

ABSTRACT
Pseudomonas nitroreducens TX1 is of special interest because of its ability to utilize 0.05% to 20% octylphenol polyethoxylates (OPEOn) as a sole source of carbon. In this study, a library containing 30,000 Tn5-insertion mutants of the wild-type strain TX1 was constructed and screened for OPEOn utilization, and 93 mutants were found to be unable to grow on OPEOn. In total, 42 separate disrupted genes were identified, and the proteins encoded by the genes were then classified into various categories, namely, information storage and processing (14.3%), cellular processes and signaling (28.6%), metabolism (35.7%), and unknown proteins (21.4%). The individual deletion of genes encoding isocitrate lyase (aceA), malate synthase (aceB), and glycolate dehydrogenase (glcE) was carried out, and the requirement for aceA and aceB but not glcE confirmed the role of the glyoxylate cycle in OPEOn degradation. Furthermore, acetaldehyde dehydrogenase and acetyl-coenzyme A (acetyl-CoA) synthetase activity levels were 13.2- and 2.1-fold higher in TX1 cells grown on OPEOn than in TX1 cells grown on succinate, respectively. Growth of the various mutants on different carbon sources was tested, and based on these findings, a mechanism involving exoscission to liberate acetaldehyde from the end of the OPEOn chain during degradation is proposed for the breakdown of OPEOn.
IMPORTANCE Octylphenol polyethoxylates belong to the alkylphenol polyethoxylate (APEOn) nonionic surfactant family. Evidence based on the analysis of intermediate metabolites suggested that the primary biodegradation of APEOn can be achieved by two possible pathways for the stepwise removal of the C2 ethoxylate units from the end of the chain. However, direct evidence for these hypotheses is still lacking. In this study, we described the use of transposon mutagenesis to identify genes critical to the catabolism of OPEOn by P. nitroreducens TX1. The exoscission of the ethoxylate chain leading to the liberation of acetaldehyde is proposed. Isocitrate lyase and malate synthase in glyoxylate cycle are required in the catabolism of ethoxylated surfactants. Our findings also provide many gene candidates that may help elucidate the mechanisms in stress responses to ethoxylated surfactants by bacteria.
INTRODUCTION
Octylphenol polyethoxylates (OPEOn, commercial name Triton X-100) is a nonionic surfactant that belongs to the alkylphenol polyethoxylate (APEOn) family. These surfactants are used in a range of industrial and household products (1, 2). APEOn structures typically consist of hydrophilic polyethoxylate chain bound to a hydrophobic moiety, such as alkylphenol or a linear primary/secondary alcohol (3). APEOn are often discharged into wastewater treatment plants or into the environment, which leads to them ultimately being degraded into shorter ethoxylate (EO) chains. Sometimes, the chains are completely removed to form nonylphenol, octylphenol, and alkylphenol monoethoxylates to triethoxylates (APEOn, n = 1 to ∼3, respectively) (4, 5). Many studies have shown that these APEOn metabolites, which have increased hydrophobicity, are more toxic than their parent compounds (6). In fact, these metabolites are able to mimic natural hormones, thus acting as endocrine disruptors when ingested by wildlife, which in turn can affect the environment and ultimately human health (7).
The fate and degradability of APEOn in the environment have received much attention (8). The biodegradation of APEOn has been studied using both pure and mixed cultures that grow solely on APEOn, and several bacterial strains have been reported as being able to degrade the EO chains of APEOn (1, 8–12). Most such isolates are proteobacteria and are often members of the genus Pseudomonas. Nguyen and Sigoillot (13) isolated four Pseudomonas strains from coastal seawater that grew on OPEOn, and these generated OPEO with 4 to 5 units of EO chain as the end products. John and White (14) reported a strain of Pseudomonas putida that grew on nonylphenol polyethoxylate (NPEOn) as the sole source of carbon, and in this case, the final accumulating metabolite was identified by gas chromatography-mass spectroscopy as NPEO2. Nishio et al. (15) isolated 11 strains of OPEOn-utilizing bacteria from paddy field soils. One strain, P. putida S-5, was shown to transform OPEOn to form OPEO2 and OPEO3, which were the dominant metabolites accumulating under aerobic conditions. Evidence based on analysis of intermediate metabolites has suggested that the primary biodegradation of APEOn under aerobic condition is achieved by a stepwise shortening of the EO chains (13, 14, 16). Two possible pathways for the stepwise removal of the C2 ethoxylate units from the end of the chain have been proposed (Fig. 1). The first is a nonoxidative hydroxyl shift mechanism using ether scission that yields hemiacetal, which then produces acetaldehyde (14, 17). The second is the cleavage of APEOn by terminal carboxylation, followed by hydrolysis to yield glycolate, which is oxidized to glyoxylate by glycolate dehydrogenase (15, 17). However, clear-cut evidence regarding the operation in organisms of these pathways during OPEOn degradation is still lacking.
FIG 1.
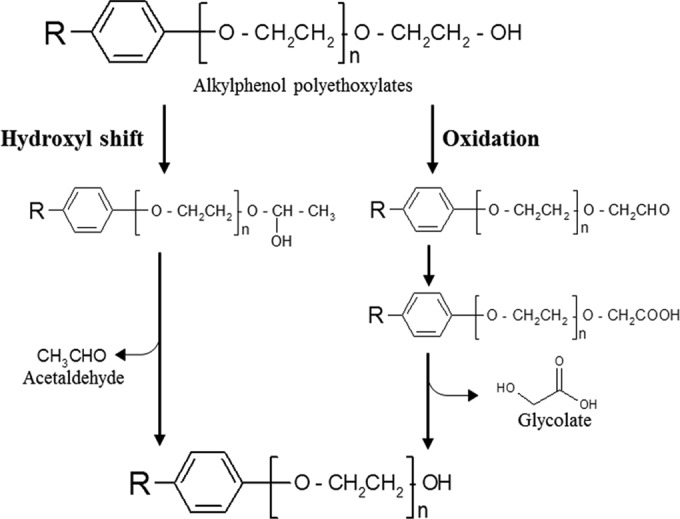
Proposed pathways for the biodegradation of alkylphenol polyethoxylates from the literature (14, 15, 17).
In our previous studies, P. nitroreducens TX1, which possesses the ability to grow on OPEOn at a wide range of concentrations, was isolated (1, 9–12, 18). The strain is able to use 0.05% to 20% OPEOn as a sole source of carbon. The liquid chromatography-mass spectrometry (LC-MS) analysis revealed that the ethoxylate chain was sequentially shortened from the hydroxyl terminal side in this strain (9). To elucidate the biodegradation mechanism of OPEOn, a library containing 30,000 mutants of strain TX1 was prepared using Tn5 insertion mutagenesis during this study. A total of 93 mutants were identified that were unable to grow on OPEOn, and these were found to have disrupted 42 individual genes. In-frame deletion of some of the target genes was then performed to confirm the roles of these genes in OPEOn degradation. The results revealed the important role of the glyoxylate cycle in OPEOn utilization by strain TX1.
MATERIALS AND METHODS
Bacterial strains and culture conditions.
The bacterial strains and plasmids used in this study are listed in Table 1. Strain TX1 was routinely grown at 30°C in Luria-Bertani (LB) or minimal salt basal (MSB) medium with appropriate sources of carbon and energy (12). In liquid culture, cells were grown in a 50-ml culture volume in 250-ml Erlenmeyer flasks with shaking in an incubator at 180 rpm. For plate culture, agar was added at a final concentration of 1.5% (wt/vol). Escherichia coli was grown in LB medium at 37°C and served as the host organism for plasmid retention. Ampicillin (20 μg · ml−1), gentamicin (20 μg · ml−1), and kanamycin (20 μg · ml−1) were used to select the transformed E. coli or TX1 cells.
