

Abstract
CD8+ cytotoxic T lymphocytes confer protection against infectious diseases caused by viruses, bacteria, and parasites. Hence, significant efforts have been invested into devising ways to generate CD8+ T cell-targeted vaccines. Generation of microbe-free protein subunit vaccines requires a thorough knowledge of protective target antigens. Such antigens are proteolytically processed peptides presented by MHC class I molecules. To induce a robust antigen-specific CD8+ T cell response through vaccination, it is essential to formulate the antigen with an effective adjuvant. Here, we describe a versatile method for generating high-frequency antigen-specific CD8+ T cells through immunization of mice using the invariant natural killer T cell agonist α-galactosylceramide as the adjuvant.
Keywords: Adjuvant, α-Galactosylceramide, Antigen-specific CD8+ T cells, Microbial protein antigens, Mouse immunization
1 Introduction
Vaccination is one of medical science’s greatest achievements; it has led to the eradication of several infectious diseases and to a reduction in morbidity and mortality from many others [1]. The majority of licensed vaccines consist of live, live-attenuated, or inactivated pathogens that were developed by empirical approaches [2]. Understanding vaccine-mediated correlates of immunity is critical for rational design of novel pathogen-free vaccines against infectious diseases for which no effective vaccine currently exists, or to improve the safety and/or efficacy of existing vaccines [2–6].
It is generally thought that the most effective vaccines confer protection via mediation of neutralizing antibodies, which are readily detectable by in vitro assays. Hence, most effort continues to be invested in designing vaccines that induce antibody-mediated protective immunity [6, 7]. In recent years, however, the focus on developing T cell-targeted vaccines has increased, as it has become clear that successful vaccination relies on both antibody- and T cell-mediated immunity (reviewed in Ref. [3, 8]).
The indispensable role of cellular immunity was observed even prior to modern advances in microbiology and immunology, from the historically recorded cases known as “experiments of nature.” As early as the nineteenth century, Jenner and colleagues observed that a smallpox-immune mother had brought forth an infant who died a few days after birth owing to an infection contracted in utero, as the baby was born with pocks/pustules [9, 10]. Given that the mother’s antibody repertoire circulates within the fetus and babies are born with underdeveloped cellular immunity [11–13], we can now surmise that protection from smallpox requires cellular immunity. Another example of the critical role of cellular immunity includes a case report for progressive vaccinia virus (VACV) infection in a subject with normal anti-viral antibody responses [14].
Immune CD8+ T cells recognize infected cells via short microbial peptide epitopes presented by the host’s major histocompatibility complex (MHC)-encoded class I molecules. Of note, MHC class I molecules present cytoplasmic antigens because their assembly with peptides predominantly occurs within the endoplasmic reticulum, though under inflammatory conditions it can occur within vacuoles as well (reviewed in Refs. [15, 16]). Recent studies have shown that CD8+ T cells play a critical role in conferring protective immunity against a variety of infectious diseases, including those caused by respiratory viruses [17–21], poxviruses [22], cytomegalovirus [23], human immunodeficiency virus (HIV; [24]), Plasmodium species [25, 26], and M. tuberculosis [27]. Hence, T cell epitope-based vaccines can complement current efforts in developing safe and effective antibody-targeted subunit vaccines.
The challenge impeding the development of T cell-targeted subunit vaccines is identifying which of the microbe’s peptide epitopes confer protective immunity amongst the many that are presented [28]. Such epitopes can be discovered by indirect approaches that can ascertain protein subunit immunogenicity and protective capability in surrogate animal models [29, 30]. Accordingly, there is a need for the design and implementation of an effective vaccination strategy that will elicit sufficiently high-frequency, antigen-specific CD8+ T cells to allow thorough characterization of the response to subunit antigens. Unlike replicative microbial vaccines, heterologous proteins and peptides are poorly immunogenic when administered alone. Therefore, to induce a robust antigen-specific CD8+ T cell response without the use of infectious agents or antigen-loaded exogenous dendritic cells (DCs) [31–33], it is essential to formulate the antigen with an effective adjuvant [34]. Successful CD8+ T cell-targeted immunization has been demonstrated for peptide epitopes that are formulated with various toll-like receptor (TLR) ligands [31, 35]. High-frequency CD8+ T cells can be induced by peptides formulated with polyinosinic-polycytidylic acid (poly(I:C); TLR3 agonist) and co-stimulatory anti-CD40 antibody [36]. Peptide immunogenicity can be improved via chemical engineering of peptides and adjuvants with amphiphilic albumin-binding tags [37]. The immunogenicity of peptides can also be modulated through engineered delivery systems; these are discussed elsewhere [38, 39] and, hence, not reviewed here.
It should be noted that full-length protein antigens are typically more desirable for vaccination studies than peptide epitopes. This is because immunization with proteins allows elicitation of both antibody- and T cell-mediated immune responses [28, 40–42]. In addition, one protein may contain several immune epitopes, which is a desirable feature for human vaccines [43]. Because MHC class I molecules present cytoplasmic antigens, the priming of naïve CD8+ T cells requires cross-presentation of exogenous antigens by antigen-presenting cells (APCs) that have taken up the administered protein. Small subsets of DCs—the tissue-resident, generally non-migratory, CD103+ as well as splenic CD8+ DCs in mice, or CD1c+ and CD141+ DCs in humans—play critical roles in cross-presentation of exogenous antigens (reviewed in Refs. [15, 16]). Antigen cross-presentation requires the activation and licensing of the appropriate DC subsets; CD4+ T cells are the classical helper cells for DC activation and licensing (reviewed in Ref. [44]). So also, CD4+ T cell help is essential for the differentiation of effector and memory CD8+ T cells (reviewed in Ref. [44]). Because MHC class II molecules—which also present processed peptide antigens generated in vacuolar compartments to control the functions of CD4+ T cells—are highly variable, vaccine design requires the identification of helper epitopes and their inclusion in CD8+ T cell-targeted vaccines. Hence, harnessing a non-polymorphic antigen-presenting molecule and invariant T cells as vaccine targets that can provide help can be a viable option for enhancing CD8+ T cell responses to subunit antigens. CD1d and the restricted semi-invariant natural killer T (NKT) cells have shown significant promise as experimental vaccine targets [44]; their biology has been recently reviewed elsewhere [45].
Immunogenicity of whole-protein antigens formulated with the NKT cell agonist α-galactosylceramide (αGalCer) as the adjuvant has demonstrated that this formulation stimulates robust antigen-specific T cell and antibody responses in mice [41, 46–48]. αGalCer is a synthetic glycolipid that closely resembles the marine sponge Agelas mauritianus-derived agelasphin 9b as well as self and bacterial glycolipid antigens (see Table 1 and references therein). Upon immunization, professional APCs, such as DCs, macrophages, and B cells, take up the administered protein antigen and αGalCer. Antigens undergo processing to short peptide epitopes that are presented by MHC class I molecules expressed by APCs for recognition via the T cell receptor (TCR) of antigen-specific CD8+ T cells. The MHC class I-like, lipid antigen-presenting CD1d molecules present αGalCer and the TCR of NKT cells recognize the co-complex. Recognition of αGalCer rapidly activates NKT cells. Activated NKT cells produce cytokine/chemokine messengers, thereby trans-activating DCs and initiating a crosstalk between cells of the innate and adaptive immune systems. Activated NKT cells and DCs together stimulate naïve antigen-specific CD8+ T cell precursors, which then proliferate and differentiate into effector and memory CD8+ T cells (Fig. 1; [28, 44, 45, 49, 50]). By virtue of its ability to communicate to virtually all cell types of the immune system, αGalCer has proven to be a potent adjuvant that enhances antibody and CD4 T cell responses to foreign antigens (reviewed in Ref. [45]).
Table 1.
Structure and properties of selected synthetic, microbial, and self NKT cell agonists
| Lipid (class)a | Chain lengthb | Structure | Agonistc | References |
|---|---|---|---|---|
| αGalCer (GSL) | C18 C24:1 |
|
IFN-γ, IL-4 Self |
[67] |
| Agel 9b (GSL) | C17 (C16-Me) phyto C24 |
|
Anti-tumour; Agelas mauritianus | [68, 69] |
| αGalCer (GSL) | C18-phyto C26 Common form used in functional studies (this is the molecule used in this chapter) |
|
Very strong Robust IFN-γ, IL-4, and other cytokines; synthetic analogue of Agel 9b (KRN7000) |
[70] |
| DB06-1 (GSL) | C18-phyto C26 (C2S) |
|
Strong; strong IFN-γ; IL-10 upon re-activation | [71] |
| α-C-GalCer (GSL) | C18-phyto C26 1,1prime;-C-glycoside |
|
Weak (mod)-to-none (hu); IFN-γ; synthetic |
[72] |
| FPh-αGalCer (GSL) | C18-phyto C10-fluoro-phenyl |
|
Very strong IFN-γ; synthetic |
[73] |
| NU-αGalCer (GSL) | C18-phyto C26 C6″-naphthylurea |
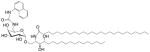
|
Very strong Robust IFN-γ; synthetic |
[74] |
| PyrC-αGalCer (GSL) | C18-phyto C26 C6″-(pyridin-4-yl)-carbamate |
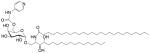
|
Very strong Robust IFN-γ, IL-12; synthetic |
[75] |
| SMC124 (GSL) | C22-C11cyclo-propyl C26 |
|
Strong IFN-γ; synthetic | [76] |
| EF77 (GSL) | C18-phyto C21:1-C10-cyclo-propyl |
|
Strong IFN-γ; synthetic | [76] |
| OCH (GSL) | C9-phyto C24 |
|
Weak (mo)-to-none (hu) IL-4 (low-to-no IFN-γ); synthetic |
[77] |
| C20-diene (GSL) | C18-phyto C20:2 |
|
Strong IL-4 (low-to-no IFN-γ); synthetic |
[78] |
| αGalACer (GSL) | C18-phyto C14 |
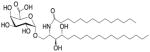
|
Weak; Sphingomonas spp. |
[79, 80] |
| Asp B (GSL) | C20:2-C9 Me C16-C2OH |
|
Weak Aspergillus fumigatus |
[81] |
Agel agelasphin, Asp B asparamide B, GalCer galactosylceramide, GalACer galacturonosylceramide, GSL glycosphingolipid, IL interleukin
Sphingosine/phytosphingosine chain length indicated first and N-acyl chain length second
Agonist strength based on Ref. [82]
Relative potency in comparison to αGalCer; mo mouse, hu human
Fig. 1.

