

ABSTRACT
The araC-ParaBAD inducible promoter system is tightly controlled and allows gene expression to be modulated over a wide range in Escherichia coli, which has led to its widespread use in other bacteria. Although anecdotal evidence suggests that araC-ParaBAD is leaky in Pseudomonas aeruginosa, neither a thorough analysis of this inducible promoter system in P. aeruginosa nor a concerted effort to identify alternatives with improved functionality has been reported. Here, we evaluated the functionality of the araC-ParaBAD system in P. aeruginosa. Using transcriptional fusions to a lacZ reporter gene, we determined that the noninduced expression from araC-ParaBAD is high and cannot be reduced by carbon catabolite repression as it can in E. coli. Modulating translational initiation by altering ribosome-binding site strength reduced the noninduced activity but also decreased the maximal induced activity and narrowed the induction range. Integrating the inducible promoter system into the posttranscriptional regulatory network that controls catabolite repression in P. aeruginosa significantly decreased the noninduced activity and increased the induction range. In addition to these improvements in the functionality of the araC-ParaBAD system, we found that the lacIq-Ptac and rhaSR-PrhaBAD inducible promoter systems had significantly lower noninduced expression and were inducible over a broader range than araC-ParaBAD. We demonstrated that noninduced expression from the araC-ParaBAD system supported the function of genes involved in antibiotic resistance and tryptophan biosynthesis in P. aeruginosa, problems that were avoided with rhaSR-PrhaBAD. rhaSR-PrhaBAD is tightly controlled, allows gene expression over a wide range, and represents a significant improvement over araC-ParaBAD in P. aeruginosa.
IMPORTANCE We report the shortcomings of the commonly used Escherichia coli araC-ParaBAD inducible promoter system in Pseudomonas aeruginosa, successfully reengineered it to improve its functionality, and show that the E. coli rhaSR-PrhaBAD system is tightly controlled and allows inducible gene expression over a wide range in P. aeruginosa.
INTRODUCTION
Pseudomonas aeruginosa is a versatile Gram-negative bacterium that inhabits a variety of different environments. It is also an opportunistic human pathogen that causes acute infections in hospitalized patients as well as chronic infections in cystic fibrosis patients. Unfortunately, P. aeruginosa infections are becoming difficult to treat because of the increasing prevalence of multidrug (antibiotic) resistance (1). To improve the treatment of these infections, we need to understand which gene functions are essential for the growth of P. aeruginosa and develop new therapeutics to inhibit them. The study of essential genes is difficult because, by definition, inactivation of an essential gene is lethal to the cell. Analysis of essential genes generally involves the construction of conditional mutants, often accomplished by controlling the expression of a gene with an inducible promoter. Inducible promoters allow the transcription of a gene to be turned on and modulated by the addition of an inducer as well as turned off when the inducer is removed. The inability to turn off gene expression can make it difficult to determine the function of a gene, particularly when low-level expression is sufficient for gene function. Inducible promoters should also allow expression over a wide range so that the induced expression can both match the native expression level and exceed it when overexpression of the gene is desired.
In the model bacterium Escherichia coli, the araC-ParaBAD inducible promoter system satisfies these criteria (2, 3). In the absence of arabinose, transcription from the araBAD promoter is repressed by the regulatory protein AraC. When AraC binds arabinose, it is repositioned at the araBAD promoter and activates transcription (4). The araBAD promoter is also controlled by carbon catabolite repression that prevents the transcription of genes necessary for the metabolism of less-preferred carbon sources (such as arabinose) when a preferred one (glucose) is available (5, 6). In the absence of glucose, the EIIA component of the glucose-specific phosphotransferase system (EIIAGlc) is phosphorylated and stimulates adenylate cyclase to produce cyclic AMP (cAMP). The transcriptional activator protein CRP (also called the catabolite activator protein, CAP) binds cAMP and activates the araBAD promoter. In the presence of glucose, EIIAGlc transfers its phosphate to glucose as it is transported across the cell envelope and into the cell. Nonphosphorylated EIIAGlc cannot stimulate adenylate cyclase, cAMP is not produced, and CRP cannot activate the araBAD promoter. As a result of these regulatory factors, gene expression from the araBAD promoter can be induced, and modulated over a wide range, by the addition of arabinose (3). In the absence of arabinose, the noninduced expression from the promoter is low, and it can be reduced even further by the addition of glucose. This tight control is arguably the most important feature of the inducible promoter system, particularly in the study of essential genes where high noninduced expression (leakiness) can prevent the determination of gene function. Promoter leakiness can also obscure the phenotype associated with any gene, whether essential or nonessential, when only a small amount of gene product is necessary for gene function.
The recognition of the tight control of the araC-ParaBAD system has prompted its use in other bacteria, including P. aeruginosa. While the basic regulatory features of AraC are preserved in P. aeruginosa, carbon catabolite repression is fundamentally different (7). Instead of utilizing a phosphotransferase system, P. aeruginosa imports glucose through an ATP-binding cassette transporter. Without EIIAGlc, adenylate cyclase is not stimulated to produce cAMP in the absence of glucose. In fact, cAMP levels do not change when P. aeruginosa is grown with different carbon sources (8), and carbon source preference, the organizing principle behind catabolite repression, is different in P. aeruginosa. P. aeruginosa prefers to catabolize amino acids and organic acids rather than sugars, such as glucose (9, 10). The regulatory factors that enforce catabolite repression in P. aeruginosa are Hfq, Crc, CbrAB, and CrcZ (7, 11). Hfq binds mRNA necessary for the assimilation of alternative carbon sources near the ribosome-binding site (RBS) and inhibits the formation of the translation initiation complex. The small regulatory RNA CrcZ modulates Hfq availability. It contains five Hfq-binding sites and can sequester Hfq. The CbrAB two-component transcriptional regulatory system controls the amount of CrcZ according to the carbon source being used (12).
