

Abstract
Arginases are enzymes that are involved in many human diseases and have been targeted for new treatments. Here a series of cinnamides was designed, synthesized and evaluated in vitro and in silico for their inhibitory activity against mammalian arginase. Using a microassay on purified liver bovine arginase (b-ARG I), (E)-N-(2-phenylethyl)-3,4-dihydroxycinnamide, also named caffeic acid phenylamide (CAPA), was shown to be slightly more active than our natural reference inhibitor, chlorogenic acid (IC50 = 6.9 ± 1.3 and 10.6 ± 1.6 µM, respectively) but it remained less active that the synthetic reference inhibitor Nω-hydroxy-nor-l-arginine nor-NOHA (IC50 = 1.7 ± 0.2 µM). Enzyme kinetic studies showed that CAPA was a competitive inhibitor of arginase with Ki = 5.5 ± 1 µM. Whereas the activity of nor-NOHA was retained (IC50 = 5.7 ± 0.6 µM) using a human recombinant arginase I (h-ARG I), CAPA showed poorer activity (IC50 = 60.3 ± 7.8 µM). However, our study revealed that the cinnamoyl moiety and catechol function were important for inhibitory activity. Docking results on h-ARG I demonstrated that the caffeoyl moiety could penetrate into the active-site pocket of the enzyme, and the catechol function might interact with the cofactor Mn2+ and several crucial amino acid residues involved in the hydrolysis mechanism of arginase. The results of this study suggest that 3,4-dihydroxycinnamides are worth being considered as potential mammalian arginase inhibitors, and could be useful for further research on the development of new arginase inhibitors.
Keywords: arginase inhibitor, cinnamide, docking, screening, structure-activity relationships
1. Introduction
l-Arginine (l-Arg) is an amino acid involved in distinct metabolic routes for the synthesis of many different compounds including proteins, urea, polyamines, proline or nitric oxide (NO), and is consequently the substrate of various enzymes. Therefore, many studies focused on the search for bioactive compounds that are able to regulate l-arginine metabolism [1]. Nitric oxide synthase (NOS) hydrolyses l-Arg to produce l-citrulline and NO, a crucial vasorelaxant factor. Arginase (amidinohydrolase, EC 3.5.3.1), by hydrolysing l-arginine into l-ornithine and urea, plays an important role in the ammonia detoxification in mammals [2]. Interestingly, it has been shown these last few years that this enzyme also plays a crucial role in the bioavailability of l-arginine for nitric oxide synthase (NOS) by substrate competition [3]. Therefore, an increased activity of arginase is associated with various diseases by reducing the supply of l-arginine needed by NOS to produce NO, and by raising production of l-ornithine resulting in vascular structural problems [4]. In fact, l-ornithine is converted into either polyamines (putrescine, spermidine and spermine) or into proline, and these downstream products can promote cell proliferation and collagen production [4,5]. Over the last few decades, interest has grown concerning the use of arginase for therapeutic uses. Indeed, the use of arginase inhibitors has proven to be beneficial in various pathophysiological states such as hypertension [6], erectile dysfunction [7], pulmonary hypertension [8], atherosclerosis [9], diabetic renal injury [10], asthma and allergic rhinitis [11], myocardial ischemia-reperfusion injury [12], wound healing [13], and cancer [14]. The first generation of arginase inhibitors comprised analogs of Nω-hydroxy-l-arginine (NOHA), the intermediate in the production of NO from l-arginine by NOS [15,16]. These inhibitors are characterized by N-hydroxy-guanidium side chains. Among them, Nω-hydroxy-nor-l-arginine (nor-NOHA) is the most potent arginase inhibitor known to date [17]. Boronic acid analogs of l-arginine formed the second generation of arginase inhibitors, among which S-(2-boronoethyl)-l-cysteine (BEC) and 2-(S)-amino-6-boronohexanoic acid (ABH) are the main representative compounds [18,19]. Some of these synthetic arginase inhibitors, such as nor-NOHA, BEC and ABH, are currently available commercially. However, the use of such molecules for therapeutic purposes has several limitations: poor bioavailability and potential toxicity (BEC, and ABH) [20], very short half-life (nor-NOHA) [21], and also high cost [22]. Considering that the development of arginase inhibitors is of great therapeutic relevance in various human diseases, investigations have been undertaken in order to develop prodrugs of NOHA [23] or optimized ABH by substituting the Cα-amino acid function [12,24,25,26] and by replacing the boronic acid function [27,28,29,30,31]. Plants have been shown to be a source of promising arginase inhibitors [32]. Nevertheless, hemisynthesis has never been used for the design of new arginase inhibitors [32]. Among the natural compounds tested as potential arginase inhibitors, we previously reported that chlorogenic acid (CGA) displayed an interesting activity on bovine liver arginase I (b-ARG I) [33,34]. We therefore considered it as a potential lead compound. CGA is an ester of caffeic and quinic acids. We showed that the caffeoyl (i.e., dihydroxy cinnamoyl) part is probably more involved in the arginase inhibition than the quinoyl moiety [33,34]. Therefore, we replaced this latter moiety, containing four asymmetric carbons, in order to improve the inhibitory activity and to simplify the chemical structure. Considering that the stability of a compound is improved when its ester bond is replaced by an amide bond [35], and that the amidification with a range of phenethylamines has already been noted for their inhibitory activity on tyrosinase (which is another binuclear metalloenzyme) [36], we decided to evaluate this kind of amides for their arginase inhibition. Finally, given that several natural polyphenolic products have significant effects on arginase [34], the functionalization has been mainly focused on methoxy and hydroxyl groups which are common in such compounds. The optimal size of compounds and the contribution of each part have been explored to identify portions of the molecule that are essential for the expected biological activity.
The present work first aimed at synthesizing new or already known cinnamide derivatives in order to test their inhibitory property on a purified bovine liver arginase (b-ARG I) (Figure 1). Then, the most active compound of this series was evaluated on a recombinant human arginase I (h-ARG I). Additionally, enzyme-kinetic and molecular docking studies were also performed on the most active compounds and compared to CGA in order to analyze their inhibition mechanism and the protein-ligand interactions, respectively.
Figure 1.

Design of cinnamide derivatives from chlorogenic acid.
2. Results and Discussion
2.1. Chemistry
Cinnamide derivatives were synthesized using phosphonium salts as condensing reagents by a procedure adapted from Okombi et al. [37]. The synthesis protocol was modified by replacing benzotriazol-1-yloxytris(dimethylamino)-phosphonium hexafluorophosphate (BOP) by benzotriazol-1-yloxytri(pyrrolidino)-phosphonium hexafluorophosphate (PyBOP) [38] to avoid the formation of the carcinogenic side-product hexamethylphosphoric acid triamide (HMPA) [39]. The amide linkage was generated by mixing the free acid with a variety of amines in the presence of PyBOP, triethylamine (Et3N) in dimethylformamide (DMF) and CH2Cl2 (Scheme 1). The by-product tripyrrolidinophosphoric acid triamide (TPPA) derived from PyBOP was more viscous and less polar than HMPA. Furthermore, its boiling point was higher, making the separation of the cinnamide derivative and TPPA more difficult and leading to low yields [39]. Cinnamide derivatives (Table 1) were obtained with yields ranging from 19% to 98%.
Scheme 1.

General procedure for the synthesis of cinnamide derivatives. Reagents and conditions: PyBOP, DMF, Et3N, CH2Cl2, 0 °C (0.5 h) then room temperature (overnight).
Table 1.
Structures of the synthesized compounds and their inhibitory potencies toward b-ARG I.
| Compound | 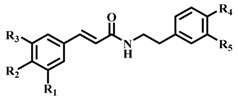 |
b-ARG I Inhibition a (%) (100 μM) | IC50 b (µM) | ||||
|---|---|---|---|---|---|---|---|
| R1 | R2 | R3 | R4 | R5 | |||
| 1 (CAPA) | OH | OH | H | H | H | 64 ± 2 | 6.9 ± 1.3 |
| 2 | OCH3 | OH | H | H | H | 37 ± 4 | n.d c |
| 3 | H | OH | H | H | H | 31 ± 4 | n.d |
| 4 | OCH3 | OCH3 | OCH3 | H | H | 35 ± 2 | n.d |
| 5 | H | H | H | H | H | 34 ± 2 | n.d |
| 6 | OH | OH | H | OH | H | 58 ± 2 | 22.1 ± 1.6 |
| 7 | OCH3 | OH | H | OH | H | 24 ± 2 | n.d |
| 8 | H | OH | H | OH | H | 40 ± 2 | n.d |
| 9 | OCH3 | OCH3 | OCH3 | OH | H | 30 ± 2 | n.d |
| 10 | H | H | H | OH | H | 34 ± 3 | n.d |
| 11 | OH | OH | H | OH | OH | 62 ± 3 | 114.9 ± 1.3 |
| 12 | OCH3 | OH | H | OH | OH | 61 ± 3 | 198.7 ± 1.4 |
| 13 | H | OH | H | OH | OH | 54 ± 1 | 170.4 ± 1.7 |
| 14 | OCH3 | OCH3 | OCH3 | OH | OH | 53 ± 1 | 193.6 ± 1.4 |
| 15 | H | H | H | OH | OH | 68 ± 3 | 39.3 ± 1.4 |
| 16 | 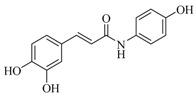 |
59 ± 1 | 35.6 ± 1.3 | ||||
| 17 d | 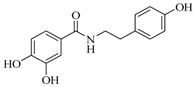 |
52 ± 2 | 175.3 ± 1.5 | ||||
| 18 | 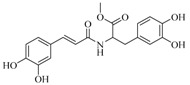 |
62 ± 1 | 41.9 ± 1.3 | ||||
| 19 | 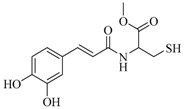 |
67 ± 1 | 37.0 ± 1.3 | ||||
| Caffeic acid | 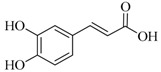 |
61 ± 4 | 86.7 ± 8.0 | ||||
| CGA | 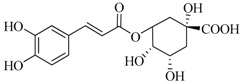 |
71 ± 1 | 10.6 ± 1.6 | ||||
| nor-NOHA e | 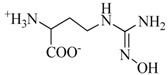 |
99 ± 1 | 1.7 ± 0.2 | ||||
a The values were obtained from three separate in-duplicate experiments; b IC50: Half maximal inhibitory concentration was obtained from three separate in-triplicate experiments; c n.d: not determined; d Compound 17 was prepared from 3,4-dihydroxybenzoic acid; e Nω-hydroxy-nor-l-arginine (nor-NOHA): used as reference inhibitor.