TABLE 1.
Bacterial strains and plasmids used in this study
| Strain or plasmid | Descriptiona | Reference or source |
|---|---|---|
| Strains | ||
| Pseudomonas nitroreducens | ||
| TX1 | Wild-type strain TX1, Ampr | Laboratory collection |
| ΔaceA mutant | Strain TX1 with in-frame deletion of aceA gene, Ampr | This study |
| ΔaceB mutant | Strain TX1 with in-frame deletion of aceB gene, Ampr | This study |
| ΔfixC mutant | Strain TX1 with in-frame deletion of fixC gene, Ampr | This study |
| ΔglcE mutant | Strain TX1 with in-frame deletion of glcE gene, Ampr | This study |
| ΔaceA(pBaceA) mutant | ΔaceA mutant harboring plasmid pBaceA, Ampr, Gmr | This study |
| ΔaceB(pBaceB) mutant | ΔaceB mutant harboring plasmid pBaceB, Ampr, Gmr | This study |
| E. coli | ||
| DH5α | F− ϕ80dlacZΔM15 Δ(lacZYA-argF)U169 deoR recA1 endA1 hsdR17(rK− mK+) phoA supE44 λ− thi-1 gyrA96 | Laboratory collection |
| DH5α/λpir | λpir lysogen of DH5α | Laboratory collection |
| BW20767 | Containing pRL27; used as donor for pRL27 conjugations | Larsen et al. (21) |
| HB101 | Containing pRK2013; used as helper for pRL27 conjugations | Figurski and Helinski (22) |
| Plasmids | ||
| pGEM-T Easy | PCR cloning vector, Ampr | Promega |
| pGaceA | Ampr, pGEM-T Easy with 2.6-kb fragment, including aceA | This study |
| pGaceB | Ampr, pGEM-T Easy with 3.2-kb fragment, including aceB | This study |
| pRL27 | Kmr, mini-Tn5 transposon (oriR6K) delivery vector | Larsen et al. (21) |
| pRK2013 | Kmr, carrying RK2 transfer genes | Figurski and Helinski (22) |
| pK18mobsacB | Kmr, oriT(RP4) sacB lacZα Plac Pmbi, mobilization and counterselection | Schafer et al. (24) |
| pKaceA | pK18mobsacB containing a gene fragment with 98% deletion of internal region of aceA | This study |
| pKaceB | pK18mobsacB containing a gene fragment with 98% deletion of internal region of aceB | This study |
| pKfixC | pK18mobsacB containing a gene fragment with 98% deletion of internal region of fixC | This study |
| pKglcE | pK18mobsacB containing a gene fragment with 98% deletion of internal region of glcE (GenBank accession no. WP_017520229.1) | This study |
| pBBR1MCS-5 | Broad-host-range cloning vector, lacZ, Gmr | Kovach et al. (26) |
| pBaceA | Gmr, pBBR1-MCS5 with 2.6-kb fragment, including aceA | This study |
| pBaceB | Gmr, pBBR1-MCS5 with 3.2-kb fragment, including aceB | This study |
| pBglcE | Gmr, pBBR1-MCS5 with 1.5-kb fragment, including glcE | This study |
Kmr, Gmr, and Ampr indicate resistance to kanamycin, gentamicin, and ampicillin, respectively.
Plasmid and chromosomal DNA isolation.
Total DNA and plasmid DNA were extracted using previously described procedures (19). For the PCR amplifications, a 50-μl PCR mixture, consisting of 0.2 mM each of the four dinucleoside triphosphates (dNTPs), 20 pmol each primer, 10 ng of extracted DNA, and 1.25 U of Taq DNA polymerase (Toyobo Co. Ltd., Japan), with an appropriate amount of reaction buffer, was used. Amplification was performed in a program temperature control system, PC-808 (Astec Co. Ltd.). Other molecular techniques were performed using standard procedures (20) or as recommended by the reagent suppliers. The oligonucleotide primers used in this study are listed in Table 2.
TABLE 2.
Oligonucleotide primers used in this study
| Primer name | Sequence (5′–3′)a | Note | Restriction site | Reference or source |
|---|---|---|---|---|
| tpnRL17-1 | AACAAGCCAGGGATGTAACG | For sequencing of transposon insertion | Larsen et al. (21) | |
| tpnRL13-2 | CAGCAACACCTTCTTCACGA | Larsen et al. (21) | ||
| AceA_FEco | GCGAATTCACGTGCTCGACTAAGCCTTC | For in-frame deletion of aceA in TX1 | EcoRI | This study |
| AceA_RXho | GCCTCGAGTGCGGACATGGCCAATCCTTC | XhoI | This study | |
| AceA_FXho | GCCTCGAGTTCCACTAAGAACTGGCTCAC | XhoI | This study | |
| AceA_RHind | GCAAGCTTCTCTTGATCAGCGGCAGTTT | HindIII | This study | |
| AceB_FEco | GCGAATTCGAAGCGAAGAACGGGTACAG | For in-frame deletion of aceB in TX1 | EcoRI | This study |
| AceB_RXho | GCCTCGAGTTCAGTCATTGCTTGCCTCAC | XhoI | This study | |
| AceB_FXho | GCCTCGAGGGTCTGTAAGCACCTCGCGCC | XhoI | This study | |
| AceB_RHind | GCAAGCTTAACCGTCTGCAGTCTTTCTGA | HindIII | This study | |
| FixC_FEco | GCGAATTCCAGTGATCCGTCGAAGGTG | For in-frame deletion of fixC in TX1 | EcoRI | This study |
| FixC_RSma | GCCCCGGGGGCGGGCATCAGAAGGGCTCC | SmaI | This study | |
| FixC_FSma | GCCCCGGGGCCAGCTGAATAGGACAAGG | SmaI | This study | |
| FixC_RHind | GCAAGCTTCAGTTCAGCTCCACCTCGAT | HindIII | This study | |
| GlcE_FEco | GCGAATTCAGTTTCGATTCGGTGGAAAA | For in-frame deletion of glcE in TX1 | EcoRI | This study |
| GlcE_RXho | GCCTCGAGATCGGCCATCAGAAACGCTCC | SmaI | This study | |
| GlcE_FXho | GCCTCGAGGAGCTCTGATGCAAACCAACC | SmaI | This study | |
| GlcE_RHind | GCAAGCTTCGTCTGCACTATGGCTTCG | HindIII | This study | |
| aceA(f)_F | GCGAATTCACGTGCTCGACTAAGCCTTC | For construction of pBaceA | EcoRI | This study |
| aceA(f)_R | GCAAGCTTCTCTTGATCAGCGGCAGTTT | HindIII | This study | |
| aceB(f)_F | GCGAATTCGAAGCGAAGAACGGGTACAG | For construction of pBaceB | EcoRI | This study |
| aceB(f)_R | GCAAGCTTAACCGTCTGCAGTCTTTCTGA | HindIII | This study | |
| glcE(f)_F | TGCCCGGGGAGCTGGAACGCGCCG | For construction of pBglcE | SmaI | This study |
| glcE(f)_R | GCACAAGCTTGATCAGGTAGATGCGCC | HindIII | This study |
Restriction enzyme sites are underlined.
Transposon mutagenesis.