Strategy by which a robust antigen-specific CD8+ T cell response is elicited in mice immunized with antigenic protein formulated with the glycolipid adjuvant αGalCer. Upon immunization, professional antigen-presenting cells, such as dendritic cells (DCs) , endocytose administered protein antigen and αGalCer. The antigen undergoes processing into short peptide epitopes that are presented by MHC class I molecules on DCs for recognition via the T cell receptor (TCR) of antigen-specific CD8+ T cells. αGalCer is presented by MHC class I-like CD1d molecules and recognized by the TCR of semi-invariant NKT cells . Recognition of αGalCer rapidly activates NKT cells, which produce cytokine/chemokine messengers, thereby trans-activating DCs and initiating a cross talk between members of the innate and adaptive immune systems. NKT cell licensing of DCs facilitates antigen cross presentation by a less well-understood mechanism(s) [42, 44, 65]. Together, this provides stimulus to naïve antigen-specific CD8+ T cell precursors that then proliferate and differentiate into effector and memory CD8+ T cells [45]
Several studies have reported that αGalCer is an effective adjuvant for the elicitation of epitope-specific CD8+ T cells against the model protein antigen ovalbumin [41, 51–53], or for enhancing antigen-specific CD8+ T cell responses upon immunization with live viral vaccines [49, 54]. We recently demonstrated the potency of αGalCer as a CD8+ T cell adjuvant for in vivo immunogenicity and protection studies which exploited pathogen-derived protein antigens ([28] and unpublished data). It is noteworthy that the activation of NKT cells by αGalCer results in the production of proinflammatory as well as immunoregulatory cytokines and chemokines (reviewed in [45]). Since DC activation and licensing as well as CD8+ T cell differentiation require proinflammatory cytokines and chemokines, efforts to synthesize and identify αGalCer analogues that elicit proinflammatory activity are being sought but are not discussed here (see Table 1; reviewed in Ref. [55]). Herein, we describe protocols for the preparation of protein antigens and αGalCer adjuvant, mouse immunization, and assessment of antigen-specific CD8+ T cell response, while analysis of antigen-specific antibody response and protective immunity is not described.
Experiments that assess immunogenicity require a large quantity of pure protein antigens, which, unlike model antigens, are not typically available commercially. Instead, the desired proteins are produced by recombinant DNA methods in Escherichia coli and then purified to a high degree of purity through engineered tags with the use of affinity resin. We designed, cloned, and produced nine recombinant VACV-derived protein subunits (rA3L62–319, rD1R565–844, rD5R330–470, rE2L26–301, rF4L1–319, rJ6R188–466, and rL4R33–249, as well as epitope-engineered subunits L4R/B8R70–79 and L4R/A34R32–90), all of which contain a C-terminal histidinetag to facilitate purification (Fig. 2; [28]). Upon immunization of mice, αGalCer-formulated antigens induced robust epitope-specific CD8+ T cell and antibody responses that were detectable in blood and in various tissues (Fig. 3a–c). Vaccination with adjuvant and microbial antigen induced protective immunity against subsequent microbial challenge (Fig. 4 and unpublished data). Our immunization approach is suitable for elicitation of antigen-specific CD8+ T cell and antibody responses to recombinant proteins that are produced as insoluble inclusion bodies (IB). Results that have emerged from the use of approaches described herein demonstrate the efficacy of the proposed immunization strategy for generating high-frequency antigen-specific CD8+ T cells against microbial protein antigens that can be exploited for immunologic and vaccine studies.
Fig. 2.

Design of recombinant VACV-derived protein antigen subunits that contain immune CD8+ T cell epitopes. Schematic chart of engineered proteins showing nine VACV antigens with amino acid positions selected for the design of a truncated recombinant subunit. Shown are the location and sequences of immune CD8+ T cell epitopes, and their N- and C-terminal tag sequences introduced to facilitate expression and purification of recombinant proteins in E. coli. Soluble protein rL4R33–249 was engineered with B8R70–79 and A34R82–90 immune epitopes to facilitate expression of soluble antigens by E. coli. All CD8+ T cell epitopes are restricted to the HLA-B*07;02 (B7.2) molecules. This figure is adapted from our previous publication with permission from the Journal of Clinical Investigation [28]
Fig. 3.

Efficient stimulation of antigen-specific immune responses is achieved through prime and boost immunization with αGalCer-formulated protein antigens. (a) Experimental design. Purified protein antigen is formulated with αGalCer and administered to mice by intraperitoneal or intranasal route as indicated. Antigen-specific immune responses were assessed ~6 days after booster immunization. (b) Representative example showing that immunization with αGalCer-formulated VACV-derived recombinant protein subunits elicited a robust epitope-specific CD8+ T cell response. Individual proteins containing HLA-B7.2 restricted CD8+ epitopes (see Fig. 2) were formulated with αGalCer and administered to groups of B7tg mice IP as in shown in (a). On d6 after boost, various tissues and blood were harvested from each group of immunized mice and assessed with B7.2 tetramer for epitope-specific CD8+ T cells. Epitope-specific CD8+ T cells were detected using a dual-fluorochrome labeling approach as described previously [28, 66]. Contour plots are gated on live CD8+ T lymphocytes; numbers indicate percent epitope-specific CD8+ T cells. Panel was reproduced from Ref. 28 with permission from the Journal of Clinical Investigation. (c) Representative example showing that immunization with αGalCer-formulated VACV-derived recombinant protein subunits elicited antigen-specific antibody responses. SDS-PAGE (left) and western blot (right) of purified VACV-derived subunits. Each lane was loaded with ~4 μg protein for SDS-PAGE and ~0.5 μg protein for immunoblot. Blots were probed with 1:500 sera dilution from mice primed and boosted with the indicated individual proteins, and developed with anti-mouse HRP-conjugated secondary antibodies. M molecular weight standards, from top to bottom: 250, 150, 100, 75, 50, 37, 25, 20, 15 kDa
Fig. 4.

Protection from lethal VACV infection upon vaccination with VACV-derived antigenic protein that contains CD8+ epitope. (a) Vaccination and challenge strategy. Three groups of mice (n = 5 mice per group) were immunized by IP route with αGalCer-formulated VACV-derived protein antigens rJ6R88–466 (contains protective CD8+ epitope), or engineered ΔrJ6R303–311/L4R37–45 (contains non-protective CD8+ epitope), or mock-vaccinated as indicated. Vaccinated mice were challenged with VACV and observed during the ensuing 18 days for severity of disease. Elicitation of J6R303–311 (protective epitope) or L4R37–45 (non-protective epitope) epitope-specific CD8+ was confirmed with corresponding tetramers in blood of vaccinated mice on d 8 after second boost. (b) Protection of protein and αGalCer-vaccinated mice from lethal respiratory VACV challenge. All mock-vaccinated mice succumbed to the disease by d8 post-infection. Both protein and αGalCer-vaccinated groups were protected from severe weight loss and death. Replacing protective CD8+ epitope J6R303–311 in rJ6R88–466 withnon-protective L4R37–45 epitope showed that protection is mediated by J6R303–311-specific CD8+ T cells with partial contribution from other immune responses against the protein antigen. ***p < 0.001; *p < 0.05 as compared to mock on d8 post challenge as defined by ANOVA with Dunnett’s post-test; mean + SEM. Symbols indicate proteins used for vaccination
2 Materials
Reagents and equipment described herein can be substituted with equivalent alternatives from other vendors.
2.1 Adjuvant Preparation
Fisherbrand™ class A clear glass threaded 6 ml vials with caps (Fisher Scientific).
αGalCer (Funakoshi, Japan); see Table 1 for structure. To dissolve αGalCer, use 5.6 % sucrose, 0.75 % l-histidine, and 0.5 % Tween 20 in sterile water for injection (WFI) for cell culture with heating at 80 °C until dissolved (see Note 1). Make a 0.2 mg/ml stock solution and store at −20 °C.
2.2 Antigen Preparation
The immunization protocol reported herein was validated with the use of vaccinia virus (VACV)-derived recombinant proteins designed and produced in our laboratory [28] and with the model antigen ovalbumin. The following materials are required to obtain recombinant protein antigens for immunizing mice:
E. coli BL21-Gold(DE3) Competent Cells (Agilent Technologies) transformed with plasmid encoding desired protein antigen.
LB broth: To 800 ml H2O add 10 g tryptone, 5 g yeast extract, and 10 g NaCl. Adjust pH to 7.5 with 0.5 M NaOH and bring the final volume to 1 L with dH2O. Sterilize the solution by autoclaving.
Kanamycin (Km): Make 50 mg/ml stock solution in sterile dH2O and store at −20 °C.
Glucose: Make 40 % (w/v) stock solution and sterilize by autoclaving.
LB agar plates containing 50 μg/ml Km and 1 % glucose.
Isopropyl β-d-thiogalactoside (IPTG): Make 1 mM stock in sterile dH2O and store at −20 °C.
Sterile phosphate-buffered saline (PBS), 1×: 137 mM NaCl, 2.7 mM KCl, 10 mM Na2HPO4, 2 mM KH2PO4.
Ni-NTA agarose.
Cell lysis buffer A: 50 mM NaH2PO4, 300 mM NaCl, 5 mM imidazole (pH 8.0). Sterilize using Complete Filtration Unit with 0.2 μm filter (VWR) and store at room temperature (RT).
Lysozyme.
Wash buffer B: 50 mM NaH2PO4, 300 mM NaCl, 10 mM imidazole (pH 8.0). Filter sterilize (see Subheading 2.2, item 9) and store at room temperature (RT).
