression of constitutively active YAP does not alter β cell functional status.
We then investigated whether YAP overexpression in human islets compromised their ability to secrete insulin in response to glucose in vitro. Glucose-stimulated insulin secretion (GSIS) assays in human islets infected with Ad-Luc or Ad-YAP showed that YAP-overexpressing islets maintained their insulin secretory capacity (Figure 3, A and B). To assess whether overexpression of YAP caused a loss of β cell identity or dedifferentiation, expression of functional genes including those encoding endocrine hormones (INS and GCG), key β cell transcription factors (PDX1, NeuroD1, MafA, Nkx2.2, Nkx6.1, and Pax4), as well as critical genes involved in glucose sensing (GCK and Slc2a2) were analyzed by RT-PCR in human islets and INS-1E cells and were fully preserved in Ad-YAP–infected or YAP-transfected cells (Figure 3, C and D). This result suggests full β cell identity and functionality upon YAP reexpression in islets. Despite the tendency of higher expression of some genes including Nkx6.1 in Ad-YAP–infected islets, statistical analysis of pooled quantification data from 4 different human islet donors shows no significant differences for all analyzed genes except for MafA (Figure 3C; P < 0.05). MafA is a well-known regulator of insulin gene expression and is required for β cell maturation (37, 38), but its modest upregulation upon YAP overexpression did not alter insulin mRNA levels in our study. This is not surprising, as insulin gene expression is known to be regulated by a complex transcriptional network involving multiple transcription factors such as PDX1, NeuroD1, and MafA (39). Thus, the slight upregulation of one (MafA) — while 2 other critical transcription factors were unchanged (NeuroD1 and PDX1) — was insufficient to enhance insulin mRNA transcription.
Figure 3. YAP overexpression does not alter β cell functional status.

(A–C) Human islets were infected with either control adenovirus expressing luciferase (Ad-Luc) or Ad-YAP for 2 days. (A) Insulin secretion during a 1-hour incubation with 2.8 mM (basal) and 16.7 mM glucose (stimulated), normalized to insulin content. (B) The insulin stimulatory index denotes the ratio of secreted insulin during the 1-hour incubation with 16.7 mM and 2.8 mM glucose. (A and B) Pooled data from 3 independent experiments from 3 different human islet donors (n = 3). (C) RT-PCR for GCK, GCG, NeuroD1, MafA, Pdx1, insulin (INS), Nkx2.2, Slc2a2, and Nkx6.1. Pooled data from 4 independent experiments from 4 different human islet donors (n = 4). (D) INS-1E cells were transfected with either control GFP or YAP plasmid for 2 days. RT-PCR for Ins1, Ins2, Pdx1, Pax4, NeuroD1, Nkx6.1, MafA, Nkx2.2, Slc2a2, and GCK. Data are pooled data from 2 independent experiments (n = 2). (C and D) For analysis, we used the Applied Biosystems Step One Real-Time PCR system with TaqMan Fast Universal PCR Master Mix for TaqMan assays. Data are the mean ± SEM. *P < 0.05 for Ad-YAP–infected compared with Ad-Luc–infected islets by 2-tailed Student’s t test.
Overexpression of constitutively active YAP protects β cells from apoptosis under diabetic conditions.
The important role of YAP-associated signaling in suppressing apoptosis led us to hypothesize that YAP activation would also protect β cells from apoptosis under diabetic conditions. INS-1E cells were transfected either with YAP or control GFP plasmid and then exposed chronically to increased glucose concentrations (glucotoxicity), its combination with the free fatty acid palmitate (glucolipotoxicity), the mixture of proinflammatory cytokines interleukin-1β (IL-1β) and interferon γ (IFN-γ; cytokine toxicity), as well as acute exposure to hydrogen peroxide (H2O2, an in vitro model of oxidative stress). Overexpression of YAP markedly reduced glucose-, glucose/palmitate-, cytokine-, as well as H2O2-induced apoptosis as represented by decreased caspase-3 and PARP cleavage (Figure 4, A–F). Glucotoxicity and glucolipotoxicity as well as proinflammatory cytokines reduced the level of transfected YAP, which inversely correlated with caspase-3 and PARP cleavage (Figure 4, A and C). In order to further corroborate the antiapoptotic impact of reexpressed YAP in diabetes, we used an in vitro model of human islets exposed to a diabetogenic milieu of the proinflammatory cytokines or glucolipotoxicity. YAP was overexpressed by adenovirus-mediated infection of human islets isolated from nondiabetic organ donors. Apoptosis triggered by proinflammatory cytokines as well as by the mixture of high glucose/palmitate was counteracted by YAP overexpression in isolated human islets, demonstrated independently by 4 human islet isolations (representative Figures shown from 3 isolations; Figure 5, A and C). Our data show that YAP overexpression significantly blocked caspase-3 activation in human islets under diabetic conditions (Figure 5, B and D). These data show that YAP functions as a prosurvival signal to ameliorate the proapoptotic effect of diabetic conditions in the β cells.
Figure 4. YAP overexpression protects β cells from apoptosis.

(A–F) INS-1E cells were transfected with either control GFP or YAP plasmid and treated with (A and B) the proinflammatory cytokines recombinant human IL-1β (2 ng/ml) and 1,000 U/ml IFN-γ (IL/IF); or (C and D) 22.2 mM glucose (Glc) or the mixture of 22.2 mM glucose and 0.5 mM palmitate (22.2Palm) for 2 days; or (E and F) 25 μM H2O2 for 5 hours. Representative Western blots (A, C, and E) and quantitative densitometry analysis (B, D, and F) of cleaved caspase-3 (Cl Casp3) and cleaved PARP (Cl PARP) protein levels are shown. (B) Cl Casp3 (n = 5) and Cl PARP (n = 3). (D) Cl Casp3 (n = 4) and Cl PARP (n = 3). (F) Cl Casp3 (n = 3) and Cl PARP (n = 3). Data are the mean ± SEM. *P < 0.05 for treated GFP-transfected compared with untreated GFP-transfected condition. **P < 0.05 for treated YAP-transfected compared with treated GFP-transfected conditions. §P = 0.06 for 22.2 mM glucose–treated YAP-transfected compared with 22.2 mM glucose–treated GFP-transfected condition. §§P = 0.07 for 22.2Palm-treated YAP-transfected compared with 22.2Palm-treated GFP-transfected condition. All by 2-tailed Student’s t test.