The plasmid pRL27, which carries the transposon Tn5 with a kanamycin (Km) resistance gene, was chosen as the vector for transferring the transposon to P. nitroreducens TX1. The introduction and subsequent transposition of the modified mini-Tn5 transposon into the P. nitroreducens TX1 genome were carried out as previously described by mating TX1 and E. coli BW20767, which carries the transposon delivery vector pRL27 (21, 22). The donor BW20767(pRL27), the helper E. coli HB101(pRK2013), and the recipient strain TX1 were mixed at a ratio of 1:1:1, spotted on an LB agar plate, and cultured at 30°C for 12 h to allow conjugational transfer of the pRL27 plasmid into the recipient cells. Transconjugants were initially selected on MSB agar medium containing 0.5% succinate, ampicillin, and kanamycin. They were then subjected to OPEOn utilization screening as follows. First, single colonies were streaked on MSB agar containing 0.5% OPEOn. The plates were then incubated in an inverted position for several days at 30°C. Mutant strains that did not show visible growth on the plate were designated OPEOn-negative mutants and were then subjected to further study.
Plasmid rescue and recovery of interrupted genes.
Larsen et al. have shown that one-step cloning of the transposon with its associated flanking DNA can be accomplished after mutation by pRL27 (21). The selected OPEOn-negative mutants were grown overnight in LB plus kanamycin and ampicillin, and then 1.5 ml of each culture was transferred to microcentrifuge tubes. Next, the cells were harvested by centrifugation at 10,000 × g for 1 min. Chromosomal DNA was isolated from the pelleted cells as described above and digested with the restriction enzyme BamHI, which does not cut within the transposon sequence of pRL27. The digested DNA was cleaned, which was followed by self-ligation using the T4 ligase at 20°C for 1 h. Material from ligated mixtures was transformed into E. coli DH5α λpir, as previously described, and plated onto LB agar-kanamycin plates in order to select for cells transformed with the ligated pRL27 plasmid that also contained the flanking P. nitroreducens TX1 DNA (23). Next, the transposon with its flanking DNA was isolated using the Midi Plus ultrapure plasmid extraction system (Viogene, Taipei, Taiwan). Primers tpnRL17-1 and tpnRL13-2, which anneal to positions within the transposon sequence in pRL27 and read outwards into flanking DNA regions, were used for sequencing (21). Sequencing of the DNA interrupted by the transposon was carried out on an automatic genetic analyzer (Applied Biosystems). The genes disrupted by the transposon were identified using the BioEdit software by a local BLAST search against the TX1 draft genome sequence and also against the GenBank database.
In-frame deletion mutagenesis.
Four genes (aceA, aceB, fixC, and glcE, which encode isocitrate lyase, malate synthase, dehydrogenase, and glycolate dehydrogenase, respectively) were inactivated by in-frame deletion to avoid any polar effect. The in-frame aceA deletion mutant of strain TX1 was constructed by allelic replacement, as previously described (24). A gene fragment containing about a 98% deletion of the internal region of aceA was created by overlap extension PCR, as described previously (25). The primers used were Ace_FEco, Ace_RXho, Ace_FXho, and Ace_RHind. The internally deleted gene fragment was cloned into a suicide vector (pK18mobsacB), which was named pKaceA. The aceA deletion mutant of TX1 was created by triple mating between strains TX1, E. coli DH5α(pKaceA), and E. coli HB101(pRK2013). The TX1 ΔaceA strain was screened as described previously (24) and confirmed by PCR. The same procedure was applied to construct the TX1 ΔaceB, TX1 ΔfixC, and TX1 ΔglcE mutants using the primers that are described in Table 2.
Complementation of the TX1 ΔaceA, TX1 ΔaceB, and TX1 ΔglcE mutants.
To determine whether the OPEOn utilization deficiency was due to an inactivated gene, the TX1 ΔaceA, TX1 ΔaceB, and TX1 ΔglcE mutants were complemented with appropriate wild-type genes expressed in a broad-host-range vector pBBR1MCS5 (26). For example, the aceA expression vector was constructed as follows: a PCR fragment (2.6 kb) obtained by amplifying the chromosomal DNA of TX1 with the primers aceA(f)_F and aceA(f)_R was ligated into a pGEM-T Easy vector (Promega Co.). The resulting vector (pGaceA) was then amplified in E. coli DH5α, purified, and digested with EcoRI and HindIII to generate the PCR product. Next, the retrieved PCR-amplified fragment was ligated into pBBR1MCS-5, which had been cut with the same restriction enzymes. The resulting plasmid (pBaceA) was then introduced into TX ΔaceA by conjugation using the helper strain E. coli HB101(pRK2013). The presence of the intact aceA gene was confirmed by DNA sequencing. The recombinant TX1 ΔaceA(pBaceA) strain was selected on MSB agar containing 0.5% OPEOn, ampicillin, and gentamicin. The same procedure was applied for complementation of the TX1 ΔaceB and TX1 ΔglcE mutants with pBaceB and pBglcE, respectively, which were constructed using the primers listed in Table 2.
Preparation of crude cell extract from strain TX1.
A 0.5-liter culture of TX1 grown on either 0.5% OPEOn-MSB or 0.5% succinate-MSB was used for the preparation of crude cell extract. The cells were collected by centrifugation (11,000 × g, 10 min, 4°C), washed with 10 mM potassium phosphate buffer (pH 7.0), and suspended in 5 ml of the same buffer. The cell suspension was subjected to sonication for 3 min with 50% pulse on ice using the Sonicator 4000 (Misonix, Farmingdale, NY) to disrupt the cells (27). In the process of cell lysis, 0.15 mM protease inhibitor (phenylmethylsulfonyl fluoride) was added. After the removal of unbroken cells and cell debris by centrifugation (11,000 × g, 10 min, 4°C), the supernatant was collected by ultracentrifugation (200,000 × g, 1 h, 4°C) and used as the crude cell extract. Protein concentrations were determined using the Bradford protein assay with bovine serum albumin as the standard.
Enzyme assays.
The acetaldehyde dehydrogenase activity assay was performed by measuring the rate of appearance of NADH at 340 nm in 1-cm-path cuvette at 25°C with a Beckman DU640 spectrophotometer (Beckman Coulter, Krefeld, Germany). The incubation mixtures contained the following constituents in a final volume of 1 ml: 10 mM potassium phosphate buffer (pH 7.0), 10 mM 2-mercaptoethanol, 2 mM NAD+, 0.5 mg of cell crude extract, and 1 mM acetaldehyde. The reaction was started by the addition of acetaldehyde. For isocitrate lyase activity, a spectrophotometric assay was used to measure isocitrate-dependent formation of glyoxylate. The standard reaction mixture (0.5 ml) contained 10 mM potassium phosphate buffer (pH 7.0), 5 mM MgCl2, 2 mM dithioerythritol, 3.5 mM phenylhydrazine, and 0.5 mg of cell crude extract. The reactions were started by the addition of 2 mM isocitrate, and the formation of the glyoxylate phenylhydrazone derivative was monitored at 324 nm. Malate synthase activity in the crude cell extracts was measured by the method of Srere et al. by tracking the glyoxylate- and acetyl-CoA-dependent release of CoA (28). Acetyl-CoA synthetase activity in the crude cell extracts was measured in the presence of ATP, acetate, and CoA, as described in a previous study (29).
RESULTS AND DISCUSSION
Transposon mutagenesis of P. nitroreducens TX1.
The availability of the draft genome sequence of P. nitroreducens TX1 provides an opportunity for investigating genes that play significant roles in OPEOn utilization (18). In order to identify genes involved in OPEOn catabolism, the Tn5 vector pRL27 (21), which has been used widely in Pseudomonas genetics, was used to construct a mutant library of strain TX1, as described in Materials and Methods. In total, 30,000 Tn5 insertion mutants of TX1 were successfully obtained. After 48 h of incubation at 30°C, these mutants were screened, and 93 (0.31%) of them failed to grow on 0.5% OPEOn-MSB but still grew on 0.5% succinate-MSB.