Elution buffer C: 50 mM NaH2PO4, 300 mM NaCl, 250 mM imidazole (pH 8.0). Filter sterilize (see Subheading 2.2, item 9) and store at RT.
Solubilization/wash buffer D: 20 mM Tris–HCl, 300 mM NaCl, 8 M urea, 10 mM imidazole (pH 8.0) (see Note 2). Filter sterilize (see Subheading 2.2, item 9) and store at RT.
Elution buffer E: 20 mM Tris–HCl, 300 mM NaCl, 8 M urea, 100 mM imidazole (pH 8.0). Filter sterilize (see Subheading 2.2, item 9) and store at RT.
10–12 % Sodium dodecyl sulfate (SDS)-polyacrylamide gel (see Note 3).
Sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) running buffer: 0.025 M Tris–HCl (pH 8.3), 0.192 M glycine, 0.1 % SDS.
Loading dye solution (5×): 0.03 M Tris–HCl (pH 6.8), 10 % SDS, 25 % 2-mercaptoethanol, 0.1 % bromophenol blue, 45 % glycerol. Store at −20 °C.
Bovine serum albumin (BSA) lyophilized powder: Prepare 0.2 mg/ml stock in 1× loading dye solution. Store at −20 °C.
Spectra Multicolor Broad Range Protein Ladder (Thermo Fisher).
GelCode Blue Stain Reagent (Thermo Fisher).
Optional: Pierce High Capacity Endotoxin Removal Spin Columns, 0.25 ml (Thermo Fisher).
Slide-A-Lyzer Dialysis Cassette, 10 K MWCO, 3 ml (Thermo Fisher).
Pierce disposable polypropylene columns, 5 ml (Thermo Fisher).
Falcon disposable polypropylene centrifuge tubes, 50 ml (Corning).
Disposable Safe-Lock Tubes, 1.5 ml (Eppendorf).
Disposable centrifuge tubes, 250 ml (Corning).
Nalgene baffled shake flask, 2000 ml (Sigma).
Disposable syringes, 5 ml (BD Biosciences).
Disposable Millex-GV Syringe Filter Unit, 0.22 μm (EMD Millipore).
Disposable 28 mm syringe filter, 0.45 μm (Corning).
Nalgene Oak Ridge High-Speed PPCO Centrifuge Tubes, 50 ml (Thermo Scientific).
Orbital shaker incubator.
550 Sonic Dismembrator (Fisher Scientific).
Precision Microprocessor-Controlled 280 Series Water Bath (Thermo Scientific).
Mini-PROTEAN 3 Mini Vertical electrophoresis system (Bio-Rad) with Power Station 300 plus power supply (Labnet International, Inc).
Eppendorf 5415D microcentrifuge.
Refrigerated Sorvall RT7 centrifuge with RTH-750 rotor.
Sorvall RC-5B refrigerated centrifuge with SS-34 rotor.
DU 530 UV/Vis Spectrophotometer (Beckman).
2.3 Immunizations
Mice: All mice used for immunization were 6–10-week-old males from the following strains: B6-K0D0;B*07;02tg (B7tg) human leukocyte antigen (HLA) class I transgenic (tg) mice were bred in house. B7tg mouse is described elsewhere [56]. C57BL/6 (B6) mice were purchased from Jackson Laboratories (Bar Harbor, ME). All animal procedures were approved by the Institutional Animal Care and Use Committee (IACUC) at Vanderbilt University.
Anesthesia: 10 mg/ml KETAVED (ketamine HCl injection, USP; Vedco) and 1 mg/ml xylazine HCl Injection (xylazine; Vanderbilt pharmacy) in sterile WFI. Ketamine/xylazine anesthesia should be approved by an IACUC at user institution.
Purified recombinant antigen or albumin from chicken egg white (ovalbumin, Ova) at 0.5–2 mg/ml in sterile PBS (see Note 4).
Sterile insulin syringes, 1 ml (BD Biosciences).
Sterile filter pipet tips, 1–200 μl (VWR).
2.4 Lymphocyte Isolation
SmartBox Auto CO2 System for rodent euthanasia (Euthanex).
Refrigerated Sorvall RT7 centrifuge with RTH-750 rotor and plate adaptors.
Hausser Scientific Hemocytometer (Fisher Scientific).
Forceps.
Surgical scissors.
Falcon sterile disposable cell strainer, white, 70 μm (Fisher Scientific).
BD sterile disposable Slip Tip syringes, 5 ml (Fisher Scientific).
Sterile disposable Falcon polypropylene centrifuge tubes, 15 ml (Fisher Scientific).
Falcon sterile disposable 60 mm × 15 mm Petri dishes (Corning).
Optional: Capillary tubes, heparinized (Fisher Scientific) or plain (Fisher Scientific).
Dimethyl sulfoxide (DMSO).
Dasatinib (LC Laboratories): Dissolve to 1 mM in DMSO and store in 0.05 ml aliquots at −20 °C.
FACS buffer: PBS containing 2 % heat-inactivated fetal bovine serum (FBS) and 50 nM dasatinib. Make and use fresh (see Note 5).
Collagenase (Sigma).
Complete RPMI medium (cRPMI): RPMI-1640 with l-glutamine and HEPES supplemented with 10 % FBS, 1× penicillin/streptomycin solution and 1.75 μl of cell culture grade 2-mercaptoethanol.
Lung digestion medium: cRPMI (see Subheading 2.4, item 15) containing 2 mg/ml collagenase and 50 nM dasatinib.
Gibco ACK lysing buffer (Thermo Fisher).
Trypan blue solution, 0.4 % in PBS (Fisher Scientific).
2.5 Tetramer Staining, Intracellular Cytokine Staining (ICCS), and Flow Cytometry
10 mM Peptide epitope (purity ≥95 %; Schufer-N, Denmark) dissolved in sterile DMSO.
Brefeldin A (Sigma). Prepare stock solution at 10 mg/ml in methanol.
BD GolgiStop Protein Transport Inhibitor (BD Biosciences).
Phorbol 12-myristate 13-acetate (PMA) (InvivoGen): Make 1 mg/ml stock solution in sterile WFI and store at −20 °C. Protect from light.
Ionomycin (Sigma): Dissolve in sterile DMSO to make 3 mM ionomycin solution and store at −20 °C.
cRPMI (see Subheading 2.4, item 15).
FACS buffer (see Subheading 2.4, item 13).
BD Cytofix/Cytoperm Fixation and Permeabilization Solution (BD Biosciences).
BD Perm/wash buffer, 10× solution (BD Biosciences).
Optional: AccuCheck counting beads (Thermo Fisher).
UltraComp eBeads (eBioscience).
Falcon 96 Well Round Bottom Cell Culture Plate (Corning).
Fisherbrand Polypropylene Microtiter Tubes, 1.2 ml (Fisher Scientific).
Refrigerated Sorvall RT7 centrifuge with RTH-750 rotor and plate adaptors.
LSR-II flow cytometer (BD Biosciences).
FlowJo flow cytometry software (Tree Star, Inc).
Tetramers, antibodies, and viability dyes: See Table 2.
Table 2.
Flow cytometry reagents
| Analysisa | Reagent | Antibody clone | Companyc |
|---|---|---|---|
| Tet | B8R70–79 (FPKNDFVSF)/B7.2-PE | –b | – |
| Tet | L4R37–45 (FPRSMLSIF)/B7.2-PE | – | – |
| Tet | A3L192–200 (SPSNHHILL)/B7.2-PE | – | – |
| Tet | D1R686–694 (HPRHYATVM)/B7.2-PE | – | – |
| Tet | D1R808–817 (RPSTRNFFEL)/B7.2-PE | – | – |
| Tet | D5R375–383 (LPKEYSSEL)/B7.2-PE | – | – |
| Tet | J6R303–311 (MPAYIRNTL)/B7.2-PE | – | – |
| Tet | Ova257–264 (SIINFEKL)/H-2Kb-PE | – | – |
| Tet | Anti-CD8α-Pacific Blue | Clone 53–6.7 | BD Biosciences |
| ICCS | Anti-CD8α-PerCP-Cy5.5 | Clone 53–6.7 | BD Biosciences |
| Tet | Anti-B220-FITC | Clone RA3-6B2 | BD Biosciences |
| ICCS | Anti-B220-APC-Cy7 | Clone RA3-6B2 | BD Biosciences |
| Tet | Anti-CD11c-FITC | Clone N418 | Tonbo Biosciences |
| ICCS | Anti-CD3ε-PE | Clone 145-2C11 | BD Biosciences |
| Tet | Anti-CD4-FITC | Clone H129.19 | BD Biosciences |
| Tet | Anti-CD11b-FITC | Clone M1/70 | Tonbo Biosciences |
| ICCS | Anti-CD107a-FITC | Clone 1D4B | BD Biosciences |
| ICCS | Anti-IFN-γ-allophycocyanin (APC) | Clone XMG1.2 | BD Biosciences |
| Tet | Propidium iodide | – | BD Biosciences |
| ICCS | Fixable Viability Dye eFluor 450 | – | eBioscience |
Tet p/MHC class I tetramer used for staining, ICCS intracellular cytokine staining
p/B7.2, H2Kb monomers and tetramers were made in-house according to published methods ([28, 83], see also “Production protocols” at http://tetramer.yerkes.emory.edu/); all tetramers are labeled with phycoerythrin (PE) and stored as 50 μg/ml stock at +4 °C
Representative vendors from which the reagents were purchased. These reagents are also available from the other vendors as well
3 Methods
Follow institutional biosafety and waste disposal regulations when working with chemicals and biologicals. Experimentation involving mice must comply with the experimenters’ IACUC regulations. The procedures described here were approved by IACUC at Vanderbilt University.
3.1 Preparation of Antigen
This section describes the preparation of recombinant protein antigen that was used to immunize mice. The sample protocol described below includes the production, purification, and formulation of a VACV protein produced as a recombinant protein by biosynthesis in Escherichia coli (E. coli). This protocol was validated for ten different recombinant proteins, three of which were produced in soluble form, with the remaining seven proteins produced in insoluble form as inclusion bodies (IB). Isolated protein antigens proved to be suitable for in vivo immunogenicity and protection studies. Each investigator should optimize this protocol to ensure optimal yield, purity, and solubility of the protein of interest.