Figure 5. YAP overexpression protects human islets from apoptosis.

(A–D) Human islets were infected with either control adenovirus expressing luciferase (Ad-Luc) or Ad-YAP and then treated with (A and B) the proinflammatory cytokines recombinant human IL-1β (2 ng/ml) and 1,000 U/ml IFN-γ (IL/IF) or (C and D) the mixture of 22.2 mM glucose (Glc) and 0.5 mM palmitate (22.2Palm) for 3 days. Representative Western blots of 3 different human islet donors (A and C) and quantitative densitometry analysis (B and D) of cleaved caspase-3 (Cl Casp3) protein levels is shown. (B and D) Pooled data from 4 independent experiments from 4 different human islet donors (n = 4). Data are the mean ± SEM. *P < 0.05 for Ad-Luc IL/IF- or 22.2Palm-treated compared with untreated Ad-Luc control condition. **P < 0.05 for Ad-YAP IL/IF- or 22.2Palm-treated compared with Ad-Luc IL/IF- or 22.2Palm-treated condition. All 2-tailed Student’s t test.
The antiapoptotic effect of YAP is mediated by redox proteins Trx1/2.
Since oxidative stress is an underlying mechanism of β cell destruction in diabetes (40–42) and ROS-associated signaling is regulated by YAP in other cell types (43, 44), we tested whether YAP overexpression would change expression of ROS-related genes. To identify YAP-regulated genes involved in the antioxidant system, we analyzed a panel of 25 ROS-related genes in the constitutively active YAP mutant–expressing or GFP-control–expressing INS-1E cells (data not shown). Our gene expression profiling revealed the components of the redox thioredoxin system including thioredoxin-1 (Trx1) and thioredoxin-2 (Trx2) as the most significantly upregulated genes in YAP-overexpressing β cells (Figure 6A). To further substantiate YAP-dependent regulation of Trx1/2, we measured the expression of Trx1/2 genes in isolated human islets. RT-PCR analysis showed that expression of Trx1 and Trx2 was higher in YAP-overexpressing human islets, compared with Ad-Luc controls (Figure 6B). Consistently, higher levels of Trx1 and Trx2 proteins were detected by Western blotting in YAP-overexpressing INS-1E cells (Figure 6, C and D) and human islets (Figure 6, E and F), confirming functional upregulation of Trx1/2 upon reexpression of YAP. Based on these observations, we sought to determine whether Trx1/2 is required for the prosurvival effect of YAP in β cells under diabetic conditions. Disruption of Trx1/2 redox equilibrium by an irreversible inhibitor of Trx1, 1-methylpropyl 2-imidazolyl disulfide (PX-12), or a chemical inhibitor of thioredoxin reductase (TrxR), auranofin, not only diminished the antiapoptotic effect of YAP but also highly exacerbated high-glucose-induced apoptosis as represented by upregulation of caspase-3 and PARP cleavage (Figure 6G). To further confirm that Trx1/2 mediates the effect of YAP on apoptosis inhibition, Trx1 and Trx2 were depleted by short interfering RNA (siRNA) in INS-1E cells. Loss of Trx1 or Trx2 potentiated glucose-induced apoptosis in the GFP control group as represented by cleavage of caspase-3 and PARP. The extent of induction (magnitude of amplification above basal high-glucose-treated and scrambled control siRNA–transfected cells, for both GFP- and YAP-transfected groups) was much lower in the YAP-transfected group; this correlates with remaining YAP present in Trx1- or Trx2-depleted cells (Figure 6H). Notably, Trx1/2 silencing reduced protein levels of exogenously expressed YAP; this correlated with massive induction of apoptosis and suggests the importance of cellular redox states for maintaining YAP protein levels (Figure 6H). Our data show a prosurvival mechanism of YAP action via Trx1/2 signaling and the functional significance of Trx1/2 as upstream effectors of YAP protein levels in the β cell.
Figure 6. Antiapoptotic effect of YAP is mediated by redox proteins Trx1/2.

(A, C, and D) INS-1E cells were transfected with either control GFP or YAP plasmid for 2 days. (A) Trx1 and Trx2 gene expression was analyzed by RT-PCR and levels normalized to actin and shown as change from GFP-transfected cells. Pooled data from 3 independent experiments (n = 3). (C and D) Representative Western blots (C) and quantitative densitometry analysis (D) of Trx1 and Trx2 protein levels are shown. Both lanes were run in the same gel but were noncontiguous. Pooled data from 4 independent experiments (n = 4). (B, E, and F) Human islets were infected with either control adenovirus expressing luciferase (Ad-Luc) or Ad-YAP for 2 days. (B) Trx1 and Trx2 gene expression was analyzed by RT-PCR and levels normalized to tubulin and shown as change from Ad-Luc–infected human islets. Pooled data from 3 independent experiments from 3 human islet donors (n = 3). (E and F) Representative Western blots (E) and quantitative densitometry analysis (F) of Trx1 and Trx2 protein levels are shown. Pooled data from 4 independent experiments from 4 different human islet donors (n = 4). Flag and GAPDH blots in E are reused from human islet number 3 in Figure 2B. (G) INS-1E cells were transfected with either control GFP or YAP plasmids and treated with 22.2 mM glucose (HG) for 2 days. PX-12 (Trx1 inhibitor) and auranofin (thioredoxin reductase inhibitor) were added for the last 24 hours. (H) INS-1E cells were transfected with either control GFP or YAP and scrambled control siRNA (siScr) or Trx1 or Trx2 siRNA and treated with 22.2 mM glucose (HG) for 2 days. All lanes were run in the same gel but were noncontiguous. Western blots show representative results from 3 independent experiments (n = 3). Data are the mean ± SEM. *P < 0.05, **P < 0.01, ***P < 0.001 for YAP compared with control GFP- or Luc- transfected/infected conditions by 2-tailed Student’s t test.
