

Abstract
Tissue cells respond to changes in tensional forces with proliferation or death through the control of RhoA. However, the response coupling mechanisms that link force with RhoA activation are poorly understood. We found that tension applied to fibronectin-coated microbeads caused recruitment of all three isoforms of the Shc adapter (p66Shc, p52Shc, and p46Shc) to adhesion complexes. The Shc PTB domain was necessary and sufficient for this recruitment, and screening studies revealed the direct interactions with the FERM domain of focal adhesion kinase (FAK) that were required for Shc translocation to adhesion complexes. The FAK/p66Shc complex specifically bound and activated the Rho guanyl exchange factors (GEFs) p115-RhoGEF and GEF-H1, leading to tension-induced RhoA activation. In contrast, the FAK/p52Shc complex bound SOS1 but not the Rho GEFs to mediate tension-induced Ras activation. Nuclear translocation and activation of the YAP/TAZ transcription factors on firm substrates required the FAK/p66Shc/Rho GEF complex, and both proliferation on firm substrates and anoikis in suspension required signaling through p66Shc and its associated Rho GEFs. These studies reveal the binary and exclusive assignment of p66Shc and p52Shc to tension-induced Rho or Ras signals, respectively, and suggest an integrated role for the two Shc isoforms in coordinating the cellular response to mechanical stimuli.
INTRODUCTION
The Rho GTPases control numerous aspects of cell fate and cytoskeletal dynamics. Accordingly, one major role for these molecular switches is to coordinate the cytoskeletal and nuclear responses to local force. Tension applied to integrin-binding microbeads, for instance, elicits a RhoA-dependent viscoelastic response leading to cell stiffening, enabling cells to respond to changes in their physical environment (1). Differentiation into osteogenic or adipogenic lines in response to constraints in cell shape also requires changes in cytoskeletal tension mediated by RhoA and its effector, ROCK (2). RhoA directs proliferation on stiff matrix beds and controls a number of large-scale developmental programs that follow physical cues, such as gastrulation, branching morphogenesis, dorsal closure, and ventralization (3–8). However, upstream events linking force-sensing structures, such as focal adhesions, to RhoA are not well understood.
The adapter proteins encoded by the highly conserved SHC1 gene link environmental signals with GTPase signaling and thus may hold clues about how cells process mechanical information. The human SHC1 gene expresses three proteins differing only in the length of their N termini (p66Shc, p52Shc, and p46Shc) (9). The most widely studied isoform, p52Shc, links upstream receptors, such as receptor tyrosine kinases, with downstream Ras signals through recruitment of Grb2 and SOS (10). Presumably because of this effect on Ras, p52Shc overexpression transforms NIH 3T3 cells (9), and the transforming activity of the polyomavirus middle T antigen requires p52Shc (11, 12). p52Shc also indirectly associates with specific integrin pairs to mediate adhesion-dependent survival and proliferation (13, 14). In platelets, which express only the p52Shc isoform and do not proliferate, recruitment of p52Shc to phosphorylated integrin β3 causes platelet activation and aggregation, indicating a more general role in outside-in integrin signaling (15, 16).
In contrast, the normal physiologic role of the p66Shc isoform is poorly understood, and the functional significance of its evolutionary link to p52Shc is even less so. Like p52Shc, p66Shc binds to activated epidermal growth factor receptor, becomes tyrosine phosphorylated, and forms stable complexes with Grb2; however, p66Shc does not activate extracellular signal-regulated kinase (ERK) and in fact blocks ERK and c-fos activation and proliferation (17, 18). p66Shc may play a role in promoting the differentiation of matrix- and tension-sensitive cells. Antisense oligonucleotides against p66Shc, for instance, block terminal differentiation of myoblasts into myotubes (19). Notably, this treatment disrupts the actin stress fiber structure and causes cell rounding, suggesting the loss of internal tensional forces. p66Shc knockdown in embryonic mouse lung explants causes the loss of epithelial cell differentiation with increased proliferation but diminished branching morphogenesis (20), and bovine zygotes microinjected with small interfering RNA for p66Shc show a marked reduction in blastocyst development with fewer mitotically arrested embryos, suggesting a role for p66Shc in promoting differentiation and enforcing cell cycle checkpoints in developmental programs at early stages (21).
Consistent with a proposed role for Shc in transmitting integrin signals, p66Shc also reports adhesion status to anchorage-dependent endothelial and epithelial cells. p66Shc localizes in part to focal adhesions, where it stimulates local RhoA activation (22).The loss of p66Shc or mutations which delocalize p66Shc from focal adhesions interfere with anoikis and impair RhoA signaling (22, 23). Notably, inhibition of cellular tension also blocks anoikis, suggesting that p66Shc participates in a mechanical test of anchorage status (22). However, the mechanism by which this pathway proceeds and its relevance to other mechanical signals have not been studied. In the study described in this report, we found that p66Shc and p52Shc respond to mechanical signals by differential recruitment of Rho and Ras guanyl exchange factors (GEFs) and that a FAK/p66Shc/Rho GEF module is required for RhoA-mediated proliferation and death responses to mechanical tension.
MATERIALS AND METHODS
Molecular cloning.
Full-length p66Shc cDNA (22) was used as a PCR template to subclone full-length p66Shc, p52Shc, and the Shc PTB domain into the EcoRI site of the double-Flag-tagged pulldown vector pIPX-C-FF-ZZ-B (a gift of Russ Carstens), and Flag-p66Shc was then subcloned into the adenoviral shuttle pDC315(io). Adenoviruses carrying the empty Flag and Flag-PTB constructs were made using the AdMax HiIQ system (Microbix). Flag-p66Shc and Flag-p52Shc were then used as PCR templates for subcloning into the lentiviral shuttle vector pCCL.PPT.hPGK.IRES.GFP.Wpre at EcoRI and ApaI sites. Green fluorescent protein (GFP)-tagged p66 lacking PTB [p66(ΔPTB)-GFP] (22) was used as a template to subclone Flag-p66(ΔPTB) into the lentiviral shuttle at EcoRI and BamHI sites. 6×His fusions were created by subcloning p66Shc, p52Shc, and p66(ΔPTB) into the bacterial expression vector pQE2 (Qiagen) at NdeI and HindIII sites. FAK cDNA was synthesized by reverse transcription-PCR of human umbilical vein endothelial cell (HUVEC) RNA. A 3× hemagglutinin (HA) tag was inserted into pCI-neo (Promega), followed by insertion of full-length FAK into the MluI and NotI sites to create HA-FAK. Overlapping primers were then used to generate deletions of the FERM domain (corresponding to amino acids [aa] 35 to 362), the kinase domain and upstream linker (aa 363 to 686), the proline-rich linker (aa 687 to 916), and the FAT domain (aa 917 to 1052). Short hairpin RNAs (shRNAs) were constructed by annealing target hairpin oligonucleotides, capturing them first in pSUPER.retro.puro (Oligoengine), and then subcloning the hairpin along with the H1 promoter into the EcoRV and XhoI sites of the lentiviral shuttle vector pCCLsin.PPT.hPGK.GFPpre. The target sequences (coding sequence numbering) were p66Shc (nucleotides [nt] 42 to 60), global Shc (nt 483 to 501 in p66 numbering), FAK (nt 1058 to 1076), FAK(2) (nt 149 to 167), GEF-H1 (nt 1724 to 1742), GEF-H1(2) (nt 277 to 295), p115-RhoGEF (nt 661 to 679), YAP (nt 543 to 561), YAP(2) (nt 551 to 569), and SOS1 (nt 1826 to 1844). RNA interference (RNAi)-resistant p66Shc and p52Shc were created by conservative mutagenesis of a subsequence targeted by the global Shc shRNA (5′-TACTTGGTTCGGTAC-3′ [wild type] → 5′-TATCTCGTACGTTAT-3′, where the mutated nucleotides are underlined). Glutathione S-transferase (GST) fusions of Raf and rhotekin receptor binding domain (RBD) sites and p66-GFP were constructed as previously described (22, 23). GST-RhoA(G17A) and GST-RhoA were kind gifts of Keith Burridge. The YAP/TAZ reporter 8× GTIIC-luciferase was from Addgene (plasmid 34615).
Transduction and knockdown.