Despite these fundamental differences in catabolite repression, the araC-ParaBAD system has been used effectively to induce the expression of several genes in P. aeruginosa (13–17). In various cases, however, we and others have noticed that araC-ParaBAD may not be as tightly controlled in P. aeruginosa as in E. coli. This motivated us to examine whether araC-ParaBAD meets the same desired criteria in P. aeruginosa that provoked its widespread use in E. coli and other bacteria. To this end, we assessed the behavior of the araC-ParaBAD system in P. aeruginosa. We found that transcription from araC-ParaBAD was high in the absence of inducer and could not be decreased by catabolite repression. We then sought to improve the functionality of the system and compare it to the lacIq-Ptac and rhaSR-PrhaBAD inducible promoter systems.
MATERIALS AND METHODS
General methods.
The strains, plasmids, and oligonucleotide primers used in this study are listed in Tables 1, 2, and 3, respectively. Strains were grown in lysogeny broth (LB; 1% tryptone, 0.5% yeast extract, 1% sodium chloride) and M9 minimal medium (48 mM sodium phosphate dibasic, 22 mM potassium phosphate monobasic, 8.6 mM sodium chloride, 19 mM ammonium chloride, 2.0 mM magnesium sulfate, 0.1 mM calcium chloride) with the indicated carbon sources. When necessary for strain construction, E. coli strains were grown in medium supplemented with 100 μg/ml ampicillin, 30 μg/ml kanamycin, or 10 μg/ml tetracycline; P. aeruginosa strains were grown in medium supplemented with 30 μg/ml gentamicin, 25 μg/ml tetracycline, or 250 μg/ml carbenicillin.
TABLE 1.
Strains
| Strain | Genotype |
|---|---|
| P. aeruginosa | |
| PA103 | Serotype O11 |
| PAO1 | Serotype O5 |
| PA14 | Serotype O10 |
| PAJM91 | PA103 attTn7::araC-ParaBAD-stRBS-lacZ frt |
| PAJM423 | PA103 attTn7::araC-ParaBAD-intRBS-lacZ frt |
| PAJM395 | PA103 attTn7::araC-ParaBAD-wkRBS-lacZ frt |
| PAJM430 | PA103 attTn7::araC-ParaBAD-5′ UTR amiE-lacZ frt |
| PAJM93 | PA103 attTn7::lacIq-Ptac-stRBS-lacZ frt |
| PAJM207 | PA103 attTn7::rhaSR-PrhaBAD-stRBS-lacZ frt |
| PAJM95 | PA103 attTn7::stRBS-lacZ frt |
| PAJM14 | PA103 attTn7::frt |
| PAJM283 | PA103 attTn7::araC-ParaBAD-stRBS-aacC1 frt |
| PAJM285 | PA103 attTn7::rhaSR-PrhaBAD-stRBS-aacC1 frt |
| PAJM290 | PA103 attCTX::tet |
| PAJM302 | PA103 attCTX::araC-ParaBAD-stRBS-aacC1 tet |
| PAJM304 | PA103 attCTX::rhaSR-PrhaBAD-stRBS-aacC1 tet |
| PAJM291 | PAO1 attCTX::tet |
| PAJM308 | PAO1 attCTX::araC-ParaBAD-stRBS-aacC1 tet |
| PAJM310 | PAO1 attCTX::rhaSR-PrhaBAD-stRBS-aacC1 tet |
| PAJM292 | PA14 attCTX::tet |
| PAJM314 | PA14 attCTX::araC-ParaBAD-stRBS-aacC1 tet |
| PAJM316 | PA14 attCTX::rhaSR-PrhaBAD-stRBS-aacC1 tet |
| PAJM259 | PA14 trpF::TnMar gm |
| PAJM297 | PA14 trpF::TnMar gm attCTX::araC-ParaBAD-trpF tet |
| PAJM326 | PA14 trpF::TnMar gm attCTX::rhaSR-PrhaBAD-trpF tet |
| PAJM258 | PA14 trpC::TnMar gm |
| PAJM295 | PA14 trpC::TnMar gm attCTX::araC-ParaBAD-trpC tet |
| PAJM324 | PA14 trpC::TnMar gm attCTX::rhaSR-PrhaBAD-trpC tet |
| PAJM235 | PA14 trpA::TnMar gm |
| PAJM293 | PA14 trpA::TnMar gm attCTX::araC-ParaBAD-trpA tet |
| PAJM322 | PA14 trpA::TnMar gm attCTX::rhaSR-PrhaBAD-trpA tet |
| E. coli | |
| DH5α | F− endA1 glnV42 thi-1 recA1 relA1 gyrA96 deoR nupG ϕ80lacZΔM15 Δ(lacZYA-argF)U169 hsdR17(rK− mK+) λ− |
| SM10 | thi-1 the leu tonA lacY supE recA::RP4-2-Tc::Mu Kmr |
| HB101 | F− mcrB mrr hsdS20(rB− mB−) recA13 leuB6 ara-14 proA2 lacY1 galK2 xyl-5 mtl-1 rpsL20(Smr) glnV44 λ− |
TABLE 2.