Plate bacteria transformed with the desired plasmid on freshly prepared LB agar plate containing 50 μg/ml Km and 1 % glucose; incubate overnight at +37 °C. Follow general rules of aseptic techniques when working with E. coli; that is, perform these procedures around the blue flame of a Bunsen burner.
Inoculate single colony into 2 ml of LB broth containing 50 μg/ml Km and 1 % glucose; incubate overnight at +37 °C (see Note 6).
Inoculate 250 ml LB broth containing 50 μg/ml Km (no glucose) with 1 ml of bacterial culture prepared as from Subheading 3.1, step 2, in 2000 ml Nalgene baffled shake flask; incubate at +37 °C with vigorous shaking (~150 rpm) to A600 nm = 0.8–1.0 (this level of growth takes ~4 h).
Induce expression of the recombinant gene by adding IPTG to 1 mM and continue incubation for an additional 4 h. Optional: Perform analysis of cellular fractions, i.e., total cell lysate as well as soluble and insoluble fractions, by 10–12 % SDS-PAGE to verify expression and solubility of the recombinant protein. Pause point: Bacterial culture can be stored overnight at +4 °C, or if the protein is produced as IB, cells can be harvested by centrifugation as in Subheading 3.1, step 5, and stored frozen at −80 °C, if necessary, for a long period of time.
Pellet cells by centrifugation at 3300 × g for 20 min at +4 °C. Discard the supernatant; carefully resuspend the pellet in 5 ml of cell lysis buffer A; then add 5 ml of the same buffer containing 4 mg/ml lysozyme; transfer to 50 ml polypropylene tube and incubate for 30 min on ice.
Sonicate with the 550 Sonic Dismembrator ten times for 10 s on ice using a setting of 5 on the Amplitude Control Knob, which is the highest setting for the micro-tip probe. Follow the manufacturer’s instructions for the instrument in use.
Transfer the cell lysate into 50 ml Nalgene Oak Ridge High-Speed PPCO Centrifuge tube, and centrifuge at 7670 × g for 20 min at +4 °C. Harvest the pellet and proceed to step 8 if protein is produced as IB; harvest supernatant and proceed to step 9 if soluble protein is produced.
To purify IB, discard supernatant and resuspend the pellet in 10 ml cell lysis buffer A without lysozyme by sonication as in Subheading 3.1, step 6, followed by centrifugation as in step 7. To solubilize protein in the IB, resuspend pellet in 0.5 ml PBS by sonication as in Subheading 3.1, step 6 first, then add 9.5 ml solubilization/wash buffer D (see Note 7).
Filter the supernatant through a 45 μm filter into a 15 ml polypropylene tube; add 2 ml Ni-NTA slurry, mix, and incubate with rotation for 2 h at +4 °C (see Note 8).
Pellet the resin at 300 × g for 5 min at +4 °C and carefully remove the supernatant (see Note 8). Resuspend resin in 14 ml of the appropriate wash buffer: for native protein purification, use wash buffer B; for solubilized denatured protein, use solubilization/wash buffer D. Centrifuge the solution at 300 × g for 5 min at +4 °C. Repeat this wash step.
Re-suspend the resin in 5 ml of the corresponding wash buffer as in Subheading 3.1, step 10 and transfer to a disposable 5 ml polypropylene column. Wash the column four times with 5 ml wash buffer B (for native protein purification) or D (for solubilized denatured protein purification).
Elute with 3 ml elution buffer C (for native protein purification) or elution buffer E (for denatured protein purification). Sterilize the eluted protein by filtration through a Millex 0.22 μm filter and store at +4 °C on ice. Use for immunization within the next 2 days.
Assess purity of isolated protein using 10–12 % SDS-PAGE. SDS-PAGE analysis has been described elsewhere [57] and, hence, is not described here. Quantify purified protein; one approach is to use a known amount of BSA protein that is separated by SDS-PAGE on the same gel as the recombinant proteins for quantification by densitometry. An average yield of purified recombinant VACV protein is ~1.5–6 mg, which corresponds to ~0.5–2 mg/ml in the eluted fraction.
Recommended: Follow vendor’s protocol to further purify protein with the use of endotoxin removal spin column (Subheading 2.2, item 21) or, alternatively, using the recently published protocol (see Note 9). Otherwise, proceed to step 15 (optional) or step 16.
Optional: Dialyze protein that was eluted under native conditions to exchange the elution buffer with PBS (see Note 10).
Remove urea from protein that was purified under denaturing conditions (see Note 11). Dialyze eluted protein against 500 ml PBS at RT; this will promote protein precipitation when urea is removed (see Note 12). Centrifuge at 10,000 × g for 5 min at RT, remove supernatant, and re-solubilize precipitate in 300 μl of 1 % SDS. This should yield ~5–20 mg/ml of protein. Incubate at +37 °C until completely dissolved. Store at RT and use for immunization over the next 2 days. Note that SDS precipitates upon thawing SDS-solubilized protein stored at −20 °C, as will the protein stored with it.
3.2 Immunizing Mice
The most common routes for immunizing a mouse are intraperitoneal (IP), subcutaneous (SQ), intravenous (IV), and intranasal (IN). SQ is commonly used for tracking CD8+ T cell responses in the draining lymph node [37]. Intramuscular (IM) administration is not generally recommended, as the muscle of the mouse is small. The route of immunization used determines trafficking properties of antigen-specific CD8+ T cells and influences the anamnestic immune response at the initial site of pathogen invasion [58]. Hence, choice of immunization route will depend on the investigator’s goal and infection model. Below, we describe protocols for IP and IN immunizations, which have been shown to elicit robust and protective CD8+ T cell responses against lethal VACV infection in mice ([28] and unpublished data). IP immunization induces a robust systemic response, resulting in antigen-specific CD8+ T cells circulating through the blood and secondary lymphoid organs, such as the spleen. IN immunization, in addition to eliciting a systemic response will stimulate a robust local mucosal immune response within the lungs [59]. Generating a robust antigen-specific CD8+ T cell response generally requires two immunizations—prime and boost. Experimentation involving mice must comply with the investigator’s IACUC regulations.
Thaw 0.2 mg/ml αGalCer stock solution at RT, and mix gently by pipetting (see Note 13).
Prepare appropriate volume of inoculum by mixing protein with αGalCer. For IN immunization, prepare 50 μl of mixture that contains 20–100 μg of protein (45 μl of 0.5–2 mg/ml solution) and 1 μg of αGalCer (5 μl of 0.2 mg/ml stock) per mouse. For IP immunization, prepare 200 μl of mixture that contains the same amount of protein and αGalCer per mouse as for IN immunization. If protein is insoluble and contains SDS, adjust SDS concentration with sterile PBS accordingly to final SDS concentration ≤ 0.05 % in the inoculum. For example, add 4 μl re-solubilized protein at 5–20 mg/ml in 1 % SDS from Subheading 3.1, step 16, and 5 μl of 0.2 mg/ml αGalCer stock to 191 μl of PBS (see Note 14). Proceed with step 3 for IP immunization, or step 4 for IN immunization.
For IP immunization, inject 200 μl of the inoculum IP into a B7.2tg mouse (see Note 15).
For IN immunization, anesthetize a B7.2tg mouse with 150 μl of ketamine-xylazine (see Note 16). After the mouse is deeply anesthetized (confirmed by the absence of foot reflexes in response to a mild pinch), gradually release 50 μl of the inoculum into both nostrils at the same time with a micropipette. Adjust the rate of release so as to allow the mouse to inhale the inoculum without forming bubbles. This should cause a rapid increase in the breathing rate. Hold the mouse in an upside down position for another couple of minutes until its breathing gradually returns to normal. Observe mice until recovery from anesthesia (see Note 17).
Boost mice 14 days after primary immunization as follows: prepare mixture of protein and αGalCer as in step 1 and inoculate mice as in step 2 (see Notes 18–20).
3.3 Isolation of Lymphocytes and Flow Cytometry
Note that all experimentation involving mice must comply with the investigator’s IACUC regulations. In addition, flow cytometry should be performed by trained personnel and in accordance with institutional regulations. Refer elsewhere for detailed overviews of flow cytometry acquisition and data analysis [60, 61].
Sacrifice mouse using SmartBox Auto CO2 System.
For each mouse, prepare two 60 mm Petri dishes and place on ice. For IP-immunized mice, remove the spleen and put on top of a 70 μm cell strainer placed inside the Petri dish. For IN-immunized mice, remove the spleen and lungs and place inside separate Petri dishes, each with a 70 μm cell strainer. Harvest spleen or lungs from non-immunized mice as negative controls. Proceed with step 3 to isolate splenic lymphocytes or step 6 to isolate lung lymphocytes (see Note 21).
To each spleen, add 2.5 ml ACK lysis buffer at RT. Using the plunger end of a 5 ml syringe, mash the tissue over the cell strainer to disperse leukocytes. Pass the cell suspension one more time though the cell strainer and transfer into a 15 ml polypropylene centrifuge tube. Incubate for 2 min at RT to lyse red blood cells (RBCs). Add 10 ml cRPMI with dasatinib to dilute ACK lysis buffer.
Centrifuge the cell suspension at 300 × g for 5 min at +4 °C. Decant supernatant.
Resuspend each splenocyte suspension by gentle pipetting in 5 ml FACS buffer and place on ice. To stain splenocytes with various fluorochrome-conjugated antibodies, proceed to step 9 (see Note 22).
Rinse the harvested lungs with PBS to remove blood. Mince tissue with a scalpel; transfer the minced lung tissue into a 15 ml polypropylene centrifuge tube containing 2 ml cRPMI supplemented with dasatinib and 2 mg/ml collagenase and let digest for 1 h at +37 °C.
Pour the digested lung slices onto a 70 μm cell strainer placed inside a Petri dish. Using the plunger end of a 5 ml syringe, mash the tissue over the cell strainer to disperse leukocytes.