(E)-N-(2-phenylethyl)-3,4-dihydroxycinnamide (compound 1, CAPA) was chosen as the basic structure (Table 1). Subsequently, compounds 2–15 were synthesized to explore the effect on arginase inhibition of hydroxyl groups at different positions on the two aromatic rings and also their substitutions by methyl groups. Compounds 16 and 17 were synthesized to help explain contribution of the double bond of caffeoyl moiety and of the single bond of the phenethylamine moiety. Compound 18 was synthesized to explore the effect of a methylcarboxylate group on the phenethylamine moiety. Finally, to evaluate the importance of the aromatic ring on the phenethylamine moiety, it was replaced by a sulfhydryl group (compound 19). The structures of these compounds were confirmed by NMR, HRMS (ESI), IR spectrum, and by comparison with previous published data.
2.2. Arginase Inhibitory Activity
Arginase inhibitory activity was first evaluated using an in vitro assay optimizing the protocol of Corraliza et al. (1994) [40] by miniaturization and by using commercially-available purified bovine arginase (b-ARG I) instead of animal tissue [34]. The most active compound was then evaluated on a recombinant human arginase (h-ARG I). Nω-hydroxy-nor-l-arginine (nor-NOHA) and CGA served as reference inhibitors.
As shown in Table 1, arginase inhibition on b-ARG I was higher than 50% for eleven target compounds at 100 μM. IC50 values of these eleven compounds were determined to better estimate their potencies and discuss the structure–activity relationships. It is worth noting that all eleven of these compounds contain a catechol group, on the cinnamoyl (compounds 1, 6, 16 and 19), benzoyl (compound 17) or phenethyl parts (compounds 12–15), or on both parts (compounds 11 and 18). In fact, suppression or methylation of one or two hydroxyl groups of the catechol function led to a severe decrease in arginase inhibition activity (compound 1 vs. 2–5). Our main result is that structural modifications led to an improvement of the activity for compound 1, caffeic acid phenylamide (CAPA), which displayed a lower IC50 value (6.9 ± 1.3 μM, Figure 3A) compared to our natural reference compound CGA (IC50 = 10.6 ± 1.6 µM). Nevertheless, this value still remains higher than that of the reference compound nor-NOHA. In terms of structure–activity relationships (SARs), functionalization of the phenethyl part of 1 by one (compound 6) or two (compound 11) hydroxyl groups decreased the activity. However, if the cinnamoyl part remained unsubstituted, inhibition was partially restored by the presence of a catechol group on the phenethyl part (compound 15 vs. 11). This result confirmed the important role played by the catechol function for the arginase inhibitory activity of this series of compounds. Reducing the length of the molecule by suppressing a bond in either the caffeoyl side or the phenethyl side also decreased activity (compound 6 vs. 16, 17). However, retaining most or all of the cinnamoyl part seemed to be of greater importance than that of the phenethyl part. Concerning the single bond of the phenylethylamine moiety, a preliminary investigation has been made involving the substitution of the ethyl linker by a methylcarboxylate group (18) and by replacing the aromatic ring by a sulfhydryl group (19). The methylcarboxylate group at position 1 of the phenethylamine moiety improved the inhibitory activity (compound 18 vs. 11), and replacing the aromatic ring of the phenethylamine moiety by the sulfhydryl group had little effect on the inhibitory activity (compound 18 vs. 19), suggesting that no specific interaction occurred between this part of the molecule and the mouth of the active site (Figure 2). Therefore, pharmacomodulations in this position could be expected to increase compounds affinity toward the active site.
Figure 3.

Kinetics of arginase inhibition by 1 (CAPA). The IC50 was determined from a sigmoidal curve (A). The competitive inhibition mechanism was determined by analysis of the: Lineweaver–Burk (B); Dixon (C); and Cornish–Bowden (D) plots. Each data point represents the mean of three in-triplicate independent experiments.
Figure 2.

Structure–activity relationships for cinnamide derivatives on b-ARG I.
Considering its effect on b-ARG I, CAPA (1) was evaluated on a recombinant h-ARG I. The reference inhibitor nor-NOHA was previously evaluated on this model and gave an IC50 value of 5.7± 0.6 µM. The IC50 value of CAPA was found to be 60.3 ± 7.8 µM. This value is higher than that determined on b-ARG I (IC50 = 6.9 ± 1.3 µM). These results illustrated the fact that the evaluation on b-ARG I as an easy-to-handle and cheap source of mammalian arginase could be used in a preliminary study in order to search new potential active compounds. However, an evaluation of the most active compounds on h-ARG I is required before further in vivo studies.
2.3. Enzyme Kinetic Studies for CAPA (1)
The mechanism of inhibition and the inhibition constant (Ki) of compound 1 (CAPA) was assessed by enzyme-kinetic studies. The kinetic data were used to generate the Lineweaver–Burk, Dixon and Cornish–Bowden plots (Figure 3B–D), showing that CAPA is a competitive inhibitor of arginase [41].
The Ki values for CAPA, CGA and nor-NOHA could be calculated on the basis of the Cheng–Prusoff equation [Ki = IC50/(1 + [S]/Km)] [42], with [S] = 14.3 mM of l-arginine and the Michaelis–Menten constant of b-ARG I Km = 55.5 ± 10.5 µM [34] (Table 2).
Table 2.
Comparison of Ki values for arginase inhibition by CAPA, CGA and nor-NOHA.
| Inhibitor | Ki (µM) | Type of Inhibition |
|---|---|---|
| CAPA (1) | 5.5 ± 1.0 | competitive |
| CGA | 8.4 ± 1.2 | competitive |
| nor-NOHA | 1.3 ± 0.1 | competitive |
The in vitro affinity of CAPA was better than that of CGA (Ki 5.5 ± 1.0 vs. 8.4 ± 1.2 µM, respectively). In the last few years, several chemical groups that target the bimanganese cluster have been identified. Here, the in vitro affinity of CAPA was similar to that described for amino imidazole derivative [28], and better than thiosemicarbazide [43], sulfamide [27], nitro [29] and aldehyde [30] derivatives, which displayed Ki greater than 50 µM. Cinnamides could be added to this list and may help to design future inhibitors.
2.4. Molecular Docking Studies
In order to gain further insight into the inhibitory mechanism, the binding modes of CAPA (1) and CGA were determined by docking simulations on h-ARG I (pdb id: 3kv2). The structure of b-ARG I is not available in the Protein Data Bank but present 100% homology with h-ARG I in the active site. In addition, the alignment of the b-ARG I and h-ARG I sequences using the stand-alone Java Web Start application, accessible from the PDB server (http://www.rcsb.org), revealed a high level of similarity between the two sequences (91% and 95%, respectively). A few residues were different, but not involved in the active site of the two arginases, whose residues are strictly conserved. The most active compound 1 (CAPA), CGA and nor-NOHA were successfully docked into the active pocket of h-ARG I by using a FlexX docking program implemented in LeadIt 2.0.2 software [44].
The results obtained with CGA confirmed that the quinoyl moiety interact with residues of the mouth of the pocket formed by the catalytic site, helping the caffeoyl moiety of chlorogenic acid to approach the bottom of the active site and interact with Asp124, Asp232, Asp234 by H-interactions and His141 via cation–π interaction (Figure 4). Interestingly, the catechol group could chelate the Mn2+ ions in the active site of arginase.
Figure 4.

3D (left); and 2D (right) binding modes of CGA inside the active site of h-ARG I. For the 3D binding mode, CGA is represented as a “ball and stick” form: C (grey), O (red), and H (white); manganese ions are represented as cyan spheres; and the crucial residues of the binding site are represented by lines. H-bonds are represented as pink dotted lines, while the metal coordination bonds are represented by cyan dotted lines. Docking simulations were performed by FlexX program implemented in LeadIt 2.0.2 software, and the pictures of docking solution were created by MOE 2008.10 software.
Since CAPA was identified as a potential lead compound of the series, interactions of this compound with the residues of the active site of the enzyme were examined more closely. We noted with interest that the caffeoyl moiety of CAPA is able to interact with the Mn2+ cofactor and can make hydrogen interactions with Asp234 and Thr246 as well as π–π interactions with His141 and His126 of h-ARG I active site. The phenethyl moiety could have hydrophobic interactions residues at the mouth of active pocket (Figure 5).