Plasmids
| Plasmid | Description or reference |
|---|---|
| pTNS3 | 46 |
| pRK2013 | 47 |
| pUC18T-miniTn7T-gm | 48 |
| pHERD20T | 15 |
| pMMB66HE | 19 |
| miniCTX1 | 22 |
| pFLP2 | 24 |
| pJM100 | pUC18T-miniTn7T-gm-araC-ParaBAD (accession no. KX787911) |
| pJM101 | pUC18T-miniTn7T-gm-lacIq-Ptac (accession no. KX782328) |
| pJM220 | pUC18T-miniTn7T-gm-rhaSR-PrhaBAD (accession no. KX777256) |
| pJM181 | pUC18T-miniTn7T-gm-stRBS-lacZ |
| pJM179 | pUC18T-miniTn7T-gm-araC-ParaBAD-stRBS-lacZ |
| pJM180 | pUC18T-miniTn7T-gm-lacIq-Ptac-stRBS-lacZ |
| pJM230 | pUC18T-miniTn7T-gm-rhaSR-PrhaBAD-stRBS-lacZ |
| pJM299 | pUC18T-miniTn7T-gm-araC-ParaBAD-intRBS-lacZ |
| pJM289 | pUC18T-miniTn7T-gm-araC-ParaBAD-wkRBS-lacZ |
| pJM301 | pUC18T-miniTn7T-gm-araC-ParaBAD-5′ UTR amiE |
| pJM302 | pUC18T-miniTn7T-gm-araC-ParaBAD-5′ UTR amiE-lacZ |
| pJM238 | pUC18T-miniTn7T-gm-araC-ParaBAD-stRBS-aacC1 |
| pJM240 | pUC18T-miniTn7T-gm-rhaSR-PrhaBAD-stRBS-aacC1 |
| pJM251 | miniCTX1-araC-ParaBAD (accession no. KX787912) |
| pJM252 | miniCTX1-lacIq-Ptac (accession no. KX782329) |
| pJM253 | miniCTX1-rhaSR-PrhaBAD (accession no. KX782327) |
| pJM259 | miniCTX1-araC-ParaBAD-stRBS-aacC1 |
| pJM260 | miniCTX1-rhaSR-PrhaBAD-stRBS-aacC1 |
| pJM256 | miniCTX1-araC-ParaBAD-PA14 trpF |
| pJM268 | miniCTX1-rhaSR-PrhaBAD-PA14 trpF |
| pJM255 | miniCTX1-araC-ParaBAD-PA14 trpC |
| pJM267 | miniCTX1-rhaSR-PrhaBAD-PA14 trpC |
| pJM254 | miniCTX1-araC-ParaBAD-PA14 trpA |
| pJM266 | miniCTX1-rhaSR-PrhaBAD-PA14 trpA |
TABLE 3.
Oligonucleotide primers
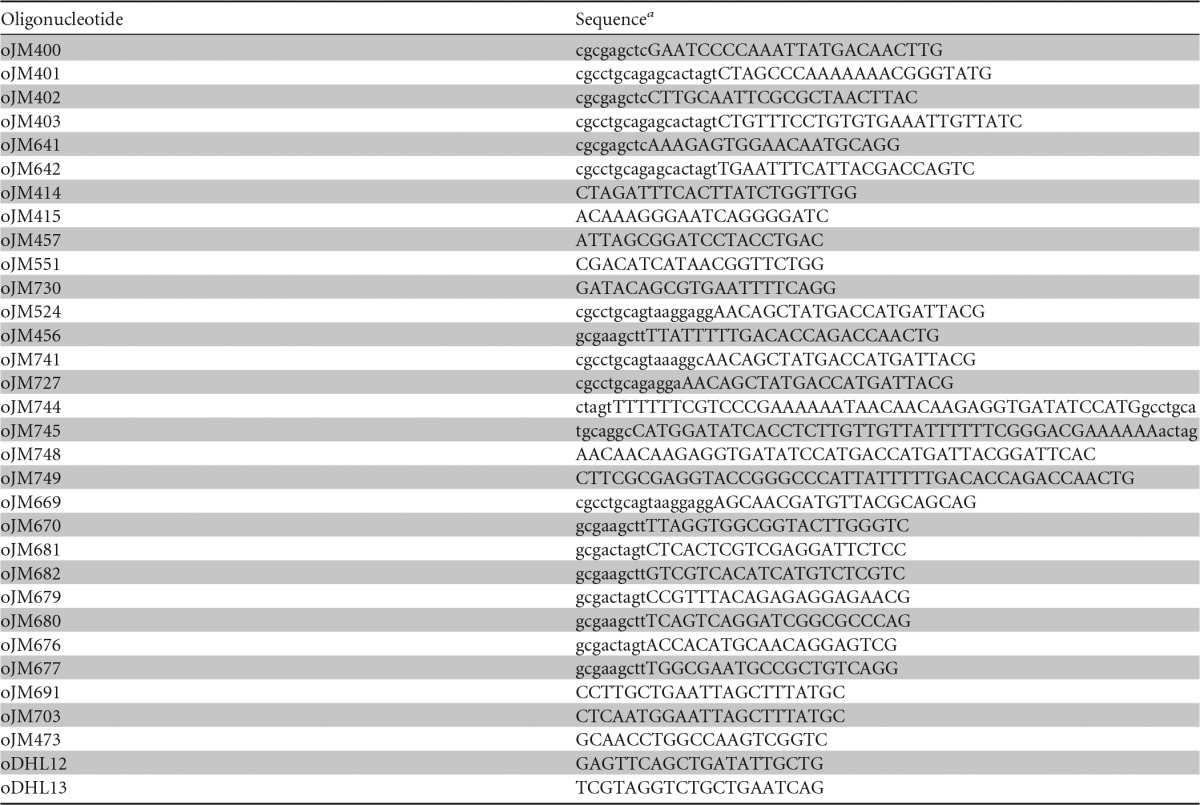
a The nucleotides shown in uppercase anneal to the PCR template, while those shown in lowercase are additional sequences that do not anneal to the template.
Plasmid construction.
Plasmid pUC18T-miniTn7T-gm (18) provided the backbone for the miniTn7 plasmids constructed in this study. The araC-ParaBAD sequence was amplified by PCR from pHERD20T (15) with oligonucleotide primers oJM400 and oJM401. The lacIq-Ptac sequence was amplified by PCR from pMMB66HE (19) with oligonucleotide primers oJM402 and oJM403. The rhaSR-PrhaBAD sequence was amplified by PCR from E. coli strain W3110 genomic DNA with oligonucleotide primers oJM641 and oJM642. The pUC18T-miniTn7T-gm plasmid and PCR products were cut with SacI and PstI and then ligated to make pUC18T-miniTn7T-gm-araC-ParaBAD (pJM100), pUC18T-miniTn7T-gm-lacIq-Ptac (pJM101), and pUC18T-miniTn7T-gm-rhaSR-PrhaBAD (pJM220), respectively. The plasmids were confirmed by PCR with oligonucleotide primers oJM414 and oJM415. DNA fragments cloned into pJM100, pJM101, or pJM220 plasmid were sequenced with oJM414 and oJM457, oJM551, or oJM730, respectively.