A total of 6,341 open reading frames (ORFs) have been putatively identified in the TX1 draft genome (18), and therefore, the Tn5 mutant library would seem to have 4.7× ORF coverage of the TX1 genome. The stability of the Tn5 transposon in the transformants was also tested using two of the mutants (C94-4 and A16-7). After 10 generations of the single-colony propagation by subculture on LB plates, the initial and final colonies were found to still retain the kanamycin resistance and, in addition, the Tn5 transposon had remained in the same genomic position on the genome, with this being determined by PCR. These findings confirm that the transposon insertion events present in the transformants from this library are stable.
The transposon insertion points of the 93 mutants defective in OPEOn utilization were identified by the plasmid rescue, followed by nucleotide sequencing using a transposon-derived primer set (21). The insertion loci harboring Tn5 were then mapped to 42 independent coding genes, three noncoding sequences, and one 23S rRNA. These results are summarized in Table 3. Of these 42 genes, 11 had been mutated multiple times to give a number of different mutant strains for a given specific gene. These were the dedA gene, which had 12 mutants; the hit gene, with 11 mutants; the aceB gene, with seven mutants; the aceA and rfaG genes, with six mutants; the rfe gene, with four mutants; the coq7 gene and a gene encoding a hypothetical protein, with three mutants; and the yfgC and fixC genes and a gene encoding a transcriptional regulator, with two mutants (Table 3). These genes with multiple mutations were used for further investigation. Among these 42 disrupted genes, 39 genes could be located within the draft genome of TX1 (accession no. AMZB01000000), while the other three genes, namely, those of the K11-8, C2-42, and T43-26 mutants, were found to be similar to genes present in the genomes of other strains, including Mycobacterium abscessus, Comamonas testosteroni, and Pseudomonas knackmussii (accession numbers WP_049232543, WP_003060809, and WP_043252236.1, respectively), but not within the draft genome of TX1 (18).
TABLE 3.
Characterization of the OPEOn-negative mutants of P. nitroreducens TX1 obtained by Tn5 insertional mutagenesis
| Mutanta | Gene | Functionb | Protein accession no. | Function group (COGs)c |
|---|---|---|---|---|
| C94-4, C100-12, N8-29, N10-35, N36-50, P30-12 | aceA | Isocitrate lyase | WP_017517106.1 | COG2224C |
| A16-7, C87-31, C90-8, K1-02, N38-2, P10-33, T16-15 | aceB | Malate synthase | WP_017520204.1 | COG2225C |
| B1-31, B1-33 | fixC | Dehydrogenase (flavoprotein) | WP_017520493.1 | COG0644C |
| A2-42 | hisP | ABC-type histidine transport system | WP_017517948.1 | COG4598E |
| C39-17 | gdh2 | NAD-specific glutamate dehydrogenase | WP_017519332.1 | COG2902E |
| C96-34 | rhtB | Putative threonine efflux protein | WP_017521173.1 | COG1280E |
| T8-50 | ilvB | Thiamine pyrophosphate-requiring enzyme | WP_017516325.1 | COG0028EH |
| N43-34 | putP | Na+/proline symporter | WP_017518917.1 | COG0591ER |
| C48-18, C77-4, C95-33, P19-3, P19-32, P38-47, P42-36, T10-24, T15-48, T57-1, T58-5 | hit | Diadenosine tetraphosphate hydrolase and other HIT family hydrolases | WP_017517344.1 | COG0537FGR |
| B3-49 | bglX | Beta-glucosidase-related glycosidase | WP_017517232.1 | COG1472G |
| P40-18 | zwf | Glucose-6-phosphate 1-dehydrogenase | WP_017520335.1 | COG0364G |
| K32-8 | gshA | Gamma-glutamylcysteine synthetase | WP_017520855.1 | COG2918H |
| A21-39 | ubiE | Methylase involved in ubiquinone biosynthesis | WP_017521165.1 | COG2226H |
| C73-29, C97-46, P24-25 | coq7 | Demethoxyubiquinone hydroxylase | WP_017517343.1 | COG2941H |
| B3-29 | fadB | 3-Hydroxyacyl-CoA dehydrogenase | WP_017519287.1 | COG1250I |
| A11-1 | yafY | Predicted transcriptional regulator | WP_017518996.1 | COG2378K |
| C4-30 | araC | AraC-type DNA-binding domain-containing protein | WP_017521913.1 | COG2207K |
| B5-50 | aRO8 | Transcriptional regulator | WP_017518869.1 | COG1167KE |
| C15-19 | hepA | Superfamily II DNA/RNA helicase | WP_017522209.1 | COG0553KL |
| K11-8 | dob10 | DEAD/DEAH box helicase | WP_049232543 | COG4581L |
| C2-42 | Tn3 | Transposase | WP_003060809 | COG1961L |
| C82-3, T44-10, T45-21, T67-19, T78-50, T77-39 | rfaG | Glycosyltransferase | WP_017520449.1 | COG0438 M |
| C85-47, T41-40, T66-4, T66-19 | rfe | UDP-N-acetylglucosamine-1-phosphate transferase | WP_017520347.1 | COG0472 M |
| N52-26 | gt2 | Glycosyltransferases | WP_017520448.1 | COG1216 M |
| P33-4 | nlpD | Membrane protein related to metalloendopeptidase | WP_017517352.1 | COG0739 M |
| P55-13 | wcaA | Glycosyltransferase involved in cell wall biogenesis | WP_017517134.1 | COG0463 M |
| T43-26 | galU | UTP-glucose-1-phosphate uridylyltransferase | WP_043252236.1 | COG1210 M |
| B7-14 | wcaG | Nucleoside diphosphate-sugar epimerase | WP_017520346.1 | COG0451MG |
| B6-16 | tar | Methyl-accepting chemotaxis protein | WP_017516259.1 | COG0840NT |
| N24-8 | anmK | Predicted molecular chaperone | WP_017517351.1 | COG2377O |
| A21-12 | COG4590 | ABC-type uncharacterized transport system | WP_017520681.1 | COG4590R |
| A7-47 | ybcL | Membrane-bound metal-dependent hydrolase | WP_017520412.1 | COG1988R |
| B2-48, B7-43 | yfgC | Putative Zn-dependent protease | WP_017519734.1 | COG4783R |
| K17-10 | roxA | Ribosomal protein L16 Arg81 hydroxylase | WP_017517102.1 | COG2850S |
| K21-12 | tauE | Cytochrome biogenesis protein | WP_017516824.1 | COG2836S |
| C3-45, C5-33, C9-50, C61-29, C63-28, C64-38, P32-21, P33-22, T11-1, T15-44, T17-21, T46-48 | dedA | Membrane protein | WP_017521718.1 | COG0586S |
| N30-8 | atoC | Response regulator | WP_017518918.1 | COG2204T |
| P36-32 | GGDEF | GGDEF domain, diguanylate cyclase | WP_017517199.1 | COG2199T |
| C12-38 | norM | Na+-driven multidrug efflux pump | WP_017518643.1 | COG0534V |
| B3-46, B4-25 | Transcriptional regulator | WP_017516487.1 | NC | |
| C11-3, T56-25, T57-21 | Hypothetical protein | WP_017520115.1 | NC | |
| A28-42 | Hypothetical protein | WP_017520049.1 | NC | |
| T12-28 | 23S rRNA | NC | ||
| A6-49 | Noncoding sequence | NC | ||
| B2-47 | Noncoding sequence | NC | ||
| P34-24 | Noncoding sequence | NC |
The representative strains (in bold) were selected for further investigation, as described in the text.
HIT, histidine triad.