HUVECs were obtained from BioWhittaker and grown to passage 4 or 5 in EGM-2 (Bio-Whittaker) medium containing 2% fetal calf serum, epidermal growth factor, fibroblast growth factor, vascular endothelial growth factor, insulin-like growth factor, ascorbic acid, hydrocortisone, heparin, and gentamicin. Cells were grown to 60 to 70% confluence prior to plasmid transfection or viral transduction. For transfection, cells were synchronized with 3.5 mM thymidine for 16 h, released from thymidine arrest, and electroporated 14 to 18 h later. For lentiviral transduction, Phoenix-293 cells were cotransfected with the transfer constructs and packaging plasmids pMD2.VSVG, pMDLg/pRRE, and pRSV-REV, and fresh supernatant was used for infection. HUVECs were typically subjected to three rounds of infection (8 h per infection) prior to testing. For adenoviral transduction, cells were exposed to adenovirus at a multiplicity of infection of 50:1 for 4 h and then recovered in medium for 24 h prior to use.
Adhesion complex isolation.
Tosyl-activated paramagnetic microbeads (diameter, 2.8 μm; Dynabeads M-280; Invitrogen) were coupled to antibodies to transferrin receptor (600 ng/10 μl beads; clone 3B82A1; Santa Cruz), bovine serum albumin (BSA; 1%), or soluble fibronectin (20 μg/10 μl beads; Sigma) using 0.1 M Na-phosphate buffer (pH 7.4) at 37°C for 24 h and then washed. The beads were sonicated to disperse them before they were added to cells grown in 35-mm culture dishes (equivalent to 10 μl original stock of beads per dish) and were allowed to settle for 40 min prior to application of a magnetic field. A ceramic permanent magnet was placed on top of the dishes 9 mm from the monolayer to apply a 5.8-pN force to the beads (measured by applying Stokes' law to free beads in suspension). Bound adhesion complexes were then extracted in ice-cold lysis buffer (150 mM NaCl, 20 mM Tris, pH 7.6, 0.1% NP-40, 2 mM MgCl2, aprotinin, leupeptin, pepstatin). Beads from five dishes were scraped and pooled per condition. Beads and associated complexes were isolated using a magnetic separation stand and washed 3 times in lysis buffer. In some experiments, cells were pretreated for 30 min with FAK inhibitor 14 (final concentration, 1 μM; Sigma-Aldrich).
Protein identification.
Lysates from HUVECs grown on five 100-mm dishes were pooled and immunoprecipitated with Flag-PTB. Coomassie-stained bands were isolated and subjected to peptide sequencing using liquid chromatography-tandem mass spectrometry.
Immunoprecipitation and protein pulldown studies.
Immunoprecipitations were performed with anti-Flag (clone M2; Sigma) or anti-Shc (BD). pQE2 plasmids were expressed in Escherichia coli strain M15(pREP4), and purified 6×His fusion proteins were incubated for 1 h at 4°C with supernatant containing the lysates of cells that had been centrifuged at 10,000 × g. Protein complexes were captured on Ni-nitrilotriacetic acid-agarose beads (Qiagen) by further incubation with the beads for 3 h at 4°C. The beads were washed three times, and complexes were extracted and analyzed by immunoblotting. In some experiments, cell lysates were treated with purified recombinant PTP-1B (catalog number P6244; Sigma-Aldrich) at 30 μg/ml in 10 mM Tris (pH 7.5), 50 mM NaCl, 2 mM dithiothreitol (DTT), 1 mM MnCl2 for 15 min at 37°C prior to pulldown.
GTPase and GEF activity.
RhoA, Ras, and Rho GEF activation was assessed using pulldown techniques. RBD-GST, RhoA-GST, RhoA(G17A)-GST, and Raf-GST were expressed in E. coli BL21-RP (Stratagene) and purified on GSH-Sepharose (GE Healthcare). Two-thirds of the lysate in the supernatant obtained after centrifugation at 10,000 × g was used for pulldown with the respective fusions and immunoblotted for RhoA, Ras, p115-RhoGEF, or GEF-H1, while one-third of the lysate was precipitated in ice-cold acetone (1:1) for assessment of total GTPase.
Immunoblotting and immunofluorescence.
For immunoblotting assays, antibodies against RhoA, phosphotyrosine, FAK, vinculin, SOS1, and p115-RhoGEF were from Santa Cruz; antibodies against pan-Ras and BiP were from BD; antibodies against FAK (pY397) and GEF-H1 were from Cell Signaling; antibody against HA was from Thermo Scientific; and antibody against actin was from Chemicon. For immunofluorescence, HUVECs were transduced with lentiviruses and then plated at a subconfluent density on fibronectin-coated glass coverslip chambers. Cells were formalin fixed for 10 min at 25°C, permeabilized in 0.2% Triton X-100 in phosphate-buffered saline (PBS) for 10 min at 25°C, and then washed thrice in PBS with 1% Tween. Cells were blocked with 3% BSA, and anti-YAP (1:100, 1 h at 25°C; catalog number sc-271134; Santa Cruz) was detected with Alexa Fluor 555-conjugated goat anti-mouse immunoglobulin antibodies (1:250, 16 h at 4°C; Molecular Probes).
Microscopy.
HUVECs were plated on fibronectin-coated 35-mm glass bottom culture dishes (Mat Tek catalog number P35G-0-14-C), and live cell microscopy was performed in a heated 37°C chamber, using a Nikon TE2000-U system equipped with a 60× oil immersion lens with a numerical aperture of 1.45. Green excitation and emission filters (492/18 nm and 530/35 nm, respectively) and red excitation and emission filters (572/23 nm and 630/60 nm, respectively) were from Chroma. For the proximity ligation assay (PLA), HUVECs were plated on fibronectin-coated chamber slides, fixed in 3% paraformaldehyde, and permeabilized with 0.2% Triton X-100. The manufacturer's protocol (catalog number DUO92101; Sigma-Aldrich) using rabbit anti-FAK (1:50; catalog number sc-558; Santa Cruz) and mouse anti-Shc (1:100, clone 30/SHC; BD) was followed. For YAP translocation, following lentiviral transduction, the background of the red channel of GFP-positive cells was subtracted, and analysis was performed by quantifying the anti-YAP signal intensity within whole-cell regions of interest as well as nuclear regions of interest, as marked by DAPI (4′,6-diamidino-2-phenylindole) staining. Nuclear translocation was quantified as the YAP nuclear signal/total YAP signal.
Cell death.
DNA fragmentation was assessed using a cell death detection enzyme-linked immunosorbent assay (Roche). Absorbance values were normalized to cell number.
Hydrogel construction and stiffness determination.
Polyacrylamide gels were prepared using 10% acrylamide and a 0.025, 0.05, 0.10, 0.20, 0.30, or 0.40% bisacrylamide cross-linker. Sulfosuccinimidyl 6-(4′-azido-2′-nitrophenylamino) hexanoate (Sulfo-SANPAH) (1 mM) was bound to the gel bed by photoactivation with UV light at a distance of 6 in. for 5 min, followed by two washes in 50 mM HEPES (pH 8.5). Type I collagen (200 μg/ml) was cross-linked overnight at 4°C prior to plating of the cells (24). Gels were saturated with culture medium for 45 min prior to plating. Young's modulus for each gel (n ≥ 3) was determined by dynamic mechanical analysis (DMA) at room temperature. DMA was performed on a Mettler-Toledo DMA 861e instrument. Gels were synthesized in a mold to produce samples for DMA with dimensions of 5.5 mm ± 0.5 mm thick and 12 mm ± 1 mm in diameter. The mode of deformation was quasistatic compression, and strain was limited to a maximum of 0.2% to keep the sample in the linear elastic regime. Each sample was tested at a linear increment of 0.5 μm/min in displacement at a frequency of 1 Hz. The sample's modulus was obtained from the slope value of the stress-strain curve from the DMA quasistatic compression test. Control conditions were performed on plastic treated with 200 μg/ml collagen.
YAP/TAZ transactivation.
HUVECs were cotransfected with 8× GTIIC-luciferase and Renilla luciferase reporters and plated at subconfluent densities. Cells were harvested 24 h later during the active growth phase, and the signals were normalized to the Renilla luciferase signal.
Statistical analysis.
All comparisons were performed using analysis of variance (ANOVA) with Tukey's post hoc intergroup comparison test.
RESULTS
Shc is recruited to adhesion complexes through its PTB domain.