Follow Subheading 3.3, steps 3–4, to lyse RBCs, prepare single-cell suspension, then resuspend cells from each lung in 2 ml of FACS buffer, and place on ice (see Note 22).
Count viable cells with a hemocytometer or counting beads. The expected recovery is ~0.5–1 × 107 leukocytes per 1 ml of cell suspension from processed lungs or spleen (see Note 23).
Add 200 μl of cells from each sample, including control non-immunized mouse leukocytes, into a 96-well round-bottom plate. Optional: If absolute count of tetramer-positive CD8+ T cells is required, we recommend using counting beads (see Note 23). Centrifuge plate at 828 × g for 2 min at +4 °C. Discard the supernatant.
Resuspend the cells in 100 μl of FACS buffer containing anti-B220-fluorescein isothiocyanate (FITC), -CD4-FITC, -CD11b-FITC, -CD11c-FITC (all at 1:200 dilution), -CD8α-Pacific Blue (1:100 dilution), and 1.5 μg/ml of corresponding R-phycoerythrin (PE)-labeled tetramer. Incubate for 1 to 2 h at +4 °C (see Note 24).
Dilute the staining reaction with 200 μl of FACS buffer and spin plate at 800 × g for 2 min at +4 °C to wash the cells. Discard the supernatant.
Resuspend the cells in 200–300 μl of FACS buffer containing 1:2000 dilution of 50 μg/ml propidium iodide stock solution, which incorporates into the nuclei of dead cells and, thereby, allowing differentiation between dead and viable cells in flow cytometry experiments. Transfer cells into 1.2 ml Microtiter Tubes; store on ice for up to 2 h before acquisition.
Prepare an acquisition template using FACSDiva software containing four fluorochromes to reflect the staining panel.
Set up compensation by using an unstained control and single stained compensation control samples.
Set up an acquisition layout containing the following plots: forward scatter (FSC)-A versus side scatter (SSC)-A. Create a gate for the lymphocyte population, and a FSC-W versus FSC-H followed by SSC-W versus SSC-H gate to discriminate singlet population. Create a histogram to display live/dead cells for propidium iodide with a gate to discriminate propidium iodide-negative live population; FITC (“dump” channel) versus CD8α-Pacific Blue to create CD8+ T cell gate; CD8α versus tetramer to create tetramer+ gate (Fig. 5).
Collect 5000–10,000 total CD8α+ events and export FCS files for the analysis in FlowJo software.
In FlowJo, to determine the frequency of antigen-specific, i.e., tetramer+, CD8+ T cells, apply gates as in Subheading 3.3, step 16. To determine background from non-specific tetramer staining, create tetramer gate for “CD8α versus tetramer” subset for cells from non-immunized control mouse, then apply it to the cells from a mouse immunized with protein and adjuvant. To determine the frequency of tetramer+ CD8+ T cells, subtract the background from each experimental value. If using counting beads, refer to vendor’s protocol to determine absolute number for the desired lymphocyte subset.
Optional: The remaining unstained cells can be cryopreserved at −80 °C in cRPMI containing 10 % DMSO for CD8+ T cell phenotyping in additional experiments if needed at a later time (see Note 25).
Fig. 5.

Analysis of epitope-specific CD8+ T cells by tetramer staining. (a) Representative example showing gating strategy and identification of epitope-specific CD8+ T cells. Cells harvested from the lungs of rL4R/B8R70–79 protein/αGalCer primed and boosted B7tg mice (IN route) were analyzed by four-color flow cytometry following staining with B8R70–79 /B7.2 tetramer. (b) Representative flow cytometry contour plots showing specificity of B8R70–79/B7.2 tetramer staining upon immunization with αGalCer-formulated protein antigen
3.4 CD8 T Cell Re-stimulation and Staining for Intracellular Interferon-γ
Staining for intracellular interferon-γ (IFN-γ) can be optionallyused as a complementary assay to validate the presence of functional antigen-specific CD8+ T cells after immunization.
Harvest spleen and isolate lymphocytes as reviewed in Subheading 3.3, steps 1–4. Work under aseptic conditions using a laminar flow hood; do not use dasatinib in any medium during tissue processing and cell incubation. Include dasatinib in FACS buffer only during the staining procedure for flow cytometry.
Resuspend cells from each spleen in 5 ml of cRPMI. Add 200 μl of cell suspension containing ~2 × 106 lymphocytes and 50 μl of anti-CD107a-FITC antibody (optional, see Note 26) to each of three wells of 96-well round-bottom plate. To the first well, add 50 μl of cRPMI containing 50 μM antigenic peptide epitope to 10 μM final. Add 50 μl of cRPMI to the second well; this will serve as a negative control for CD8+ T cell re-stimulation. Add 50 μl of cRPMI containing PMA and ionomycin to a final concentration of 50 ng/ml PMA plus 2 μg/ ml ionomycin to the third well; this will serve as a positive control for CD8+ T cell stimulation.
Incubate for 2 h at +37 °C in CO2 incubator (5 % CO2), then add 50 μl cRPMI containing a combination of vesicular transport inhibitors, such as brefeldin A (10 μg/ml final) and GolgiStop (1:1500); incubate for an additional 4 h, for a total of 6 h. These inhibitors will allow for intracellular accumulation of secreted proteins including cytokines such as IFN-γ at levels that are readily detectable by flow cytometry.
Spin plate at 828 × g for 2 min at +4 °C. Discard the supernatant, resuspend in 250 μl of PBS, and spin again at 828 × g for 2 min at +4 °C to wash the cells. Discard the supernatant.
Resuspend the cells in 100 μl of PBS containing eFluor 450 amino-reactive viability dye (1:2000). Incubate 30 min at +4 °C.
Add 200 μl of FACS buffer and centrifuge plate at 828 × g for 2 min at +4 °C to wash the cells. Discard the supernatant.
Resuspend the cells in 100 μl of FACS buffer supplemented with dasatinib and containing anti-B220-APC-Cy7 (allophycocyanin-cyanine7 tandem dye) (1:100), -CD3ε-PE (1:100) and -CD8α-PerCP-Cy5.5 (peridinin chlorophyll-cyanine5.5 tandem dye) (1:200) antibodies (see Note 27). Incubate for 1 h at +4 °C.
Add 200 μl of FACS buffer and spin plate at 828 × g for 2 min at +4 °C to wash the cells. Discard the supernatant.
Resuspend the cells in 100 μl of BD Cytofix/Cytoperm and incubate for 10 min at +4 °C.
Wash the cells twice by adding 200 μl of 1× BD Perm/wash and centrifuging at 828 × g for 2 min at +4 °C. Discard the supernatant.
Resuspend the cells in 100 μl of 1× BD Perm/wash containing anti-IFN-γ-allophycocyanin (1:200) antibody. Incubate for 1 h at +4 °C.
Add 200 μl of 1× BD Perm/wash and centrifuge plate at 828×g for 2 min at +4 °C to wash the cells. Discard the supernatant, resuspend in 200–300 μl of FACS buffer, and transfer cells into 1.5 ml microtubes. Stained cells can be stored on ice for up to 4 h before data acquisition. Alternatively, cells can be fixed as in Subheading 3.4, step 9, washed with FACS buffer as in step 8, resuspended in 200–300 μl of FACS buffer, and stored at +4 °C up to 16 h before data acquisition. Longer storage times have not been tested.
Prepare an acquisition template for six fluorochromes and set up compensation as detailed in Subheading 3.3, steps 14–15.
Set up an acquisition layout containing the following plots: forward scatter (FSC)-A versus side scatter (SSC)-A. Create a gate for the lymphocyte population and an SSC-W versus SSC-H gate to identify the singlet population. Create a histogram to display live/dead cells for eFluor450 with a gate to identify live cells; B220-APC-Cy7 versus CD3ε-PE to create T cell gate: CD8α-PerCP-Cy5.5 versus CD3ε-PE to create CD8+ T cell gate; CD107a-FITC versus IFN-γ-allophycocyanin to create CD107a+IFN-γ+ CD8+ T cell gate (Fig. 6).
Analyze sample as detailed in Subheading 3.3, steps 17–18. To determine frequency of CD107a+IFN-γ+ CD8+ T cells, subtract the background from a well without antigenic peptide epitope from each experimental value.
Fig. 6.

Analysis of epitope-specific CD8+ T cells by intracellular cytokine staining (ICCS). (a) Representative example showing gating strategy and identification of epitope-specific CD8+ T cells. Cells harvested from the spleen of protein/αGalCer primed and boosted mice (IP route) were re-stimulated in vitro with the indicated antigenic peptide epitope, then stained with the indicated antibodies and analyzed by six-color flow cytometry. (b) Representative flow cytometry plots showing that IFN-γ was specifically induced by a subset of CD8+ T cells after peptide epitope re-stimulation
Acknowledgments
Supported by Vanderbilt University Discovery Grant as well as VA Merit Award (BX001444) and NIH Contracts (AI040079), Research (AI042284, HL121139), Core (CA068485, DK058404), and Center (CA068485) grants.
Footnotes
For preparation and storage of αGalCer, use only glass vials to minimize leaching of impurities out of a plastic container and adsorption of the lipid to plastic (see Subheading 2.1, item 1).
All urea-containing buffers should be made fresh. At physiological pH, urea produces cyanate that can cause carbamylation of proteins by reacting with amino, carboxyl, and sulfhydryl groups, which may alter their stability and function.
Refer to methods described elsewhere [57] for making protein electrophoresis gel and performing SDS-PAGE.
Proteins purified from IBs may contain up to 0.05 % SDS for solubility.
Low concentrations of dasatinib were reported to substantially improve CD8+ T cell detection with tetramers by preventing T cell receptor internalization [62]. Dasatinib is a reversible inhibitor that targets a wide variety of protein kinase C family enzymes [63]. Hence, we routinely use dasatinib in FACS buffer instead of sodium azide to maintain the TCR at the cell surface and for improved staining and cell viability.
Use freshly transformed bacterial colonies grown on plates; or alternatively, plate transformed bacteria from frozen glycerol stock.
Prior resuspension of IB in PBS through vigorous sonication facilitates further solubilization of proteins within IB by using urea-containing solubilization/wash buffer D. Centrifuge cells at 7670 × g for 20 min at +4 °C and harvest the supernatant.