The proteins were classified into functional categories according to the clusters of orthologous groups (COG). The functional categories are information storage and processing, including COG categories K (transcription) and L (replication, recombination, and repair); cellular processes and signaling, including O (posttranslational modification, protein turnover, and chaperones), M (cell wall/membrane/envelope biogenesis), N (cell motility), T (signal transduction mechanisms), and V (defense mechanisms); metabolism, including C (energy production and conversion), G (carbohydrate transport and metabolism), E (amino acid transport and metabolism), F (nucleotide transport and metabolism), H (coenzyme transport and metabolism), and I (lipid transport and metabolism); poorly characterized, including R (general function prediction only) and S (function unknown). NC, proteins not classified into a COG.
Functional characterization of the mutant genes.
The proteins encoded by the 42 unique genes identified in this study (OPEOn growth-associated proteins) were grouped into functional classes using the clusters of orthologous groups (COG), as shown in Table 3. When this was done, six proteins (14.3%) were classified into the information storage- and processing-related categories (K and L). Furthermore, the cellular process- and signaling-related categories (O, M, N, T, and V) consisted of 12 proteins (28.6%). Fifteen proteins (35.7%) were present in the metabolism-related categories (C, G, E, F, H, and I). Two poorly characterized COG groups (R and S) were found to contain six proteins (14.3%). In addition, for the category “no related COGs” (the protein is not predicted to belong to any of the currently defined COGs), there were found to be three proteins (7.1%).
Overall, 12 (28.5%) genes were predicted to encode catabolic enzymes. Three proteins (isocitrate lyase, malate synthase, and FixC dehydrogenase) are known to be involved in energy production and conversion, whereas nine proteins (diadenosine tetraphosphate hydrolase, NAD+-specific glutamate dehydrogenase, thiamine pyrophosphate-requiring enzyme, β-glucosidase-related glycosidase, glucose-6-phosphate 1-dehydrogenase, γ-glutamylcysteine synthetase, UbiE methylase involved in ubiquinone biosynthesis, demethoxyubiquinone hydroxylase, and 3-hydroxyacyl-CoA dehydrogenase) are known to be involved in the metabolism of amino acids, carbohydrates, coenzymes, and lipids. Of these, diadenosine tetraphosphate hydrolase (hit) is a key enzyme controlling the in vivo concentration of the dinucleotide diadenosine tetraphosphate that has been proposed to play a range of roles in processes, such as control of DNA replication and repair, signaling in stress response, and apoptosis (30).
OPEOn is one of the most widely used nonionic surfactants; it is used in biology to lyse cells to allow the extraction of protein and other cellular organelles. Previous studies have suggested that OPEOn affects the cell membrane by disrupting its structural integrity and functioning, which results in increased membrane fluidity and permeabilization (31). In our previous work, we found that P. nitroreducens TX1 is able to tolerate up to 20% OPEOn in MSB medium. Therefore, the membrane of TX1 might have special structural features that allow OPEOn to be tolerated. In this context, it should be noted that seven (16.6%) out of the 42 identified proteins, namely, three glycosyltransferases (rfaG, gt2, and wcaA), a UDP-N-acetylglucosamine-1-phosphate transferase (rfe), a membrane protein related to metalloendopeptidase (nlpD), a UTP-glucose-1-phosphate uridylyltransferase (galU), and a nucleoside-diphosphate-sugar epimerase (wcaG), are known to be linked to cell wall/membrane/envelope biogenesis. For example, the deletion of ssg, which encodes a glycosyltransferase in Pseudomonas alkylphenolia KL28, has been shown to cause the loss of lipopolysaccharide O antigen, which alters the composition of the exopolysaccharide. Furthermore, this mutant strain was found to have reduced surface spreading, reduced pellicle formation, and reduced biofilm formation; these were probably due to the cumulative effects of lipopolysaccharide truncation and alterations to the cell's exopolysaccharide composition (23). In some bacterial species, UDP-N-acetylglucosamine-1-phosphate transferase has been shown to play an important role in the biosynthesis of various polymers within the bacterial cell wall, such as common antigen and lipopolysaccharide O antigen (32). UTP-glucose-1-phosphate uridylyltransferase is an enzyme associated with glycogenesis. This enzyme synthesizes UDP-glucose from glucose-1-phosphate. UDP-glucose has been reported to be involved in galactose utilization, in glycogen synthesis, and in the synthesis of various carbohydrate moieties, such as glycolipids, glycoproteins, and proteoglycans (33). Interestingly, yfgC codes for a putative Zn-dependent protease, and although its function is unknown, the chemical and genetic data suggest that it also plays a role in outer membrane protein biogenesis (34). Furthermore, five (11.9%) out of the 42 genes were predicted to encode transport proteins, among which two ABC transporters were identified. Such transport proteins were likely to play important roles in the transport of OPEOn into the cell. For the functions of the DedA membrane protein, the recent genetic approaches have revealed important roles in membrane homeostasis. Bacterial DedA family mutants display such intriguing phenotypes as cell division defects, temperature sensitivity, altered membrane lipid composition, elevated envelope-related stress responses, and loss of proton motive force (35). Finally, there are a significant number of mutants that fall within the hypothetical, unknown, and unclassified gene categories, which suggests that there is still a large number of unknown genes across the P. nitroreducens genome potentially involved in OPEOn metabolism that remain to be explored, including the three not found in the TX1 draft genome. Further studies on these genes using complementation and other strategies will improve our understanding of the strain TX1 and its tolerance and degradation of OPEOn.
Role of the glyoxylate cycle in OPEOn degradation by TX1.
Isocitrate lyase and malate synthase, which are both key enzymes in the glyoxylate cycle, were frequently detected during screening of mutants unable to grow on 0.5% OPEOn. In the glyoxylate cycle, two molecules of acetyl-CoA are converted into oxaloacetate, thus bypassing the reactions of the tricarboxylic acid (TCA) cycle in which CO2 is released. The glyoxylate cycle is essential when cells are growing on C2 compounds, because this pathway allows the synthesis of all cellular compounds with three or more carbon atoms; examples include the biosynthesis of amino acids and nucleotides. To confirm the role of these two enzymes in OPEOn utilization, in-frame deletion mutants lacking the aceA and aceB genes were created from strain TX1. As expected from the transposon mutagenesis results, the TX1 ΔaceA and TX1 ΔaceB mutants were unable to grow on OPEOn. The in-frame deletion mutants were then used for complementation analysis. Two plasmids, pBaceA and pBaceB, which carry the aceA and aceB genes, respectively, were constructed and transferred independently from E. coli into the TX1 ΔaceA and TX1 ΔaceB mutant strains by conjugation. In both cases, the introduction of the wild-type copy of the gene resulted in a return to the wild-type growth pattern, confirming the role of the aceA and aceB in OPEOn utilization. In addition, isocitrate lyase and malate synthase activities in cell extract of wild-type cells grown on OPEOn were 277.6 ± 5 (mean ± standard deviation) and 15.1 ± 5 nmol · min−1 · mg−1, respectively. These activities were downregulated 8.8- and 1.7-fold in cells grown on succinate (31.3 ± 6.5 and 8.7 ± 2 nmol · min−1 · mg−1, respectively). These results clearly reveal that the glyoxylate cycle plays a critical role in OPEOn degradation by strain TX1.
Growth of the mutants on different carbon sources.