To construct miniTn7 plasmids with lacZ transcriptional fusions, the lacZ sequence was amplified by PCR from miniCTX-lacZ (20) with oligonucleotide primers oJM524 and oJM456. oJM524 introduces a strong ribosome-binding site (stRBS; 5′-TAAGGAGG-3′) with a 7-bp spacer sequence between the RBS and start codon. The lacZ PCR product, as well as pUC18T-miniTn7T-gm, pJM100, pJM101, and pJM220 plasmids, were cut with PstI and HindIII and then ligated to make pUC18T-miniTn7T-gm-stRBS-lacZ (pJM181), pUC18T-miniTn7T-gm-araC-ParaBAD-stRBS-lacZ (pJM179), pUC18T-miniTn7T-gm-lacIq-Ptac-stRBS-lacZ (pJM180), and pUC18T-miniTn7T-gm-rhaSR-PrhaBAD-stRBS-lacZ (pJM230), respectively. To build derivatives with reduced RBS strength, pUC18T-miniTn7T-gm-araC-ParaBAD-intRBS-lacZ (pJM299) and pUC18T-miniTn7T-gm-araC-ParaBAD-stRBS-lacZ (pJM289), the lacZ sequence was amplified by PCR with oligonucleotide primers oJM741 or oJM727, respectively, and with oJM456. oJM741 introduces an intermediate-strength RBS (intRBS) with an 8-bp spacer sequence between the RBS and start codon. oJM727 introduces a weak RBS (wkRBS) with a 7-bp spacer sequence between the RBS and start codon. Both PCR products were cut with PstI and HindIII and then ligated with pJM100 as described above.
To build a miniTn7-araC-ParaBAD plasmid with the 5′ untranslated region (UTR) of amiE, oligonucleotides oJM744 and oJM745 were annealed and then ligated with pJM100 that had been cut with SpeI and PstI to make pUC18T-miniTn7T-gm-araC-ParaBAD-5′ UTR amiE (pJM301). The annealed oJM744-oJM745 DNA fragment has an NcoI site that overlaps with the amiE start codon to simplify the construction of translational fusions. To construct a derivative with a lacZ translational fusion, the lacZ sequence was amplified by PCR from miniCTX-lacZ with oligonucleotide primers oJM748 and oJM749. The PCR product was then ligated into pJM301 that had been cut with NcoI and HindIII by isothermal assembly (21) to make pUC18T-miniTn7T-gm-araC-ParaBAD-5′ UTR amiE-lacZ (pJM302).
To build miniTn7 plasmids with aacC1 (gm, gentamicin acetyltransferase) transcriptional fusions, the aacC1 sequence was amplified by PCR from pUC18T-miniTn7T-gm with oligonucleotide primers oJM669 and oJM670. oJM679 introduces a strong RBS (as described above). The aacC1 PCR product, as well as pJM100 and pJM220 plasmids, were cut with PstI and HindIII and then ligated to make pUC18T-miniTn7T-gm-araC-ParaBAD-stRBS-aacC1 (pJM238) and pUC18T-miniTn7T-gm-rhaSR-PrhaBAD-stRBS-aacC1 (pJM240), respectively.
To make the miniCTX derivatives of each of these plasmids, miniCTX1 plasmid (22) as well as pJM100, pJM101, pJM220, pJM238, and pJM240 plasmids were cut with SacI and HindIII. The SacI-HindIII miniCTX1 and released DNA fragments were then ligated to produce miniCTX1-araC-ParaBAD (pJM251), miniCTX1-lacIq-Ptac (pJM252), miniCTX1-rhaSR-PrhaBAD (pJM253), miniCTX1-araC-ParaBAD-stRBS-aacC1 (pJM259), and miniCTX1-rhaSR-PrhaBAD-stRBS-aacC1 (pJM260), respectively.
To make miniCTX plasmids for complementation of trp mutants, the trpF, trpC, and trpA sequences were amplified by PCR from P. aeruginosa strain PA14 genomic DNA with oligonucleotide primers oJM681 and oJM682, oJM679 and oJM680, and oJM676 and oJM677, respectively. Each PCR product contains the native RBS from each trp gene. The PCR products, as well as pJM251 and pJM253 plasmids, were cut with SpeI and HindIII and then ligated to make miniCTX1-araC-ParaBAD-PA14 trpF (pJM256), miniCTX1-rhaSR-PrhaBAD-PA14 trpF (pJM268), miniCTX1-araC-ParaBAD-PA14 trpC (pJM255), miniCTX1-rhaSR-PrhaBAD-PA14 trpC (pJM267), miniCTX1-araC-ParaBAD-PA14 trpA (pJM254), and miniCTX1-rhaSR-PrhaBAD-PA14 trpA (pJM266). DNA fragments cloned into miniCTX plasmids were confirmed with oligonucleotide primers oJM691 and oJM703. The cloned DNA fragments were sequenced with oJM703 and oJM457 (miniCTX1-araC-ParaBAD, pJM251) or oJM730 (miniCTX1-rhaSR-PrhaBAD, pJM253).
Conjugations.
P. aeruginosa recipient strains, as well as E. coli donor and helper strains, were grown in 3 ml LB (with antibiotic when appropriate) at 37°C with rolling for about 8 h. One milliliter of each culture was centrifuged at 8,000 × g for 2 min in microcentrifuge tubes. The culture supernatants were aspirated, cell pellets were resuspended in 1 ml LB, and cell suspensions were centrifuged. Aspiration, resuspension, and centrifugation were repeated. The supernatant was aspirated and cell pellets were resuspended in 35 μl LB. Cell suspensions were spotted onto LB agar and incubated at 37°C overnight. The cells were scraped off and resuspended in LB and serially diluted 10-fold, and 100 μl of each dilution was spread on Vogel-Bonner minimal medium (VBMM; 10 mM sodium citrate tribasic, 9.5 mM citric acid, 57 mM potassium phosphate dibasic, 17 mM sodium ammonium phosphate, 1 mM magnesium sulfate, 0.1 mM calcium chloride, pH 7.0) agar with antibiotic (gentamicin or tetracycline) and incubated at 37°C overnight. Chromosomal integration of miniTn7 was confirmed by PCR with oligonucleotide primers oJM473 and oJM414, while miniCTX integration was confirmed with oDHL12 and oDHL13.