Figure 5.

3D (left); and 2D (right) binding modes of CAPA (1) inside the active site of h-ARG I. For the 3D binding mode, CAPA is represented as a “ball and stick” form: C (grey), O (red), and H (white); manganese ions are represented as cyan spheres; and the crucial residues of the binding site are represented by lines. H-bonds are represented as pink dotted lines, while the metal coordination bonds are represented by cyan dotted lines. Docking simulations were performed by FlexX program implemented in LeadIt 2.0.2 software, and the pictures of docking solution were created by MOE 2008.10 software.
In addition, it is worth noting that compound 15 took a position that was opposite to that of CAPA, the cinnamoyl moiety staying at the mouth whereas the phenethylamine with a catechol group approached the active pocket of b-ARG I (data not shown). These opposite binding modes confirmed the hypothesis of the crucial role played by the catechol group in arginase inhibition, and clearly explained why the activity was recovered by compound 15 despite the fact the cinnamoyl moiety was unfunctionalized.
3. Materials and Methods
3.1. Chemistry
All reagents were purchased from Sigma-Aldrich and used without further purification, except for the purified liver bovine arginase I (b-ARG I) which was from MP Biomedicals (One unit (1 U) of b-ARG I is defined by this manufacturer as the amount of enzyme that converted 1 µmole of l-arginine to urea and l-ornithine per minute at pH 9.5 and 37 °C), and for the recombinant human arginase I (h-ARG I) (BXC572/n P0408M10) which was from Interchim (Number of units is not defined by the manufacturer and thus a preliminary study (data not shown) allowed us to choose the concentration of enzyme leading to an absorbance of about 1). Solvents (methanol, ethyl acetate, cyclohexane and dichloromethane) were supplied by Carlo Erba Reagents and VWR Chemicals companies. Reactions were monitored by thin-layer chromatography (TLC) on pre-coated silica gel aluminum plates (Macherey-Nagel) and visualized under UV light (254 and 365 nm). Flash column chromatography was carried out using EasyVarioFlash® DL cartridges (Merck, Fontenay-sous-Bois, France) for dry loading, eluting with CH2Cl2 and MeOH. 1H-NMR at 300 MHz and 13C-NMR at 75 MHz (only for compounds lacking analytical data from literature) were acquired using a Bruker AC300 spectrometer (Bruker BioSpin, Wissembourg, France). Chemical shifts (δ) were reported in parts per million (ppm) relative to the residual solvent signals. Coupling constants (J) were reported in hertz (Hz). Data were presented as follows: chemical shift (δ, ppm), multiplicity (s, singlet; bs, broad singlet; d, doublet; dd, doublet of doublet; t, triplet; q, quadruplet; m, multiplet), coupling constant (J, Hz), and integration. High-resolution electrospray ionisation mass spectra (HRMS-ESI) were analyzed by using the SCA Illkirch QToF instrument.
3.2. General Procedure for the Synthesis of Amide Derivatives 1−19
A solution of substituted cinnamic acid or 3,4-dihydroxybenzoic acid (1.66 mmol) and trimethylamine (Et3N) (0.35 mL, 2.49 mmol, 1.5 equivalent (eq)) in dimethylformamide (DMF) (3.5 mL) was cooled in an ice bath. A corresponding amine (1.66 mmol, 1 eq) was added and followed by addition of a solution of benzotriazolyloxy-tris(pyrrolidino)-phosphonium hexafluorophosphate (PyBOP) (864 mg, 1.66 mmol, 1 eq) in dichloromethane (CH2Cl2) (3.5 mL). The mixture was stirred for 30 min at 0 °C and left overnight at room temperature before CH2Cl2 was evaporated under vacuum. Then, 30 mL of water was added to the remaining solution and the resulting mixture was extracted with ethyl acetate (EtOAc) (3 × 75 mL). The organic phase was successively washed with 100 mL of 1 M HCl solution, 100 mL of water, 100 mL of 1 M NaHCO3 solution and 100 mL of brine, then dried over Na2SO4, filtered and evaporated under vacuum. The crude product was purified by silica gel flash chromatography (eluent: CH2Cl2/MeOH 98:2 to 80:20) to afford the desired compound [37].
(E)-N-(2-Phenylethyl)-3,4-dihydroxycinnamide (1). General procedure using 3,4-dihydroxycinnamic acid (300 mg, 1.66 mmol) and 2-phenylethylamine (209 µL, 1.66 mmol) afforded compound 1 (213 mg, 45%) as a white solid. Rf = 0.2 (CH2Cl2/MeOH 95:5); IR (ATR) γ cm−1: 3259, 3087, 3061, 3027, 2969, 2935, 2870, 1702, 1652, 1590, 1513; 1H-NMR (Acetone-d6, 300 MHz): δ 8.23 (s, 2H), 7.41 (d, J = 15.6 Hz, 1H), 7.31–7.17 (m, 6H), 7.06 (s, 1H), 6.93 (d, J = 8.0 Hz, 1H), 6.83 (d, J = 8.0 Hz, 1H), 6.43 (d, J = 15.6 Hz, 1H), 3.57–3.50 (m, 2H), 2.90–2.82 (m, 2H); HRMS (ESI) m/z calcd for C17H18NO3 [M + H]+: 284.1208, found: 284.1288. The analytical data were in line with previously described data [45].
(E)-N-(2-Phenylethyl)-3-methoxy-4-hydroxycinnamide (2). General procedure using 3-methoxy-4-hydroxycinnamic acid (322 mg, 1.66 mmol) and 2-phenylethylamine (209 µL, 1.66 mmol) afforded compound 2 (388 mg, 79%) as a white solid. Rf = 0.33 (Cyclohexane/EtOAc 4:6); IR (ATR) γ cm−1: 3270, 3063, 3027, 2935, 1652, 1585, 1510; 1H-NMR (MeOD-d4, 300 MHz): δ 7.44 (d, J = 15.7 Hz, 1H), 7.32–7.17 (m, 5H), 7.12 (s, 1H), 7.03 (d, J = 8.4 Hz, 1H), 6.79 (d, J = 8.4 Hz, 1H), 6.40 (d, J = 15.7 Hz, 1H), 3.88 (s, 3H), 3.52 (t, J = 7.2 Hz, 2H), 2.86 (t, J = 7.2 Hz, 2H); HRMS (ESI) m/z calcd for C18H20NO3 [M + H]+: 298.1365, found: 298.1442. The analytical data were in line with previously described data [46].
(E)-N-(2-Phenylethyl)-4-hydroxycinnamide (3). General procedure using 4-hydroxycinnamic acid (272 mg, 1.66 mmol) and 2-phenylethylamine (209 µL, 1.66 mmol) afforded compound 3 (288 mg, 65%) as a colorless syrup. Rf = 0.56 (CH2Cl2/MeOH 95:5); IR (ATR) γ cm−1: 3400, 3263, 3085, 3064, 3026, 2934, 2813, 2750, 2687, 2608, 1651, 1600, 1579, 1511; 1H-NMR (MeOD-d4, 300 MHz): δ 7.47–7.38 (m, 3H), 7.31–7.17 (m, 5H), 6.78 (d, J = 8.4 Hz, 2H), 6.38 (d, J = 15.7 Hz, 1H), 3.51 (t, J = 7.2 Hz, 2H), 2.85 (t, J = 7.2 Hz, 2H); HRMS (ESI) m/z calcd for C17H18NO2 [M + H]+: 268.1259, found: 268.1336. The analytical data were in line with previously described data [47].
(E)-N-(2-Phenylethyl)-3,4,5-trimethoxycinnamide (4). General procedure using 3,4,5-trimethoxycinnamic acid (395 mg, 1.66 mmol) and 2-phenylethylamine (209 µL, 1.66 mmol) afforded compound 4 (511 mg, 90%) as a white solid. Rf = 0.56 (CH2Cl2/MeOH 95:5); IR (ATR) γ cm−1: 3305, 3087, 3062, 3030, 3005, 2967, 2933, 2862, 2835, 1653, 1615, 1581, 1536, 1506; 1H-NMR (CDCl3, 300 MHz): δ 7.53 (d, J = 15.5 Hz, 1H), 7.36–7.22 (m, 5H), 6.71 (s, 2H), 6.22 (d, J = 15.5 Hz, 1H), 5.55 (bs, 1H), 3.88 (s, 6H), 3.87 (s, 3H), 3.71–3.65 (m, 2H), 2.89 (t, J = 6.8 Hz, 2H); 13C-NMR (CDCl3, 75 MHz): δ 165.9, 153.5, 141.1, 139.6, 138.9, 130.5, 128.9, 128.8, 126.7, 120.0, 104.9, 61.1, 56.2, 40.9, 35.7; HRMS (ESI) m/z calcd for C20H24NO4 [M + H]+: 342.1627, found: 342.1709.