Eleven mutants (TX1 ΔaceA, TX1 ΔaceB, TX1 ΔfixC, C48-18, C73-29, C82-3, C85-47, B2-48, C3-45, B3-46, and C11-3), which are representatives of the 11 genes present in the Tn5 library as multiple events, TX1 ΔglcE, and the three complemented strains [TX1 ΔaceA(pBaceA), TX1 ΔaceB(pBaceB), and TX1 ΔglcE(pBglcE)] were tested for their growth on 0.1% OPEOn, 0.1% NPEOn, 0.1% dodecyl octaethoxylate (AEO8), 0.1% ethanol, 0.1% acetate, 0.1% glycolate, or 0.1% pyruvate as the sole source of carbon (Table 4). The glcE gene was deleted to test the cleavage of OPEOn by terminal carboxylation, followed by hydrolysis to yield glycolate in strain TX1 (Fig. 1, oxidation). Wild-type TX1 is unable to grow on polyethylene glycol 400 (PEG 400), polyethylene glycol 1000 (PEG 1000), acetaldehyde, or glyoxylate; therefore, these compounds were not used for growth testing of the mutants. The generation time of wild-type TX1 was much longer when grown on glycolate (10.8 to 11.8 h) than on OPEOn, NPEOn, AEO8, ethanol, acetate, or pyruvate (1.5 to 4.8 h) (Table 4). All mutants were able to grow on pyruvate, with generation times ranging from 3.6 to 4 h. When NPEOn or AEO8 was used as the sole source of carbon, five mutants, TX1 ΔfixC, TX1 ΔglcE, TX1 ΔaceA(pBaceA), TX1 ΔaceB(pBaceB), and TX1 ΔglcE(pBglcE), showed growth on these compounds, with generation times ranging from 1.5 to 3.7 h, whereas the rest of mutants failed to grow at all. When ethanol or acetate was used as the sole source of carbon, the two glyoxylate cycle mutants (TX1 ΔaceA and TX1 ΔaceB) failed to grow, but others were able to grow on ethanol or acetate, with generation times ranging from 2.7 to 4.8 h. In addition, the two mutants TX1 ΔaceB and TX1 ΔglcE failed to grow on 0.1% glycolate, but the other mutants were able to grow on it, with generation times ranging from 10.8 to 11.8 h.
TABLE 4.
Growth of TX1 mutants on various carbon sources
| Straina | Description | Generation time (mean ± SD) (h)b |
||||||
|---|---|---|---|---|---|---|---|---|
| OPEOn | NPEOn | AEO8 | Ethanol | Acetate | Pyruvate | Glycolate | ||
| TX1 | Wild type | 1.5 ± 0.1 | 3.2 ± 0.1 | 3.7 ± 0.2 | 4.8 ± 0.2 | 2.7 ± 0.2 | 3.6 ± 0.4 | 11.3 ± 3.1 |
| TX1 ΔaceA | Deletion of isocitrate lyase gene | − | − | − | − | − | 3.6 ± 0.4 | 11.5 ± 2.5 |
| TX1 ΔaceB | Deletion of malate synthase gene | − | − | − | − | − | 3.6 ± 0.4 | − |
| TX1 ΔfixC | Deletion of dehydrogenase gene | 1.5 ± 0.1 | 3.1 ± 0.1 | 3.7 ± 0.1 | 4.8 ± 0.2 | 2.7 ± 0.2 | 3.6 ± 0.4 | 10.8 ± 2.2 |
| TX1 ΔglcE | Deletion of glycolate dehydrogenase gene | 1.5 ± 0.1 | 3.2 ± 0.1 | 3.7 ± 0.1 | 4.7 ± 0.1 | 2.9 ± 0.3 | 4 ± 0.2 | − |
| C48-18 | Mutation at diadenosine tetraphosphate hydrolase gene | − | − | − | 4.7 ± 0.2 | 2.7 ± 0.2 | 4 ± 0.2 | 11.2 ± 4.2 |
| C73-29 | Mutation at demethoxyubiquinone hydroxylase gene | − | − | − | 4.7 ± 0.2 | 2.7 ± 0.2 | 4 ± 0.2 | 11.3 ± 3.3 |
| C82-3 | Mutation at glycosyltransferase gene | − | − | − | 4.7 ± 0.2 | 2.9 ± 0.3 | 4 ± 0.2 | 11.5 ± 2.8 |
| C85-47 | Mutation at UDP-N-acetylglucosamine-1-phosphate transferase gene | − | − | − | 4.7 ± 0.1 | 2.7 ± 0.2 | 4 ± 0.2 | 11.7 ± 2.5 |
| B2-48 | Mutation at putative Zn-dependent protease gene | − | − | − | 4.8 ± 0.2 | 2.9 ± 0.3 | 4 ± 0.2 | 10.9 ± 3.2 |
| C3-45 | Mutation at gene encoding membrane protein | − | − | − | 4.7 ± 0.2 | 2.9 ± 0.3 | 4 ± 0.2 | 11.6 ± 2.7 |
| B3-46 | Mutation at gene encoding transcriptional regulator | − | − | − | 4.8 ± 0.1 | 2.9 ± 0.3 | 4 ± 0.2 | 11.8 ± 2.8 |
| C11-3 | Mutation at gene encoding unknown protein | − | − | − | 4.7 ± 0.1 | 2.9 ± 0.3 | 4 ± 0.2 | 11.7 ± 3.1 |
| TX1 ΔaceA(pBaceA) | Complementation of isocitrate lyase gene | 1.5 ± 0.1 | 3.2 ± 0.1 | 3.6 ± 0.2 | 4.7 ± 0.1 | 2.9 ± 0.3 | 4 ± 0.2 | 11.1 ± 3.2 |
| TX1 ΔaceB(pBaceB) | Complementation of malate synthase gene | 1.5 ± 0.1 | 3.1 ± 0.1 | 3.7 ± 0.1 | 4.7 ± 0.2 | 2.9 ± 0.3 | 4 ± 0.2 | 10.8 ± 2.6 |
| TX1 ΔglcE(pBglcE) | Complementation of glycolate dehydrogenase gene | 1.5 ± 0.1 | 3.1 ± 0.1 | 3.7 ± 0.1 | 4.8 ± 0.2 | 2.9 ± 0.3 | 4 ± 0.2 | 11.2 ± 2.8 |
Each strain was cultured in 5 ml of MSB plus 0.1% of the relevant carbon source.
−, no growth; OPEOn, Triton X-100; NPEOn, Triton N-101; AEO8, dodecyl octaethoxylate. Data are the results from three independent experiments.
Proposed mechanism for the degradation of OPEOn by P. nitroreducens TX1.
The biodegradation of long-chain APEOn has been studied using isolated bacterial strains that grow solely on OPEOn or NPEOn. Most isolates are from the genus Pseudomonas, which belongs to the Gammaproteobacteria (9, 13, 14). Based on the results of metabolite analyses, the initial degradation reaction of APEOn has been found to be a shortening of the ethoxylate chain by exoscission of the EO chains from the hydroxyl terminal side (13, 14, 16) (Fig. 1). Our group isolated Pseudomonas strain TX1, which has the ability to grow on OPEOn at a wide range of concentrations (9). LC-MS analysis of the intermediate metabolites revealed the presence of a gradual shortening process that affected the EO chains during OPEOn utilization. The results suggested that the same exo-type biodegradation process is occurring in TX1 as in other isolates.