Discussion
Diabetes results from an insufficient functional β cell mass. The identification of signaling molecules that foster β cell mass regeneration by simultaneous blockade of β cell apoptosis and augmentation of β cell proliferation would be highly important and most suitable for a β cell–directed therapy in diabetes. As regenerating β cells are similarly susceptible to apoptotic attack in general and even further under diabetes-relevant stressors such as inflammation and glucolipotoxicity, targets that induce β cell proliferation should ideally be combined with apoptosis-blocking strategies in order to maintain a functional β cell mass and thus to correct impaired glucose homeostasis. Here we show that the Hippo terminal effector YAP not only induced adult human β cell proliferation but also protected β cells from apoptosis induced by a diabetic milieu, suggesting that YAP is a potent factor involved in both β cell replication and survival.
Stimulating cell cycle reentry and proliferation of adult human β cells has proven to be very difficult and challenging (45–49). Processes that mediate the postnatal and age-dependent decline in proliferative and regenerative capacity of β cells remain largely unknown (50–53). Hypothetically, the loss of YAP signals right at the onset of pancreatic neurogenin-3 (Ngn3) expression and development of the endocrine pancreas (54) would reduce the ability of adult β cells to reenter the cell cycle and thus limit the regenerative potential of adult β cells. Overexpression of the YAP canonical target gene encoding connective tissue growth factor (CTGF) in a β cell regeneration model of partial pancreatectomy or treatment of human islets with recombinant human CTGF induces β cell proliferation (55), demonstrating that a YAP target acts directly on islets to promote β cell replication. Recent research has provided further insights into the importance of YAP in tissue regeneration. A functional consequence of the experimental elevation of YAP activity is the entry of nondividing or hardly dividing cells into the cell cycle, seen in both liver (56, 57) and heart (31, 58, 59). Together, these lines of evidence show that YAP transcriptional activity is enhanced and required for initiating cell proliferation and for complete tissue recovery in response to different types of injury. Hence, transient elevation of YAP activity might be useful for β cells that normally do not undergo proliferation and for augmenting the regenerative capability of damaged β cells in the context of diabetes.
Functional interaction of YAP with many proliferation-associated signaling networks contributes to its growth-promoting activity and is related to multiple physiological processes such as the regulation of cell cycle and proliferation (19–24, 32). YAP-dependent human β cell proliferation was accompanied and regulated by increased expression of transcription factor FOXM1. FOXM1 is a master regulator of cellular proliferation by controlling G1/S transition, S-phase progression, as well as G2/M transition. Gannon’s lab pioneered establishing the critical role of FOXM1 in postnatal β cell proliferation and mass expansion under conditions of increased β cell stress and demand in vivo (60, 61). Mice with pancreas-specific deletion of FOXM1 showed progressive β cell loss and overt diabetes (62). Conversely, FOXM1 overexpression alone is sufficient to induce β cell proliferation in mouse and human primary islets, highlighting the central role of FOXM1 in β cell proliferation (34). Consistent with the YAP-dependent FOXM1 upregulation in our study, Mizuno et al. reported that the YAP-TEAD complex directly binds to the FOXM1 promoter and induces transcriptional upregulation of FOXM1 in a cancer model of malignant mesothelioma (32). As YAP is a potential oncoprotein, interventions to increase β cell proliferation raise concerns about the possibility of neoplastic growth. Thus, YAP-based therapeutic approaches to restore lost or damaged β cells should be combined with strategies designed to limit YAP overactivation and uncontrolled cell proliferation. The acute and targeted manipulation of YAP may keep its levels physiological, tightly controlled, and ideally sufficient for modest β cell proliferation, mass expansion, and restoration of glucose homeostasis.
Our data show that overexpression of the constitutively active form of YAP effectively diminished β cell death triggered by multiple diabetic conditions in vitro including proinflammatory cytokines, gluco- and lipotoxicity, and oxidative stress. The prosurvival effect of YAP reexpression in stressed β cells induced by different diabetic stimuli raises the possibility that the antiapoptotic outcome of YAP comes from coordinating and regulating a common antiapoptotic gene expression program that promotes β cell protection. By gene expression profiling and subsequent protein analysis, we demonstrate that YAP induced profound upregulation of Trx system components in β cells. In line with our data, 2 independent studies showed YAP-induced upregulation of Trx components either through liver-specific deletion of MST1/2 and subsequent YAP activation or direct activation of YAP (44, 63). The Trx system consists of NADPH, TrxR, and Trx and is a redox-sensitive signaling system that protects cells from oxidative stress. It has emerged as a key element linking redox regulation to the pathogenesis of several diseases (64). Oxidative stress has been recognized as one of the common underlying mechanisms of β cell failure in the pathogenesis of both types of diabetes (40–42). In vivo β cell–specific overexpression of Trx1 protects mice from development of diabetes in both T1D and T2D rodent models, demonstrating the efficacy of Trx1 to halt the progressive β cell demise (65, 66). Trx1 also functions as the molecular sensor for the redox-dependent regulation of MST1 activity; Trx1 inhibits stress-induced MST1 activation by directly compromising MST1 homodimerization and autophosphorylation (67). MST1 hyperactivity is a major underlying mechanism of β cell apoptosis and the consequent decrease in β cell mass in the diabetic state (28). Thus, Trx1 might function as a YAP-dependent molecular signal to turn off the prodiabetic milieu–induced MST1 activation and subsequent apoptosis as part of compensatory negative-feedback loop within the Hippo signaling. Further detailed investigation is required to support such a possibility. As highlighted, YAP overexpression induced Trx1/2 at mRNA and protein levels in both human islets and INS-1E cells in control (Figure 6, A–F) and diabetic states (Figure 6H). On the other hand, Trx1/2 silencing itself reduced exogenously expressed YAP protein (Figure 6H); this suggests that the Trx1/2 system — or the cellular redox state in general — controls the level of YAP. Thus, shifting the cellular redox environment to pro-oxidant states by loss of Trx1 or Trx2 promotes YAP degradation and reverses the prosurvival effect of YAP. Together, YAP-induced Trx1/2 upregulation and Trx1/2-mediated YAP regulation by MST1 (as speculated above) form complex regulatory signaling loops (see scheme in Figure 7), which control redox state and cell survival.
Figure 7. A model for how β cell proliferation and apoptosis are regulated by exogenously introduced YAP.