(E)-N-(2-Phenylethyl)cinnamide (5). General procedure using cinnamic acid (246 mg, 1.66 mmol) and 2-phenylethylamine (209 µL, 1.66 mmol) afforded compound 5 (330 mg, 79%) as a white solid. Rf = 0.7 (CH2Cl2/MeOH 95:5); IR (ATR) γ cm−1: 3265, 3062, 3030, 2971, 2944, 2862, 1959, 1895, 1819, 1660, 1651, 1603, 1579, 1544; 1H-NMR (CDCl3, 300 MHz): δ 7.62 (d, J = 15.5 Hz, 1H), 7.50–7.47 (m, 2H), 7.40–7.31 (m, 5H), 7.25–7.22 (m, 3H), 6.32 (d, J = 15.5 Hz, 1H), 5.58 (bs, 1H), 3.71–3.65 (m, 2H), 2.90 (t, J = 6.6, 2H); HRMS (ESI) m/z calcd for C17H18NO [M + H]+: 252.1310, found : 252.1386. The analytical data were in line with previously described data [48].
(E)-N-(2-(4-Hydroxyphenyl)ethyl)-3,4-dihydroxycinnamide (6). General procedure using 3,4-dihydroxycinnamic acid (300 mg, 1.66 mmol) and 2-(4-hydroxyphenyl)ethylamine (228 mg, 1.66 mmol) afforded compound 6 (358 mg, 72%) as a slight yellow solid. Rf = 0.64 (Cyclohexane/EtOAc 1:9); IR (ATR) γ cm−1: 3349, 3167, 3030, 2960, 2930, 2877, 1727, 1645, 1602, 1578, 1535, 1514; 1H-NMR (DMSO-d6, 300 MHz): δ 9.23 (bs, 3H), 8.02 (bs, 1H), 7.21 (d, J = 15.6 Hz, 1H), 7.0 (d, J = 8.2 Hz, 2H), 6.93 (s, 1H), 6.82 (d, J = 8.0 Hz, 1H), 6.73 (d, J = 8.0 Hz, 1H), 6.67 (d, J = 8.2 Hz, 2H), 6.31 (d, J = 15.6 Hz, 1H), 3.34–3.28 (m, 2H), 2.63 (t, J = 7.3 Hz, 2H); HRMS (ESI) m/z calcd for C17H18NO4 [M + H]+: 300.1236, found: 300.1235. The analytical data were in line with previously described data [49].
(E)-N-(4-Hydroxyphenethyl)-3-methoxy-4-hydroxycinnamide (7). General procedure using 3-methoxy-4-hydroxycinnamic acid (322 mg, 1.66 mmol) and 4-hydroxyphenethylamine (228 mg, 1.66 mmol) afforded compound 7 (298 mg, 57%) as a white solid. Rf = 0.22 (Cyclohexane/EtOAc 5:5); IR (ATR) γ cm−1: 3287, 3015, 2936, 1651, 1586, 1510; 1H-NMR (MeOD-d4, 300 MHz): δ 7.43 (d, J = 16.0 Hz, 1H), 7.12 (s, 1H), 7.07–7.01 (m, 3H), 6.79 (d, J = 8.1 Hz, 1H), 6.71 (d, J = 8.0 Hz, 2H), 6.40 (d, J = 16.0 Hz, 1H), 3.88 (s, 3H), 3.47 (t, J = 6.9 Hz, 2H), 2.76 (t, J = 6.9 Hz, 2H); HRMS (ESI) m/z calcd for C18H20NO4 [M + H]+: 314.1314, found: 314.1396. The analytical data were in line with previously described data [50].
(E)-N-(4-Hydroxyphenethyl)-4-hydroxycinnamide (8). General procedure using 4-hydroxycinnamic acid (272 mg, 1.66 mmol) and 4-hydroxyphenethylamine (228 mg, 1.66 mmol) afforded compound 8 (233 mg, 50%) as a white solid. Rf = 0.30 (Cyclohexane/EtOAc 5:5); IR (ATR) γ cm−1: 3431, 3171, 3024, 2942, 1895, 1660, 1622, 1602, 1590, 1530, 1510; 1H-NMR (MeOD-d4, 300 MHz): δ 7.46–7.38 (m, 3H), 7.05 (d, J = 8.1 Hz, 2H), 6.78 (d, J = 8.3 Hz, 2H), 6.71 (d, J = 8.1 Hz, 2H), 6.38 (d, J = 15.6 Hz, 1H), 3.45 (t, J = 7.3 Hz, 2H), 2.75 (t, J = 7.3 Hz, 2H); HRMS (ESI) m/z calcd for C17H18NO3 [M + H]+: 284.1208, found: 284.1285. The analytical data were in line with previously described data [51].
(E)-N-(4-Hydroxyphenethyl)-3,4,5-trimethoxycinnamide (9). General procedure using 3,4,5-trimethoxycinnamic acid (395 mg, 1.66 mmol) and 4-hydroxyphenethylamine (228 mg, 1.66 mmol) afforded compound 9 (586 mg, 98%) as a white solid. Rf = 0.26 (Cyclohexane/EtOAc 5:5); IR (ATR) γ cm−1: 3323, 3012, 2963, 2940, 2909, 2888, 2847, 2824, 2605, 1994, 1652, 1605, 1580, 1544, 1516, 1504; 1H-NMR (MeOD-d4, 300 MHz): δ 7.44 (d, J = 15.7 Hz, 1H), 7.06 (d, J = 8.1 Hz, 2H), 6.86 (s, 2H), 6.72 (d, J = 8.1 Hz, 2H), 6.51 (d, J = 15.7 Hz, 1H), 3.87 (s, 6H), 3.78 (s, 3H), 3.48 (t, J = 7.2 Hz, 2H), 2.76 (t, J = 7.2 Hz, 2H); 13C-NMR (MeOD-d4 + 10% CDCl3, 75 MHz): δ 168.5, 156.8, 154.5, 141.6, 140.4, 132.1, 131.1, 130.7, 121.2, 116.2, 106.1, 61.3, 56.7, 42.5, 35.7; HRMS (ESI) m/z calcd for C20H24NO5 [M + H]+: 358.1576, found: 358.1657.
(E)-N-(4-Hydroxyphenethyl)-cinnamide (10). General procedure using cinnamic acid (246 mg, 1.66 mmol) and 4-hydroxyphenethylamine (228 mg, 1.66 mmol) afforded compound 10 (359 mg, 81%) as a white solid. Rf = 0.44 (Cyclohexane/EtOAc 5:5); IR (ATR) γ cm−1: 3429, 3268, 3059, 3022, 2941, 2922, 2858, 1889, 1665, 1624, 1611, 1592, 1532, 1512; 1H-NMR (MeOD-d4, 300 MHz): δ 7.56–7.49 (m, 3H), 7.42–7.33 (m, 3H), 7.06 (d, J = 8.2 Hz, 2H), 6.72 (d, J = 8.2 Hz, 2H), 6.57 (d, J = 16.0 Hz, 1H), 3.47 (t, J = 6.9 Hz, 2H), 2.76 (t, J = 6.9 Hz, 2H); HRMS (ESI) m/z calcd for C17H18NO2 [M + H]+: 268.1259, found: 268.1337. The analytical data were in line with previously described data [52].
(E)-N-(3,4-Dihydroxyphenethyl)-3,4-dihydroxycinnamide (11). General procedure using 3,4-dihydroxycinnamic acid (300 mg, 1.66 mmol) and 3,4-dihydroxyphenethylamine hydrochloride (315 mg, 1.66 mmol) afforded compound 11 (229 mg, 44%) as a slight yellow solid. Rf = 0.45 (CH2Cl2/MeOH 85:15); IR (ATR) γ cm−1: 3214, 2938, 2723, 1695, 1650, 1586, 1514; 1H-NMR (Acetone-d6, 300 MHz): δ 8.15 (bs, 4H), 7.43 (s, 1H), 7.42 (d, J = 15.6 Hz, 1H), 7.06 (s, 1H), 6.89 (d, J = 8.1 Hz, 1H), 6.8 (d, J = 8.2 Hz, 1H), 6.72 (s, 1H), 6.69 (d, J = 8.2 Hz, 1H), 6.52 (d, J = 8.1 Hz, 1H), 6.44 (d, J = 15.6 Hz, 1H), 3.47 (m, 2H), 2.67 (t, J = 6.9 Hz, 2H); HRMS (ESI) m/z calcd for C17H18NO5 [M + H]+: 316.1185; found: 316.1181. The analytical data were in line with previously described data [53].
(E)-N-(3,4-Dihydroxyphenethyl)-3-methoxy-4-hydroxycinnamide (12). General procedure using 3-methoxy-4-hydroxycinnamic acid (322 mg, 1.66 mmol) and 3,4-dihydroxyphenethylamine hydrochloride (315 mg, 1.66 mmol) afforded compound 12 (164 mg, 30%) as a white solid. Rf = 0.14 (CH2Cl2/MeOH 95:5); IR (ATR) γ cm−1: 3321, 2969, 2937, 2841, 2720, 1651, 1585, 1510; 1H-NMR (MeOD-d4, 300 MHz): δ 7.43 (d, J = 16.0 Hz, 1H), 7.12 (s, 1H), 7.02 (d, J = 8.0 Hz, 1H), 6.79 (d, J = 8.3 Hz, 1H), 6.71–6.67 (m, 2H), 6.55 (d, J = 8.0 Hz, 1H), 6.40 (d, J = 16.0 Hz, 1H), 3.88 (s, 3H), 3.46 (t, J = 7.0 Hz, 2H), 2.70 (t, J = 7.0 Hz, 2H). 13C-NMR (MeOD-d4, 75 MHz): δ 169.3, 149.9, 149.4, 146.4, 144.9, 142.2, 132.2, 128.4, 123.4, 121.2, 118.9, 117.0, 116.6, 116.5, 111.6, 56.5, 42.7, 36.2; HRMS (ESI) m/z calcd for C18H20NO5 [M + H]+: 330.1263; found: 330.1342.