The terminal oxidation model for the biodegradation of PEG under aerobic conditions has been reviewed previously (36, 37). In this model, the biodegradation of the PEG molecules is initiated through the oxidation of the terminal EO unit to a carboxyl group, which is followed by the liberation of glycolate. The carboxyl is converted to glyoxylate by glycolate dehydrogenase (GlcE). Tasaki et al. (38) isolated the adh1 gene from P. putida S-5 and expressed this gene in E. coli. By measuring the reduction of 2,6-dichlorophenolindophenol (DCPIP) spectrophotometrically at 600 nm using a cell crude extract, alcohol dehydrogenase (Adh1) was shown to have activity against OPEOn of various EO chain lengths (38). However, its relevance to the ether cleavage step in OPEOn biodegradation remains unknown. The use of redox dye-based assay with crude extracts can be misleading, because crude cell extract may contain multiple activities that are able to utilize DCPIP as an electron acceptor (17). Furthermore, the detection of glycolate and an increase in GlcE activity have not been reported; such findings would support the terminal oxidation model. In our study, a fixC gene encoding a dehydrogenase was found among the mutants. However, an in-frame deletion mutant of the fixC gene retained the ability to grow on OPEOn in a manner similar to the wild type. These results suggest that the transposon insertion into fixC has a polar effect, and the fixC gene product itself may not play an important role in OPEOn utilization. In addition, TX1 grows only slowly on glycolate but does not grow at all on glyoxylate, PEG 400, or PEG 1000 as the sole source of carbon. In order to determine whether glycolic acid is involved in the metabolism of OPEOn by strain TX1, a glcE in-frame deletion mutant was constructed. As shown in Table 4, TX1 ΔglcE grew on OPEOn to a level comparable to that of the wild type. Interestingly, TX1 ΔglcE and TX1 ΔaceB were unable to grow on glycolate, whereas TX1 ΔaceA was able to grow. Interestingly, one isocitrate lyase gene was found in the TX1 genome (18). In addition, the isocitrate lyase activity in the cell extract of TX1 ΔaceA cells grown on glycolate or succinate was not detectable. These results suggest that the degradation of glycolate does not involve the glyoxylate cycle, because the isocitrate lyase mutant was able to grow on glycolate in a manner similar to the wild-type TX1 (Fig. 2). Considering the results, we conclude that the degradation of OPEOn by strain TX1 does not demonstrate an oxidative pathway involving the liberation of glycolate.
FIG 2.

Proposed pathway and mechanism (black lines) for the degradation of OPEOn by P. nitroreducens TX1.
In the nonoxidative biodegradation model, biodegradation of the EO chain proceeds via the shift of a hydroxyl group, followed by the liberation of acetaldehyde, which is then quickly transformed to acetate by dehydrogenases that are ubiquitously distributed throughout the cell (39). The pathway for acetate degradation is well known and involves the conversion of acetate into acetyl-CoA, which then enters in the central carbon metabolism of the cell (39). In the glyoxylate cycle, the input of two molecules of acetyl-CoA results in a net synthesis of one molecule of succinate, which is then available for biosynthetic purposes (40). As shown in Table 4, TX1 was able to grow on either ethanol or acetate as the sole source of carbon, with generation times ranging from 2.7 to 4.8 h, but TX1 ΔaceA and TX1 ΔaceB were unable to grow on these carbon sources. These results suggest that the glyoxylate cycle plays a critical role in ethanol or acetate catabolism. The ethanol oxidation product acetate must first be activated to acetyl-CoA so it can be used as a carbon source and for the replenishment of intermediates within the TCA cycle; this would occur via the glyoxylate bypass in strain TX1. Our results suggest that nonoxidative biodegradation with the liberation of acetaldehyde is the most likely pathway for OPEOn utilization in strain TX1 (Fig. 2). In addition, acetaldehyde dehydrogenase and acetyl-CoA synthetase activities were upregulated 13.1- and 2.1-fold in cells grown on OPEOn (211 ± 14 and 68.8 ± 1.1 nmol · min−1 · mg−1, respectively) versus TX1 grown on succinate (16 ± 12 and 32.6 ± 1.2 nmol · min−1 · mg−1, respectively). Further studies on the mechanism used for the liberation of acetaldehyde will be needed in order to improve our understanding of the degradation of OPEOn by strain TX1.
ACKNOWLEDGMENTS
This work was supported by the Ministry of Science and Technology, Taiwan (grant NSC102-2628-B-008-001-MY3) and the NRF-NSC cooperation program by the National Research Foundation of Korea (grant NRF-2013K2A1B8053138) and Ministry of Science and Technology, Taiwan (grant NSC 102-2923-B-008-001-MY2).
REFERENCES
- 1.Chen HJ, Tseng DH, Huang SL. 2005. Biodegradation of octylphenol polyethoxylate surfactant Triton X-100 by selected microorganisms. Bioresour Technol 96:1483–1491. doi: 10.1016/j.biortech.2004.11.013. [DOI] [PubMed] [Google Scholar]
- 2.Saito I, Onuki A, Seto H. 2004. Indoor air pollution by alkylphenols in Tokyo. Indoor Air 14:325–332. doi: 10.1111/j.1600-0668.2004.00250.x. [DOI] [PubMed] [Google Scholar]
- 3.Pagano M, Lopez A, Volpe A, Mascolo G, Ciannarella R. 2008. Oxidation of nonionic surfactants by Fenton and H2O2/UV processes. Environ Technol 29:423–433. doi: 10.1080/09593330801983862. [DOI] [PubMed] [Google Scholar]
- 4.Giger W, Brunner PH, Schaffner C. 1984. 4-Nonylphenol in sewage sludge: accumulation of toxic metabolites from nonionic surfactants. Science 225:623–625. doi: 10.1126/science.6740328. [DOI] [PubMed] [Google Scholar]
- 5.Huang SL, Tuan NN, Lee K. 2016. Occurrence, human intake and biodegradation of estrogen-like nonylphenols and octylphenols. Curr Drug Metab 17:293–302. doi: 10.2174/1389200217666151210124821. [DOI] [PubMed] [Google Scholar]
- 6.Tanenbaum DM, Wang Y, Williams SP, Sigler PB. 1998. Crystallographic comparison of the estrogen and progesterone receptor's ligand binding domains. Proc Natl Acad Sci U S A 95:5998–6003. doi: 10.1073/pnas.95.11.5998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Sahambi SK, Pelland A, Cooke GM, Schrader T, Tardif R, Charbonneau M, Krishnan K, Haddad S, Cyr DG, Devine PJ. 2010. Oral p-tert-octylphenol exposures induce minimal toxic or estrogenic effects in adult female Sprague-Dawley rats. J Toxicol Environ Health A 73:607–622. doi: 10.1080/15287390903566682. [DOI] [PubMed] [Google Scholar]
- 8.Ying GG, Williams B, Kookana R. 2002. Environmental fate of alkylphenols and alkylphenol ethoxylates—a review. Environ Int 28:215–226. doi: 10.1016/S0160-4120(02)00017-X. [DOI] [PubMed] [Google Scholar]
- 9.Lin YW, Guo GL, Hsieh HC, Huang SL. 2010. Growth of Pseudomonas sp. TX1 on a wide range of octylphenol polyethoxylate concentrations and the formation of dicarboxylated metabolites. Bioresour Technol 101:2853–2859. [DOI] [PubMed] [Google Scholar]
- 10.Chen HJ, Guo GL, Tseng DH, Cheng CL, Huang SL. 2006. Growth factors, kinetics and biodegradation mechanism associated with Pseudomonas nitroreducens TX1 grown on octylphenol polyethoxylates. J Environ Manage 80:279–286. doi: 10.1016/j.jenvman.2005.09.009. [DOI] [PubMed] [Google Scholar]