Electroporations.
Recipient strains were grown in 3 ml LB in duplicate at 37°C with rolling for about 8 h. The two 3-ml cultures were pooled and then dispensed into four microcentrifuge tubes. The cultures were centrifuged at 8,000 × g for 2 min. Each cell pellet was resuspended in 1 ml 300 mM sucrose and centrifuged twice (23). The four cell pellets were resuspended and pooled in a total of 300 μl of 300 mM sucrose. One hundred microliters of each suspension was transferred to 1-mm-gap-width electroporation cuvettes. One hundred nanograms of pFLP2 plasmid was added to each suspension. Cells were electroporated at 1,800 V in an Eppendorf electroporator 2510. Nine hundred microliters of LB was added to each electroporation. Recovery cultures were incubated at 37°C with rolling for 1 h. Cultures were serially diluted 10-fold, spread on LB agar with antibiotic (carbenicillin), and incubated at 37°C overnight.
Excision of antibiotic resistance cassette by Flp-FRT recombination.
Recipient strains containing chromosomal gentamicin resistance cassette flanked by FRT recombination sites were electroporated with pFLP2 plasmid (24). Transformants were streaked on LB with carbenicillin, as well as on LB with gentamicin, to screen for excision of the gentamicin resistance cassette by Flp recombination. Gentamicin-sensitive transformants were streaked from LB with carbenicillin to LB with 5% sucrose. Strains that have the pFLP2 plasmid are sucrose sensitive, while those that have lost the plasmid are sucrose resistant. Sucrose-resistant colonies were streaked on LB, LB with gentamicin, and LB with carbenicillin to confirm both excision of the gentamicin resistance cassette and loss of the pFLP2 plasmid.
β-Galactosidase assays.
Strains were grown in 3 ml medium (LB, M9-glucose, M9-Casamino Acids, or M9-succinate) at 37°C with rolling until cell density reached an optical density at 600 nm (OD600) of about 0.5. Cultures were diluted 50-fold into the 3 ml medium with or without inducer (l-arabinose, isopropyl-β-d-thiogalactopyranoside [IPTG], or l-rhamnose) and grown at 37°C with rolling until an OD600 of about 0.5 (mid-exponential growth phase). One milliliter of each culture was centrifuged at 10,000 × g in a microcentrifuge tube for 1 min. Cell pellets were stored at −20°C. Cell pellets were thawed on ice and resuspended in 1 ml cold Z buffer (60 mM sodium phosphate dibasic, 40 mM sodium phosphate monobasic, 10 mM potassium chloride, 1 mM magnesium sulfate, pH 7.0, with 50 mM β-mercaptoethanol). One hundred microliters of each cell suspension was added to microcentrifuge tubes containing 900 μl Z buffer, 100 μl chloroform, and 50 μl 0.1% SDS. Reaction mixtures were vortexed and incubated at 30°C for 10 min. Two hundred microliters of 4 mg/ml ortho-nitrophenyl-β-galactoside (in 0.1 M phosphate buffer; 60 mM sodium phosphate dibasic, 40 mM sodium phosphate monobasic, pH 7.0) was added to each sample. Reactions were vortexed briefly and incubated at 30°C for 10 to 20 min. Four hundred microliters of 1 M sodium carbonate was added to each sample to terminate the reactions. Reaction mixtures were vortexed and centrifuged to remove cell debris. One milliliter of each reaction supernatant was transferred to disposable cuvettes. The absorbance of each reaction mixture was measured at 420 nm (A420). β-Galactosidase activity (in Miller units) was calculated as (1,000 × A420)/(reaction time in minutes × cell suspension volume in ml × OD600).
Gentamicin resistance assays.
For broth culture assays, strains were grown in 3 ml LB with or without inducer (0.2% l-arabinose or 0.03125% l-rhamnose) at 37°C with rolling until an OD600 of approximately 0.5. Five microliters of each culture was added to 150 μl LB or LB with gentamicin (80, 20, 5, 1.25, 0.31, or 0.078 μg/ml) in a 96-well plate. Five microliters of each culture was also added to 150 μl LB with inducer (0.2% l-arabinose or 0.03125% l-rhamnose) and gentamicin (80, 20, 5, 1.25, 0.31, or 0.078 μg/ml). Each condition was tested in triplicate. Cultures were grown at 37°C with shaking in a BioTek Synergy H1 hybrid plate reader for 8 h, and the OD600 was measured every 15 min. Relative growth of culture after 300 min (transition state-early stationary phase for cultures without gentamicin) was calculated (average of triplicates with gentamicin divided by average of triplicates without gentamicin). For spot dilution plate assays, strains were grown in 3 ml LB with or without inducer (0.2% l-arabinose or 0.03125% l-rhamnose) at 37°C with rolling until an OD600 of approximately 0.5, as described above. The cultures were then serially diluted 10-fold in LB, 5 μl of each dilution was spotted on LB agar and LB agar with gentamicin, and cells were grown at 37°C overnight.
Complementation of tryptophan auxotrophy.
Strains were grown in 3 ml M9 minimal medium supplemented with 50 mM succinate, as well as with 1 mM l-tryptophan or 0.003125% l-rhamnose when indicated. Cultures were grown at 37°C with rolling for about 15 h and the OD600 was measured. Strains were also grown in 200 μl M9 with 50 mM succinate, as well as with l-rhamnose (0.006, 0.003, 0.0015, 0.0008, 0.0004, and 0.0002%) or l-tryptophan (1.0, 0.25, 0.0625, and 0.0156 mM), as indicated. Each condition was tested in triplicate. These cultures were grown at 37°C with shaking in a BioTek Synergy H1 hybrid plate reader for 8 h, and the OD600 was measured every 15 min.