Save an aliquot of the supernatant for SDS-PAGE analysis.
LPS is a TLR4 ligand that could potentiate T cell and antibody responses and, hence, we recommend its removal. Nonetheless, we did not find trace amounts of endotoxin contaminants and other impurities co-purifying with the protein preparations to contribute substantially to the elicitation of a CD8+ T cell response in the presence of αGalCer [28]. In addition, LPS removal in the past has resulted in dramatic losses of VACV protein. Should it become necessary to remove co-purifying endotoxins, use the recently described endotoxin clearance method by including a non-ionic detergent wash step during metal chelation affinity purification of the protein antigen [64].
This step is optional because components of elution buffer C do not affect the performance of the immunization protocol or antigen-specific immune responses.
Proteins purified under denaturing conditions contain a high concentration of urea, which is incompatible with the immunization protocol. Removal of urea by common laboratory approaches may result in protein aggregation. Stimulation of antigen-specific T cell responses in vivo does not require native conformation of protein antigen, but does require the protein to be in a reasonably soluble form. Although soluble urea-free protein can be obtained through in vitro refolding, such a procedure is laborious and requires optimization for each protein. We found that a low SDS concentration (0.01–0.05 %) is compatible with IP immunization protocol. This residual low concentration of SDS can promote protein solubility after the removal of urea by dialysis.
Some proteins can be refolded successfully and remain soluble after dialysis.
αGalCer solution should be clear; if appears cloudy, re-heat at +80 °C until dissolved. Repeated freeze and thaw does not affect the reagent’s performance as long as the solution upon thawing remains clear and has not changed color.
Prepare mixture of protein with αGalCer right before immunization; usage of SDS-containing protein for IN immunization has not been validated.
For the first experiment, we recommend setting up a control wherein the mouse is inoculated IP with the same formulation but without protein antigen. Leukocytes harvested from this mouse after immunization will serve as a control to determine the background of the tetramer staining assay. An additional control for the performance of the reagents and immunization protocol that can be considered is the inclusion of a C57BL/6 mouse immunized by either the IP or IN route with 1 μg of αGalCer and 50 μg of ovalbumin (Ova) as detailed in the main protocol. Ova-specific CD8+ should be readily detected with the readily available H2Kb/SIINFEKL tetramer (NIH Tetramer Core, Emory University, Atlanta, USA; http://tetramer.yerkes.emory.edu/) in the spleen on day 6–14 after boost.
Depending on the weight of the mouse, the amount of anesthesia can vary from 125 to 165 μl.
For the first experiment, we recommend setting up a control wherein the mouse is inoculated IN with the same formulation containing PBS and αGalCer but without protein antigen. Lymphocytes harvested from this mouse after immunization will serve as a control to determine the background of the tetramer staining assay.
Usually, antigen-specific CD8+ T cells can be detected at low frequency on days 8–14 after primary immunization. At least one booster immunization substantially increases their frequency, which is more desirable for immunological and vaccination studies.
For the first time, before the mouse is sacrificed and leukocytes harvested, it may be necessary to confirm that immunization has induced antigen-specific CD8+ T cells of detectable frquency. This can be accomplished by assessing mouse blood leukocytes. A robust antigen-specific CD8+ T cell response should be readily detected systemically the blood on day 6–14 after boost.
Although it is not reviewed in the protocol here, protein antigen and αGalCer immunization also induces antigen-specific antibody responses that can be assessed in mouse serum (but see Fig. 3C).
Harvested organs can be left on ice while additional mice are being processed or reagents are being prepared for up to 24 h. Longer storage times have not been tested.
Cell suspensions can be left on ice for up to 8 h while additional tissues are being processed. Longer storage times have not been tested.
We found that precise counting of CD8+ T cells in the lungs requires counting beads. Counting beads are added directly to staining solution to account for cell losses, assuming proportional bead and cell losses during washes. When working with counting beads, follow vendor’s protocol at https://tools.thermofisher.com/content/sfs/manuals/PCB100_accu-check_beads_man.pdf.
The investigator should determine optimal staining conditions, such as concentration of tetramer, incubation temperature, and time; each of these parameters will differentially influence experimental outcome.
Cryopreservation does not affect staining performance but may affect viability. An expected lymphocyte viability after thawing is 80 % or higher.
CD107a is a marker for degranulation activity of immune effector cells including CD8+ T cells. Staining with anti-CD107a antibody is optional and performed during re-stimulation of cells.
Upon in vitro re-stimulation, epitope-specific CD8+ T cells markedly down-regulate their T cell receptor from the cell surface and, hence, stain poorly with the complex of peptide/ MHC tetramers.
References
- 1.Plotkin SA, Plotkin SL. The development of vaccines: how the past led to the future. Nat Rev Microbiol. 2011;9:889–893. doi: 10.1038/nrmicro2668. [DOI] [PubMed] [Google Scholar]
- 2.Zepp F. Principles of vaccine design-lessons from nature. Vaccine. 2010;28(Suppl 3):C14–C24. doi: 10.1016/j.vaccine.2010.07.020. [DOI] [PubMed] [Google Scholar]
- 3.Amanna IJ, Slifka MK. Wanted, dead or alive: new viral vaccines. Antiviral Res. 2009;84:119–130. doi: 10.1016/j.antiviral.2009.08.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bevan MJ. Understand memory, design better vaccines. Nat Immunol. 2011;12:463–465. doi: 10.1038/ni.2041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Pulendran B, Ahmed R. Immunological mechanisms of vaccination. Nat Immunol. 2011;131:509–517. doi: 10.1038/ni.2039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Koff WC, Gust ID, Plotkin SA. Toward a human vaccines project. Nat Immunol. 2014;15:589–592. doi: 10.1038/ni.2871. [DOI] [PubMed] [Google Scholar]
- 7.Amanna IJ, Slifka MK. Contributions of humoral and cellular immunity to vaccine-induced protection in humans. Virology. 2011;411:206–215. doi: 10.1016/j.virol.2010.12.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Moss B. Smallpox vaccines: targets of protective immunity. Immunol Rev. 2011;239:8–26. doi: 10.1111/j.1600-065X.2010.00975.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Rees AR. The antibody molecule: from antitoxins to therapeutic antibodies. 1. Oxford Medical Histories/Oxford University Press; Oxford: 2015. p. 384. [Google Scholar]
- 10.Jenner E. Two cases of Small-Pox Infection communicated to the Foetus in Utero under peculiar circumstances, with additional remarks. Med Chir Trans. 1809;1:271–277. [PMC free article] [PubMed] [Google Scholar]
- 11.Adkins B, Leclerc C, Marshall-Clarke S. Neonatal adaptive immunity comes of age. Nat Rev Immunol. 2004;4:553–564. doi: 10.1038/nri1394. [DOI] [PubMed] [Google Scholar]
- 12.Swamy GK, Wheeler SM. Neonatal pertussis, cocooning and maternal immunization. Expert Rev Vaccines. 2014;13:1107–1114. doi: 10.1586/14760584.2014.944509. [DOI] [PubMed] [Google Scholar]
- 13.Verhasselt V. Is infant immunization by breastfeeding possible? Philos Trans R Soc Lond B Biol Sci. 2015 doi: 10.1098/rstb.2014.0139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.O’Connell CJ, Karzon DT, Barron AL, Plaut ME, Ali VM. Progressive vaccinia with normal antibodies. A case possibly due to deficient cellular immunity. Ann Intern Med. 1964;60:282–28. doi: 10.7326/0003-4819-60-2-282. [DOI] [PubMed] [Google Scholar]
- 15.Blum JS, Wearsch PA, Cresswell P. Pathways of antigen processing. Annu Rev Immunol. 2013;31:443–473. doi: 10.1146/annurev-immunol-032712-095910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Segura E, Amigorena S. Cross-presentation in mouse and human dendritic cells. Adv Immunol. 2015;127:1–31. doi: 10.1016/bs.ai.2015.03.002. [DOI] [PubMed] [Google Scholar]
- 17.Thomas PG, Keating R, Hulse-Post DJ, Doherty PC. Cell-mediated protection in influenza infection. Emerg Infect Dis. 2006;12:48–54. doi: 10.3201/eid1201.051237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Brown LE, Kelso A. Prospects for an influenza vaccine that induces cross-protective cytotoxic T lymphocytes. Immunol Cell Biol. 2009;87:300–308. doi: 10.1038/icb.2009.16. [DOI] [PubMed] [Google Scholar]
- 19.Kohlmeier JE, Woodland DL. Immunity to respiratory viruses. Annu Rev Immunol. 2009;27:61–82. doi: 10.1146/annurev.immunol.021908.132625. [DOI] [PubMed] [Google Scholar]