We used fibronectin-bound paramagnetic microbeads to induce adhesion complexes on the dorsal surface of endothelial cells, which are known to be highly mechanosensitive. This technique allows both application of tension and recovery of adhesion complexes for analysis (25). These complexes were characterized by recruitment of vinculin but not BiP, which is found exclusively on the interior of cells (Fig. 1A). All three splice isoforms of Shc (p66Shc, p52Shc, and p46Shc) were associated with adhesion complexes and underwent modest but distinct further recruitment upon application of magnetic tension (Fig. 1A). Beads coated with BSA or antibodies against transferrin receptor, an untargeted plasma membrane protein, did not appreciably bind vinculin or Shc, indicating specific recruitment of endogenous Shc to adhesion complexes under tension (Fig. 1A). In support of the findings of these magnetic separation studies, we directly visualized p66-GFP translocation to fibronectin-coated microbeads, particularly under tension (Fig. 1B). Flag-p66Shc lacking the central PTB domain did not translocate to adhesion complexes upon application of tension, whereas full-length Flag-p66Shc and the isolated PTB domain did (Fig. 1C and D). In addition, ectopic expression of the isolated PTB domain competed with all three endogenous Shc forms for association with adhesion complexes (Fig. 1E), indicating that direct interactions of the Shc PTB domain with an adhesion complex-associated binding partner are necessary and sufficient for tension-induced Shc recruitment.
FIG 1.

Shc is recruited to adhesion complexes through its PTB domain. (A) (Left) Paramagnetic beads covalently bound to antitransferrin receptor antibodies (anti-TfR), BSA, or fibronectin were allowed to settle on HUVECs for 40 min and were then subjected to a magnetic field for the indicated times. After lysis, the beads were recovered and both bead-associated protein complexes (top) and lysates (bottom) were immunoblotted with the indicated antibodies. (Right) The bar graph shows the relative recruitment of Shc isoforms to fibronectin-coated beads (mean ± SEM, n = 6). Rel Abs, relative absorbance. (B) Cells were transfected with p66-GFP and fibronectin-coated beads that were allowed to settle for 40 min (left) and then subjected to a magnetic field for 5 to 10 min (center and right). Fluorescence and differential inference contrast (DIC) images are shown for each field. (C) HUVECs were transduced with Flag-tagged full-length (FL) p66Shc or p66Shc from which the PTB domain was deleted, treated with microbeads coated with either BSA or fibronectin (Fn), and then subjected to a magnetic field for 5 min. Adhesion complexes and lysates were immunoblotted (IB) for Flag. (D) Cells were transduced with Flag or Flag-PTB, treated with fibronectin-bound microbeads, and then exposed to a magnetic field for 5 min. Adhesion complexes and lysates were immunoblotted for Flag. (E) Cells were treated as described in the legend to panel D, and adhesion complexes and lysates were immunoblotted for Shc. The data in panels C to E are representative of those from 3 separate experiments. The numbers to the left of the gels in panels C to E are molecular masses (in kilodaltons).
FAK recruits Shc to adhesion complexes.
We next studied the mechanism by which Shc associates with adhesion complexes. Shc has a number of known binding partners; thus, we used the isolated PTB domain to limit candidate partners. This domain has distinct binding surfaces which bind either phosphoinositide lipids or phosphotyrosine peptides (26). We previously found that specific disruption of the PTB phosphotyrosine binding pocket delocalizes p66Shc from native focal adhesions (22). Therefore, we examined tyrosine-phosphorylated proteins that coimmunoprecipitated with the PTB domain and found a prominent band migrating at 120 to 125 kDa (Fig. 2A). Using liquid chromatography-tandem mass spectrometry, we identified the major component of this band to be FAK. FAK coimmunoprecipitated with Shc, demonstrating the association of the endogenous proteins (Fig. 2B). Further, His-tagged p66Shc and the isolated PTB domain specifically pulled down FAK, whereas p66Shc lacking the PTB domain did not, supporting a direct interaction between proteins (Fig. 2C).
FIG 2.

The Shc PTB domain binds the FAK FERM domain. (A) Cells transduced with either Flag or Flag-PTB were immunoprecipitated with anti-Flag and immunoblotted with antiphosphotyrosine (pTyr) or anti-Flag. HC, Ig heavy chain, LC, Ig light chain. (B) Cell lysates were immunoprecipitated with protein G-Sepharose (PGS), control Ig, or anti-Shc and immunoblotted for FAK and Shc. The bottom panel shows the results obtained with the lysate after immunoprecipitation, showing substantial capture of Shc. The results are representative of those from 3 immunoprecipitation experiments. Shc (IP), Shc in the immunoprecipitate; Irrel, irrelevant antibody. (C) Bacterially expressed His fusions of full-length p66Shc, p66Shc from which the PTB domain was deleted, or the isolated PTB domain were used to pull down endogenous FAK from cell lysates. The relative expression of His-Shc baits is 1:2.1:0.7. The results are representative of those from 2 experiments. (D) (Top) The cartoons depict serial deletions of the HA-tagged FAK transfected into HUVECs. His-tagged p66Shc was used to pull down cell lysates. Input and pulldown fractions for FAK and His-p66 bait are shown. The relative expression of the HA-FAK proteins is 1:1.1:1.1:1.4:1.6. (E) Cells transfected with full-length (FL) or FERM domain-only FAK-HA fusions were pulled down by His-p66. Pretreatment of lysates with PTP-1B is marked. Input and pulldown fractions were blotted for HA and Src. (F) Lysates of untransfected cells were used for pulldown by His-tagged wild-type p66 or p66 R285Q. Input and pulldown fractions for endogenous FAK are shown. (G) Fibronectin-coated beads were added to untreated or FAK inhibitor 14-treated (1 μM for 30 min) cells, and tension was applied. Adhesion complex and lysate fractions were blotted for Shc and FAK. The results in panels E to G are representative of those from 2 experiments. The numbers to the left and right of the gels in panels A to G are molecular masses (in kilodaltons). (H) HUVECs were plated on fibronectin, and PLA was performed. Primary antibodies (Ab) for FAK and Shc, Shc only, or FAK only were omitted for 3 negative controls, as shown. Bars, 20 μm.
We next expressed HA-tagged deletion mutants lacking each of the four distinct regions of FAK (the N-terminal FERM domain, the central catalytic domain, the C-terminal FAT domain, and an unstructured proline-rich domain between the catalytic and FAT domains). Deletion of the FERM domain but not the other domains abrogated association with p66Shc (Fig. 2D). This domain docks with the catalytic domain to inhibit kinase activity but is known to control FAK signaling through interaction with other proteins (27). Indeed, His-p66 pulled down the isolated FERM domain, demonstrating that the FERM domain is necessary and sufficient for FAK-Shc interactions (Fig. 2E). Of note, Src was also pulled down by His-p66. However, Src does not appear to be an adapter between Shc and FAK, as it was also pulled down with the isolated FERM domain, which lacks Src binding sites. To further demonstrate the binding of the Shc PTB domain to a phosphotyrosine motif, lysates were also treated with the tyrosine phosphatase PTP-1B, which abrogated p66Shc binding to both full-length FAK and the isolated FERM domain (Fig. 2E). In addition, we introduced an Arg285Gln mutation into p66Shc, which specifically disrupts the phosphotyrosine binding pocket of the PTB surface by preventing Arg285 coordination with the anionic phosphate group. This mutation completely abrogated pulldown of endogenous FAK (Fig. 2F), confirming the requirement of the Shc PTB domain in binding a FAK phosphotyrosine motif. As FAK is a tyrosine kinase capable of autophosphorylation, we also pretreated cells with FAK inhibitor 14. However, inhibition of FAK had no effect on the recruitment of Shc, indicating that this recruitment is independent of FAK activity (Fig. 2G). Finally, to further demonstrate direct FAK-Shc interactions, we used a proximity ligation assay (PLA) to visualize endogenous protein interactions in cells plated on fibronectin. Strong PLA signals were demonstrated in punctate ventral regions of the cell, suggesting the association of endogenous FAK and Shc proteins at native adhesion sites (Fig. 2H).
As expected, FAK was recruited to fibronectin-coated beads, and phosphorylation of Y397 increased upon application of tension (Fig. 3A). To determine the sequence of recruitment, we used two different shRNAs to knock down FAK. The knockdown of FAK blocked the basal and tension-induced association of Shc with adhesion complexes (Fig. 3B). In contrast, the knockdown of all Shc isoforms had no effect on the appearance of FAK on adhesion complexes, and expression of the isolated PTB domain also had no effect on FAK translocation (Fig. 3C and D). Taken together, the findings of these studies support the model whereby the docking of FAK on integrin-based structures precedes and is required for Shc recruitment.