Accession number(s).
The annotated nucleotide sequences of plasmids pJM100, pJM101, pJM220, pJM251, pJM252, and pJM253 were submitted to GenBank under accession numbers KX787911, KX782328, KX777256, KX787912, KX782329, and KX782327, respectively.
RESULTS
Gene expression from araC-ParaBAD is leaky in the absence of arabinose in P. aeruginosa.
Our initial analysis of the araC-ParaBAD inducible promoter system was motivated by two important questions. First, how much gene expression occurs from the araBAD promoter in the absence of arabinose? In other words, how tightly controlled is the inducible promoter system? Second, is this noninduced gene expression decreased by catabolite repression in the presence of glucose? To address these questions, we constructed a miniTn7 delivery plasmid with the araC-ParaBAD inducible promoter system (pUC18T-miniTn7T-gm-araC-ParaBAD). This new plasmid differs from the previous version built in our laboratory (17) in that the new plasmid contains the araBAD transcriptional initiation sequences but lacks any translational initiation sequences. This new design allowed maximum flexibility in the manipulation and use of the inducible promoter system. We then built a transcriptional fusion of the araBAD promoter with the E. coli lacZ gene that encodes the enzyme β-galactosidase. We engineered the transcriptional fusion to contain a strong ribosome-binding site to allow us to accurately measure low-level lacZ gene expression. The miniTn7-araC-ParaBAD-lacZ reporter was integrated in single copy into the chromosome of P. aeruginosa strain PA103. The strain bearing the reporter was grown to mid-exponential growth phase in rich medium (lysogeny broth [LB]) and β-galactosidase activity was measured. As shown in Fig. 1A, we observed an increase in β-galactosidase activity in the presence of increasing concentrations of arabinose between 0.003 and 0.8%. These results met the expectation that the araC-ParaBAD system would be inducible over a range of inducer concentrations. In the absence of arabinose, however, we still observed significant β-galactosidase activity. While the maximal inducible activity measured was about 1,800 Miller units (MU), the noninduced activity was about 270 MU. This noninduced activity is similar to the amount of activity we have observed with some constitutive promoters (data not shown). Therefore, it appears that there is leaky expression from the araC-ParaBAD system in the absence of inducer in P. aeruginosa.
FIG 1.

High noninduced expression from araC-ParaBAD cannot be decreased through catabolite repression in P. aeruginosa. The PA103 attTn7::araC-ParaBAD-stRBS-lacZ frt strain was grown to mid-exponential growth phase in LB or LB with increasing concentrations of arabinose (A), LB with or without 0.2% arabinose and with or without 0.5% glucose (B), or with LB, LB–0.5% glucose, M9–0.5% glucose, M9–0.5% Casamino Acids (CAA), or M9–50 mM succinate (C). β-Galactosidase activity was measured in Miller units (MU). The mean activity of three biological replicates is shown, and standard deviations are presented with error bars.
To determine if gene expression from the araC-ParaBAD inducible promoter system is subjected to catabolite repression in P. aeruginosa, we began by testing if the addition of glucose can decrease lacZ expression. The strain containing the lacZ reporter was grown in rich medium with and without 0.5% glucose. If the araBAD promoter was controlled by catabolite repression as it is in E. coli, then β-galactosidase activity should be decreased in the presence of glucose. Instead, we observed no difference in reporter activity in the presence of glucose compared to that in its absence (Fig. 1B). To test if the addition of glucose could decrease the induced expression, we also grew the reporter strain with and without 0.5% glucose in rich medium with 0.2% arabinose. Again, we observed the same reporter activity in the presence and absence of glucose (Fig. 1B). To rule out the possibility that catabolite repression by glucose was suppressed in rich medium, we also assessed noninduced expression in M9 minimal medium supplemented with 0.5% glucose. As observed in rich medium, glucose did not change the noninduced expression (Fig. 1C). P. aeruginosa preferentially utilizes amino acids and organic acids as carbon sources over sugars. Accordingly, amino acids and organic acids (such as succinate and citrate) elicit strong catabolite repression. Glucose, on the other hand, stimulates weak catabolite repression. We considered the possibility that growth with amino acids and organic acids could impose catabolite repression on the araBAD promoter. Growth in M9 minimal medium supplemented with 0.5% Casamino Acids or 50 mM succinate had no effect on the noninduced expression from the araBAD promoter (Fig. 1C). These data show that there is leaky expression from the araC-ParaBAD inducible promoter system in the absence of arabinose in P. aeruginosa, and that this noninduced activity cannot be decreased through catabolite repression.
Modulating translational initiation affects both noninduced and maximal induced expression from araC-ParaBAD.
