

ABSTRACT
Previously, a collection of virulent phages infecting Pseudomonas aeruginosa was isolated from open water reservoirs and residual waters. Here, we described the comparative genomics of a set of five related phages from the collection, the physical structure of the genome, the structural proteomics of the virion, and the transcriptional program of archetypal phage PaMx41. The phage genomes were closely associated with each other and with those of two other P. aeruginosa phages, 119X and PaP2, which were previously filed in the databases. Overall, the genomes were approximately 43 kb, harboring 53 conserved open reading frames (ORFs) and three short ORFs in indel regions and containing 45% GC content. The genome of PaMx41 was further characterized as a linear, terminally redundant DNA molecule. A total of 16 ORFs were associated with putative functions, including nucleic acid metabolism, morphogenesis, and lysis, and eight virion proteins were identified through mass spectrometry. However, the coding sequences without assigned functions represent 70% of the ORFs. The PaMx41 transcription program was organized in early, middle, and late expressed genomic modules, which correlated with regions containing functionally related genes. The high genomic conservation among these distantly isolated phages suggests that these viruses undergo selective pressure to remain unchanged. The 119X lineage represents a unique set of phages that corresponds to a novel phage group. The features recognized in the genomes and the broad host range of clinical strains suggest that these phages are candidates for therapy applications.
IMPORTANCE Pseudomonas aeruginosa is an opportunistic pathogen that causes stubborn nosocomial infections that are frequently resistant to multiple antibiotics. Bacterial viruses (bacteriophages or phages) represent a natural mechanism for pathogenic bacterial control. Here, a group of virulent phages, previously shown to infect a broad range of clinical P. aeruginosa strains, was characterized at the genomic and molecular levels. These phages belong to a unique and tightly related group. In addition, we conducted a transcriptional study of an archetypal phage of this group to characterize the role of many unknown coding sequences based on expression temporalities. These results contribute to our knowledge of 119X-like phages and, in general, provide information concerning P. aeruginosa podophage diversity and lytic cycles.
INTRODUCTION
The renewed interest in the study of genetic diversity in phage populations has been prompted by the accessibility of massive DNA sequencing platforms, which has radically increased the number of phage genome sequences reported in the data banks (1). Presently, genome sequencing is an invaluable tool in phage characterization, which is fundamental at the first stages of the functional annotation of new coding sequences (2). Homologous sequences or structures in data banks provide accurate clues concerning the potential function of coding sequences. In addition, the genome sequence is the most important criterion used to understand relationships between phage groups (3). Pseudomonas aeruginosa is a Gram-negative bacterium that is globally distributed in a wide range of environments and is one of the most common opportunistic human pathogens (4, 5). Phages that infect this bacterium are frequently isolated and characterized because of their potential application as therapeutic tools (6). Pseudomonas tailed phages are the most abundant group deposited in databases. Among these organisms, 38% are podoviruses, which show icosahedral heads and short tails (7). The International Committee on Taxonomy of Viruses classified virulent podophages that infect P. aeruginosa into three genera and the phage 119X as an unassigned species.
Several studies have focused on a more comprehensive characterization at the genomic, transcriptional, and structural proteomics levels of the three podophage genera, Phikmvvirus (8, 9), Luz24virus (10, 11), and Lit1virus (12, 13). At the genomic level, the sizes ranged from 41.6 to 74.9 kb, and the GC content was typically lower (52% to 62%) than the average of many genomes of P. aeruginosa strains (66%). Direct terminal repeats are common in the three groups, ranging from 184 bp in Luz24 (10) to 665 bp in Lit1 phages (12). Genomes are organized into coding blocks harboring functionally related genes that typically correlate with the temporal nature of genome transcription observed during the lytic cycle (9, 11, 13).
There is little available information concerning 119X and its relatives. Comparative genomic analysis (14) shows that 119X is similar to PaP2, a phage that was isolated in China (15). In addition, in a previous study, Sepulveda-Robles et al. (16) reported the isolation of 68 virulent P. aeruginosa phages in Mexico. Only 13 of those phages were assigned to the Podoviridae family using electron microscopy. Based on infection, restriction, and DNA hybridization patterns, all of these phages were clustered in a single group, and a partial sequence of one of these phages (PaMx32) suggested that these organisms are similar to 119X (16). Interestingly, these podophages showed the broadest host range among Mexican strains, productively infecting 30% to 60% of the more than 100 clinical isolates tested (16); however, these isolates were unable to infect the type strains PAO1 and PA14.
To obtain knowledge about this group of phages, the genomes of five independent isolates were sequenced. This work describes and compares these five isolates, revealing a closely related group that includes 119X and PaP2, which are different from phages recognized previously. Because of their general similarity, we used phage PaMx41 as a model to describe the virion genome topology, protein composition, lytic cycle, and transcriptional timing of the functionally different gene clusters of the group. Differences at the genomic and transcriptional levels between P. aeruginosa podophages are discussed.
MATERIALS AND METHODS
Bacterial strains and phage propagation.
Bacteriophages PaMx33, PaMx35, PaMx41, PaMx43, and PaMx46 were isolated from open water reservoirs and residual waters (16). A clinical strain, Pseudomonas aeruginosa Ps26, was used as the host bacterium because these phages were unable to adequately infect the type strains PAO1 or PA14 and the sequenced strains LESB58 and 39016. Host bacteria were grown overnight with shaking at 37°C in lysogeny broth (LB) medium. The phages were propagated using a standard agar overlay method, eluted from the agar, precipitated in 1.4 M NaCl and 16% (wt/vol) polyethylene glycol 8000 (PEG 8000) at 4°C, and subsequently purified through CsCl gradient centrifugation as previously described (17). Virion particles were stable for over 1 year of storage at 4°C.
Genome sequencing and assembly.
Bacteriophage genomes were determined using high-throughput DNA sequencing using SOLiD technology (Life Technologies, Grand Island, NY, USA). The reads were preprocessed using the Applied Biosystems de novo assembly accessories. Phage genomes were de novo assembled from 50-bp sequencing reads using Velvet v1.1 (18).