(E)-N-(3,4-Dihydroxyphenethyl)-4-hydroxycinnamide (13). General procedure using 4-hydroxycinnamic acid (272 mg, 1.66 mmol) and 3,4-dihydroxyphenethylamine hydrochloride (315 mg, 1.66 mmol) afforded compound 13 (204 mg, 41%) as a white solid. Rf = 0.15 (CH2Cl2/MeOH 95:5); IR (ATR) γ cm−1: 3270, 3025, 2938, 2817, 2701, 1696, 1648, 1577, 1511; 1H-NMR (MeOD-d4, 300 MHz): δ 7.44 (d, J = 16.0 Hz, 1H), 7.40 (d, J = 8.4 Hz, 2H), 6.79 (d, J = 8.4 Hz, 2H), 6.70–6.67 (m, 2H), 6.55 (d, J = 8.0 Hz, 1H), 6.38 (d, J = 16.0 Hz, 1H), 3.45 (t, J = 7.0 Hz, 2H), 2.70 (t, J = 7.0 Hz, 2H); HRMS (ESI) m/z calcd for C17H18NO4 [M + H]+: 300.1158; found: 300.1233. The analytical data were in line with previously described data [54].
(E)-N-(3,4-Dihydroxyphenethyl)-3,4,5-trimethoxycinnamide (14). General procedure using 3,4,5-trimethoxycinnamic acid (395 mg, 1.66 mmol) and 3,4-dihydroxyphenethylamine hydrochloride (315 mg, 1.66 mmol) afforded compound 14 (436 mg, 70%) as a white solid. Rf = 0.26 (CH2Cl2/MeOH 95:5); IR (ATR) γ cm−1: 3337, 3105, 3040, 2963, 2944, 2843, 2728, 1706, 1658, 1613, 1605, 1585, 1544, 1504; 1H-NMR (MeOD-d4, 300 MHz): δ 7.44 (d, J = 15.7 Hz, 1H), 6.87 (s, 2H), 6.71–6.67 (m, 2H), 6.57–6.48 (m, 2H), 3.87 (s, 6H), 3.78 (s, 3H), 3.47 (t, J = 7.2 Hz, 2H), 2.71 (t, J = 7.2 Hz, 2H); HRMS (ESI) m/z calcd for C20H24NO6 [M + H]+: 374.1525; found: 374.1605. The analytical data were in line with previously described data [55].
(E)-N-(3,4-Dihydroxyphenethyl)cinnamide (15). General procedure using cinnamic acid (246 mg, 1.66 mmol) and 3,4-dihydroxyphenethylamine hydrochloride (315 mg, 1.66 mmol) afforded compound 15 (293 mg, 62%) as a colorless syrup. Rf = 0.29 (CH2Cl2/MeOH 95:5); IR (ATR) γ cm−1: 3265, 2927, 1702, 1655, 1599, 1519; 1H-NMR (MeOD-d4, 300 MHz): δ 7.56–7.49 (m, 3H), 7.42–7.35 (m, 3H), 6.71–6.67 (m, 2H), 6.60–6.54 (m, 2H), 3.47 (t, J = 7.2 Hz, 2H), 2.71 (t, J = 7.2 Hz, 2H); HRMS (ESI) m/z calcd for C17H18NO3 [M + H]+: 284.1208; found: 284.1283. The analytical data were in line with previously described data [54].
(E)-N-(4-Hydroxyphenyl)-3,4-dihydroxycinnamide (16). General procedure using 3,4-dihydroxycinnamic acid (300 mg, 1.66 mmol) and 4-hydroxyphenylamine (181 mg, 1.66 mmol) afforded compound 16 (84 mg, 19%) as a brown solid. Rf = 0.65 (Cyclohexane/EtOAc 1:9); IR (ATR) γ cm−1: 3210, 1696, 1648, 1588, 1535, 1508; 1H-NMR (DMSO-d6, 300 MHz): δ 9.83 (s, 1H), 9.46 (bs, 1H), 9.21 (bs, 2H), 7.46 (d, J = 8.6 Hz, 2H), 7.33 (d, J = 15.6 Hz, 1H), 6.98 (s, 1H), 6.88 (d, J = 8.3 Hz, 1H), 6.76 (d, J = 8.3 Hz, 1H), 6.70 (d, J = 8.6 Hz, 2H), 6.49 (d, J = 15.6 Hz, 1H); HRMS (ESI) m/z calcd for C15H14NO4 [M + H]+: 272.0923, found: 272.0918. The analytical data were in line with previously described data [56].
N-(4-Hydoxyphenethyl)-3,4-dihydroxybenzamide (17). General procedure using 3,4-dihydroxybenzoic acid (256 mg, 1.66 mmol) and 4-hydroxyphenethylamine (228 mg, 1.66 mmol) afforded compound 19 (53 mg, 12%) as a colorless syrup. Rf = 0.19 (CH2Cl2/MeOH 95:5); IR (ATR) γ cm−1: 3177, 2970, 2948, 2869, 1698, 1632, 1589, 1553, 1510; 1H-NMR (MeOD-d4, 300 MHz): δ 7.24 (d, J = 2.0 Hz, 1H), 7.15 (dd, J1 = 8.2 Hz, J2 = 2.0 Hz, 2H), 7.06 (d, J = 8.3 Hz, 2H), 6.78 (d, J = 8.2 Hz, 1H), 6.71 (d, J = 8.3 Hz, 2H), 3.49 (t, J = 7.3 Hz, 2H), 2.78 (t, J = 7.3 Hz, 2H). HRMS (ESI) m/z calcd for C15H16NO4 [M + H]+: 274.1001, found: 274.1072. The analytical data were in line with previously described data [57].
3-(3,4-Dihydroxyphenyl)-2-[3-(3,4-dihydroxyphenyl)-acryloylamino]-propionic acid methyl ester (18). General procedure using 3,4-dihydroxycinnamic acid (300 mg, 1.66 mmol) and l-3,4-dihydroxyphenylalanine methyl ester (315 mg, 1.66 mmol, previously prepared following [58]) afforded compound 18 (332 mg, 54%) as a yellow solid. Rf = 0.5 (CH2Cl2/MeOH 85:15); IR (ATR) γ cm−1: 3308, 2953, 2838, 2725, 1722, 1650, 1594, 1514; 1H-NMR (DMSO-d6, 300 MHz): δ 8.35 (d, J = 6.9 Hz, 1H), 7.20 (d, J = 15.6 Hz, 1H), 6.93 (s, 1H), 6.83 (d, J = 8.1 Hz, 1H), 6.73 (d, J = 8.1 Hz, 1H), 6.61–6.59 (m, 2H), 6.45 (d, J = 8.1 Hz, 1H), 6.38 (d, J = 15.7 Hz, 1H), 4.46 (m, 1H), 3.60 (s, 3H), 2.89–2.72 (m, 2H); HRMS (ESI) m/z calcd for C19H20NO7 [M + H]+: 374.1240; found: 374.1240. The analytical data were in line with previously described data [59].
(E)-N-(3,4-Dihydroxycinnamoyl)-l-cysteine methyl ester (19). General procedure using 3,4-dihydroxycinnamic acid (300 mg, 1.66 mmol) and l-cysteine methyl ester hydrochloride (285 mg, 1.66 mmol) afforded compound 19 ( 50.8 mg, 10%) as a white solid. Rf = 0.19 (CH2Cl2/MeOH 95:5); IR (ATR) γ cm−1: 3472, 3400, 3318, 3163, 2952, 2551, 1730, 1654, 1592, 1524; 1H-NMR (Acetone-d6, 300 MHz): δ 8.31 (bs, 2H), 7.52 (d, J = 7.6 Hz, 1H), 7.44 (d, J = 15.6 Hz, 1H), 7.09 (d, J = 1.8 Hz, 1H), 6.97 (dd, J1 = 1.8 Hz, J2 = 8.2 Hz, 1H), 6.84 (d, J = 8.2 Hz, 1H), 6.62 (d, J = 15.6 Hz, 1H), 4.87–4.80 (m, 1H), 3.73 (s, 3H), 3.05–2.93 (m, 2H), 1.96 (t, J = 8.5 Hz, 1H); HRMS (ESI) m/z calcd for C13H16NO5S [M + H]+: 298.0671; found: 298.0750. The analytical data were in line with previously described data [53].