FIG 3.

FAK recruits Shc to adhesion complexes. (A) (Left) Microbeads coupled to anti-transferrin receptor antibodies, BSA, or fibronectin were allowed to settle on cells for 40 min, and a magnetic field was applied for the indicated times. Adhesion complexes and lysates were immunoblotted for FAK and FAK (pY397). (Right) The bar graphs show the means ± SEMs from 6 (for FAK) or 5 [for FAK (pY397)] determinations. P was <0.05 at 5 min (FAK) and 10 min [FAK and FAK (pY397)]. (B) Cells were transduced with control or two different FAK shRNAs [FAK and FAK(2)], and fibronectin-coated microbeads were subjected to magnetic tension for 5 min and assessed for recruitment of Shc to adhesion complexes. FAK was knocked down ∼92% [FAK] and ∼70% [FAK(2)]. (C) Cells were transduced with control or Shc shRNA and treated as described in the legend to panel B to assess the recruitment of FAK to adhesion complexes. Shc was knocked down ∼93%. (D) Cells were transduced with either the empty Flag construct or Flag-PTB and assessed for FAK recruitment to adhesion complexes. The results in panels B to D are representative of those from 2 to 4 experiments. The numbers to the left of the gels are molecular masses (in kilodaltons).
The FAK/p66Shc complex mediates tension-induced RhoA activation.
We previously demonstrated that p66Shc mediates RhoA activation in adherent cells (23). RhoA activation following matrix adhesion is associated with matrix ligation and spreading, as well as the initiation of tension against the matrix and the cellular response to its stiffness. To distinguish the role of p66Shc in responding to integrin ligation versus tension, we assessed RhoA activity in cells before and during the application of external force to fibronectin-coated beads. RhoA activation was observed within 3 min of tension and peaked at 5 min, whereas knockdown of p66Shc completely prevented force-induced RhoA activation (Fig. 4A). RhoA activation by tension was also blocked by expression of the isolated PTB domain, indicating that docking of p66Shc onto adhesion complexes was required for RhoA activity (Fig. 4B). In support of this model, knockdown of its binding partner, FAK, with two different shRNAs also blocked tension-induced RhoA activation (Fig. 4C). To demonstrate that this effect was specific to p66Shc, we knocked down all Shc isoforms by targeting the common PTB domain and expressed an RNAi-resistant p66Shc cDNA, essentially abrogating only p52Shc and p46Shc expression (Fig. 4D). Tension-induced RhoA activation was restored by reexpression of p66Shc in the absence of p52Shc and p46Shc (Fig. 4E), indicating that mechanical tension specifically requires p66Shc to initiate RhoA signaling and validating the specificity of the knockdown approach.
FIG 4.

p66Shc mediates RhoA activation. (A) (Left) Cells expressing control (cont) or p66Shc shRNA were exposed to fibronectin-coated beads, and magnetic force was applied for the indicated times. RhoA activity is shown. (Right) Specificity of p66Shc knockdown on Shc expression. The p66Shc knockdown efficiency was ∼93%. (B) Cells transduced with Flag alone or Flag-PTB were exposed to fibronectin-coated beads, magnetic force was applied for 5 min, and then RhoA activity was assessed. (C) Cells expressing control or two different FAK shRNAs [FAK and FAK(2)] were exposed to fibronectin-coated beads, magnetic force was applied for 5 min, and then RhoA activity was assessed. FAK knockdown efficiencies were ∼70 to 90%. (D) Cells were transduced with an RNAi-resistant Flag-p66Shc (p66 mutant [p66mut]) and/or shRNA targeting all Shc isoforms. (E) The effect of p52Shc/p46Shc knockdown on RhoA activation is shown. Magnetic force was applied for 5 min, as in the assays whose results are presented in panels A to D. The results in panels A to E are representative of those from 2 or 3 separate experiments. The numbers to the left of the gels are molecular masses (in kilodaltons).
Tension induces GEF-H1 and p115-RhoGEF translocation to the FAK/p66Shc complex.
We reasoned that mechanical force triggers RhoA pathways through the recruitment and activation of specific Rho GEFs to the FAK/p66Shc complex. To narrow the field from over 80 human Rho GEFs, we screened the expression profile of unstimulated HUVECs to determine the expression of Rho GEFs and found that these cells express significant levels of AHRGEF1 (p115-RhoGEF), AHRGEF2 (GEF-H1), AHRGEF11 (PDZ-RhoGEF), and AHRGEF23 (TRIO) (data not shown). Both PDZ-RhoGEF and TRIO have been shown to interact with FAK and localize to focal adhesions (28, 29). However, only low levels of the PDZ-RhoGEF protein were detected by immunoblotting, and while TRIO was detected in cell lysates, it did not translocate to adhesion complexes at baseline or under tension (not shown). In contrast, both p115-RhoGEF and GEF-H1 were specifically recruited to fibronectin-bound microspheres upon application of tension, whereas no association was seen with BSA-bound microspheres (Fig. 5A). In addition, knockdown of either FAK or p66Shc completely blocked recruitment of both p115-RhoGEF and GEF-H1, consistent with a requirement for the FAK/p66Shc complex in Rho GEF trafficking (Fig. 5B to E). Further, His-tagged p66Shc pulled down both p115-RhoGEF and GEF-H1, as well as FAK, suggesting that the FAK/p66Shc complex may directly recruit these GEFs (Fig. 5F). To demonstrate specificity, we used His-tagged p52Shc, which pulled down FAK but did not pull down either p115-RhoGEF or GEF-H1 (Fig. 5F). Finally, removal of FAK from the pulldown complex by use of p66Shc from which PTB was deleted or by FAK knockdown also prevented the recovery of p115-RhoGEF and GEF-H1 (Fig. 5F and G). Thus, FAK/p66Shc but not FAK/p52Shc is capable of recruiting GEFs active for RhoA.
FIG 5.

p66Shc cooperates with FAK to recruit Rho GEFs. (A) HUVECs were exposed to microbeads bound to either BSA or fibronectin (Fn), and a magnetic field was applied for 5 min. The recruitment of p115-RhoGEF and GEF-H1 to adhesion complexes is shown. (B to E) Cells were transduced with control shRNA or shRNA directed against FAK (B, C) or p66Shc (D, E). Magnetic force was applied to fibronectin-bound microbeads for 5 min, and adhesion complexes were isolated to assess recruitment of p115-RhoGEF (B, D) or GEF-H1 (C, E). (F) Solid-phase His fusions of full-length p66Shc, p52Shc, or p66Shc from which PTB was deleted were used to pull down cell lysates. The captured proteins are indicated. (G) His-p66Shc was used to pull down lysates in cells transduced with control or FAK shRNA. The results in panels A to G are representative of those from 2 to 4 separate experiments. The numbers to the left of the gels are molecular masses (in kilodaltons).
The FAK/p66Shc complex activates RhoA through GEF-H1 and p115-RhoGEF.
To determine the activation state of GEFs, we employed a pulldown technique which takes advantage of the high affinity of active Rho GEFs for the essentially nucleotide-free mutant RhoA(G17A) (30). The magnetic force applied to fibronectin-coated beads for 5 min caused the activation of both p115-RhoGEF and GEF-H1 in whole-cell lysates (Fig. 6A and B). Wild-type RhoA was used as a negative control for the pulldown assays. Knockdown of either p66Shc or FAK completely blocked activation of both p115-RhoGEF and GEF-H1 (Fig. 6C to F). In addition, knockdown of either p115-RhoGEF or GEF-H1 prevented the tension-induced activation of RhoA (Fig. 6G and H). In total, these data demonstrate that following application of force to adhesion complexes, a FAK/p66Shc complex is required for both the recruitment and activation of p115-RhoGEF and GEF-H1, thus activating RhoA in response to tension.
FIG 6.