Genome composition and functional annotation.
The genes were predicted using GeneMark v1.1 (19), and ribosome binding sites were identified using rbs_finder.pl (20). Putative functions were assigned to predicted open reading frames (ORFs) based on the results of (i) blastp searches against GenBank nonredundant protein databases, (ii) structural similarities using HHpred (21) and I-TASSER (4), and (iii) a search of the protein conserved domains using InterPro (22). Putative P. aeruginosa promoter sequences (TTGaCc-N15:18-TAtAA [nucleotides in uppercase are present in more than 40% of the sequences; nucleotides in lowercase are present in more than 30% but less than 40% of the sequences]) (23) were predicted using Pattern Locator (24), PromoterHunter (25), and Neural Network Promoter Prediction (26) tools. Only those sequences detected using three tools were accepted. The E-CAI server (27) was used to calculate the global genomic GC content and to determine the average percentage of codons ending in A3+T3.
Phage genome comparative analysis.
The homology among the five newly reported phages and with 119X (GenBank accession number NC_007807.1) and PaP2 (GenBank accession number NC_005884.1) was observed via blastn against the nucleotide collection of NCBI. Pairwise genome comparisons at the nucleotide level were performed using MUMmer v3.0 (28) with a break length of 120 nucleotides. Single nucleotide polymorphisms (SNPs) were identified based on the global comparison of the seven phages using the progressiveMauve algorithm (29). The protein sequences of predicted ORFs were compared with all of the bacteriophage protein sequences deposited in the NCBI nonredundant protein database using blastp v2.2.30. An ORF was classified as a homolog between genomes when its blastp E value was lower than 1e−3 and the length of the shortest ORF among homologs was at least 35% of the longest sequence.
Analysis of virion structural proteins.
PaMx41 structural proteins from CsCl-purified virions were separated using Tris-glycine sodium dodecyl sulfate-12% polyacrylamide gel electrophoresis (SDS-PAGE) using Precision Plus Protein Dual Xtra prestained protein standards (Bio-Rad, Hercules, CA) as molecular weight markers. The gel bands were excised and destained, and an in-gel protein digestion was performed using trypsin. The peptides were extracted and analyzed using a nanoscale liquid chromatography-electrospray ionization-tandem mass spectrometry (NanoLC-ESI-MS/MS) system. Protein identification against the virus nonredundant NCBI protein database (v1.6b9; Matrix Science, London, United Kingdom) was conducted using Mascot search algorithms. The search parameters were set to monoisotopic mass values, allowing one missed cleavage, with a peptide mass tolerance of 1.2 Da and a fragment mass tolerance of 0.6 Da. The modifications were deamidation (NQ), oxidation (M), and propionamide (C).
Analysis of genome ends.
Sequence coverage was assessed after mapping the sequencing reads on the PaMx41 genome using the SHRiMP package (30) and visualization using Tablet software (31). The in silico restriction pattern of PaMx41 was predicted using NEBcutter (32). Virion DNA of phage PaMx41 was digested using the Bal 31 exonuclease. The samples were removed at 0, 15, 30, and 45 min after the addition of the enzyme. The reaction was stopped with the addition of 50 mM EGTA and incubation at 70°C for 10 min. The samples were purified through phenol-chloroform extraction and resuspended in 10 μl of water. The DNA samples were digested with BglI endonuclease at 37°C for 2 h. The restriction pattern was visualized using 1% agarose gel electrophoresis.
One-step growth curve.
To determine the phage growth characteristics, three independent single-step growth experiments were performed. Briefly, the host strain, Pseudomonas aeruginosa Ps26, was grown in liquid LB medium at 37°C until an optical density at 600 nm (OD600) of 0.4 (approximately 1 ×108 cells) was reached. One milliliter of culture was concentrated and suspended in 0.1 ml of LB that was supplemented with 10 mM MgCl2, and an aliquot containing 1 × 107 phages was added to the culture, corresponding to a multiplicity of infection (MOI) of 0.1. Subsequently, the phages were adsorbed for 10 min at room temperature. The sample was diluted 1:1,000 in LB and incubated at 37°C for 120 min. The samples were collected at different times after infection for phage quantification. The samples were split in two, and one fraction was immediately plated on lawns of host bacteria, while the second fraction was treated with chloroform to kill the bacterial cells, infected or not, and the phage was quantified on lawns of host bacteria. The burst size was determined as the phage titer at 85 min (titer of directly plated samples) divided by the titer of infected cells at 20 min of infection (titer of directly plated samples minus titer of chloroform-treated samples at 20 min).
RNA and Northern blot methods.
RNA was extracted from Ps26 cells that were infected with phage PaMx41 at an MOI of 10. Briefly, 50 ml of host culture at an OD600 of 0.4 was harvested and resuspended in 1 ml of LB with 10 mM MgCl2. An aliquot containing 5 × 1010 phages was added to the cell suspension, and phages were adsorbed for 10 min. The cells were washed with fresh LB, resuspended in 100 ml of LB, and incubated at 37°C. The samples were collected at 0, 15, 30, 45, 60, and 75 min after infection, supplemented with 0.3 mg/ml of rifampin and 0.5 mg/ml of chloramphenicol, and harvested on ice. RNA was obtained from the bacterial pellets through phenol-chloroform extraction and treated with RNase-free DNase I according to the manufacturer's recommendations (Thermo Fisher Scientific, Waltham, MA). Total RNA was resuspended in diethyl pyrocarbonate (DEPC)-treated water and stored at −80°C until further use. A total of 15 μg of each RNA sample was separated on a 1.2% agarose electrophoresis gel supplemented with 20 mM guanidine thiocyanate using Tris-borate-EDTA (TBE) as a running buffer. Formamide was used as a loading buffer. Subsequently, the samples were blotted onto Hybond-N+ membranes (GE Healthcare Biosciences, Little Chalfont, Buckinghamshire, United Kingdom) as previously described (33). RNA was fixed to the membrane through UV cross-linking for 5 min. The DNA fragments that were obtained after digesting PaMx41 genomic DNA were used as probes. Probes A and B were obtained using BglI; probes C, F, and G were obtained using BamHI; probes D and J were obtained with NcoI; probe E was obtained with BclI; probe H was obtained with AgeI; and probe K was obtained with SpeI. The restriction fragments were purified using QIAquick gel extraction kit (Qiagen, Germany). The DNA fragments were used as the templates for radiolabeling reactions with [α-32P]dATP (PerkinElmer, Boston, MA) using the random primer method (Random primer labeling kit; Amersham, Little Chalfont, Buckinghamshire, United Kingdom). The radiolabeled probes were purified using Centri-Sep spin columns (Life Technologies, Carlsbad, CA). Prehybridizing and hybridizing procedures were conducted in 7% SDS–0.5 M NaH2PO4 (pH 7.2)–2% dextran sulfate (34). The membranes were washed twice at 60°C with 1× SSC (1× SSC is 0.15 M NaCl plus 0.015 M sodium citrate)/0.1% SDS, dried, and exposed. The film was developed after overnight exposure with an intensifying screen at −80°C.