Two important features of an inducible promoter system are low noninduced expression and high maximum induced expression, resulting in a wide range within which the expression of a gene can be modulated. Once we determined that the noninduced expression from the araC-ParaBAD system cannot be decreased by the addition of glucose or preferred carbon sources, such as Casamino Acids or succinate, we explored ways to mitigate the noninduced activity. One way to minimize the manifestation of the noninduced expression is to decrease translational initiation by reducing ribosome-binding site (RBS) strength. We reasoned that we might be able to offset high transcription of lacZ with low translation of the mRNA and limit the amount of the protein gene product synthesized in the absence of inducer. A caveat of this approach, however, is that reducing translational initiation could decrease both the noninduced and the maximal induced activities. If the noninduced activity decreased more than maximal induced activity, then there might be an RBS that restricts the noninduced activity and preserves the maximal induced activity. To attempt to find such an RBS, we constructed lacZ transcriptional fusions with three different RBS: strong (CTGCAGTAAGGAGGAACAGCTATG), intermediate (CTGCAGAGGAAACAGCTATG), and weak (CTGCAGTAAGGCAACAGCTATG) (where boldface indicates the mRNA ribosome-binding site nucleotides that base pair with the 16S rRNA, as well as the start codon nucleotides). The strong RBS, the same as the one used in the transcriptional fusion described above, is complementary to the 3′ end of the P. aeruginosa 16S rRNA (UGGAUCACCUCCUUA-3′ [where boldface indicates the nucleotides that base pair with the ribosome-binding site]), the region of the 30S ribosomal subunit that recognizes mRNA and drives the formation of the translation initiation complex. These sequences have a Gibbs free energy of binding (ΔG) of −13.82 kcal/mol (calculated using OligoAnalyzer 3.1 from Integrated DNA Technologies). The intermediate and weak RBS have ΔG of −6.24 and −4.67 kcal/mol, respectively. The intermediate RBS slightly diminished the noninduced β-galactosidase activity compared to that of the strong RBS, while the weak RBS decreased the noninduced activity more than 5-fold (Fig. 2A). As anticipated, however, both the intermediate and weak RBS also caused a reduction in the maximal induced activity (Fig. 2B). In fact, the maximal induced activity decreased more than the noninduced activity, leading to decreased induction ratios (the ratio of the maximal induced activity to the noninduced activity) (Fig. 2C). Therefore, reducing RBS strength incompletely achieved our goals of decreasing the noninduced expression and widening the range within which gene expression can be induced. Although the effects of the noninduced activity were reduced, it came at the cost of a lower maximal induced level and contracted the range within which gene expression could be modulated.
FIG 2.
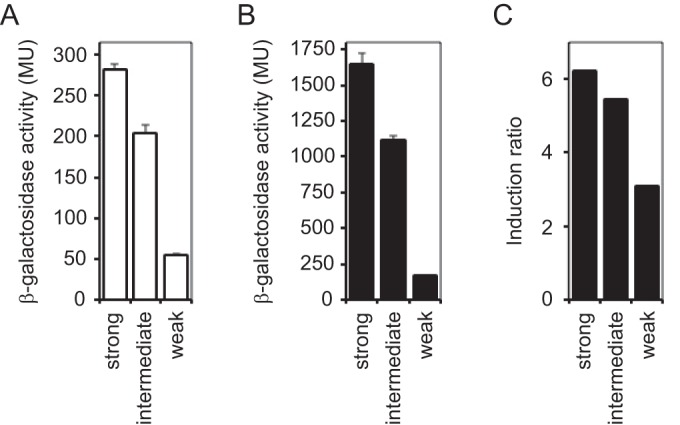
Decreased translational initiation decreased both noninduced and maximally induced expression from araC-ParaBAD. (A) PA103 attTn7::araC-ParaBAD-stRBS-lacZ frt (strong), PA103 attTn7::araC-ParaBAD-intRBS-lacZ frt (intermediate), and PA103 attTn7::araC-ParaBAD-wkRBS-lacZ frt (weak) were grown to mid-exponential growth phase in LB without arabinose. β-Galactosidase activity was measured. (B) Cells were grown to mid-exponential growth phase in LB with 0.8% arabinose. (C) Comparison of induction ratios with strong RBS, intermediate RBS, and weak RBS. Induction ratio is maximum induced activity to noninduced activity.
The 5′ UTR of P. aeruginosa amiE improved the functionality of araC-ParaBAD.
Another approach we envisioned to minimize the appearance of the high-level noninduced expression of the araC-ParaBAD system was to integrate it into the regulatory network that governs catabolite repression in P. aeruginosa. During growth with a preferred carbon source, P. aeruginosa blocks translation of mRNA gene products needed to catabolize nonpreferred carbon sources (7). In this form of catabolite repression, the RNA-binding protein Hfq binds short sequences, called CA (for catabolite activity) motifs, in the 5′ UTR of target mRNA and blocks translation by occluding nearby RBS (11). During growth with a nonpreferred carbon source, such as glucose, the small RNA CrcZ sequesters Hfq and allows translation of target mRNA (12). We thought that if we appended the 5′ UTR of such a target gene to the lacZ reporter gene, then perhaps we could subject lacZ to catabolite repression and decrease noninduced araC-ParaBAD activity without affecting the maximal induced activity. To this end, we inserted a portion of the 5′ UTR of the well-studied catabolite-repressed gene amiE into our miniTn7-araC-ParaBAD-lacZ reporter between the ParaBAD transcriptional start site and the lacZ start codon (Fig. 3A). It is worth noting that the RBS in the 5′ UTR amiE should be considered approximately intermediate in strength (the sequence is AACAAGAGGTGATATCCATG [where boldface indicates the mRNA ribosome-binding site nucleotides that base pair with the 16S rRNA, as well as the start codon nucleotides]; ΔG of −7.58 kcal/mol, with an 8-nucleotide spacing between RBS and start codon). The P. aeruginosa amiEBCRS operon encodes enzymes necessary to metabolize aliphatic amides such as acetamide (25). The amiE gene is preceded by the 134-bp untranslated region. The amiE-distal portion of the 5′ UTR contains a transcriptional terminator (amiL) that prevents read-through transcription of the ami operon in the absence of aliphatic amides. To eliminate that regulation, we used the amiE-proximal 40 bp of the 5′ UTR. This removes the amiL transcriptional terminator (ΔG of −29.10 kcal/mol) but preserves a putative 7-bp stem-loop (TTTTTCGTCCCGAAAAA; ΔG of −3.80 kcal/mol) and the CA motif. It is not known if this putative stem-loop is important for Hfq binding and catabolite repression. If this portion of the 5′ UTR amiE is sufficient for catabolite repression, then growth with amino acids (preferred carbon source, strong catabolite repression) should restrict lacZ translation and result in low noninduced β-galactosidase activity with araC-ParaBAD (Fig. 3B). Growth with glucose (nonpreferred carbon source, weak catabolite repression), on the other hand, should allow lacZ translation and lead to higher noninduced activity as well as high maximal induced activity (Fig. 3C).
FIG 3.