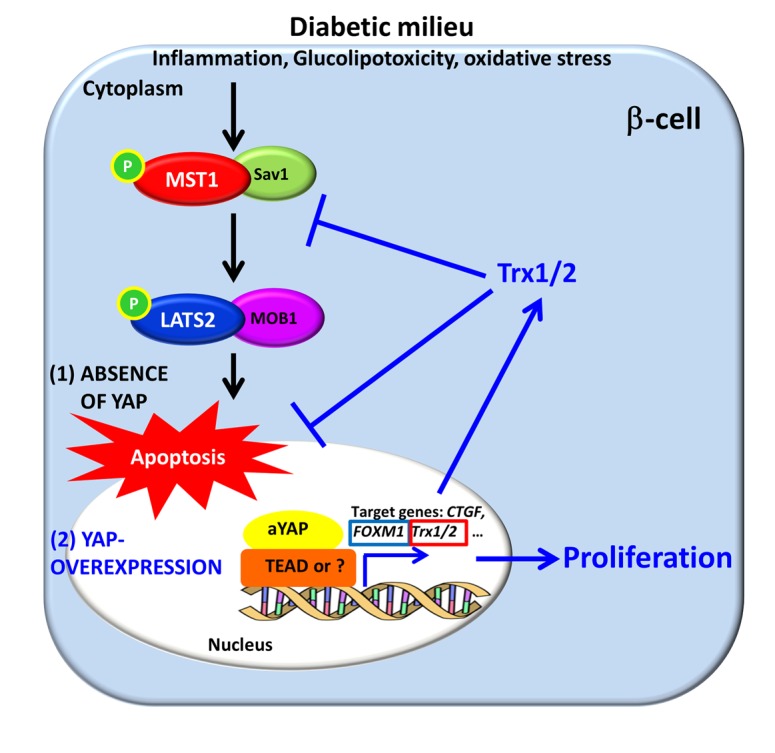
Various diabetic stimuli lead to activation of central Hippo signaling kinases MST1/LATS2. (1) In the absence of YAP, this activation results in collective transcriptional and posttranscriptional events leading to β cell apoptosis and impaired proliferation (black arrows). (2) Reexpression of the constitutively active form of YAP, which is absent in the mature β cell and functionally insensitive to the inhibitory action of MST1/LATS2 signaling, induces expression of a plethora of genes such as Trx1/2 and FOXM1 with antiapoptotic and proproliferative output, which protects β cells from diabetogenic attack (blue arrows). YAP-induced Trx1/2 upregulation and Trx1/2-mediated YAP regulation (through MST1) forms a complex regulatory signaling loop, which controls redox state and cell survival.
In full agreement with our study, George et al. (54) also reported the robust induction of human β cell proliferation by YAP activation with preservation of insulin-secretory function. In this study, Ngn3-dependent specification during development of the endocrine pancreas is sufficient for loss of YAP and decreased proliferation. Also, mechanistic (or mammalian) target of rapamycin (mTOR) signaling was proposed as an underlying mechanism of YAP-induced β cell proliferation (54). We have not directly assessed whether mTOR activity is regulated in YAP-overexpressing human islets, but FOXM1 might function as a part of mTOR-associated signaling in YAP-hyperactivated cells. This link between mTOR and FOXM1 was previously identified for epidermal growth factor receptor (EGFR) signaling–mediated β cell proliferation in response to overnutrition (68). Consistent with George et al. (54), most of the BrdU- and insulin-positive or Ki67- and insulin-positive cells were YAP positive, suggesting mostly a cell-autonomous proproliferative action of YAP. However, a non–cell autonomous effect of YAP overexpression cannot entirely be ruled out. For example, CTGF, an established canonical YAP target gene, is highly upregulated upon YAP overexpression in islets, as shown by George et al. (54) as well as in our study in human islets (data not shown). CTGF is a secreted growth factor that is capable of promoting human β cell proliferation and β cell regeneration in a model of partial pancreatectomy (55). Thus, highly produced CTGF and/or other uncharacterized secreted proteins upon YAP reconstitution may trigger proliferation in YAP-negative cells by a paracrine mechanism. Further studies are required to reveal such a possibility.
Conclusion.
Insufficient and dysfunctional β cell mass is a central feature of both types of diabetes. Thus, strategies aiming to regenerate pancreatic β cell mass by blocking β cell apoptosis as well as promoting proliferation are urgently needed. Considering the remarkable antiapoptotic and proproliferative output of YAP reexpression in β cells, manipulation of YAP may serve as a therapeutic tool for restoring functional β cell mass in diabetes. To warrant this, further mechanistic studies as well as the in vivo preclinical assessment of efficacy and side effects of YAP overexpression in β cells are required. Here, owing to its oncogenic potential, YAP activity must be strictly regulated.
Methods
Cell culture, treatment, and islet isolation.
Human islets were isolated from pancreases of healthy organ donors at the University of Chicago and at ProdoLabs and cultured on extracellular matrix–coated (ECM-coated) dishes (Novamed) as described previously (69). The clonal rat β cell line INS-1E was provided by Claes Wollheim, the University of Geneva, Geneva, Switzerland and the University of Lund, Lund, Sweden. Human islets were cultured in complete CMRL-1066 (Invitrogen) medium at 5.5 mM glucose and INS-1E cells in complete RPMI-1640 (Invitrogen) medium at 11.1 mM glucose as described previously (28). Islets and INS-1E were exposed to complex diabetogenic conditions (28). In some experiments, cells were additionally cultured with 10 μM FOXM1 inhibitor thiostrepton (Calbiochem), 1–5 μM selective Trx1/2 inhibitor PX-12, and 0.1–1 μM TrxR inhibitor auranofin (both Cayman Chemical). BrdU was added to the culture media at 10 μM for 2 days to label proliferating cells.
Transfections.
To knock down Trx1 and Trx2 in INS-1E cells, SMARTpool technology from Dharmacon was used. A mix of ON-TARGETplus siRNAs directed against the following sequences: in rat Txn1, GCAAAAUGAUCAAGCCCUU, GGAUGUUGCUGCAGACUGU, AGCUUGUGGUAGUGGACUU, CCUUGAAGUAGGACGUGGAU; and rat Txn2, GAUCCUAGGACCUCGGUUA, UAGCCAAACAGCACGGGAA, CAAAGUGGACAUUGACGAU, GAAGCUAAUUGGCUGACAA (100 nM, Dharmacon) was transiently transfected into INS-1E cells as described previously (28). GFP and YAP (pCMV-flag YAPS127A, a gift from Kunliang Guan [Addgene plasmid number 27370]) plasmids were used to overexpress these proteins in INS-1E cells.