Accession number(s).
The complete genome sequences of the five phages were deposited in GenBank under accession numbers KU884561 (PaMx33), KU884562 (PaMx35), KU884563 (PaMx41), KU884564 (PaMx43), and KU884565 (PaMx46).
RESULTS AND DISCUSSION
Genomic characterization.
Phages PaMx33, PaMx35, PaMx41, PaMx43, and PaMx46 are members of a podovirus group whose genomes displayed similar restriction and cross-hybridization patterns (16). Each phage lysed 30 to 44 strains of a collection of approximately 100 clinical P. aeruginosa isolates (16). To compare their genomic features, the complete sequences of the five genomes were assembled and annotated (see Materials and Methods). Three homologous phage genomes were identified, 119X, PaP2 (14), and PaMx32 (16). The genomic sequence of 119X was deposited in GenBank in 2006 (accession number NC_007807.1), but the phage was isolated in Australia approximately 50 years ago (7). PaP2 was isolated in China and its genomic sequence was deposited in GenBank in 2004 (accession number NC_005884.1), and PaMx32 is a member of the present group whose genome was partially sequenced (GenBank accession number JQ067091.1). The seven completely sequenced phage genomes were compared and analyzed. The genomes were approximately 43 kb long (Table 1) and contained 56 different ORFs (Table 1). Fifty-three ORFs were common to all phages, with five in insertions in various genomes (Fig. 1B). These sequences contained 45% GC content (Table 1 and Fig. 1A), which was in contrast with the approximately 66% GC content of P. aeruginosa strains (35). These differences impact the bias of codon usage in 119X-like phages, which contain 59% A3+T3-ending codons versus 23% A3+T3-ending codons for various other P. aeruginosa strains (see Materials and Methods). The consequences of this difference need further investigation.
TABLE 1.
Genome sizes and nucleotide compositions of the five sequenced phages and two homologs in the GenBank database
| Phage | GenBank accession no. | Length (bp) | GC content (%) | C3+G3 (%)a | No. of ORFs |
|---|---|---|---|---|---|
| PaMx33 | KU884561 | 43,265 | 45.25 | 41.37 | 54 |
| PaMx35 | KU884562 | 43,733 | 45.04 | 40.87 | 56 |
| PaMx41 | KU884563 | 43,490 | 45.21 | 41.52 | 55 |
| PaMx43 | KU884564 | 43,223 | 45.33 | 41.96 | 54 |
| PaMx46 | KU884565 | 43,266 | 45.24 | 41.41 | 54 |
| PaP2 | NC_005884.1 | 43,783 | 45.36 | 42.78 | 55 |
| 119X | NC_007807.1 | 43,365 | 44.89 | 40.67 | 53 |
C3+G3 refers to codons ending in G+C.
FIG 1.

Consensus genome map with functional annotation of 119X-like phages and transcription program of PaMx41. (A) GC content plot for PaMx41 genome. (B) Fifty three conserved ORFs, present in all seven genomes of the group, are numbered and indicated as color-coded arrows: light blue, morphogenesis; dark blue, virion structural proteins; yellow, lysis cassette; green, nucleotide DNA and RNA metabolism; and gray, unknown function. The enlisted putative functions below the map were assigned to the ORF numbers by predicted amino acid sequence, structural similarities, or through mass spectrometry (MS) analysis of the virion proteins. The pink blocks in the map represent indel regions. Phage 119X lacks a DNA segment of 175 bp containing ORFa (a) and harbors a noncoding 300-bp insertion between ORF24 and ORF25. PAP2 shows an insertion of 547 bp that duplicates ORF31 (31). An insertion of 303 bp encoding ORFb (b) in PaMx41 and another insertion of 506 bp in PaMx35 encode ORFb and ORFc (c). The functions of these additional ORFs are unknown. (C) DNA fragments from PaMx41 used as probes for mRNA detection through Northern blot assays are placed below the map according to their positions. Below each probe, the viral mRNAs identified as early (E), middle (M), or late (L), followed by progressive numbers according to the position on the map and their estimated sizes in parenthesis, are indicated in kilobases (see Fig. S1 in the supplemental material). The three temporally regulated gene expression classes and the assigned functions to the ORFs in each coding module are indicated at the bottom. The mRNAs below overlapping regions of probes C and D were identified using both probes, and the mRNAs shown below overlapped a region of probes I, J, and K were identified using the three probes.
Comparative genomics.
The phage genome sequences were compared to evaluate their level of similarity. The results showed a high degree of conservation, even for phages isolated from distant global locations and time periods (from 99.95% to 89.5% identity). High levels of conservation have been reported in genomes of Pseudomonas (36) and Brucella (37) virulent phages isolated from distant places. Using progressiveMauve (29) and the genome of PaMx43 as a reference, we observed five large indels: three insertions (including one duplication) and one deletion (Fig. 1B; see also Table S2 in the supplemental material).