3.3. Biological Evaluation
3.3.1. In Vitro Enzymatic Assay
Arginase catalyzes the hydrolysis of l-arginine into l-ornithine and urea. Urea amount produced by this reaction can be detected using a colorimetric assay. We adapted the Corraliza et al. (1994) method [40] by miniaturizing this colorimetric assay and by using a purified bovine liver arginase or a recombinant human arginase I instead of cell lysates [34], as described below. In each well of a microplate, the following solutions were added in this order: (1) 10 µL of a buffer containing and Tris-HCl (50 mM, pH 7.5) 0.1% of Bovine Serum Albumin (TBSA buffer), with or without (control) arginase (0.025 U/µL for b-ARGI or 3.125 µg/mL for h-ARGI); (2) 30 µL of Tris-HCl solution (50 mM, pH 7.5) containing MnCl2 10 mM as a co-factor; (3) 10 µL of a solution containing an inhibitor or its solvent (as a control—see Section 3.3.2); and (4) 20 µL of l-arginine (pH 9.7, 0.05 M). The microplate was covered with a plastic sealing film and incubated for 60 min in a 37 °C water bath. In the case of recombinant h-ARGI the enzyme was activated for 10 min at 55 °C prior to Step (2). The reaction was stopped by adding 120 µL of H2SO4/H3PO4/H2O (1:3:7) after placing the microplate on ice. Thereafter, 10 µL of alpha-isonitrosopropiophenone (5% in absolute ethanol) was added and the microplate was covered with an aluminum sealing film and heated in an oven at a temperature of 100 °C for 45 min. The microplate was kept in the dark until reading. After 5 min centrifugation and cooling for another 10 min, the microplate was shaken for 2 min and the absorbance was read at 550 nm and 25 °C using a spectrophotometer (Synergy HT BioTeck, Winooski, VT, USA). The level of arginase activity was expressed as relative to the “100% arginase activity” (see Section 3.3.2). Each experimental condition (e.g., various inhibitor concentrations) was repeated three times per microplate.
3.3.2. Determination of IC50 Values and Percentages of Arginase Inhibition
For each tested compound, a stock solution (70 mM) was prepared in DMSO and stored at −26 °C. Just before the assay, these stock solutions were successively diluted in ultra-pure water to get the following concentrations: 7000, 2100, 700, 210, 70, 21, 7, 2.1 and 0.7 µM (i.e., final concentrations in the wells of 1000, 300, 100, 30, 10, 3, 1, 0.3, and 0.1 µM, respectively). For a first screening, compounds were only tested at 100 µM (final concentration). Each dilution was incubated with arginase for one hour, as described above. The resulting absorbance was converted into percentage of arginase inhibition, i.e., relative to the absorbance of controls with solvent (“100% arginase activity”), and plotted on a semi-logarithmic scale. The IC50 values were estimated with Prism® (GraphPad Software, version 5.0.3, La Jolla, CA, USA) by non-linear sigmoidal curve-fitting.
3.3.3. Enzyme Kinetic Study and Determination of Ki Values
The type of inhibition was determined with the same experimental approach with three concentrations of CAPA (10, 20 and 30 μM) and a control under increasing l-arginine concentrations (2.86, 7.15, 14.30 mM). The kinetics data were analyzed using Lineweaver–Burk plot (i.e., 1/velocity vs. 1/[l-arginine] (Figure 2B), Dixon plot (i.e., 1/velocity vs. [inhibitor]) (Figure 2C) and Cornish–Bowden plot (i.e., [l-arginine]/velocity vs. [inhibitor]) (Figure 2D). The type of inhibition was determined following the graphical method described by Cornish–Bowden [41]. The Ki values of competitive inhibitors were calculated based on the Cheng–Prusoff equation [Ki = IC50/(1 + [S]/Km)] [42].
3.4. Molecular Docking Study
3.4.1. Preparation of Target Protein
The 3D crystal structure of h-ARG I complexed with co-ligand nor-NOHA (pdb id: 3kv2) was retrieved from Protein Data Bank (http://www.rcsb.org). The receptor was prepared following the standard procedure of LeadIt 2.0.2 software. The binding site was defined as all the amino acid residues enclosed within 16.0 Å radius sphere centered by the co-ligand, nor-NOHA. All water molecules were removed.
3.4.2. Preparation of Ligands
Ligands were built and prepared by using Sybyl-X 2.0 software [60]. Hydrogen were added to the 3D structure, which was optimized by energy minimization using Conj Grad Method with convergence criterion set at 0.0001 kcal/(Åmol), max iterations (10,000) and with Gasteiger Hükel charge assigned in Tripos force field; other parameter values were kept at default. Then, the structures were subjected to a simulated annealing run and further optimized to obtain the lowest-energy conformers, which were stored in the database for the docking simulation.
3.4.3. Docking Simulation
The docking simulations were performed using the FlexX docking program [44,61] implemented in LeadIt 2.0.2 software with number of poses set at 10 for analyzing, maximum number of solutions per iteration set at 1000 and maximum number of solutions per fragmentation set at 200, other docking parameters were kept at default setting. The molecular docking technique used in this study was a flexible one in which the ligand binding process was an induced-fit process. The 3D and 2D binding modes of docking solutions were generated by using MOE 2008.10 software [62].
4. Conclusions
In summary, a series of nineteen cinnamide derivatives were synthesized and first evaluated for their arginase inhibitory capacities on mammalian arginase I (b-ARG I), together with the two natural caffeic and chlorogenic acids. Among them, chlorogenic acid and six of the synthesized compounds showed an IC50 lower than 50 µM. Caffeic acid phenethylamide (CAPA, compound 1) was the most active of this series, with an IC50 value of 6.9 μM. Whereas the activity of nor-NOHA was retained (IC50 = 5.7 ± 0.6 µM) on an assay using a human recombinant arginase (h-ARG I), CAPA showed poorer activity (IC50 = 60.3 ± 7.8 µM) on h-ARG I compared to b-ARG-I. Therefore, although preliminary studies on b-ARG I, which is much cheaper than h-ARG I, could be useful to identify new mammalian arginase inhibitors, the most active compounds should also be tested on h-ARG I before going further with in vivo experiments. Enzyme kinetic studies, performed on b-ARG I, identified CAPA as a competitive inhibitor with Ki = 5.5 ± 1.0 μM. SARs and docking studies indicated the crucial role of caffeoyl moiety, which could penetrate into the active site of ARG-I in order to chelate the Mn2+ cofactor and interact with the important preserved residues (Asp124, His126, His141, Asp232, Asp234, and Thr246) involved in the catalytic site of arginase, thus disturbing the enzymatic activity. Overall, our results identified for the first time the important role of the caffeoyl moiety in binding the arginase active site. Considering the high IC50 value obtained with CAPA against h-ARG I, cinnamide derivatives could constitute potential new lead compounds for the development of arginase inhibitors with potential therapeutic applications. For instance, the phenethylamine moiety could be replaced by a part displaying specific interactions on the top of the arginase active site in order to design a new generation of cinnamides.
Acknowledgments
The authors gratefully acknowledge the Ministère Français de l’Enseignement supérieur et de la Recherche for awarding a PhD fellowship to Thanh-Nhat Pham and the Université de Bourgogne Franche-Comté for giving a travel grant to have research collaborations in Vietnam. Thanks are also due to Maude Nappey and Andy Zedet for technical assistance and Stéphanie Boullanger for NMR measurements. The molecular modeling work was supported by the Vietnam's National Foundation for Science and Technology Development-NAFOSTED (Grant # 106-YS.05-2015.31 to Khac-Minh Thai).
Author Contributions
Ideas and experiments design: Corine Girard-Thernier and Khac-Minh Thai. Computational development: Thanh-Nhat Pham and Khac-Minh Thai. Chemistry and biology: Thanh-Nhat Pham, Simon Bordage, Marc Pudlo. Analysis and data interpretation: Thanh-Nhat Pham, Marc Pudlo, Céline Demougeot and Corine Girard-Thernier. Writing and review of the paper: all authors. Study supervision: Corine Girard-Thernier and Khac-Minh Thai.
Conflicts of Interest
The authors declare no conflict of interest.