- 20.Kremer M, Suezer Y, Volz A, Frenz T, Majzoub M, Hanschmann KM, Lehmann MH, Kalinke U, Sutter G. Critical role of perforin-dependent CD8+ T cell immunity for rapid protective vaccination in a murine model for human smallpox. PLoS Pathog. 2012;8:e1002557. doi: 10.1371/journal.ppat.1002557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Goulding J, Bogue R, Tahiliani V, Croft M, Salek-Ardakani S. CD8+ T cells are essential for recovery from a respiratory vaccinia virus infection. J Immunol. 2012;189:2432–2440. doi: 10.4049/jimmunol.1200799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Gordon SN, Cecchinato V, Andresen V, Heraud JM, Hryniewicz A, Parks RW, Venzon D, Chung HK, Karpova T, McNally J, Silvera P, Reimann KA, Matsui H, Kanehara T, Shinmura Y, Yokote H, Franchini G. Smallpox vaccine safety is dependent on T cells and not B cells. J Infect Dis. 2011;203:1043–1053. doi: 10.1093/infdis/jiq162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Li CR, Greenberg PD, Gilbert MJ, Goodrich JM, Riddell SR. Recovery of Hla-restricted cytomegalovirus (Cmv)-specific T-cell responses after allogeneic bone-marrow transplant - correlation with Cmv disease and effect of ganciclovir prophylaxis. Blood. 1994;83:1971–1979. [PubMed] [Google Scholar]
- 24.Goulder PJR, Watkins DI. Impact of MHC class I diversity on immune control of immunodeficiency virus replication. Nat Rev Immunol. 2008;8:619–630. doi: 10.1038/nri2357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Schmidt NW, Butler NS, Badovinac VP, Harty JT. Extreme CD8+ T cell requirements for anti-malarial liver-stage immunity following immunization with radiation attenuated sporozoites. PLoS Pathog. 2010;6:e1000998. doi: 10.1371/journal.ppat.1000998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Epstein JE, Tewari K, Lyke KE, Sim BKL, Billingsley PF, Laurens MB, Gunasekera A, Chakravarty S, James ER, Sedegah M, Richman A, Velmurugan S, Reyes S, Li M, Tucker K, Ahumada A, Ruben AJ, Li T, Stafford R, Eappen AG, Tamminga C, Bennett JW, Ockenhouse CF, Murphy JR, Komisar J, Thomas N, Loyevsky M, Birkett A, Plowe CV, Loucq C, et al. Live attenuated malaria vaccine designed to protect through hepatic CD8(+) T cell immunity. Science. 2011;334:475–480. doi: 10.1126/science.1211548. [DOI] [PubMed] [Google Scholar]
- 27.Woodworth JSM, Behar SM. Mycobacterium tuberculosis-specific CD8(+) T cells and their role in immunity. Crit Rev Immunol. 2006;26:317–352. doi: 10.1615/critrevimmunol.v26.i4.30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Gilchuk P, Spencer CT, Conant SB, Hill T, Gray JJ, Niu X, Zheng M, Erickson JJ, Boyd KL, McAfee KJ, Oseroff C, Hadrup SR, Bennink JR, Hildebrand W, Edwards KM, Crowe JE, Jr, Williams JV, Buus S, Sette A, Schumacher TN, Link AJ, Joyce S. Discovering naturally processed antigenic determinants that confer protective T cell immunity. J Clin Invest. 2013;123:1976–1987. doi: 10.1172/JCI67388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Gerdts V, Littel-van den Hurk SV, Griebel PJ, Babiuk LA. Use of animal models in the development of human vaccines. Future Microbiol. 2007;2:667–675. doi: 10.2217/17460913.2.6.667. [DOI] [PubMed] [Google Scholar]
- 30.Gerdts V, Wilson HL, Meurens F, van Drunen Littel-van den Hurk S, Wilson D, Walker S, Wheler C, Townsend H, Potter AA. Large animal models for vaccine development and testing. ILAR J. 2015;56:53–62. doi: 10.1093/ilar/ilv009. [DOI] [PubMed] [Google Scholar]
- 31.van Hall T, van der Burg SH. Mechanisms of peptide vaccination in mouse models: tolerance, immunity, and hyperreactivity. Adv Immunol. 2012;114:51–76. doi: 10.1016/B978-0-12-396548-6.00003-2. [DOI] [PubMed] [Google Scholar]
- 32.Koup RA, Douek DC. Vaccine design for CD8+ T lymphocyte responses. Cold Spring Harb Perspect Med. 2011;1:a007252. doi: 10.1101/cshperspect.a007252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Remakus S, Rubio D, Ma X, Sette A, Sigal LJ. Memory CD8+ T cells specific for a single immunodominant or subdominant determinant induced by peptide-dendritic cell immunization protect from an acute lethal viral disease. J Virol. 2012;86:9748–9759. doi: 10.1128/JVI.00981-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Coffman RL, Sher A, Seder RA. Vaccine adjuvants: putting innate immunity to work. Immunity. 2010;33:492–503. doi: 10.1016/j.immuni.2010.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Duthie MS, Windish HP, Fox CB, Reed SG. Use of defined TLR ligands as adjuvants within human vaccines. Immunol Rev. 2011;239:178–196. doi: 10.1111/j.1600-065X.2010.00978.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Cho HI, Celis E. Optimized peptide vaccines eliciting extensive CD8+ T-cell responses with therapeutic antitumor effects. Cancer Res. 2009;69:9012–9019. doi: 10.1158/0008-5472.CAN-09-2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Liu H, Moynihan KD, Zheng Y, Szeto GL, Li AV, Huang B, Van Egeren DS, Park C, Irvine DJ. Structure-based programming of lymph-node targeting in molecular vaccines. Nature. 2014;507:519–522. doi: 10.1038/nature12978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Saroja C, Lakshmi P, Bhaskaran S. Recent trends in vaccine delivery systems: a review. Int J Pharm Invest. 2011;1:64–74. doi: 10.4103/2230-973X.82384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Moon JJ, Huang B, Irvine DJ. Engineering nano- and microparticles to tune immunity. Adv Mater. 2012;24:3724–3746. doi: 10.1002/adma.201200446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Hermans IF, Silk JD, Gileadi U, Salio M, Mathew B, Ritter G, Schmidt R, Harris AL, Old L, Cerundolo V. NKT cells enhance CD4+ and CD8+ T cell responses to soluble antigen in vivo through direct interaction with dendritic cells. J Immunol. 2003;171:5140–5147. doi: 10.4049/jimmunol.171.10.5140. [DOI] [PubMed] [Google Scholar]
- 41.Singh N, Hong S, Scherer DC, Serizawa I, Burdin N, Kronenberg M, Koezuka Y, Van Kaer L. Activation of NK T cells by CD1d and alpha-galactosylceramide directs conventional T cells to the acquisition of a Th2 phenotype. J Immunol (Cutting Edge) 1999;163:2373–2377. [PubMed] [Google Scholar]
- 42.Semmling V, Lukacs-Kornek V, Thaiss CA, Quast T, Hochheiser K, Panzer U, Rossjohn J, Perlmutter P, Cao J, Godfrey DI, Savage PB, Knolle PA, Kolanus W, Forster I, Kurts C. Alternative cross-priming through CCL17-CCR4-mediated attraction of CTLs toward NKT cell-licensed DCs. Nat Immunol. 2010;11:313–320. doi: 10.1038/ni.1848. [DOI] [PubMed] [Google Scholar]
- 43.Oseroff C, Peters B, Pasquetto V, Moutaftsi M, Sidney J, Panchanathan V, Tscharke DC, Maillere B, Grey H, Sette A. Dissociation between epitope hierarchy and immunoprevalence in CD8+ responses to vaccinia virus western reserve. J Immunol. 2008;180:7193–7202. doi: 10.4049/jimmunol.180.11.7193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Gottschalk C, Mettke E, Kurts C. The role of invariant natural killer T cells in dendritic cell licensing, cross-priming, and memory CD8(+) T cell generation. Front Immunol. 2015;6:379. doi: 10.3389/fimmu.2015.00379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Hill TM, Bezbradica JS, Van Kaer L, Joyce S. CD1d-restricted natural killer T cells. Encyclopaedia Live Sci. 2016 doi: 10.1002/9780470015902.a0020180.pub2. [DOI] [Google Scholar]
- 46.Fujii S, Shimizu K, Smith C, Bonifaz L, Steinman RM. Activation of natural killer T cells by alpha-galactosylceramide rapidly induces the full maturation of dendritic cells in vivo and thereby acts as an adjuvant for combined CD4 and CD8 T cell immunity to a coadministered protein. J Exp Med. 2003;198:267–279. doi: 10.1084/jem.20030324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Galli G, Pittoni P, Tonti E, Malzone C, Uematsu Y, Tortoli M, Maione D, Volpini G, Finco O, Nuti S, Tavarini S, Dellabona P, Rappuoli R, Casorati G, Abrignani S. Invariant NKT cells sustain specific B cell responses and memory. Proc Natl Acad Sci U S A. 2007;104:3984–3989. doi: 10.1073/pnas.0700191104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Kamijuku H, Nagata Y, Jiang X, Ichinohe T, Tashiro T, Mori K, Taniguchi M, Hase K, Ohno H, Shimaoka T, Yonehara S, Odagiri T, Tashiro M, Sata T, Hasegawa H, Seino KI. Mechanism of NKT cell activation by intranasal coadministration of alpha-galactosylceramide, which can induce cross-protection against influenza viruses. Mucosal Immunol. 2008;1:208–218. doi: 10.1038/mi.2008.2. [DOI] [PubMed] [Google Scholar]
- 49.Reilly EC, Thompson EA, Aspeslagh S, Wands JR, Elewaut D, Brossay L. Activated iNKT cells promote memory CD8+ T cell differentiation during viral infection. PLoS One. 2012;7:e37991. doi: 10.1371/journal.pone.0037991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Carreno LJ, Kharkwal SS, Porcelli SA. Optimizing NKT cell ligands as vaccine adjuvants. Immunotherapy. 2014;6:309–320. doi: 10.2217/imt.13.175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Ko SY, Ko HJ, Chang WS, Park SH, Kweon MN, Kang CY. alpha-galactosylceramide can act as a nasal vaccine adjuvant inducing protective immune responses against viral infection and tumor. J Immunol. 2005;175:3309–3317. doi: 10.4049/jimmunol.175.5.3309. [DOI] [PubMed] [Google Scholar]