Tension activates Rho GEFs through p66Shc and FAK. For the assays who results are presented in panels A to G, cells were exposed to fibronectin-coated microbeads and a magnetic field was applied for 5 min. (A, B) Total cellular GEF activity of p115-RhoGEF (A) and GEF-H1 (B) obtained by pulldown with RhoA(G17A) is shown. GST-RhoA (wild type) was included as a negative control. (C to F) GEF activity for p115-RhoGEF (C, E) and GEF-H1 (D, F) is shown following knockdown of p66Shc (C, D) or FAK (E, F). (G) (Left) RhoA activation in response to tension is shown in cells transduced with control (con), GEF-H1, or p115-RhoGEF shRNA. (Center and right) Protein levels after knockdown. The knockdown efficiency was ∼69% (GEF-H1) and 92% (p115). (H) Cells were transduced with separate shRNA for GEF-H1, and RhoA activity was assessed. The knockdown efficiency was ∼79% [GEF-H1(2)]. The results in panels A to H are representative of those from 2 to 4 separate experiments. The numbers to the left of the gels are molecular masses (in kilodaltons).
The FAK/p52Shc complex activates Ras through SOS1.
p52Shc contains an intact PTB domain, and indeed, His-p52Shc is capable of binding to FAK (Fig. 5F). However, p52Shc neither recruits Rho GEFs nor supports RhoA activation (Fig. 4A and E and 5F). When it is associated with growth factor receptors, p52Shc is known to facilitate Ras activation; thus, we asked whether p52Shc also supports Ras activation in response to mechanical force. Similarly to RhoA, Ras was activated within 3 min of the application of tension to fibronectin-coated microbeads, peaking at 5 min and decreasing by 10 min (Fig. 7A). Knockdown of p66Shc had no effect on Ras activity; however, global Shc knockdown with reexpression of p66Shc (depleting p52Shc and p46Shc) effectively prevented Ras activation in response to tension (Fig. 7B). Conversely, global Shc knockdown with reexpression of p52Shc (Fig. 7C) restored tension-induced Ras activity (Fig. 7D). Thus, p52Shc is necessary and sufficient for Ras activation but not RhoA activation. His-tagged p52Shc but not p66Shc or p52Shc lacking the PTB domain pulled down both FAK and the Ras GEF SOS1 (Fig. 7E), and knockdown of SOS1 completely blocked tension-induced Ras activation (Fig. 7F). The different Shc isoforms therefore bifurcate mechanotransduction through exclusive FAK/p66Shc → RhoA and FAK/p52Shc → Ras pathways through differential GEF recruitment.
FIG 7.

p52Shc cooperates with FAK to recruit SOS. (A) Cells transduced with control or p66Shc shRNA were exposed to fibronectin-coated microbeads, and a magnetic field was applied for the indicated times. Ras activity is shown. (B) Cells expressing RNAi-resistant Flag-p66Shc (p66 mutant [p66mut]) and/or shRNA targeting all Shc isoforms were subjected to magnetic tension, as described in the legend to panel A. Ras activity is shown. (C) Cells were transduced with RNAi-resistant p52Shc (p52 mutant [p52mut]) and/or global Shc shRNA. (D) Cells expressing the p52 mutant with Shc shRNA or controls were subjected to magnetic tension, and Ras activity was assessed. (E) 6×His fusions of full-length p66Shc, p66Shc from which PTB was deleted, or p52Shc were used to pull down lysates, and the recovered proteins are indicated. (F) The effect of SOS1 knockdown on tension-induced Ras activation is shown. SOS1 knockdown efficiency was ∼89%. The results in panels A to F are representative of those from 2 to 4 separate experiments. The numbers to the left of the gels are molecular masses (in kilodaltons).
p115-RhoGEF and GEF-H1 mediate proliferation and death responses to tension.
In prior studies, we demonstrated that p66Shc, working through RhoA, is required to initiate anoikis upon detachment of endothelial or epithelial cells from solid environments and that knockdown of either p66Shc or RhoA, loss of focal adhesion targeting of p66Shc, or inhibition of actinomyosin tension prevents anoikis (22, 23). These studies suggest that loss of anchorage is sensed as load failure of the tension applied against candidate anchorage points. We found that knockdown of either p115-RhoGEF or GEF-H1 also significantly decreased cell death in endothelial cells forced to float for 16 h (Fig. 8A), suggesting that the same FAK/p66Shc complex which recruits Rho GEFs in response to tension also relays mechanical signals reporting detachment.
FIG 8.

p115-RhoGEF and GEF-H1 mediate anchorage signals. (A) HUVECs were transduced with the indicated shRNAs, and cell death was measured after 16 h of adhesion or floating conditions. Significant differences between conditions are shown at the top of the bar graphs. Means ± SEMs from 4 determinations are shown. Knockdown efficiencies for all GEFs were between 70 and 87%. Abs/cell, absorbance per cell. (B) Cell counts after 48 h on hydrogels of the indicated stiffness (Young's modulus [E]), showing a progressive effect on proliferation. (C) Phase-contrast images of HUVECs plated on collagen-coated hydrogels showing the morphological response to changes in matrix bed stiffness. Bars, 50 μm. (D) Cells expressing shRNA against the control (con), p66Shc, or FAK were plated on hydrogels (approximately 17.8 kPa) or plastic, each of which was bound to collagen, and cell counts were determined after 48 h. (E, F) Cell counts under the conditions described in the legend to panel D, except that cells were transduced with shRNA against the control, p115-RhoGEF, GEF-H1, or SOS1. Significant differences are shown above the bar graphs. The results in panels D to F are means ± SEMs from 4 determinations. Knockdown efficiencies were between 70 and 90% for all GEFs. NS, not significant.
To further test whether the FAK/p66Shc/Rho GEF complex acts as a substrate mechanosensor, we studied the proliferative response of endothelial cells to matrix stiffening. Blood vessel walls are known to have high stiffness, with human carotid arteries estimated to have Young's moduli of 700 to 900 kPa in vivo (31, 32). Endothelial cells plated on collagen-coated polyacrylamide hydrogels demonstrated progressive flattening and proliferation rates on matrix beds with Young's moduli of between 13.6 and 95.1 kPa in addition to essentially nondeformable plastic (Fig. 8B and C). At both extremes of stiffness, the morphologies were consistent with those in prior studies on a variety of different cell types, which demonstrated flattening and spreading on stiff environments and rounding with a loss of cell-cell interactions on very soft matrices. On intermediate-stiffness gels, the endothelial cells had features of both soft and firm morphologies. At 25.4 kPa, for instance, limited cell-cell contacts formed within multicellular islands and the cells did not have the ability to form confluent monolayers. Within multicellular aggregates, the cells did not flatten on soft gels, leading to the appearance of irregular islands of cells molded around each other. In addition, the proliferative response to being plated on collagen-coated firm plastic dishes was substantially inhibited by knockdown of either p66Shc or FAK (Fig. 8D). Proliferation on plastic was also decreased by knockdown of either p115-RhoGEF or GEF-H1 (Fig. 8E and F) but not SOS1, supporting the involvement of the tension-responsive FAK/p66Shc complex in sensing matrix stiffness. In the absence of a functional complex, proliferation defaulted back to rates consistent with that on softer gels.
The proliferative response to increases in matrix stiffness is known to be mediated by the YAP/TAZ transcription factors, which require RhoA to initiate their activation and nuclear translocation (33). As with other cell types, HUVECs displayed stronger YAP nuclear localization on plastic than on soft gels (Fig. 9A). Further, YAP knockdown by two different shRNAs decreased the proliferation of cells plated on firm plastic, consistent with the importance of YAP in the proliferative response to matrix stiffness (Fig. 9B). To gain further evidence that the FAK/p66Shc complex acts as a relevant mechanosensor, we tracked YAP translocation in endothelial cells plated on a rigid surface (matrix-coated glass coverslips) following infection with lentiviruses expressing both shRNAs and GFP. Because YAP was found to different extents in both the cytosol and the nucleus, we estimated the percentage of nuclear YAP per cell under different conditions. Using GFP to mark shRNA-transduced cells, we found that control shRNA had no effect on nuclear YAP translocation (Fig. 9C). However, knockdown of p66Shc or FAK significantly reduced the nuclear translocation of YAP, an effect seen only in GFP-positive shRNA-expressing cells (Fig. 9C). In addition, YAP/TAZ transactivation in response to increasing substrate stiffness, as assessed by use of a luciferase reporter, was significantly decreased by knockdown of p66Shc or its associated GEFs, p115-RhoGEF and GEF-H1 (Fig. 9D and E). In contrast, knockdown of p52Shc/p46Shc (global Shc knockdown with reexpression of p66Shc) or knockdown of the p52Shc-associated SOS1 had no effect on YAP/TAZ transactivation on stiff surfaces (Fig. 9E and F). These studies confirm that p66Shc is a mechanoresponsive protein upstream of RhoA and the YAP/TAZ transcription factors (Fig. 10).