References
- 1.Maccallini C., Fantacuzzi M., Amoroso R. Amidine-based bioactive compounds for the regulation of arginine metabolism. Mini Rev. Med. Chem. 2013;13:1305–1310. doi: 10.2174/1389557511313090005. [DOI] [PubMed] [Google Scholar]
- 2.Dimski D.S. Ammonia metabolism and the urea cycle: Function and clinical implications. J. Vet. Intern. Med. Am. Coll. Vet. Int. Med. 1994;8:73–78. doi: 10.1111/j.1939-1676.1994.tb03201.x. [DOI] [PubMed] [Google Scholar]
- 3.Morris S.M. Regulation of enzymes of the urea cycle and arginine metabolism. Annu. Rev. Nutr. 2002;22:87–105. doi: 10.1146/annurev.nutr.22.110801.140547. [DOI] [PubMed] [Google Scholar]
- 4.Caldwell R.B., Toque H.A., Narayanan S.P., Caldwell R.W. Arginase: An old enzyme with new tricks. Trends Pharmacol. Sci. 2015;36:395–405. doi: 10.1016/j.tips.2015.03.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Jenkinson C.P., Grody W.W., Cederbaum S.D. Comparative properties of arginases. Comp. Biochem. Physiol. B Biochem. Mol. Biol. 1996;114:107–132. doi: 10.1016/0305-0491(95)02138-8. [DOI] [PubMed] [Google Scholar]
- 6.Bagnost T., Ma L., da Silva R.F., Rezakhaniha R., Houdayer C., Stergiopulos N., André C., Guillaume Y., Berthelot A., Demougeot C. Cardiovascular effects of arginase inhibition in spontaneously hypertensive rats with fully developed hypertension. Cardiovasc. Res. 2010;87:569–577. doi: 10.1093/cvr/cvq081. [DOI] [PubMed] [Google Scholar]
- 7.Segal R., Hannan J.L., Liu X., Kutlu O., Burnett A.L., Champion H.C., Kim J.H., Steppan J., Berkowitz D.E., Bivalacqua T.J. Chronic oral administration of the arginase inhibitor 2(S)-amino-6-boronohexanoic acid (ABH) improves erectile function in aged rats. J. Androl. 2012;33:1169–1175. doi: 10.2164/jandrol.111.015834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Grasemann H., Dhaliwal R., Ivanovska J., Kantores C., McNamara P.J., Scott J.A., Belik J., Jankov R.P. Arginase inhibition prevents bleomycin-induced pulmonary hypertension, vascular remodeling, and collagen deposition in neonatal rat lungs. Am. J. Physiol. Lung Cell. Mol. Physiol. 2015;308:L503–L510. doi: 10.1152/ajplung.00328.2014. [DOI] [PubMed] [Google Scholar]
- 9.Olivon V.C., Fraga-Silva R.A., Segers D., Demougeot C., de Oliveira A.M., Savergnini S.S., Berthelot A., de Crom R., Krams R., Stergiopulos N., et al. Arginase inhibition prevents the low shear stress-induced development of vulnerable atherosclerotic plaques in ApoE−/− mice. Atherosclerosis. 2013;227:236–243. doi: 10.1016/j.atherosclerosis.2012.12.014. [DOI] [PubMed] [Google Scholar]
- 10.Morris S.M., Gao T., Cooper T.K., Kepka-Lenhart D., Awad A.S. Arginase-2 mediates diabetic renal injury. Diabetes. 2011;60:3015–3022. doi: 10.2337/db11-0901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Meurs H., Zaagsma J., Maarsingh H., van Duin M. Use of Arginase Inhibitors in the Treatment of Asthma and Allergic Rhinitis. 20150164930 A1. US. 2010 Mar 4; doi: 10.1111/all.13770. [DOI] [PMC free article] [PubMed]
- 12.Van Zandt M.C., Whitehouse D.L., Golebiowski A., Ji M.K., Zhang M., Beckett R.P., Jagdmann G.E., Ryder T.R., Sheeler R., Andreoli M., et al. Discovery of (R)-2-Amino-6-borono-2-(2-(piperidin-1-yl)ethyl)hexanoic acid and congeners as highly potent inhibitors of human arginases I and II for treatment of myocardial reperfusion injury. J. Med. Chem. 2013;56:2568–2580. doi: 10.1021/jm400014c. [DOI] [PubMed] [Google Scholar]
- 13.Kavalukas S.L., Uzgare A.R., Bivalacqua T.J., Barbul A. Arginase inhibition promotes wound healing in mice. Surgery. 2012;151:287–295. doi: 10.1016/j.surg.2011.07.012. [DOI] [PubMed] [Google Scholar]
- 14.Singh R., Pervin S., Karimi A., Cederbaum S., Chaudhuri G. Arginase Activity in human breast cancer cell lines: Nω-Hydroxy-l-arginine selectively inhibits cell proliferation and induces apoptosis in MDA-MB-468 cells. Cancer Res. 2000;60:3305–3312. [PubMed] [Google Scholar]
- 15.Boucher J.L., Custot J., Vadon S., Delaforge M., Lepoivre M., Tenu J.P., Yapo A., Mansuy D. Nω-Hydroxy-l-arginine, an intermediate in the l-arginine to nitric oxide pathway, is a strong inhibitor of liver and macrophage arginase. Biochem. Biophys. Res. Commun. 1994;203:1614–1621. doi: 10.1006/bbrc.1994.2371. [DOI] [PubMed] [Google Scholar]
- 16.Custot J., Boucher J.-L., Vadon S., Guedes C., Dijols S., Delaforge M., Mansuy D. Nω-Hydroxyamino-α-amino acids as a new class of very strong inhibitors of arginases. JBIC J. Biol. Inorg. Chem. 1996;1:73–82. doi: 10.1007/s007750050025. [DOI] [Google Scholar]
- 17.Custot J., Moali C., Brollo M., Boucher J.L., Delaforge M., Mansuy D., Tenu J.P., Zimmermann J.L. The new α-amino acid Nω-hydroxy-nor-l-arginine: A high-affinity inhibitor of arginase well adapted to bind to its manganese cluster. J. Am. Chem. Soc. 1997;119:4086–4087. doi: 10.1021/ja970285o. [DOI] [Google Scholar]
- 18.Kim N.N., Cox J.D., Baggio R.F., Emig F.A., Mistry S.K., Harper S.L., Speicher D.W., Morris S.M., Ash D.E., Traish A., et al. Probing erectile function: S-(2-Boronoethyl)-l-cysteine binds to arginase as a transition state analogue and enhances smooth muscle relaxation in human penile corpus cavernosum. Biochemistry. 2001;40:2678–2688. doi: 10.1021/bi002317h. [DOI] [PubMed] [Google Scholar]
- 19.Baggio R., Emig F.A., Christianson D.W., Ash D.E., Chakder S., Rattan S. Biochemical and functional profile of a newly developed potent and isozyme-selective arginase inhibitor. J. Pharmacol. Exp. Ther. 1999;290:1409–1416. [PubMed] [Google Scholar]
- 20.Ivanenkov Y.A., Chufarova N. V. Small-molecule arginase inhibitors. Pharm. Pat. Anal. 2013;3:65–85. doi: 10.4155/ppa.13.75. [DOI] [PubMed] [Google Scholar]
- 21.Havlinova Z., Babicova A., Hroch M., Chladek J. Comparative pharmacokinetics of Nω-hydroxy-nor-l-arginine, an arginase inhibitor, after single-dose intravenous, intraperitoneal and intratracheal administration to brown Norway rats. Xenobiotica. 2013;43:886–894. doi: 10.3109/00498254.2013.780672. [DOI] [PubMed] [Google Scholar]
- 22.Morris S.M., Jr. Recent advances in arginine metabolism: Roles and regulation of the arginases. Br. J. Pharmacol. 2009;157:922–930. doi: 10.1111/j.1476-5381.2009.00278.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Schade D., Kotthaus J., Klein N., Kotthaus J., Clement B. Prodrug design for the potent cardiovascular agent Nω-hydroxy-l-arginine (NOHA): Synthetic approaches and physicochemical characterization. Org. Biomol. Chem. 2011;9:5249–5259. doi: 10.1039/c0ob01117g. [DOI] [PubMed] [Google Scholar]
- 24.Ilies M., di Costanzo L., Dowling D.P., Thorn K.J., Christianson D.W. Binding of α,α-disubstituted amino acids to arginase suggests new avenues for inhibitor design. J. Med. Chem. 2011;54:5432–5443. doi: 10.1021/jm200443b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Golebiowski A., Beckett R.P., van Zandt M., Ji M.K., Whitehouse D., Ryder T.R., Jagdmann E., Andreoli M., Mazur A., Padmanilayam M., et al. 2-Substituted-2-amino-6-boronohexanoic acids as arginase inhibitors. Bioorg. Med. Chem. Lett. 2013;23:2027–2030. doi: 10.1016/j.bmcl.2013.02.024. [DOI] [PubMed] [Google Scholar]
- 26.Golebiowski A., Whitehouse D., Beckett R.P., van Zandt M., Ji M.K., Ryder T.R., Jagdmann E., Andreoli M., Lee Y., Sheeler R., et al. Synthesis of quaternary α-amino acid-based arginase inhibitors via the Ugi reaction. Bioorg. Med. Chem. Lett. 2013;23:4837–4841. doi: 10.1016/j.bmcl.2013.06.092. [DOI] [PubMed] [Google Scholar]
- 27.Cama E., Shin H., Christianson D.W. Design of amino acid sulfonamides as transition-state analogue inhibitors of arginase. J. Am. Chem. Soc. 2003;125:13052–13057. doi: 10.1021/ja036365b. [DOI] [PubMed] [Google Scholar]
- 28.Ilies M., di Costanzo L., North M.L., Scott J.A., Christianson D.W. 2-Aminoimidazole amino acids as inhibitors of the binuclear manganese metalloenzyme human arginase I. J. Med. Chem. 2010;53:4266–4276. doi: 10.1021/jm100306a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Zakharian T.Y., di Costanzo L., Christianson D.W. S-2-Amino-6-nitrohexanoic acid binds to human arginase I through multiple nitro−metal coordination interactions in the binuclear manganese cluster. J. Am. Chem. Soc. 2008;130:17254–17255. doi: 10.1021/ja807702q. [DOI] [PubMed] [Google Scholar]