Additionally, 35 indels of 1 to up to 23 nucleotides were identified (see Table S1 in the supplemental material). Most of the 1-bp insertions and deletions were located in noncoding regions, and those located within coding regions generated translational frameshifting and were positioned close to the 5′ and 3′ ends. The 5′ indels shifted the beginning of the protein to the next start codon, whereas the 3′ indels produced a premature stop codon, and both cases resulted in shorter proteins but might conserve function because the core sequence remained unchanged. In addition, short indels that were longer than 1 bp within ORFs were in-frame variants that predictably exerted little effect on the putative proteins. Additionally, a total of 3,956 single nucleotide polymorphisms (SNPs) were observed in the global genome comparison; however, the number of SNPs in pairwise alignments varied from 25 to 2,270 (see Table S2 in the supplemental material). Notably, the PaMx43 genome had the lowest number of SNPs (610) relative to that of PaP2 from China (see Table S2). Thus, except for the ORF31 duplication in PaP2, this phage was more related to PaMx43 than to any other Mexican phage described here. The observed variations suggest a strong preference of the genomes to conserve functions despite their geographical or temporal isolation.
Although at the nucleotide level no other sequence showed similarity to the genomes of 119X-like phages, distant relationships were observed at the protein level (38). By comparing the amino acid sequences of the putative proteins encoded in the analyzed genomes, it was found that only seven sequences were shared between different phages (see Materials and Methods): large terminase, endolysin, MazG-like protein, DNA polymerase, helicase, ORF24, and ORF35. No more than two homologous proteins were shared with the same phage. However, several proteins that were encoded in 119X-like phages matched the hypothetical proteins observed in large contigs from metagenome data (see Table S3 in the supplemental material) that were obtained from sludge and groundwater (39, 40), suggesting that related phages might be observed in a wide range of water reservoirs. The unique nature of most of the coding sequences from 119X-like phages facilitated the discrimination of this group.
Genome functional annotation.
We observed 53 ORFs conserved in each of the seven genomes analyzed (Table 1) and only three accessory ORFs, namely, a, b and c, in various genomes (Fig. 1B). Ribosome binding sites were observed for all of the predicted ORFs, except for ORF15, ORF36, and ORF47. Indeed, the start codons of ORF15 and ORF36 overlapped with the stop codons of the upstream ORFs (ORF14 and ORF37, respectively). The assignment of functions to the recognized ORFs was based on the predicted amino acid sequences and structural similarities that were shared with characterized proteins (see Materials and Methods). A total of 27 ORFs were not homologous to any other sequence, and 13 ORFs resembled proteins without assigned functions from metagenomics data and other phage and bacterial origins. Only 16 ORFs possessed functionally conserved domains that were similar to proteins with a known function. The identified functions belong to nucleotide, DNA, and RNA metabolism, packaging, morphogenesis, and lysis modules (Fig. 1B shows the putative assigned functions, and Table S3 in the supplemental material summarizes the criteria that were used for function assignation).
Transcription program and regulatory sequences of the PaMx41 genome.
The lytic cycle of viruses is an orderly gene expression program that is coordinated with the availability of specific proteins needed at different times of infection (41). The diversity of strategies used by different groups of phages to achieve successful propagation makes it hard to predict the transcriptional timing of a specific phage (42). To obtain knowledge concerning the timing of the lytic cycle and improve the annotation of 119X-like phages, we evaluated the temporal pattern of gene expression for PaMx41. One-step growth curve assays of phage PaMx41 (see Fig. S1 in the supplemental material) showed that the eclipse period (phase of infection spanning from adsorption to the detection of phage particles after chloroform treatment) lasted for 60 min. Cell lysis was initiated 15 min later at 75 min after infection, and the burst size was approximately 50 viral particles per infected cell. We also examined viral mRNA synthesis at different times after infection through Northern blot analyses using 11 restriction fragments as probes scattered throughout the genome (Fig. 1; see also Fig. S2 in the supplemental material).
Based on the timing of when each viral mRNA appeared after the infection, the transcripts were assigned to one of three temporally regulated expression groups: early (E) transcripts that were observed at 15 min of infection, middle (M) mRNAs at 30 min, and late (L) mRNAs at 45 min. In all cases, the uninfected sample was free of phage-associated signals. We assumed that all identified signals corresponded to mRNAs that were oriented in the same manner as the ORFs that corresponded to each probe. However, there is evidence for the transcription of the antisense strand in N4-like Pseudomonas phages (13).
Early mRNAs were just two short transcripts, E1 and E2, that were revealed with probe A (Fig. 1C; see also Fig. S2 in the supplemental material), which corresponded to the gene cluster next to the left terminus (ORF1, ORFa, ORF2, and ORF3). The products of the four early ORFs lack similarity with other known proteins but may be related to host gene shutoff transcriptional regulation or antitermination (43, 44). Interestingly, E1 but not E2 was a transient transcript observed only at 15 min of infection (see Fig. S2). Phage early mRNA shutdown has been reported in T7 (45) and in the phage PhiKZ; early mRNA shutdown is observed after the early stage of infection (46). Probes G, H, I, J, and K detected 11 middle mRNAs covering the leftward-oriented cluster in the right arm of the genome (Fig. 1B and C), expressing functions associated with DNA replication and nucleic acid metabolism. As the 2-kb early region is duplicated at both ends of the genome, it is likely that middle transcription may be physically linked to early transcription from right-end duplication.
The late transcripts included seven mRNAs that were revealed using probes B to F (Fig. 1C) and comprise morphogenesis, virion structural, and lysis ORFs (Fig. 1; see also Fig. S2 in the supplemental material). A short signal was observed at a position corresponding to approximately 0.9 to 0.7 kb in almost all of the autoradiograms, irrespective of the probe used, and was considered to be nonspecific, so it was not taken into account. Most of the phage mRNAs remained detectable until the end of the infection, but during lysis at 75 min, most of the signals disappeared (see Fig. S2), which likely reflects mRNA degradation at the start of host cell lysis.