FIG 9.

p66Shc acts upstream of YAP/TAZ transactivation. (A) (Left) HUVECs were plated on either plastic or hydrogels (∼17.8 kPa) and immunostained for endogenous YAP. Bars, 20 μm. (Right) The proportion of nuclear YAP to total cellular YAP (mean ± SEM for 30 cells) was quantified. (B) (Top) Cells were transduced with either control shRNA (Con) or two different shRNAs against YAP [YAP and YAP(2)] and plated on plastic at equal densities. Cell counts were performed 48 h later (data are means ± SEMs from 4 experiments). (Bottom) Efficiency of knockdown [YAP, ∼85%; YAP(2), ∼90%]. (C) (Left) HUVECs were transduced with shRNA as indicated and stained for YAP (red, top) and DAPI (blue, bottom). GFP expression (green, bottom) marks lentivirus-transduced cells expressing shRNAs. Arrows, GFP-expressing cells. Bars, 20 μm. (Right) YAP nuclear signal intensity of GFP-negative and GFP-positive cells, following transduction with shRNAs against the control, p66Shc, or FAK. Data are means ± SEMs for 18 to 27 cells. (D) YAP/TAZ transactivation was measured by use of a luciferase reporter in cells expressing control or p66Shc shRNA plated on plastic or hydrogels of the indicated elastic moduli. Data are means ± SEMs from 4 determinations. (E) YAP/TAZ transactivation in cells expressing the indicated shRNAs and plated on plastic or hydrogels of the indicated elastic moduli. Data are means ± SEMs from 3 determinations. (F) YAP/TAZ activation of cells expressing control shRNA or pan-Shc shRNA plus RNAi-resistant p66Shc (p66 mutant [p66mut]) plated on soft gels or plastic. Data are means ± SEMs from 3 to 6 determinations. Significant differences are shown above the bar graphs. NS, not significant. Knockdown efficiencies for p66, p52, and FAK were >90%, and those for GEFs were 70 to 90%.
FIG 10.
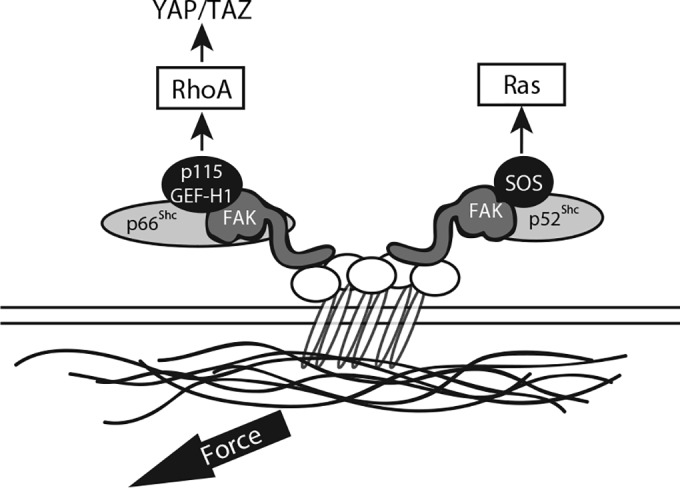
Proposed signaling pathways. The schematic shows the bifurcation of RhoA and Ras activation in response to tension. p66Shc and p52Shc each associate with FAK but recruit distinct GEFs to transduce mechanical signals to RhoA or Ras, respectively.
DISCUSSION
RhoA plays a dominant role in the cellular response to mechanical forces. Here, we identify p66Shc to be critical for RhoA activation following application of tension to adhesion complexes. Prior studies suggest that the Shc proteins in general may respond to mechanical stimuli. Application of shear stress to endothelial cells, for instance, induces the association of all three Shc isoforms with β1 and αvβ3 integrins, and abnormal shear patterns in mouse vasculature stimulate Shc translocation to junctional mechanosensory complexes (34, 35). Conditional deletion of all Shc isoforms in cardiomyocytes leads to impaired mechanical coupling, and knock-in mutants of Shc prevent the formation of muscle spindles, mechanosensory organs within skeletal muscle (36, 37). In this study, we link Shc with tension-induced RhoA activity, providing a potential mechanism for such observations and for multiple cellular processes related to RhoA-linked force transduction.
Our data specifically implicate the p66Shc isoform in the activation of RhoA through the selective recruitment of p115-RhoGEF and GEF-H1. The existence of over 80 human Rho GEFs reflects the importance of accurately coupling Rho GTPases to a large number of upstream events in different biological contexts and speaks to the importance of identifying specific GEFs responsible for RhoA activation downstream of mechanical force. p115-RhoGEF and GEF-H1 in particular have been previously linked to mechanical signals. Adhesion of fibroblasts to solid-phase fibronectin, for instance, activates RhoA through p115-RhoGEF and LARG, leading to stress fiber and focal adhesion formation (38). The application of tension to matrix-coated microbeads similarly initiates RhoA-dependent cytoskeletal reinforcement through GEF-H1 and LARG (25). Finally, while the manuscript was under review, a further study identified GEF-H1 and p115-RhoGEF to be mediators of tension downstream of the adhesion protein JAM-A (39).
We further demonstrate that recruitment and activation of p115-RhoGEF and GEF-H1 require cooperative interactions of p66Shc with FAK. FAK is well-known as a mechanosensitive protein, and FAK autophosphorylation has been shown to promote transient coimmunoprecipitation with Shc (40). However, FAK and Shc are thought to mediate adhesion-dependent ERK activation as well as motility through independent pathways, perhaps reflecting the existence of distinct as well as shared functions for FAK and Shc (41, 42). The exact nature of Rho GEF binding to the FAK/p66Shc complex is unclear at this point, though FAK is known to bind multiple GEFs as well as GTPase-activating proteins (GAPs) (43). It is unlikely that p66Shc directly binds these GEFs, as p66Shc lacking only its PTB domain does not pull down Rho GEFs, and p52Shc, which contains an intact PTB domain, instead associates with SOS1. Thus, it is possible that p66Shc and p52Shc differentially modify the binding properties of FAK, leading to the recruitment of different GEFs or GEF-specific adapters.
The FAK/p66Shc/Rho GEF complex is also required to generate RhoA signals upstream of the YAP/TAZ transcription factors, demonstrating its broad functional relevance. YAP and TAZ relay proliferative, death, and differentiation signals that arise from mechanosensory testing of the cell's environment, a pathway which requires RhoA-dependent tension (33). The loss of TAZ, for example, leads to disorders of mechanosensation, such as polycystic kidney disease and emphysema in mice and the loss of anchorage-independent growth in transformed cells, whereas TAZ overexpression blocks anoikis (44–46). Interestingly, we found that disruption of the FAK/p66Shc/Rho GEF complex impairs both proliferative signals from anchorage to a rigid environment and death signals following detachment from a rigid environment. While the former is linked to increases and the latter is linked to decreases in YAP/TAZ activity (33, 47), both require a RhoA-dependent tension test of the environment, presumably initiated by the same signaling complex. The importance of p66Shc in such environmental sampling may also be reflected in its epigenetic silencing, which disables the tension test in anchorage-independent normal blood cells and abnormal metastatic cancer cells (23, 48). We speculate that the differential regulation of p66Shc expression in different cell types may allow titration of the RhoA response in order to modulate lineage-specific interactions with the cellular environment. Taken together, our data suggest that p66Shc may play a key role in mechanosensation, lending physical context to RhoA signals.
Finally, we observed that p52Shc and p66Shc simultaneously respond to applied tension but do so through recruitment of distinct classes of GEFs (Fig. 10). p66Shc recruits p115-RhoGEF and GEF-H1, which are specific for certain Rho subfamily GTPases (RhoA/B/C but not Ras, Rac, or Cdc42), whereas p52Shc recruits SOS1, which is Ras and Rac specific (49–51). This observation highlights a novel cooperation between the two principal Shc isoforms and suggests an evolutionarily conserved function—the reporting of environmental conditions to GTPase-linked pathways—that may explain their common genetic ancestry. Our data support this general model and provide insight into the complementary roles that p66Shc and p52Shc play in sorting mechanical signals to different GTPases.