- 52.Courtney AN, Thapa P, Singh S, Wishahy AM, Zhou D, Sastry J. Intranasal but not intravenous delivery of the adjuvant alpha-galactosylceramide permits repeated stimulation of natural killer T cells in the lung. Eur J Immunol. 2011;41:3312–3322. doi: 10.1002/eji.201041359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Nishimura T, Kitamura H, Iwakabe K, Yahata T, Ohta A, Sato M, Takeda K, Okumura K, Van Kaer L, Kawano T, Taniguchi M, Nakui M, Sekimoto M, Koda T. The interface between innate and acquired immunity: glycolipid antigen presentation by CD1d-expressing dendritic cells to NKT cells induces the differentiation of antigen-specific cytotoxic T lymphocytes. Int Immunol. 2000;12:987–994. doi: 10.1093/intimm/12.7.987. [DOI] [PubMed] [Google Scholar]
- 54.Guillonneau C, Mintern JD, Hubert FX, Hurt AC, Besra GS, Porcelli S, Barr IG, Doherty PC, Godfrey DI, Turner SJ. Combined NKT cell activation and influenza virus vaccination boosts memory CTL generation and protective immunity. Proc Natl Acad Sci U S A. 2009;106:3330–3335. doi: 10.1073/pnas.0813309106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Duwaerts CC, Gregory SH. Targeting the diverse immunological functions expressed by hepatic NKT cells. Expert Opin Ther Targets. 2011;15:973–988. doi: 10.1517/14728222.2011.584874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Alexander J, Oseroff C, Sidney J, Sette A. Derivation of HLA-B*0702 transgenic mice: functional CTL repertoire and recognition of human B*0702-restricted CTL epitopes. Hum Immunol. 2003;64:211–223. doi: 10.1016/s0198-8859(02)00786-3. [DOI] [PubMed] [Google Scholar]
- 57.Green M, Sambrook J. A laboratory manual. 4. Vol. 3. Cold Spring Harbor Laboratory Press; 2012. Molecular cloning. p Protocol 8. [Google Scholar]
- 58.Belyakov IM, Ahlers JD. What role does the route of immunization play in the generation of protective immunity against mucosal pathogens? J Immunol. 2009;183:6883–6892. doi: 10.4049/jimmunol.0901466. [DOI] [PubMed] [Google Scholar]
- 59.Li AV, Moon JJ, Abraham W, Suh H, Elkhader J, Seidman MA, Yen M, Im EJ, Foley MH, Barouch DH, Irvine DJ. Generation of effector memory T cell-based mucosal and systemic immunity with pulmonary nanoparticle vaccination. Sci Transl Med. 2013;5:204ra130. doi: 10.1126/scitranslmed.3006516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Sharrow SO. Overview of flow cytometry. Curr Protoc Immunol. 2002;Chapter 5(Unit 5):1. doi: 10.1002/0471142735.im0501s50. [DOI] [PubMed] [Google Scholar]
- 61.Roederer M. Multiparameter FACS analysis. Curr Protoc Immunol. 2002;Chapter 5(Unit 5):8. doi: 10.1002/0471142735.im0508s49. [DOI] [PubMed] [Google Scholar]
- 62.Lissina A, Ladell K, Skowera A, Clement M, Edwards E, Seggewiss R, van den Berg HA, Gostick E, Gallagher K, Jones E, Melenhorst JJ, Godkin AJ, Peakman M, Price DA, Sewell AK, Wooldridge L. Protein kinase inhibitors substantially improve the physical detection of T-cells with peptide-MHC tetramers. J Immunol Methods. 2009;340:11–24. doi: 10.1016/j.jim.2008.09.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Montero JC, Seoane S, Ocana A, Pandiella A. Inhibition of SRC family kinases and receptor tyrosine kinases by dasatinib: possible combinations in solid tumors. Clin Cancer Res. 2011;17:5546–5552. doi: 10.1158/1078-0432.CCR-10-2616. [DOI] [PubMed] [Google Scholar]
- 64.Zimmerman T, Petit Frere C, Satzger M, Raba M, Weisbach M, Dohn K, Popp A, Donzeau M. Simultaneous metal chelate affinity purification and endotoxin clearance of recombinant antibody fragments. J Immunol Methods. 2006;314:67–73. doi: 10.1016/j.jim.2006.05.012. [DOI] [PubMed] [Google Scholar]
- 65.Wagner CS, Grotzke JE, Cresswell P. Intracellular events regulating cross-presentation. Front Immunol. 2012;3:138. doi: 10.3389/fimmu.2012.00138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Hadrup SR, Bakker AH, Shu CJ, Andersen RS, van Veluw J, Hombrink P, Castermans E, Thor Straten P, Blank C, Haanen JB, Heemskerk MH, Schumacher TN. Parallel detection of antigen-specific T-cell responses by multidimensional encoding of MHC multimers. Nat Methods. 2009;6:520–526. doi: 10.1038/nmeth.1345. [DOI] [PubMed] [Google Scholar]
- 67.Kain L, Webb B, Anderson BL, Deng S, Holt M, Costanzo A, Zhao M, Self K, Teyton A, Everett C, Kronenberg M, Zajonc DM, Bendelac A, Savage PB, Teyton L. The identification of the endogenous ligands of natural killer T cells reveals the presence of mammalian alpha-linked glycosylceramides. Immunity. 2014;41:543–554. doi: 10.1016/j.immuni.2014.08.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Morita M, Motoki K, Akimoto K, Natori T, Sakai T, Sawa E, Yamaji K, Koezuka Y, Kobayashi E, Fukushima H. Structure-activity relationship of alpha-galactosylceramides against B16-bearing mice. J Med Chem. 1995;38:2176–2187. doi: 10.1021/jm00012a018. [DOI] [PubMed] [Google Scholar]
- 69.Natori T, Koezuka Y, Higa T. Agelasphins, Novel a-galactosylceramides from the marine sponge Agelas mauritianus. Tetrahedron Lett. 1993;34:5591–5592. [Google Scholar]
- 70.Kawano T, Cui J, Koezuka Y, Toura I, Kaneko Y, Motoki K, Ueno H, Nakagawa R, Sato H, Kondo E, Koseki H, Taniguchi M. CD1d-restricted and TCR-mediated activation of valpha14 NKT cells by glycosylceramides. Science. 1997;278:1626–1629. doi: 10.1126/science.278.5343.1626. [DOI] [PubMed] [Google Scholar]
- 71.Birkholz AM, Girardi E, Wingender G, Khurana A, Wang J, Zhao M, Zahner S, Illarionov PA, Wen X, Li M, Yuan W, Porcelli SA, Besra GS, Zajonc DM, Kronenberg M. A novel glycolipid antigen for NKT cells that preferentially induces IFN-gamma production. J Immunol. 2015;195:924–933. doi: 10.4049/jimmunol.1500070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Schmieg J, Yang G, Franck RW, Tsuji M. Superior protection against malaria and melanoma metastases by a C-glycoside analogue of the natural killer T cell ligand alpha-Galactosylceramide. J Exp Med. 2003;198:1631–1641. doi: 10.1084/jem.20031192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Li X, Fujio M, Imamura M, Wu D, Vasan S, Wong CH, Ho DD, Tsuji M. Design of a potent CD1d-binding NKT cell ligand as a vaccine adjuvant. Proc Natl Acad Sci U S A. 2010;107:13010–13015. doi: 10.1073/pnas.1006662107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Aspeslagh S, Li Y, Yu ED, Pauwels N, Trappeniers M, Girardi E, Decruy T, Van Beneden K, Venken K, Drennan M, Leybaert L, Wang J, Franck RW, Van Calenbergh S, Zajonc DM, Elewaut D. Galactose-modified iNKT cell agonists stabilized by an induced fit of CD1d prevent tumour metastasis. EMBO J. 2011;30:2294–2305. doi: 10.1038/emboj.2011.145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Aspeslagh S, Nemcovic M, Pauwels N, Venken K, Wang J, Van Calenbergh S, Zajonc DM, Elewaut D. Enhanced TCR footprint by a novel glycolipid increases NKT-dependent tumor protection. J Immunol. 2013;191:2916–2925. doi: 10.4049/jimmunol.1203134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Tyznik AJ, Farber E, Girardi E, Birkholz A, Li Y, Chitale S, So R, Arora P, Khurana A, Wang J, Porcelli SA, Zajonc DM, Kronenberg M, Howell AR. Glycolipids that elicit IFN-gamma-biased responses from natural killer T cells. Chem Biol. 2011;18:1620–1630. doi: 10.1016/j.chembiol.2011.10.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Miyamoto K, Miyake S, Yamamura T. A synthetic glycolipid prevents autoimmune encephalomyelitis by inducing TH2 bias of natural killer T cells. Nature. 2001;413:531–534. doi: 10.1038/35097097. [DOI] [PubMed] [Google Scholar]
- 78.Yu KO, Im JS, Molano A, Dutronc Y, Illarionov PA, Forestier C, Fujiwara N, Arias I, Miyake S, Yamamura T, Chang YT, Besra GS, Porcelli SA. Modulation of CD1d-restricted NKT cell responses by using N-acyl variants of alpha-galactosylceramides. Proc Natl Acad Sci U S A. 2005;102:3383–3388. doi: 10.1073/pnas.0407488102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Kinjo Y, Wu D, Kim G, Xing GW, Poles MA, Ho DD, Tsuji M, Kawahara K, Wong CH, Kronenberg M. Recognition of bacterial glycosphingolipids by natural killer T cells. Nature. 2005;434:520–525. doi: 10.1038/nature03407. [DOI] [PubMed] [Google Scholar]
- 80.Mattner J, Debord KL, Ismail N, Goff RD, Cantu C, 3rd, Zhou D, Saint-Mezard P, Wang V, Gao Y, Yin N, Hoebe K, Schneewind O, Walker D, Beutler B, Teyton L, Savage PB, Bendelac A. Exogenous and endogenous glycolipid antigens activate NKT cells during microbial infections. Nature. 2005;434:525–529. doi: 10.1038/nature03408. [DOI] [PubMed] [Google Scholar]
- 81.Albacker LA, Chaudhary V, Chang YJ, Kim HY, Chuang YT, Pichavant M, DeKruyff RH, Savage PB, Umetsu DT. Invariant natural killer T cells recognize a fungal glycosphingolipid that can induce airway hyperreactivity. Nat Med. 2013;19:1297–1304. doi: 10.1038/nm.3321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Joyce S, Girardi E, Zajonc DM. NKT cell ligand recognition logic: molecular basis for a synaptic duet and transmission of inflammatory effectors. J Immunol. 2011;187:1081–1089. doi: 10.4049/jimmunol.1001910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Rodenko B, Toebes M, Hadrup SR, van Esch WJ, Molenaar AM, Schumacher TN, Ovaa H. Generation of peptide-MHC class I complexes through UV-mediated ligand exchange. Nat Protoc. 2006;1:1120–1132. doi: 10.1038/nprot.2006.121. [DOI] [PubMed] [Google Scholar]