The three temporal classes of mRNAs that are observed above suggest a classical phage transcriptional program. However, because the mRNA ends were not identified, it was difficult to infer the location of the promoters originating the transcripts. Therefore, we conducted a search of P. aeruginosa σ70 promoter consensus sequences. Only one putative promoter that was recognized by the host RNA polymerase was identified in the negative strand of the PaMx41 genome upstream of ORF3 (at 1,915 to 1,950 bp; AGGCTTGACTTCTAACTAGGCCAGACTAGAATGACC; −35 and −10 are shown in italics). Although, the transcription results represent a specific case of PaMx41, the putative promoter was conserved in the seven compared phages. Thus, 119X-like phages may depend on the host RNA polymerase to initiate the transcription of early genes. σ70 promoter sequences were not identified in the rest of the genome, and an RNA polymerase homolog was not observed among the predicted ORFs. We hypothesized that the specificity of the host RNA polymerase is changed by an early phage function to transcribe phage middle and late promoter regions (47).
The three temporalities of gene expression and bacterial σ70 promoters have been observed in other P. aeruginosa phages but under different expression regimens. Phage LUZ19 encodes its own RNA polymerase and harbors three σ70 consensus sequences that may activate early transcription (9). Seven σ70 promoters spread throughout the genome of LUZ24, suggesting that transcription of the different temporal expression modules relies on the host RNA polymerase (10). Although the genomic analysis of PaMx41 showed a σ70 promoter, it did not reveal the promoters leading to middle and late transcription. Thus, despite the global transcriptional similarities among phages infecting P. aeruginosa, strategies for the regulation of host machinery may be specific for each phage.
Identification of the virion genome termini.
The sequencing reads of the five sequenced genomes may be assembled circularly. However, in tailed phages, this topology may also result from direct terminal redundancy of a linear genomic DNA segment, where the sequencing coverage of the redundant segment is twice that of the nonredundant portion (48). Indeed, we detected a 2-kb segment showing approximately twice the sequencing coverage as that of the rest of the genome (Fig. 2A). The coverage values suggested that the duplication of approximately 2 kb might represent the chromosome ends of PaMx41, and the hypothesized model is depicted in Fig. 2B.
FIG 2.
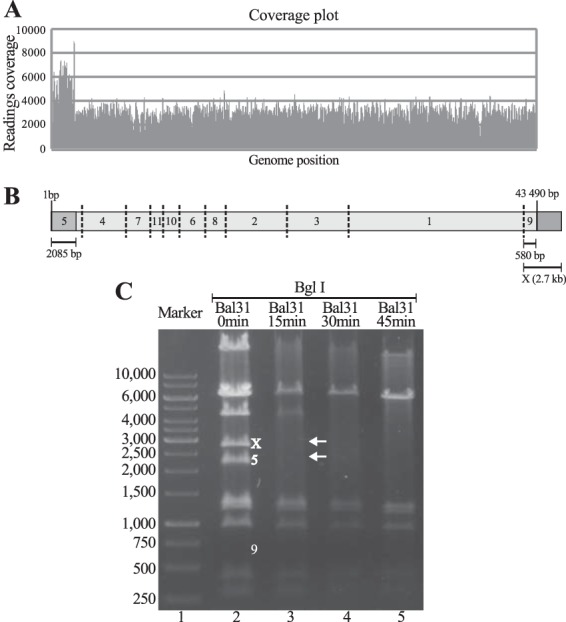
Direct terminal redundancy and identification of phage PaMx41 genome ends. (A) Genome coverage plot showing the left end region of 2,085 bp, with approximately twice the reading values than the rest of the DNA, arbitrarily assigned to the left end. (B) Proposed terminal redundant topology and expected restriction pattern of the PaMx41 genome with BglI. The numbers assigned to the expected fragments correlate with size: 1 is the largest and 11 is the shortest. In light gray is the nonredundant genome allotment; in dark gray is the 2,085 bp showing duplicated coverage values. Fragment 9 of 580 bp was expected in the absence of terminal redundancy. (C) BglI restriction pattern of the PaMx41 genome before and after treatment with exonuclease Bal 31 for the indicated times (see Materials and Methods). The bands in lane 2 correspond to the predicted fragments in B except for the absence of band 9 and the additional band X. The arrows in lane 3 identify the restriction fragments that disappeared first with Bal 31-treated DNA, corresponding to the ends of the linear genome.
To examine the suggested genome topology, we conducted restriction assays of the genomic PaMx41 DNA using the endonuclease BglI. The model shown in Fig. 2B indicates that 11 bands are expected from a linear topology. The restriction fragment corresponding to the right end of the terminally redundant genome should be approximately 2.7 kb instead of the 580 bp expected in the linear nonredundant genome (Fig. 2B). The restriction assay (Fig. 2C) showed a total of 11 bands, including the left-end and right-end fragments (5 and X in Fig. 2C, lane 2), which was consistent with the direct terminal redundancy topology. Moreover, a time course treatment of the PaMx41 DNA with exonuclease Bal 31, followed by BglI restriction, resulted in the rapid loss of the end fragments and sequential reduction of the internal fragments (Fig. 2C, lanes 3 to 5). The outcome of such treatment was consistent with the linear topology for the virion genome. Taken together, these results showed direct terminal redundancy of approximately 5% of the genome. Four hypothetical ORFs (1, a, 2, and 3) were encoded in the duplicated region. Whether this feature is relevant for the transcriptional phage program and/or for circularization of the intracellular phage genome needs further investigation.
Virion structural proteins.