Glucose-stimulated insulin secretion.
Glucose-stimulated insulin secretion in human islets was performed as described previously (28).
Immunohistochemistry.
Human islets cultured on ECM dishes were fixed, permeabilized, blocked, and incubated overnight at 4°C with anti–human Ki67 (catalog 08-1192; Invitrogen), mouse anti-BrdU (catalog NA61; Calbiochem), rabbit anti-Flag (catalog F7425; Sigma-Aldrich) and anti-insulin (catalog A0546; Dako) antibodies followed by fluorescein isothiocyanate (FITC)-, Cy3-, or AMCA-conjugated secondary antibodies (Jackson ImmunoResearch Laboratories). Slides were mounted with Vectashield with 4′,6-diamidino-2-phenylindole (DAPI; Vector Labs). Fluorescence was analyzed using a Nikon MEA53200 microscope and images were acquired using NIS-Elements software (Nikon).
Western blot analysis.
Western blotting was performed as described previously (28). Membranes were incubated overnight at 4°C with rabbit anti–cleaved caspase-3 (catalog 9664), rabbit anti-Trx1 (catalog 2429), rabbit anti-Trx2 (catalog 14907), rabbit anti-YAP (catalog 4912), rabbit anti-PARP (catalog 9532), rabbit anti–cleaved PARP (rat specific, catalog 9545), rabbit anti-tubulin (catalog 2146), rabbit anti-GAPDH (catalog 2118), rabbit anti–β-actin (catalog 4967) (all Cell Signaling Technology), rabbit anti-FOXM1 (catalog ab55006; Abcam), and rabbit anti-Flag (catalog F7425; Sigma-Aldrich), followed by horseradish peroxidase–linked anti–rabbit IgG (Jackson ImmunoResearch Laboratories). Membranes were developed using a chemiluminescence assay system (Pierce) and analyzed using DocIT LS image acquisition 6.6a (UVP BioImaging Systems). Densitometric analysis of the immunoblots was performed using Vision Works LS Image Acquisition and Analysis software Version 6.8 (UVP BioImaging Systems).
RNA extraction and RT-PCR analysis.
Total RNA was isolated from cultured human islets and INS-1E cells using TriFast (PEQLAB Biotechnologie), and RT-PCR performed as described previously (28). For analysis, we used the StepOne Real-Time PCR system with TaqMan Fast Universal PCR Master Mix for TaqMan assays (both Applied Biosystems). TaqMan Gene Expression Assays were used for PDX1 (Hs00426216_m1), SLC2A2 (Hs01096905_m1), GCK (Hs01564555_m1), INS (Hs02741908_m1), GCG (Hs01031536_s1), Nkx2.2 (Hs00159616_m1), MAFA (Hs01651425_s1), Nkx6.1 (Hs00232555_m1), NeuroD1 (HS01922995_s1), FoxM1 (Hs01073586_m1), and PPIA (Hs99999904_m1) for human and PDX1 (Rn00755591_m1), SLC2A2 (Rn00563565_m1), GCK (Rn00688285_m1), INS1 (Rn02121433_g1), INS2 (Rn01774648_g1), Nkx2.2 (Rn04244749_m1), Nkx6.1 (Rn01450076_m1), NeuroD1 (Rn00824571_s1), MafA (Rn00845206_s1), Pax4 (Rn00582529_m1) and PPIA (Rn00690933_m1) for rat or the SYBR Green Real-Time PCR Kit (Applied Biosystems) for mRNA expression of ROS-regulated genes with actin or tubulin as housekeeping controls for all experiments. The following primers were used: rat Txn1 Fw 5′-GTCAAATGCATGCCGACCTT-3′ and Rev 5′-AGTGGCTTCGAGCTTTTCCTT-3′; rat Txn2 Fw 5′-AAGCCTCCTCAAGGTGTGTG-3′ and Rev 5′-AGACCACCAGCATTGTACGG-3′; human TXN1 Fw 5′-GATGTGGATGACTGTCAGGATGT-3′ and Rev 5′-TCACCCACCTTTTGTCCCTTC-3′; human TXN2 Fw 5′-AGAGAAGATGGTGGCCAAGC -3′ and Rev 5′-CTCAATGGCGAGGTCTGTGT-3′.
Adenovirus infection.
Isolated human islets were infected with Ad-Luc as a control (provided by Allan E. Karlsen, Novo Nordisk A/S, Denmark) or active YAP (Ad-YAPS127A with an N-terminal triple FLAG epitope tag, provided by William Pu, Harvard University, Cambridge, Massachusetts, USA) at a multiplicity of infection (MOI) of 100 for 4 hours. Adenovirus was washed off with PBS and replaced by fresh complete medium. Staining, GSIS, and RNA and protein isolations were performed at 48, 72, or 96 hours after infection.
Statistics.
Samples (human islets) were evaluated in a randomized manner by 2 investigators (TY, ZA) who were blinded to the treatment conditions. All values were expressed as means ± SEM with the number of independent individual experiments presented in the figure legends. The different groups were compared by 2-tailed Student’s t test unless otherwise mentioned. A P value less than 0.05 was considered statistically significant.
Study approval.
Ethical approval for the use of human islets had been granted by the Ethics Committee of the University of Bremen.
Author contributions
AA conceived the project. TY and AA designed the project. TY, SR, ZA, BL, SA, KG, and AA preformed experiments and analyzed data. JO contributed new reagents or analytic tools. TY, KM, and AA wrote the paper. AA and KM supervised the project.
Supplementary Material
Acknowledgments
This work was supported by the China Scholarship Council, the German Research Foundation (DFG), the European Research Council, the Juvenile Diabetes Research Foundation (JDRF), and the German Diabetes Center grant (DZD) from the German Federal Ministry of Education and Research. Human islets were provided through the Integrated Islet Distribution Program supported by the NIH and the JDRF.