ACKNOWLEDGMENTS
We thank Manasvini Ammanamanchi and Melanie Maurer for their assistance in mechanical testing of the hydrogels.
We acknowledge the University of Texas Southwestern for institutional support for the Proteomics Core Facility.
REFERENCES
- 1.Matthews BD, Overby DR, Mannix R, Ingber DE. 2006. Cellular adaptation to mechanical stress: role of integrins, Rho, cytoskeletal tension and mechanosensitive ion channels. J Cell Sci 119:508–518. doi: 10.1242/jcs.02760. [DOI] [PubMed] [Google Scholar]
- 2.McBeath R, Pirone DM, Nelson CM, Bhadriraju K, Chen CS. 2004. Cell shape, cytoskeletal tension, and RhoA regulate stem cell lineage commitment. Dev Cell 6:483–495. doi: 10.1016/S1534-5807(04)00075-9. [DOI] [PubMed] [Google Scholar]
- 3.Ewald AJ, Brenot A, Duong M, Chan BS, Werb Z. 2008. Collective epithelial migration and cell rearrangements drive mammary branching morphogenesis. Dev Cell 14:570–581. doi: 10.1016/j.devcel.2008.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Farge E. 2003. Mechanical induction of Twist in the Drosophila foregut/stomodeal primordium. Curr Biol 13:1365–1377. doi: 10.1016/S0960-9822(03)00576-1. [DOI] [PubMed] [Google Scholar]
- 5.Harden N, Ricos M, Ong YM, Chia W, Lim L. 1999. Participation of small GTPases in dorsal closure of the Drosophila embryo: distinct roles for Rho subfamily proteins in epithelial morphogenesis. J Cell Sci 112(Pt 3):273–284. [DOI] [PubMed] [Google Scholar]
- 6.Moore KA, Polte T, Huang S, Shi B, Alsberg E, Sunday ME, Ingber DE. 2005. Control of basement membrane remodeling and epithelial branching morphogenesis in embryonic lung by Rho and cytoskeletal tension. Dev Dyn 232:268–281. doi: 10.1002/dvdy.20237. [DOI] [PubMed] [Google Scholar]
- 7.Nakaya Y, Sukowati EW, Wu Y, Sheng G. 2008. RhoA and microtubule dynamics control cell-basement membrane interaction in EMT during gastrulation. Nat Cell Biol 10:765–775. doi: 10.1038/ncb1739. [DOI] [PubMed] [Google Scholar]
- 8.Zhao XH, Laschinger C, Arora P, Szaszi K, Kapus A, McCulloch CA. 2007. Force activates smooth muscle alpha-actin promoter activity through the Rho signaling pathway. J Cell Sci 120:1801–1809. doi: 10.1242/jcs.001586. [DOI] [PubMed] [Google Scholar]
- 9.Pelicci G, Lanfrancone L, Grignani F, McGlade J, Cavallo F, Forni G, Nicoletti I, Pawson T, Pelicci PG. 1992. A novel transforming protein (SHC) with an SH2 domain is implicated in mitogenic signal transduction. Cell 70:93–104. doi: 10.1016/0092-8674(92)90536-L. [DOI] [PubMed] [Google Scholar]
- 10.Rozakis-Adcock M, McGlade J, Mbamalu G, Pelicci G, Daly R, Li W, Batzer A, Thomas S, Brugge J, Pelicci PG, Schlessinger J, Pawson T. 1992. Association of the Shc and Grb2/Sem5 SH2-containing proteins is implicated in activation of the Ras pathway by tyrosine kinases. Nature 360:689–692. doi: 10.1038/360689a0. [DOI] [PubMed] [Google Scholar]
- 11.Nicholson PR, Empereur S, Glover HR, Dilworth SM. 2001. ShcA tyrosine phosphorylation sites can replace ShcA binding in signalling by middle T-antigen. EMBO J 20:6337–6346. doi: 10.1093/emboj/20.22.6337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ong SH, Dilworth S, Hauck-Schmalenberger I, Pawson T, Kiefer F. 2001. ShcA and Grb2 mediate polyoma middle T antigen-induced endothelial transformation and Gab1 tyrosine phosphorylation. EMBO J 20:6327–6336. doi: 10.1093/emboj/20.22.6327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wary KK, Mainiero F, Isakoff SJ, Marcantonio EE, Giancotti FG. 1996. The adaptor protein Shc couples a class of integrins to the control of cell cycle progression. Cell 87:733–743. doi: 10.1016/S0092-8674(00)81392-6. [DOI] [PubMed] [Google Scholar]
- 14.Wary KK, Mariotti A, Zurzolo C, Giancotti FG. 1998. A requirement for caveolin-1 and associated kinase Fyn in integrin signaling and anchorage-dependent cell growth. Cell 94:625–634. doi: 10.1016/S0092-8674(00)81604-9. [DOI] [PubMed] [Google Scholar]
- 15.Cowan KJ, Law DA, Phillips DR. 2000. Identification of Shc as the primary protein binding to the tyrosine-phosphorylated beta 3 subunit of alpha IIbbeta 3 during outside-in integrin platelet signaling. J Biol Chem 275:36423–36429. doi: 10.1074/jbc.M004068200. [DOI] [PubMed] [Google Scholar]
- 16.Deshmukh L, Gorbatyuk V, Vinogradova O. 2010. Integrin {beta}3 phosphorylation dictates its complex with the Shc phosphotyrosine-binding (PTB) domain. J Biol Chem 285:34875–34884. doi: 10.1074/jbc.M110.159087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Migliaccio E, Mele S, Salcini AE, Pelicci G, Lai KM, Superti-Furga G, Pawson T, Di Fiore PP, Lanfrancone L, Pelicci PG. 1997. Opposite effects of the p52shc/p46shc and p66shc splicing isoforms on the EGF receptor-MAP kinase-fos signalling pathway. EMBO J 16:706–716. doi: 10.1093/emboj/16.4.706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Xi G, Shen X, Clemmons DR. 2010. p66shc inhibits insulin-like growth factor-I signaling via direct binding to Src through its polyproline and Src homology 2 domains, resulting in impairment of Src kinase activation. J Biol Chem 285:6937–6951. doi: 10.1074/jbc.M109.069872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Natalicchio A, Laviola L, De Tullio C, Renna LA, Montrone C, Perrini S, Valenti G, Procino G, Svelto M, Giorgino F. 2004. Role of the p66Shc isoform in insulin-like growth factor I receptor signaling through MEK/Erk and regulation of actin cytoskeleton in rat myoblasts. J Biol Chem 279:43900–43909. doi: 10.1074/jbc.M403936200. [DOI] [PubMed] [Google Scholar]
- 20.Lee MK, Smith SM, Banerjee MM, Li C, Minoo P, Volpe MV, Nielsen HC. 2014. The p66Shc adapter protein regulates the morphogenesis and epithelial maturation of fetal mouse lungs. Am J Physiol Lung Cell Mol Physiol 306:L316–L325. doi: 10.1152/ajplung.00062.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Favetta LA, Madan P, Mastromonaco GF, St John EJ, King WA, Betts DH. 2007. The oxidative stress adaptor p66Shc is required for permanent embryo arrest in vitro. BMC Dev Biol 7:132. doi: 10.1186/1471-213X-7-132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ma Z, Myers DP, Wu RF, Nwariaku FE, Terada LS. 2007. p66Shc mediates anoikis through RhoA. J Cell Biol 179:23–31. doi: 10.1083/jcb.200706097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ma Z, Liu Z, Wu RF, Terada LS. 2010. p66Shc restrains Ras hyperactivation and suppresses metastatic behavior. Oncogene 29:5559–5567. doi: 10.1038/onc.2010.326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wang YL, Pelham RJ Jr. 1998. Preparation of a flexible, porous polyacrylamide substrate for mechanical studies of cultured cells. Methods Enzymol 298:489–496. doi: 10.1016/S0076-6879(98)98041-7. [DOI] [PubMed] [Google Scholar]