To identify the proteins making up the virions, we analyzed a CsCl-purified stock of PaMx41 virions using SDS-PAGE and mass spectrometry. A total of eight proteins were identified (Fig. 3), expressed from the positive-strand gene cluster that occupied most of the middle genome region of PaMx41 (Fig. 1). Based on structural similarities, we identified the specific function of two virion proteins, the portal and major capsid proteins (Fig. 1B; see also Table S3 in the supplemental material). The particular functions of the other six structural proteins (gene products [gp] 10, 13, 20, 21, 22, and 23) could not be assigned because of the lack of homology. Only three coding sequences showed amino acid variations between PaMx33 and PaMx46—those for portal protein (gp 7), gp 10, and the putative DNA helicase—despite differences in host range observed between these phages (16). These sequence differences may be associated with specific differences in bacterial cell receptor recognition. To our knowledge, portal proteins or recombination proteins have never been associated with receptor recognition; thus, gp 10 variations may be responsible for the host range differences between these phages.
FIG 3.

PaMx41 virion structural proteins. The protein components of PaMx41 purified virions were resolved using 12% SDS-PAGE and stained with Coomassie brilliant blue. Observed bands were excised from the gel and MS-MS treated, and the data were analyzed and correlated with the predicted amino acid composition encoded by phage genes. Sequence coverage is the percentage of the protein amino acid sequence covered by the matching peptides. Molecular mass (kDa) was calculated from the gene sequence. The Mascot score is −10log10 the probability of matching the mass value calculated for the corresponding gene.
Conclusion.
The genomes of the five phages reported here were almost identical at the nucleotide level to those of phages 119X and PaP2. Together, these phages form a unique group of organisms that do not share more than two coding sequences with other phages in the databases. Most of the putative proteins that are associated with a function, or even the experimentally identified virion proteins, shared only structural similarity or conserved domains with characterized proteins. Up to 70% of the ORFs in the genomes of the phages remain without an assigned function. The transcriptional study of this group revealed a classical gene expression program organized into early, middle, and late genomic modules. The uniqueness of the early genes and lack of recognizable middle and late promoter sequences in this group of phages suggested that these sequences possess particular elements of the phage-bacteria interaction. The results facilitated the integration of 119X-like phages as a unique lineage within the Podoviridae family.
Supplementary Material
ACKNOWLEDGMENTS
We thank Omar Sepúlveda-Robles for providing the phages reported here, Víctor Flores for assembling the genome sequences, Rosaura Hernández-Rivas for her technical assistance with the Northern blot assays, and the late Guillermo Mendoza for the MS analysis of the virion proteins. We also thank Rosa Bermudez, Norma Oviedo, Luis Kameyama, and Javier Hernandez for valuable comments and discussions during the experimental study. We are especially grateful to Donald Court for carefully reviewing the manuscript and an anonymous reviewer for valuable suggestions.
This work was financially supported through grant 166814 to G.G. and graduate fellowships to I.C.-P. (CVU: 263643) and A.C. (CVU: 233018) from Consejo Nacional de Ciencia y Tecnología (CONACyT).
Footnotes
Supplemental material for this article may be found at http://dx.doi.org/10.1128/AEM.01415-16.
REFERENCES
- 1.Sime-Ngando T. 2014. Environmental bacteriophages: viruses of microbes in aquatic ecosystems. Front Microbiol 5:355. doi: 10.3389/fmicb.2014.00355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Light S, Basile W, Elofsson A. 2014. Orphans and new gene origination, a structural and evolutionary perspective. Curr Opin Struct Biol 26:73–83. doi: 10.1016/j.sbi.2014.05.006. [DOI] [PubMed] [Google Scholar]
- 3.Pope WH, Bowman CA, Russell DA, Jacobs-Sera D, Asai DJ, Cresawn SG, Jacobs WR, Hendrix RW, Lawrence JG, Hatfull GF, Science Education Alliance Phage Hunters Advancing Genomics and Evolutionary Science, Phage Hunters Integrating Research and Education, Mycobacterial Genetics Course . 2015. Whole genome comparison of a large collection of mycobacteriophages reveals a continuum of phage genetic diversity. eLife 4:. doi: 10.7554/eLife.06416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Pirnay JP, Bilocq F, Pot B, Cornelis P, Zizi M, Van Eldere J, Deschaght P, Vaneechoutte M, Jennes S, Pitt T, De Vos D. 2009. Pseudomonas aeruginosa population structure revisited. PLoS One 4:. doi: 10.1371/journal.pone.0007740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Tummler B, Wiehlmann L, Klockgether J, Cramer N. 2014. Advances in understanding Pseudomonas. F1000Prime Rep 6:9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Henry M, Bobay LM, Chevallereau A, Saussereau E, Ceyssens PJ, Debarbieux L. 2015. The search for therapeutic bacteriophages uncovers one new subfamily and two new genera of Pseudomonas-infecting Myoviridae. PLoS One 10:. doi: 10.1371/journal.pone.0117163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Pires DP, Vilas Boas D, Sillankorva S, Azeredo J. 2015. Phage therapy: a step forward in the treatment of Pseudomonas aeruginosa infections. J Virol 89:7449–7456. doi: 