Footnotes
Conflict of interest: The authors declare that no conflict of interest exists.
Reference information:JCI Insight. 2016;1(18):e86326. doi:10.1172/jci.insight.86326.
Contributor Information
Ting Yuan, Email: ting.yuan830@gmail.com.
Sahar Rafizadeh, Email: rafizadeh_s@yahoo.com.
Zahra Azizi, Email: zahra.azizivarzaneh@gmail.com.
Blaz Lupse, Email: blazpikalupse@gmail.com.
Kanaka Durga Devi Gorrepati, Email: durga.kpb@googlemail.com.
Sushil Awal, Email: sush01wal@gmail.com.
Jose Oberholzer, Email: Jober@uic.edu.
Kathrin Maedler, Email: kmaedler@uni-bremen.de.
Amin Ardestani, Email: ardestani.amin@gmail.com.
References
- 1.Kurrer MO, Pakala SV, Hanson HL, Katz JD. Beta cell apoptosis in T cell-mediated autoimmune diabetes. Proc Natl Acad Sci U S A. 1997;94(1):213–218. doi: 10.1073/pnas.94.1.213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Mathis D, Vence L, Benoist C. beta-Cell death during progression to diabetes. Nature. 2001;414(6865):792–798. doi: 10.1038/414792a. [DOI] [PubMed] [Google Scholar]
- 3.Butler AE, Janson J, Bonner-Weir S, Ritzel R, Rizza RA, Butler PC. Beta-cell deficit and increased beta-cell apoptosis in humans with type 2 diabetes. Diabetes. 2003;52(1):102–110. doi: 10.2337/diabetes.52.1.102. [DOI] [PubMed] [Google Scholar]
- 4.Rhodes CJ. Type 2 diabetes-a matter of beta-cell life and death? Science. 2005;307(5708):380–384. doi: 10.1126/science.1104345. [DOI] [PubMed] [Google Scholar]
- 5.Vetere A, Choudhary A, Burns SM, Wagner BK. Targeting the pancreatic β-cell to treat diabetes. Nat Rev Drug Discov. 2014;13(4):278–289. doi: 10.1038/nrd4231. [DOI] [PubMed] [Google Scholar]
- 6.Tuttle RL, et al. Regulation of pancreatic beta-cell growth and survival by the serine/threonine protein kinase Akt1/PKBalpha. Nat Med. 2001;7(10):1133–1137. doi: 10.1038/nm1001-1133. [DOI] [PubMed] [Google Scholar]
- 7.Bernal-Mizrachi E, et al. Defective insulin secretion and increased susceptibility to experimental diabetes are induced by reduced Akt activity in pancreatic islet beta cells. J Clin Invest. 2004;114(7):928–936. doi: 10.1172/JCI20016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Elghazi L, Balcazar N, Bernal-Mizrachi E. Emerging role of protein kinase B/Akt signaling in pancreatic beta-cell mass and function. Int J Biochem Cell Biol. 2006;38(2):157–163. doi: 10.1016/j.biocel.2005.08.017. [DOI] [PubMed] [Google Scholar]
- 9.Subramanian SL, Hull RL, Zraika S, Aston-Mourney K, Udayasankar J, Kahn SE. cJUN N-terminal kinase (JNK) activation mediates islet amyloid-induced beta cell apoptosis in cultured human islet amyloid polypeptide transgenic mouse islets. Diabetologia. 2012;55(1):166–174. doi: 10.1007/s00125-011-2338-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Mokhtari D, Myers JW, Welsh N. MAPK kinase kinase-1 is essential for cytokine-induced c-Jun NH2-terminal kinase and nuclear factor-kappaB activation in human pancreatic islet cells. Diabetes. 2008;57(7):1896–1904. doi: 10.2337/db07-1670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Shu L, et al. TCF7L2 promotes beta cell regeneration in human and mouse pancreas. Diabetologia. 2012;55(12):3296–3307. doi: 10.1007/s00125-012-2693-z. [DOI] [PubMed] [Google Scholar]
- 12.Figeac F, Uzan B, Faro M, Chelali N, Portha B, Movassat J. Neonatal growth and regeneration of beta-cells are regulated by the Wnt/beta-catenin signaling in normal and diabetic rats. Am J Physiol Endocrinol Metab. 2010;298(2):E245–E256. doi: 10.1152/ajpendo.00538.2009. [DOI] [PubMed] [Google Scholar]
- 13.Goulev Y, Fauny JD, Gonzalez-Marti B, Flagiello D, Silber J, Zider A. SCALLOPED interacts with YORKIE, the nuclear effector of the hippo tumor-suppressor pathway in Drosophila. Curr Biol. 2008;18(6):435–441. doi: 10.1016/j.cub.2008.02.034. [DOI] [PubMed] [Google Scholar]
- 14.Wu S, Liu Y, Zheng Y, Dong J, Pan D. The TEAD/TEF family protein Scalloped mediates transcriptional output of the Hippo growth-regulatory pathway. Dev Cell. 2008;14(3):388–398. doi: 10.1016/j.devcel.2008.01.007. [DOI] [PubMed] [Google Scholar]
- 15.Zhao B, Li L, Lei Q, Guan KL. The Hippo-YAP pathway in organ size control and tumorigenesis: an updated version. Genes Dev. 2010;24(9):862–874. doi: 10.1101/gad.1909210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Harvey KF, Zhang X, Thomas DM. The Hippo pathway and human cancer. Nat Rev Cancer. 2013;13(4):246–257. doi: 10.1038/nrc3458. [DOI] [PubMed] [Google Scholar]
- 17.Halder G, Johnson RL. Hippo signaling: growth control and beyond. Development. 2011;138(1):9–22. doi: 10.1242/dev.045500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Harvey K, Tapon N. The Salvador-Warts-Hippo pathway - an emerging tumour-suppressor network. Nat Rev Cancer. 2007;7(3):182–191. doi: 10.1038/nrc2070. [DOI] [PubMed] [Google Scholar]
- 19.Tremblay AM, et al. The Hippo transducer YAP1 transforms activated satellite cells and is a potent effector of embryonal rhabdomyosarcoma formation. Cancer Cell. 2014;26(2):273–287. doi: 10.1016/j.ccr.2014.05.029. [DOI] [PubMed] [Google Scholar]
- 20.Lau AN, et al. Tumor-propagating cells and Yap/Taz activity contribute to lung tumor progression and metastasis. EMBO J. 2014;33(5):468–481. doi: 10.1002/embj.201386082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Mori M, et al. Hippo signaling regulates microprocessor and links cell-density-dependent miRNA biogenesis to cancer. Cell. 2014;156(5):893–906. doi: 10.1016/j.cell.2013.12.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Jiao S, et al. A peptide mimicking VGLL4 function acts as a YAP antagonist therapy against gastric cancer. Cancer Cell. 2014;25(2):166–180. doi: 10.1016/j.ccr.2014.01.010. [DOI] [PubMed] [Google Scholar]