- 11.Chen HJ, Huang SL, Tseng DH. 2004. Aerobic biotransformation of octylphenol polyethoxylate surfactant in soil microcosms. Environ Technol 25:201–210. doi: 10.1080/09593330409355453. [DOI] [PubMed] [Google Scholar]
- 12.Tuan NN, Hsieh HC, Lin YW, Huang SL. 2011. Analysis of bacterial degradation pathways for long-chain alkylphenols involving phenol hydroxylase, alkylphenol monooxygenase and catechol dioxygenase genes. Bioresour Technol 102:4232–4240. doi: 10.1016/j.biortech.2010.12.067. [DOI] [PubMed] [Google Scholar]
- 13.Nguyen MH, Sigoillot JC. 1996. Isolation from coastal sea water and characterization of bacterial strains involved in non-ionic surfactant degradation. Biodegradation 7:369–375. [DOI] [PubMed] [Google Scholar]
- 14.John DM, White GF. 1998. Mechanism for biotransformation of nonylphenol polyethoxylates to xenoestrogens in Pseudomonas putida. J Bacteriol 180:4332–4338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Nishio E, Ichiki Y, Tamura H, Morita S, Watanabe K, Yoshikawa H. 2002. Isolation of bacterial strains that produce the endocrine disruptor, octylphenol diethoxylates, in paddy fields. Biosci Biotechnol Biochem 66:1792–1798. doi: 10.1271/bbb.66.1792. [DOI] [PubMed] [Google Scholar]
- 16.Maki H, Masuda N, Fujiwara Y, Ike M, Fujita M. 1994. Degradation of alkylphenol ethoxylates by Pseudomonas sp. strain TR01. Appl Environ Microbiol 60:2265–2271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.White GF, Russell NJ, Tidswell EC. 1996. Bacterial scission of ether bonds. Microbiol Rev 60:216–232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Huang SL, Chen H, Hu A, Tuan NN, Yu CP. 2014. Draft genome sequence of Pseudomonas nitroreducens strain TX1, which degrades nonionic surfactants and estrogen-like alkylphenols. Genome Announc 2(1):. doi: 10.1128/genomeA.01262-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ausubel FM, Brent R, Kingston RE, Moore DD, Seidman JG, Smith JA, Struhl K (ed). 2003. Current protocols in molecular biology, ringbou ed. John Wiley & Sons, Charlottesville, VA. [Google Scholar]
- 20.Sambrook J, Fritsch EF, Maniatis T. 1989. Molecular cloning: a laboratory manual, 2nd ed Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY. [Google Scholar]
- 21.Larsen RA, Wilson MM, Guss AM, Metcalf WW. 2002. Genetic analysis of pigment biosynthesis in Xanthobacter autotrophicus Py2 using a new, highly efficient transposon mutagenesis system that is functional in a wide variety of bacteria. Arch Microbiol 178:193–201. doi: 10.1007/s00203-002-0442-2. [DOI] [PubMed] [Google Scholar]
- 22.Figurski DH, Helinski DR. 1979. Replication of an origin-containing derivative of plasmid RK2 dependent on a plasmid function provided in trans. Proc Natl Acad Sci U S A 76:1648–1652. doi: 10.1073/pnas.76.4.1648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Veeranagouda Y, Lee K, Cho AR, Cho K, Anderson EM, Lam JS. 2011. Ssg, a putative glycosyltransferase, functions in lipo- and exopolysaccharide biosynthesis and cell surface-related properties in Pseudomonas alkylphenolia. FEMS Microbiol Lett 315:38–45. doi: 10.1111/j.1574-6968.2010.02172.x. [DOI] [PubMed] [Google Scholar]
- 24.Schäfer A, Tauch A, Jager W, Kalinowski J, Thierbach G, Puhler A. 1994. Small mobilizable multi-purpose cloning vectors derived from the Escherichia coli plasmids pK18 and pK19: selection of defined deletions in the chromosome of Corynebacterium glutamicum. Gene 145:69–73. doi: 10.1016/0378-1119(94)90324-7. [DOI] [PubMed] [Google Scholar]
- 25.Ho SN, Hunt HD, Horton RM, Pullen JK, Pease LR. 1989. Site-directed mutagenesis by overlap extension using the polymerase chain reaction. Gene 77:51–59. doi: 10.1016/0378-1119(89)90358-2. [DOI] [PubMed] [Google Scholar]
- 26.Kovach ME, Elzer PH, Hill DS, Robertson GT, Farris MA, Roop RM Jr, Peterson KM. 1995. Four new derivatives of the broad-host-range cloning vector pBBR1MCS, carrying different antibiotic-resistance cassettes. Gene 166:175–176. doi: 10.1016/0378-1119(95)00584-1. [DOI] [PubMed] [Google Scholar]
- 27.Tuan NN, Lin YW, Huang SL. 2013. Catabolism of 4-alkylphenols by Acinetobacter sp. OP5: genetic organization of the oph gene cluster and characterization of alkylcatechol 2, 3-dioxygenase. Bioresour Technol 131:420–428. [DOI] [PubMed] [Google Scholar]
- 28.Srere PA, Brazil H, Gonen L. 1963. The citrate condensing enzyme of pigeon breast muscle and moth flight muscle. Acta Chem Scand 17:129–134. [Google Scholar]
- 29.Gardner JG, Grundy FJ, Henkin TM, Escalante-Semerena JC. 2006. Control of acetyl-coenzyme A synthetase (AcsA) activity by acetylation/deacetylation without NAD(+) involvement in Bacillus subtilis. J Bacteriol 188:5460–5468. doi: 10.1128/JB.00215-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Szurmak B, Wyslouch-Cieszynska A, Wszelaka-Rylik M, Bal W, Dobrzanska M. 2008. A diadenosine 5′,5″-P1P4 tetraphosphate (Ap4A) hydrolase from Arabidopsis thaliana that is activated preferentially by Mn2+ ions. Acta Biochim Pol 55:151–160. [PubMed] [Google Scholar]
- 31.Koley D, Bard AJ. 2010. Triton X-100 concentration effects on membrane permeability of a single HeLa cell by scanning electrochemical microscopy (SECM). Proc Natl Acad Sci U S A 107:16783–16787. doi: 10.1073/pnas.1011614107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Al-Dabbagh B, Mengin-Lecreulx D, Bouhss A. 2008. Purification and characterization of the bacterial UDP-GlcNAc:undecaprenyl-phosphate GlcNAc-1-phosphate transferase WecA. J Bacteriol 190:7141–7146. doi: 10.1128/JB.00676-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Thoden JB, Holden HM. 2007. The molecular architecture of glucose-1-phosphate uridylyltransferase. Protein Sci 16:432–440. doi: 10.1110/ps.062626007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Oh E, Becker AH, Sandikci A, Huber D, Chaba R, Gloge F, Nichols RJ, Typas A, Gross CA, Kramer G, Weissman JS, Bukau B. 2011. Selective ribosome profiling reveals the cotranslational chaperone action of trigger factor in vivo. Cell 147:1295–1308. doi: 10.1016/j.cell.2011.10.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Doerrler WT, Sikdar R, Kumar S, Boughner LA. 2013. New functions for the ancient DedA membrane protein family. J Bacteriol 195:3–11. doi: 10.1128/JB.01006-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kawai F, Kimura T, Fukaya M, Tani Y, Ogata K, Ueno T, Fukami H. 1978. Bacterial oxidation of polyethylene glycol. Appl Environ Microbiol 35:679–684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kawai F. 2002. Microbial degradation of polyethers. Appl Microbiol Biotechnol 58:30–38. doi: 10.1007/s00253-001-0850-2. [DOI] [PubMed] [Google Scholar]
- 38.Tasaki Y, Yoshikawa H, Tamura H. 2006. Isolation and characterization of an alcohol dehydrogenase gene from the octylphenol polyethoxylate degrader Pseudomonas putida S-5. Biosci Biotechnol Biochem 70:1855–1863. doi: 10.1271/bbb.60009. [DOI] [PubMed] [Google Scholar]
- 39.Alber BE, Spanheimer R, Ebenau-Jehle C, Fuchs G. 2006. Study of an alternate glyoxylate cycle for acetate assimilation by Rhodobacter sphaeroides. Mol Microbiol 61:297–309. doi: 10.1111/j.1365-2958.2006.05238.x. [DOI] [PubMed] [Google Scholar]
- 40.Kornberg HL, Krebs HA. 1957. Synthesis of cell constituents from C2-units by a modified tricarboxylic acid cycle. Nature 179:988–991. doi: 10.1038/179988a0. [DOI] [PubMed] [Google Scholar]