10.1128/JVI.00385-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ceyssens PJ, Glonti T, Kropinski NM, Lavigne R, Chanishvili N, Kulakov L, Lashkhi N, Tediashvili M, Merabishvili M. 2011. Phenotypic and genotypic variations within a single bacteriophage species. Virol J 8:134. doi: 10.1186/1743-422X-8-134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lavigne R, Lecoutere E, Wagemans J, Cenens W, Aertsen A, Schoofs L, Landuyt B, Paeshuyse J, Scheer M, Schobert M, Ceyssens PJ. 2013. A multifaceted study of Pseudomonas aeruginosa shutdown by virulent podovirus LUZ19. mBio 4:. doi: 10.1128/mBio.00061-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ceyssens PJ, Hertveldt K, Ackermann HW, Noben JP, Demeke M, Volckaert G, Lavigne R. 2008. The intron-containing genome of the lytic Pseudomonas phage LUZ24 resembles the temperate phage PaP3. Virology 377:233–238. doi: 10.1016/j.virol.2008.04.038. [DOI] [PubMed] [Google Scholar]
- 11.Zhao X, Chen C, Shen W, Huang G, Le S, Lu S, Li M, Zhao Y, Wang J, Rao X, Li G, Shen M, Guo K, Yang Y, Tan Y, Hu F. 2016. Global transcriptomic analysis of interactions between Pseudomonas aeruginosa and bacteriophage PaP3. Sci Rep 6:19237. doi: 10.1038/srep19237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ceyssens PJ, Brabban A, Rogge L, Lewis MS, Pickard D, Goulding D, Dougan G, Noben JP, Kropinski A, Kutter E, Lavigne R. 2010. Molecular and physiological analysis of three Pseudomonas aeruginosa phages belonging to the “N4-like viruses”. Virology 405:26–30. doi: 10.1016/j.virol.2010.06.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wagemans J, Blasdel BG, Van den Bossche A, Uytterhoeven B, De Smet J, Paeshuyse J, Cenens W, Aertsen A, Uetz P, Delattre AS, Ceyssens PJ, Lavigne R. 2014. Functional elucidation of antibacterial phage ORFans targeting Pseudomonas aeruginosa. Cell Microbiol 16:1822–1835. doi: 10.1111/cmi.12330. [DOI] [PubMed] [Google Scholar]
- 14.Kwan T, Liu J, Dubow M, Gros P, Pelletier J. 2006. Comparative genomic analysis of 18 Pseudomonas aeruginosa bacteriophages. J Bacteriol 188:1184–1187. doi: 10.1128/JB.188.3.1184-1187.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Tan Y, Zhang K, Rao X, Jin X, Huang J, Zhu J, Chen Z, Hu X, Shen X, Wang L, Hu F. 2007. Whole genome sequencing of a novel temperate bacteriophage of P. aeruginosa: evidence of tRNA gene mediating integration of the phage genome into the host bacterial chromosome. Cell Microbiol 9:479–491. doi: 10.1111/j.1462-5822.2006.00804.x. [DOI] [PubMed] [Google Scholar]
- 16.Sepulveda-Robles O, Kameyama L, Guarneros G. 2012. High diversity and novel species of Pseudomonas aeruginosa bacteriophages. Appl Environ Microbiol 78:4510–4515. doi: 10.1128/AEM.00065-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Carlson K. 2005. Working with bacteriophages: common techniques and methodological approaches, p 437–449. In Kutter E, Sulakvelidze A (ed), Bacteriophages: biology and applications. CRC Press, Boca Raton, FL. [Google Scholar]
- 18.Zerbino DR, Birney E. 2008. Velvet: algorithms for de novo short read assembly using de Bruijn graphs. Genome Res 18:821–829. doi: 10.1101/gr.074492.107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Borodovsky M, Lomsadze A, Ivanov N, Mills R. 2003. Eukaryotic gene prediction using GeneMark.hmm. Curr Protoc Bioinformatics 4.6:4.6.1–4.6.12. doi: 10.1002/0471250953.bi0406s01. [DOI] [PubMed] [Google Scholar]
- 20.Suzek BE, Ermolaeva MD, Schreiber M, Salzberg SL. 2001. A probabilistic method for identifying start codons in bacterial genomes. Bioinformatics 17:1123–1130. doi: 10.1093/bioinformatics/17.12.1123. [DOI] [PubMed] [Google Scholar]
- 21.Soding J, Biegert A, Lupas AN. 2005. The HHpred interactive server for protein homology detection and structure prediction. Nucleic Acids Res 33:W244–W248. doi: 10.1093/nar/gki408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hunter S, Jones P, Mitchell A, Apweiler R, Attwood TK, Bateman A, Bernard T, Binns D, Bork P, Burge S, de Castro E, Coggill P, Corbett M, Das U, Daugherty L, Duquenne L, Finn RD, Fraser M, Gough J, Haft D, Hulo N, Kahn D, Kelly E, Letunic I, Lonsdale D, Lopez R, Madera M, Maslen J, McAnulla C, McDowall J, McMenamin C, Mi H, Mutowo-Muellenet P, Mulder N, Natale D, Orengo C, Pesseat S, Punta M, Quinn AF, Rivoire C, Sangrador-Vegas A, Selengut JD, Sigrist CJ, Scheremetjew M, Tate J, Thimmajanarthanan M, Thomas PD, Wu CH, Yeats C, Yong SY. 2012. InterPro in 2011: new developments in the family and domain prediction database. Nucleic Acids Res 40:D306–D312. doi: 10.1093/nar/gkr948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Potvin E, Sanschagrin F, Levesque RC. 2008. Sigma factors in Pseudomonas aeruginosa. FEMS Microbiol Rev 32:38–55. doi: 10.1111/j.1574-6976.2007.00092.x. [DOI] [PubMed] [Google Scholar]
- 24.Mrazek J, Xie S. 2006. Pattern locator: a new tool for finding local sequence patterns in genomic DNA sequences. Bioinformatics 22:3099–3100. doi: 10.1093/bioinformatics/btl551. [DOI] [PubMed] [Google Scholar]
- 25.Klucar L, Stano M, Hajduk M. 2010. phiSITE: database of gene regulation in bacteriophages. Nucleic Acids Res 38:D366–D370. doi: 10.1093/nar/gkp911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Reese MG. 2001. Application of a time-delay neural network to promoter annotation in the Drosophila melanogaster genome. Comput Chem 26:51–56. doi: 10.1016/S0097-8485(01)00099-7. [DOI] [PubMed] [Google Scholar]