- 23.Ma B, et al. Hypoxia regulates Hippo signalling through the SIAH2 ubiquitin E3 ligase. Nat Cell Biol. 2015;17(1):95–103. doi: 10.1038/ncb3073. [DOI] [PubMed] [Google Scholar]
- 24.Lin Z, et al. Cardiac-specific YAP activation improves cardiac function and survival in an experimental murine MI model. Circ Res. 2014;115(3):354–363. doi: 10.1161/CIRCRESAHA.115.303632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Gao T, et al. Hippo signaling regulates differentiation and maintenance in the exocrine pancreas. Gastroenterology. 2013;144(7):e86326. doi: 10.1053/j.gastro.2013.02.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.George NM, Day CE, Boerner BP, Johnson RL, Sarvetnick NE. Hippo signaling regulates pancreas development through inactivation of Yap. Mol Cell Biol. 2012;32(24):5116–5128. doi: 10.1128/MCB.01034-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Cebola I, et al. TEAD and YAP regulate the enhancer network of human embryonic pancreatic progenitors. Nat Cell Biol. 2015;17(5):615–626. doi: 10.1038/ncb3160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ardestani A, et al. MST1 is a key regulator of beta cell apoptosis and dysfunction in diabetes. Nat Med. 2014;20(4):385–397. doi: 10.1038/nm.3482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ardestani A, Maedler K. MST1: a promising therapeutic target to restore functional beta cell mass in diabetes. Diabetologia. 2016;59(9):1843–1849. doi: 10.1007/s00125-016-3892-9. [DOI] [PubMed] [Google Scholar]
- 30.Zhang ZW, Men T, Feng RC, Li YC, Zhou D, Teng CB. miR-375 inhibits proliferation of mouse pancreatic progenitor cells by targeting YAP1. Cell Physiol Biochem. 2013;32(6):1808–1817. doi: 10.1159/000356614. [DOI] [PubMed] [Google Scholar]
- 31.von Gise A, et al. YAP1, the nuclear target of Hippo signaling, stimulates heart growth through cardiomyocyte proliferation but not hypertrophy. Proc Natl Acad Sci U S A. 2012;109(7):2394–2399. doi: 10.1073/pnas.1116136109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Mizuno T, et al. YAP induces malignant mesothelioma cell proliferation by upregulating transcription of cell cycle-promoting genes. Oncogene. 2012;31(49):5117–5122. doi: 10.1038/onc.2012.5. [DOI] [PubMed] [Google Scholar]
- 33.Cao X, Pfaff SL, Gage FH. YAP regulates neural progenitor cell number via the TEA domain transcription factor. Genes Dev. 2008;22(23):3320–3334. doi: 10.1101/gad.1726608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Davis DB, et al. FoxM1 is up-regulated by obesity and stimulates beta-cell proliferation. Mol Endocrinol. 2010;24(9):1822–1834. doi: 10.1210/me.2010-0082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kwok JM, Myatt SS, Marson CM, Coombes RC, Constantinidou D, Lam EW. Thiostrepton selectively targets breast cancer cells through inhibition of forkhead box M1 expression. Mol Cancer Ther. 2008;7(7):2022–2032. doi: 10.1158/1535-7163.MCT-08-0188. [DOI] [PubMed] [Google Scholar]
- 36.Hegde NS, Sanders DA, Rodriguez R, Balasubramanian S. The transcription factor FOXM1 is a cellular target of the natural product thiostrepton. Nat Chem. 2011;3(9):725–731. doi: 10.1038/nchem.1114. [DOI] [PubMed] [Google Scholar]
- 37.Olbrot M, Rud J, Moss LG, Sharma A. Identification of beta-cell-specific insulin gene transcription factor RIPE3b1 as mammalian MafA. Proc Natl Acad Sci U S A. 2002;99(10):6737–6742. doi: 10.1073/pnas.102168499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Zhang C, et al. MafA is a key regulator of glucose-stimulated insulin secretion. Mol Cell Biol. 2005;25(12):4969–4976. doi: 10.1128/MCB.25.12.4969-4976.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Docherty HM, Hay CW, Ferguson LA, Barrow J, Durward E, Docherty K. Relative contribution of PDX-1, MafA and E47/beta2 to the regulation of the human insulin promoter. Biochem J. 2005;389(Pt 3):813–820. doi: 10.1042/BJ20041891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Watson D, Loweth AC. Oxidative and nitrosative stress in beta-cell apoptosis: their contribution to beta-cell loss in type 1 diabetes mellitus. Br J Biomed Sci. 2009;66(4):208–215. doi: 10.1080/09674845.2009.11730278. [DOI] [PubMed] [Google Scholar]
- 41.Lenzen S. Oxidative stress: the vulnerable beta-cell. Biochem Soc Trans. 2008;36(Pt 3):343–347. doi: 10.1042/BST0360343. [DOI] [PubMed] [Google Scholar]
- 42.Robertson RP, Harmon J, Tran PO, Poitout V. Beta-cell glucose toxicity, lipotoxicity, and chronic oxidative stress in type 2 diabetes. Diabetes. 2004;53 Suppl 1:S119–S124. doi: 10.2337/diabetes.53.2007.s119. [DOI] [PubMed] [Google Scholar]