- 25.Guilluy C, Swaminathan V, Garcia-Mata R, O'Brien ET, Superfine R, Burridge K. 2011. The Rho GEFs LARG and GEF-H1 regulate the mechanical response to force on integrins. Nat Cell Biol 13:724–729. doi: 10.1038/ncb2254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Zhou MM, Ravichandran KS, Olejniczak EF, Petros AM, Meadows RP, Sattler M, Harlan JE, Wade WS, Burakoff SJ, Fesik SW. 1995. Structure and ligand recognition of the phosphotyrosine binding domain of Shc. Nature 378:584–592. doi: 10.1038/378584a0. [DOI] [PubMed] [Google Scholar]
- 27.Lietha D, Cai X, Ceccarelli DF, Li Y, Schaller MD, Eck MJ. 2007. Structural basis for the autoinhibition of focal adhesion kinase. Cell 129:1177–1187. doi: 10.1016/j.cell.2007.05.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Iwanicki MP, Vomastek T, Tilghman RW, Martin KH, Banerjee J, Wedegaertner PB, Parsons JT. 2008. FAK, PDZ-RhoGEF and ROCKII cooperate to regulate adhesion movement and trailing-edge retraction in fibroblasts. J Cell Sci 121:895–905. doi: 10.1242/jcs.020941. [DOI] [PubMed] [Google Scholar]
- 29.Medley QG, Buchbinder EG, Tachibana K, Ngo H, Serra-Pages C, Streuli M. 2003. Signaling between focal adhesion kinase and trio. J Biol Chem 278:13265–13270. doi: 10.1074/jbc.M300277200. [DOI] [PubMed] [Google Scholar]
- 30.Garcia-Mata R, Wennerberg K, Arthur WT, Noren NK, Ellerbroek SM, Burridge K. 2006. Analysis of activated GAPs and GEFs in cell lysates. Methods Enzymol 406:425–437. doi: 10.1016/S0076-6879(06)06031-9. [DOI] [PubMed] [Google Scholar]
- 31.Khamdaeng T, Luo J, Vappou J, Terdtoon P, Konofagou EE. 2012. Arterial stiffness identification of the human carotid artery using the stress-strain relationship in vivo. Ultrasonics 52:402–411. doi: 10.1016/j.ultras.2011.09.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Riley WA, Barnes RW, Evans GW, Burke GL. 1992. Ultrasonic measurement of the elastic modulus of the common carotid artery. The Atherosclerosis Risk in Communities (ARIC) Study. Stroke 23:952–956. [DOI] [PubMed] [Google Scholar]
- 33.Dupont S, Morsut L, Aragona M, Enzo E, Giulitti S, Cordenonsi M, Zanconato F, Le Digabel J, Forcato M, Bicciato S, Elvassore N, Piccolo S. 2011. Role of YAP/TAZ in mechanotransduction. Nature 474:179–183. doi: 10.1038/nature10137. [DOI] [PubMed] [Google Scholar]
- 34.Chen KD, Li YS, Kim M, Li S, Yuan S, Chien S, Shyy JY. 1999. Mechanotransduction in response to shear stress. Roles of receptor tyrosine kinases, integrins, and Shc. J Biol Chem 274:18393–18400. [DOI] [PubMed] [Google Scholar]
- 35.Liu Y, Sweet DT, Irani-Tehrani M, Maeda N, Tzima E. 2008. Shc coordinates signals from intercellular junctions and integrins to regulate flow-induced inflammation. J Cell Biol 182:185–196. doi: 10.1083/jcb.200709176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hardy WR, Li L, Wang Z, Sedy J, Fawcett J, Frank E, Kucera J, Pawson T. 2007. Combinatorial ShcA docking interactions support diversity in tissue morphogenesis. Science 317:251–256. doi: 10.1126/science.1140114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Vanderlaan RD, Hardy WR, Kabir MG, Pasculescu A, Jones N, deTombe PP, Backx PH, Pawson T. 2011. The ShcA phosphotyrosine docking protein uses distinct mechanisms to regulate myocyte and global heart function. Circ Res 108:184–193. doi: 10.1161/CIRCRESAHA.110.233924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Dubash AD, Wennerberg K, Garcia-Mata R, Menold MM, Arthur WT, Burridge K. 2007. A novel role for Lsc/p115 RhoGEF and LARG in regulating RhoA activity downstream of adhesion to fibronectin. J Cell Sci 120:3989–3998. doi: 10.1242/jcs.003806. [DOI] [PubMed] [Google Scholar]
- 39.Scott DW, Tolbert CE, Burridge K. 2016. Tension on JAM-A activates RhoA via GEF-H1 and p115 RhoGEF. Mol Biol Cell 27:1420–1430. doi: 10.1091/mbc.E15-12-0833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Schlaepfer DD, Jones KC, Hunter T. 1998. Multiple Grb2-mediated integrin-stimulated signaling pathways to ERK2/mitogen-activated protein kinase: summation of both c-Src- and focal adhesion kinase-initiated tyrosine phosphorylation events. Mol Cell Biol 18:2571–2585. doi: 10.1128/MCB.18.5.2571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Barberis L, Wary KK, Fiucci G, Liu F, Hirsch E, Brancaccio M, Altruda F, Tarone G, Giancotti FG. 2000. Distinct roles of the adaptor protein Shc and focal adhesion kinase in integrin signaling to ERK. J Biol Chem 275:36532–36540. doi: 10.1074/jbc.M002487200. [DOI] [PubMed] [Google Scholar]
- 42.Gu J, Tamura M, Pankov R, Danen EH, Takino T, Matsumoto K, Yamada KM. 1999. Shc and FAK differentially regulate cell motility and directionality modulated by PTEN. J Cell Biol 146:389–403. doi: 10.1083/jcb.146.2.389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Schaller MD. 2010. Cellular functions of FAK kinases: insight into molecular mechanisms and novel functions. J Cell Sci 123:1007–1013. doi: 10.1242/jcs.045112. [DOI] [PubMed] [Google Scholar]
- 44.Chan SW, Lim CJ, Guo K, Ng CP, Lee I, Hunziker W, Zeng Q, Hong W. 2008. A role for TAZ in migration, invasion, and tumorigenesis of breast cancer cells. Cancer Res 68:2592–2598. doi: 10.1158/0008-5472.CAN-07-2696. [DOI] [PubMed] [Google Scholar]
- 45.Tian Y, Kolb R, Hong JH, Carroll J, Li D, You J, Bronson R, Yaffe MB, Zhou J, Benjamin T. 2007. TAZ promotes PC2 degradation through a SCFbeta-Trcp E3 ligase complex. Mol Cell Biol 27:6383–6395. doi: 10.1128/MCB.00254-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Zhou Z, Hao Y, Liu N, Raptis L, Tsao MS, Yang X. 2011. TAZ is a novel oncogene in non-small cell lung cancer. Oncogene 30:2181–2186. doi: 10.1038/onc.2010.606. [DOI] [PubMed] [Google Scholar]
- 47.Zhao B, Li L, Wang L, Wang CY, Yu J, Guan KL. 2012. Cell detachment activates the Hippo pathway via cytoskeleton reorganization to induce anoikis. Genes Dev 26:54–68. doi: 10.1101/gad.173435.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Li X, Xu Z, Du W, Zhang Z, Wei Y, Wang H, Zhu Z, Qin L, Wang L, Niu Q, Zhao X, Girard L, Gong Y, Ma Z, Sun B, Yao Z, Minna JD, Terada LS, Liu Z. 2014. Aiolos promotes anchorage independence by silencing p66(Shc) transcription in cancer cells. Cancer Cell 25:575–589. doi: 10.1016/j.ccr.2014.03.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Glaven JA, Whitehead IP, Nomanbhoy T, Kay R, Cerione RA. 1996. Lfc and Lsc oncoproteins represent two new guanine nucleotide exchange factors for the Rho GTP-binding protein. J Biol Chem 271:27374–27381. doi: 10.1074/jbc.271.44.27374. [DOI] [PubMed] [Google Scholar]
- 50.Jaiswal M, Gremer L, Dvorsky R, Haeusler LC, Cirstea IC, Uhlenbrock K, Ahmadian MR. 2011. Mechanistic insights into specificity, activity, and regulatory elements of the regulator of G-protein signaling (RGS)-containing Rho-specific guanine nucleotide exchange factors (GEFs) p115, PDZ-RhoGEF (PRG), and leukemia-associated RhoGEF (LARG). J Biol Chem 286:18202–18212. doi: 10.1074/jbc.M111.226431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Nimnual AS, Yatsula BA, Bar-Sagi D. 1998. Coupling of Ras and Rac guanosine triphosphatases through the Ras exchanger Sos. Science 279:560–563. doi: 10.1126/science.279.5350.560. [DOI] [PubMed] [Google Scholar]