- 27.Puigbo P, Bravo IG, Garcia-Vallve S. 2008. E-CAI: a novel server to estimate an expected value of Codon Adaptation Index (eCAI). BMC Bioinformatics 9:65. doi: 10.1186/1471-2105-9-65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kurtz S, Phillippy A, Delcher AL, Smoot M, Shumway M, Antonescu C, Salzberg SL. 2004. Versatile and open software for comparing large genomes. Genome Biol 5:R12. doi: 10.1186/gb-2004-5-2-r12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Darling AE, Mau B, Perna NT. 2010. progressiveMauve: multiple genome alignment with gene gain, loss and rearrangement. PLoS One 5:. doi: 10.1371/journal.pone.0011147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Rumble SM, Lacroute P, Dalca AV, Fiume M, Sidow A, Brudno M. 2009. SHRiMP: accurate mapping of short color-space reads. PLoS Comput Biol 5:. doi: 10.1371/journal.pcbi.1000386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Milne I, Stephen G, Bayer M, Cock PJ, Pritchard L, Cardle L, Shaw PD, Marshall D. 2013. Using Tablet for visual exploration of second-generation sequencing data. Brief Bioinformatics 14:193–202. doi: 10.1093/bib/bbs012. [DOI] [PubMed] [Google Scholar]
- 32.Vincze T, Posfai J, Roberts RJ. 2003. NEBcutter: a program to cleave DNA with restriction enzymes. Nucleic Acids Res 31:3688–3691. doi: 10.1093/nar/gkg526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Chomczynski P. 1992. One-hour downward alkaline capillary transfer for blotting of DNA and RNA. Anal Biochem 201:134–139. doi: 10.1016/0003-2697(92)90185-A. [DOI] [PubMed] [Google Scholar]
- 34.Kyes S, Pinches R, Newbold C. 2000. A simple RNA analysis method shows var and rif multigene family expression patterns in Plasmodium falciparum. Mol Biochem Parasitol 105:311–315. doi: 10.1016/S0166-6851(99)00193-0. [DOI] [PubMed] [Google Scholar]
- 35.Mathee K, Narasimhan G, Valdes C, Qiu X, Matewish JM, Koehrsen M, Rokas A, Yandava CN, Engels R, Zeng E, Olavarietta R, Doud M, Smith RS, Montgomery P, White JR, Godfrey PA, Kodira C, Birren B, Galagan JE, Lory S. 2008. Dynamics of Pseudomonas aeruginosa genome evolution. Proc Natl Acad Sci U S A 105:3100–3105. doi: 10.1073/pnas.0711982105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ceyssens PJ, Miroshnikov K, Mattheus W, Krylov V, Robben J, Noben JP, Vanderschraeghe S, Sykilinda N, Kropinski AM, Volckaert G, Mesyanzhinov V, Lavigne R. 2009. Comparative analysis of the widespread and conserved PB1-like viruses infecting Pseudomonas aeruginosa. Environ Microbiol 11:2874–2883. doi: 10.1111/j.1462-2920.2009.02030.x. [DOI] [PubMed] [Google Scholar]
- 37.Flores V, Lopez-Merino A, Mendoza-Hernandez G, Guarneros G. 2012. Comparative genomic analysis of two brucellaphages of distant origins. Genomics 99:233–240. doi: 10.1016/j.ygeno.2012.01.001. [DOI] [PubMed] [Google Scholar]
- 38.Hatfull GF. 2012. The secret lives of mycobacteriophages. Adv Virus Res 82:179–288. doi: 10.1016/B978-0-12-394621-8.00015-7. [DOI] [PubMed] [Google Scholar]
- 39.Skennerton CT, Angly FE, Breitbart M, Bragg L, He S, McMahon KD, Hugenholtz P, Tyson GW. 2011. Phage encoded H-NS: a potential Achilles heel in the bacterial defence system. PLoS One 6:. doi: 10.1371/journal.pone.0020095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Wrighton KC, Thomas BC, Sharon I, Miller CS, Castelle CJ, VerBerkmoes NC, Wilkins MJ, Hettich RL, Lipton MS, Williams KH, Long PE, Banfield JF. 2012. Fermentation, hydrogen, and sulfur metabolism in multiple uncultivated bacterial phyla. Science 337:1661–1665. doi: 10.1126/science.1224041. [DOI] [PubMed] [Google Scholar]
- 41.Doron S, Fedida A, Hernandez-Prieto MA, Sabehi G, Karunker I, Stazic D, Feingersch R, Steglich C, Futschik M, Lindell D, Sorek R. 2016. Transcriptome dynamics of a broad host-range cyanophage and its hosts. ISME J 10:1437–1455. doi: 10.1038/ismej.2015.210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Guzina J, Djordjevic M. 2015. Inferring bacteriophage infection strategies from genome sequence: analysis of bacteriophage 7-11 and related phages. BMC Evol Biol 15(Suppl):S1. doi: 10.1186/1471-2148-15-S1-S1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.McAllister WT, Barrett CL. 1977. Roles of the early genes of bacteriophage T7 in shutoff of host macromolecular synthesis. J Virol 23:543–553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Das A. 1992. How the phage lambda N gene product suppresses transcription termination: communication of RNA polymerase with regulatory proteins mediated by signals in nascent RNA. J Bacteriol 174:6711–6716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Pfennig-Yeh ML, Ponta H, Hirsch-Kauffmann M, Rahmsdorf HJ, Herrlich P, Schweiger M. 1978. Early T7 gene expression: rates of RNA synthesis and degradation, protein kinase dependent termination of transcription, and efficiency of translation. Mol Gen Genet 166:127–140. doi: 10.1007/BF00285915. [DOI] [PubMed] [Google Scholar]
- 46.Ceyssens PJ, Minakhin L, Van den Bossche A, Yakunina M, Klimuk E, Blasdel B, De Smet J, Noben JP, Blasi U, Severinov K, Lavigne R. 2014. Development of giant bacteriophage ϕKZ is independent of the host transcription apparatus. J Virol 88:10501–10510. doi: 10.1128/JVI.01347-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Rabussay D, Geiduschek EP. 1977. Phage T4-modified RNA polymerase transcribes T4 late genes in vitro. Proc Natl Acad Sci U S A 74:5305–5309. doi: 10.1073/pnas.74.12.5305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Li S, Fan H, An X, Fan H, Jiang H, Chen Y, Tong Y. 2014. Scrutinizing virus genome termini by high-throughput sequencing. PLoS One 9:. doi: 10.1371/journal.pone.0085806. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.