- 43.Shao D, et al. A functional interaction between Hippo-YAP signalling and FoxO1 mediates the oxidative stress response. Nat Commun. 2014;5:e86326. doi: 10.1038/ncomms4315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Wu H, et al. The Ets transcription factor GABP is a component of the hippo pathway essential for growth and antioxidant defense. Cell Rep. 2013;3(5):1663–1677. doi: 10.1016/j.celrep.2013.04.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Saisho Y, Butler AE, Manesso E, Elashoff D, Rizza RA, Butler PC. β-cell mass and turnover in humans: effects of obesity and aging. Diabetes Care. 2013;36(1):111–117. doi: 10.2337/dc12-0421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Gregg BE, et al. Formation of a human β-cell population within pancreatic islets is set early in life. J Clin Endocrinol Metab. 2012;97(9):3197–3206. doi: 10.1210/jc.2012-1206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kulkarni RN, Mizrachi EB, Ocana AG, Stewart AF. Human β-cell proliferation and intracellular signaling: driving in the dark without a road map. Diabetes. 2012;61(9):2205–2213. doi: 10.2337/db12-0018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Bernal-Mizrachi E, Kulkarni RN, Scott DK, Mauvais-Jarvis F, Stewart AF, Garcia-Ocaña A. Human β-cell proliferation and intracellular signaling part 2: still driving in the dark without a road map. Diabetes. 2014;63(3):819–831. doi: 10.2337/db13-1146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Stewart AF, et al. Human β-cell proliferation and intracellular signaling: part 3. Diabetes. 2015;64(6):1872–1885. doi: 10.2337/db14-1843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Chen H, et al. PDGF signalling controls age-dependent proliferation in pancreatic β-cells. Nature. 2011;478(7369):349–355. doi: 10.1038/nature10502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Teta M, Long SY, Wartschow LM, Rankin MM, Kushner JA. Very slow turnover of beta-cells in aged adult mice. Diabetes. 2005;54(9):2557–2567. doi: 10.2337/diabetes.54.9.2557. [DOI] [PubMed] [Google Scholar]
- 52.Krishnamurthy J, et al. p16INK4a induces an age-dependent decline in islet regenerative potential. Nature. 2006;443(7110):453–457. doi: 10.1038/nature05092. [DOI] [PubMed] [Google Scholar]
- 53.Tschen SI, Dhawan S, Gurlo T, Bhushan A. Age-dependent decline in beta-cell proliferation restricts the capacity of beta-cell regeneration in mice. Diabetes. 2009;58(6):1312–1320. doi: 10.2337/db08-1651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.George NM, Boerner BP, Mir SU, Guinn Z, Sarvetnick NE. Exploiting expression of Hippo effector, Yap, for expansion of functional islet mass. Mol Endocrinol. 2015;29(11):1594–1607. doi: 10.1210/me.2014-1375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Riley KG, et al. Connective tissue growth factor modulates adult β-cell maturity and proliferation to promote β-cell regeneration in mice. Diabetes. 2015;64(4):1284–1298. doi: 10.2337/db14-1195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Dong J, et al. Elucidation of a universal size-control mechanism in Drosophila and mammals. Cell. 2007;130(6):1120–1133. doi: 10.1016/j.cell.2007.07.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Camargo FD, et al. YAP1 increases organ size and expands undifferentiated progenitor cells. Curr Biol. 2007;17(23):2054–2060. doi: 10.1016/j.cub.2007.10.039. [DOI] [PubMed] [Google Scholar]
- 58.Xin M, et al. Hippo pathway effector Yap promotes cardiac regeneration. Proc Natl Acad Sci U S A. 2013;110(34):13839–13844. doi: 10.1073/pnas.1313192110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Heallen T, et al. Hippo pathway inhibits Wnt signaling to restrain cardiomyocyte proliferation and heart size. Science. 2011;332(6028):458–461. doi: 10.1126/science.1199010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Zhang H, et al. Gestational diabetes mellitus resulting from impaired beta-cell compensation in the absence of FoxM1, a novel downstream effector of placental lactogen. Diabetes. 2010;59(1):143–152. doi: 10.2337/db09-0050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Ackermann Misfeldt A, Costa RH, Gannon M. Beta-cell proliferation, but not neogenesis, following 60% partial pancreatectomy is impaired in the absence of FoxM1. Diabetes. 2008;57(11):3069–3077. doi: 10.2337/db08-0878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Zhang H, et al. The FoxM1 transcription factor is required to maintain pancreatic beta-cell mass. Mol Endocrinol. 2006;20(8):1853–1866. doi: 10.1210/me.2006-0056. [DOI] [PubMed] [Google Scholar]
- 63.Nagaraj R, et al. Control of mitochondrial structure and function by the Yorkie/YAP oncogenic pathway. Genes Dev. 2012;26(18):2027–2037. doi: 10.1101/gad.183061.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Yoshihara E, Masaki S, Matsuo Y, Chen Z, Tian H, Yodoi J. Thioredoxin/Txnip: redoxisome, as a redox switch for the pathogenesis of diseases. Front Immunol. 2014;4:e86326. doi: 10.3389/fimmu.2013.00514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Hotta M, et al. Pancreatic beta cell-specific expression of thioredoxin, an antioxidative and antiapoptotic protein, prevents autoimmune and streptozotocin-induced diabetes. J Exp Med. 1998;188(8):1445–1451. doi: 10.1084/jem.188.8.1445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Yamamoto M, et al. Transgenic expression of antioxidant protein thioredoxin in pancreatic beta cells prevents progression of type 2 diabetes mellitus. Antioxid Redox Signal. 2008;10(1):43–49. doi: 10.1089/ars.2007.1586. [DOI] [PubMed] [Google Scholar]
- 67.Chae JS, Gil Hwang S, Lim DS, Choi EJ. Thioredoxin-1 functions as a molecular switch regulating the oxidative stress-induced activation of MST1. Free Radic Biol Med. 2012;53(12):2335–2343. doi: 10.1016/j.freeradbiomed.2012.10.527. [DOI] [PubMed] [Google Scholar]
- 68.Zarrouki B, et al. Epidermal growth factor receptor signaling promotes pancreatic β-cell proliferation in response to nutrient excess in rats through mTOR and FOXM1. Diabetes. 2014;63(3):982–993. doi: 10.2337/db13-0425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Schulthess FT, et al. CXCL10 impairs beta cell function and viability in diabetes through TLR4 signaling. Cell Metab. 2009;9(2):125–139. doi: 10.1016/j.cmet.2009.01.003. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.