- 30.Shin H., Cama E., Christianson D.W. Design of amino acid aldehydes as transition-state analogue inhibitors of arginase. J. Am. Chem. Soc. 2004;126:10278–10284. doi: 10.1021/ja047788w. [DOI] [PubMed] [Google Scholar]
- 31.Zakharian T.Y., di Costanzo L., Christianson D.W. Synthesis of (2S)-2-amino-7,8-epoxyoctanoic acid and structure of its metal-bridging complex with human arginase I. Org. Biomol. Chem. 2008;6:3240–3243. doi: 10.1039/b811797g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Girard-Thernier C., Pham T.-N., Demougeot C. The promise of plant-derived substances as inhibitors of arginase. Mini Rev. Med. Chem. 2015;15:798–808. doi: 10.2174/1389557515666150511153852. [DOI] [PubMed] [Google Scholar]
- 33.Pham T., Guglielmetti A., Fimbel S., Demougeot C., Girard-Thernier C. Arginase inhibitory activity of several natural polyphenols using a novel in vitro test on purified bovine arginase. Planta Med. 2014;80:P1L9. doi: 10.1055/s-0034-1394667. [DOI] [Google Scholar]
- 34.Bordage S., Pham T.-N., Zedet A., Gugglielmetti A.-S., Nappey M., Demougeot C., Girard-Thernier C. Investigation of mammal arginase inhibitory properties of natural ubiquitous polyphenols by using an optimized colorimetric microplate assay. Planta Med. 2016 doi: 10.1055/s-0042-118711. accepted. [DOI] [PubMed] [Google Scholar]
- 35.Wang L.-N., Wang W., Hattori M., Daneshtalab M., Ma C.-M. Synthesis, Anti-HCV, antioxidant and reduction of intracellular reactive oxygen species generation of a chlorogenic acid analogue with an amide bond replacing the ester bond. Molecules. 2016;21:1656. doi: 10.3390/molecules21060737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Le Mellay-Hamon V., Criton M. Phenylethylamide and phenylmethylamide derivatives as new tyrosinase inhibitors. Biol. Pharm. Bull. 2009;32:301–303. doi: 10.1248/bpb.32.301. [DOI] [PubMed] [Google Scholar]
- 37.Okombi S., Rival D., Boumendjel A., Mariotte A.-M., Perrier E. Para-Coumaric Acid or Para-Hydroxycinnamic Acid Derivatives and Their Use in Cosmetic or Dermatological Compositions. 8481593 B2. US. 2007 Aug 9;
- 38.Coste J., Le-Nguyen D., Castro B. PyBOP: A new peptide coupling reagent devoid of toxic by-product. Tetrahedron Lett. 1990;31:205–208. doi: 10.1016/S0040-4039(00)94371-5. [DOI] [Google Scholar]
- 39.Berndt M., Hölemann A., Niermann A., Bentz C., Zimmer R., Reissig H.-U. Replacement of HMPA in Samarium Diiodide Promoted Cyclizations and Reactions of Organolithium Compounds. Eur. J. Org. Chem. 2012;2012:1299–1302. doi: 10.1002/ejoc.201101830. [DOI] [Google Scholar]
- 40.Corraliza I.M., Campo M.L., Soler G., Modolell M. Determination of arginase activity in macrophages: A micromethod. J. Immunol. Methods. 1994;174:231–235. doi: 10.1016/0022-1759(94)90027-2. [DOI] [PubMed] [Google Scholar]
- 41.Cornish-Bowden A. A simple graphical method for determining the inhibition constants of mixed, uncompetitive and non-competitive inhibitors (Short Communication) Biochem. J. 1974;137:143–144. doi: 10.1042/bj1370143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Yung-Chi C., Prusoff W.H. Relationship between the inhibition constant (KI) and the concentration of inhibitor which causes 50 per cent inhibition (I50) of an enzymatic reaction. Biochem. Pharmacol. 1973;22:3099–3108. doi: 10.1016/0006-2952(73)90196-2. [DOI] [PubMed] [Google Scholar]
- 43.Di Costanzo L., Pique M.E., Christianson D.W. Crystal structure of human arginase I complexed with thiosemicarbazide reveals an unusual thiocarbonyl μ-sulfide ligand in the binuclear manganese cluster. J. Am. Chem. Soc. 2007;129:6388–6389. doi: 10.1021/ja071567j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.BioSolveIT–GmbH . Germany: 2012. [(accessed on 18 March 2016)]. FlexX (Protein–Ligand Docker) user & technical reference as part of LeadIT 2.0; p. 15. Available online: https://www.biosolveit.de/FlexX. [Google Scholar]
- 45.Weng Y.-C., Chuang C.-F., Chuang S.-T., Chiu H.-L., Kuo Y.-H., Su M.-J. KS370G, a synthetic caffeamide derivative, improves left ventricular hypertrophy and function in pressure-overload mice heart. Eur. J. Pharmacol. 2012;684:108–115. doi: 10.1016/j.ejphar.2012.03.029. [DOI] [PubMed] [Google Scholar]
- 46.Shi Z.-H., Li N.-G., Shi Q.-P., Hao-Tang B.S.P., Tang Y.-P., Wei-Li B.S.P., Lian-Yin B.S.P., Yang J.-P., Duan J.-A. Design, synthesis and biological evaluation of ferulic acid amides as selective matrix metalloproteinase inhibitors. Med. Chem. 2013;9:947–954. doi: 10.2174/1573406411309070008. [DOI] [PubMed] [Google Scholar]
- 47.Yamazaki Y., Kawano Y., Uebayasi M. Induction of adiponectin by natural and synthetic phenolamides in mouse and human preadipocytes and its enhancement by docosahexaenoic acid. Life Sci. 2008;82:290–300. doi: 10.1016/j.lfs.2007.11.016. [DOI] [PubMed] [Google Scholar]
- 48.Yao H., Tang Y., Yamamoto K. Metal-free oxidative amide formation with N-hydroxysuccinimide and hypervalent iodine reagents. Tetrahedron Lett. 2012;53:5094–5098. doi: 10.1016/j.tetlet.2012.07.024. [DOI] [Google Scholar]
- 49.Hong S.S., Jeong W., Kwon J.G., Choi Y.-H., Ahn E.-K., Ko H.-J., Seo D.-W., Oh J.S. Phenolic Amides from the Fruits of Tribulus terrestris and Their Inhibitory Effects on the Production of Nitric Oxide. Bull. Korean Chem. Soc. 2013;34:3105–3108. doi: 10.5012/bkcs.2013.34.10.3105. [DOI] [Google Scholar]
- 50.Kan S., Chen G., Han C., Chen Z., Song X., Ren M., Jiang H. Chemical constituents from the roots of Xanthium sibiricum. Nat. Prod. Res. 2011;25:1243–1249. doi: 10.1080/14786419.2010.539182. [DOI] [PubMed] [Google Scholar]
- 51.Lin C.-F., Lay H.-L., Ni C.-L., Chen C.-C. Phenolic Components of Dendrobium antennatum. Chem. Nat. Compd. 2013;49:520–522. doi: 10.1007/s10600-013-0657-2. [DOI] [Google Scholar]
- 52.Tamiz A.P., Cai S.X., Zhou Z.-L., Yuen P.-W., Schelkun R.M., Whittemore E.R., Weber E., Woodward R.M., Keana J.F.W. Structure−Activity relationship of N-(Phenylalkyl)cinnamides as novel NR2B subtype-selective NMDA receptor antagonists. J. Med. Chem. 1999;42:3412–3420. doi: 10.1021/jm990199u. [DOI] [PubMed] [Google Scholar]
- 53.Son S., Lewis B.A. Free radical scavenging and antioxidative activity of caffeic acid amide and ester analogues: Structure−activity relationship. J. Agric. Food Chem. 2001;50:468–472. doi: 10.1021/jf010830b. [DOI] [PubMed] [Google Scholar]
- 54.Wu Z., Zheng L., Li Y., Su F., Yue X., Tang W., Ma X., Nie J., Li H. Synthesis and structure–activity relationships and effects of phenylpropanoid amides of octopamine and dopamine on tyrosinase inhibition and antioxidation. Food Chem. 2012;134:1128–1131. doi: 10.1016/j.foodchem.2012.02.152. [DOI] [PubMed] [Google Scholar]
- 55.Michalet S., Cartier G., David B., Mariotte A.-M., Dijoux-franca M.-G., Kaatz G. W., Stavri M., Gibbons S. N-Caffeoylphenalkylamide derivatives as bacterial efflux pump inhibitors. Bioorg. Med. Chem. Lett. 2007;17:1755–1758. doi: 10.1016/j.bmcl.2006.12.059. [DOI] [PubMed] [Google Scholar]
- 56.Shi Z.-H., Li N.-G., Shi Q.-P., Tang H., Tang Y.-P., Li W., Yin L., Yang J.-P., Duan J.-A. Design, synthesis and biological evaluation of caffeic acid amides as selective MMP-2 and MMP-9 inhibitors. Drug Dev. Res. 2012;73:343–351. doi: 10.1002/ddr.21038. [DOI] [Google Scholar]
- 57.Chou S.-C., Su C.-R., Ku Y.-C., Wu T.-S. The constituents and their bioactivities of Houttuynia cordata. Chem. Pharm. Bull. 2009;57:1227–1230. doi: 10.1248/cpb.57.1227. [DOI] [PubMed] [Google Scholar]
- 58.Larsson R., Blanco N., Johansson M., Sterner O. Synthesis of C-1 indol-3-yl substituted tetrahydroisoquinoline derivatives via a Pictet–Spengler approach. Tetrahedron Lett. 2012;53:4966–4970. doi: 10.1016/j.tetlet.2012.07.017. [DOI] [Google Scholar]
- 59.Lee S.U., Shin C.-G., Lee C.-K., Lee Y.S. Caffeoylglycolic and caffeoylamino acid derivatives, halfmers of l-chicoric acid, as new HIV-1 integrase inhibitors. Eur. J. Med. Chem. 2007;42:1309–1315. doi: 10.1016/j.ejmech.2007.02.016. [DOI] [PubMed] [Google Scholar]
- 60.SYBYL-X, Tripos International. [(accessed on 18 March 2016)]. Available online: https://www.certara.com.
- 61.Rarey M., Kramer B., Lengauer T., Klebe G. A fast flexible docking method using an incremental construction algorithm. J. Mol. Biol. 1996;261:470–489. doi: 10.1006/jmbi.1996.0477. [DOI] [PubMed] [Google Scholar]
- 62.Chemical Computing Group Inc. Molecular Operating Environment (MOE) [(accessed on 18 March 2016)]. Available online: https://www.chemcomp.com.