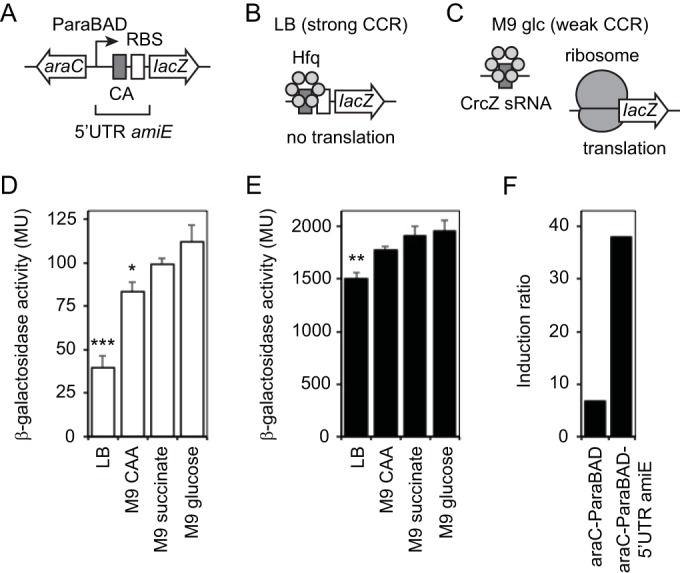
5′ UTR of P. aeruginosa amiE decreased noninduced expression from araC-ParaBAD through carbon catabolite repression. (A) Design of araC-ParaBAD inducible promoter system with 5′ UTR of P. aeruginosa amiE and lacZ reporter. (B) Inhibition of lacZ mRNA translation by strong carbon catabolite repression (CCR) in LB medium. Hfq binds CA motif in 5′ UTR amiE and blocks formation of translation initiation complex. (C) Translation of lacZ mRNA under weak carbon catabolite repression (CCR) in M9 glucose. CrcZ sRNA sequesters Hfq and allows formation of translation initiation complex. (D) PA103 attTn7::araC-ParaBAD-5′ UTR amiE-lacZ frt strain was grown to mid-exponential growth phase in LB, M9–0.5% Casamino Acids (CAA), M9–50 mM succinate, and M9–0.5% glucose without arabinose. β-Galactosidase activity was measured. Statistical significance was determined using one-way analysis of variance followed by Dunnett's test comparing each to M9 glucose (*, P ≤ 0.05; **, P ≤ 0.01; ***, P ≤ 0.001). (E) Cells were grown to mid-exponential growth phase with 0.8% arabinose in LB, M9–0.5% Casamino Acids (CAA), M9–50 mM succinate, and M9–0.5% glucose. Statistical significance was determined as described above. (F) Comparison of induction ratios without 5′ UTR amiE and with 5′ UTR amiE in LB. Induction ratio is maximum induced activity to noninduced activity.
To determine if the 5′ UTR amiE could decrease the noninduced expression of the araC-ParaBAD system through catabolite repression, the araC-ParaBAD-5′ UTR amiE-lacZ reporter strain was grown to mid-exponential growth phase in LB and M9 minimal medium with either Casamino Acids, succinate, or glucose, and then β-galactosidase activity was measured. As shown in Fig. 3D, the lowest noninduced activity was observed when the strain was grown in LB, a medium rich in amino acids. The noninduced activity progressively increased in M9 minimal medium supplemented with Casamino Acids, succinate, and glucose. In contrast, the noninduced expression from the araBAD promoter without the 5′ UTR amiE was not reduced in the presence of amino acids or succinate (as shown in Fig. 1C). Therefore, the 40-bp amiE-proximal portion of the 5′ UTR amiE appeared to allow catabolite repression of the lacZ reporter. Although the 5′ UTR amiE-lacZ reporter activity was affected in a manner consistent with catabolite repression, the magnitude of the effect was lower than that expected based on previous studies with amiE (12).
Irrespective of the mild catabolite repression, inclusion of the 5′ UTR amiE did diminish the noninduced araC-ParaBAD activity while only modestly reducing the maximal induced activity. When the araC-ParaBAD-5′UTR amiE-lacZ reporter strain was grown with 0.8% arabinose (maximal induction) in LB and M9 Casamino Acids, succinate, or glucose, the maximal induced activity was affected in a way reflective of mild catabolite repression. As observed for the noninduced activity, the maximal induced activity was the lowest in LB and progressively increased in M9 Casamino Acids, M9 succinate, and M9 glucose (Fig. 3E). When grown in LB, the maximal induced activity was reduced only about 17% with the 5′ UTR of amiE compared to without it (1,500 and 1,800 MU, respectively; compare Fig. 1A and 3E). The net effect of decreased noninduced activity and largely unaffected maximal induced activity was an increased induction ratio with the 5′ UTR amiE compared to that without it (Fig. 3F). The induction ratio was 5.6-fold higher with the 5′ UTR amiE than without it when grown in LB.
The lacIq-Ptac and rhaSR-PrhaBAD inducible promoter systems are tightly controlled in P. aeruginosa.
While we explored modifications to improve the araC-ParaBAD system, an alternative was to simply use different inducible promoter systems. Perhaps the most commonly used inducible promoter system is the IPTG-inducible E. coli lacI-Plac. A less commonly used but tightly controlled inducible promoter system is the rhamnose-inducible E. coli rhaSR-PrhaBAD (26, 27). Although Valvano and coworkers showed that the E. coli rhaSR-PrhaBAD system is tightly controlled in Burkholderia cenocepacia (28) and used it to identify and study essential genes (29–32), the functionality of this system in P. aeruginosa has not been reported. We constructed and tested two alternative inducible promoter systems for use in P. aeruginosa, miniTn7-lacIq-Ptac and miniTn7-rhaSR-PrhaBAD. The tac promoter is a hybrid promoter derived from the E. coli trp and lacUV5 promoters (33, 34). This hybrid promoter has low noninduced expression and higher maximal induced expression than the wild-type lac promoter in E. coli. With the same lacZ transcriptional reporter gene (with a strong RBS) used to evaluate araC-ParaBAD (Fig. 1), gene expression from both lacIq-Ptac and rhaSR-PrhaBAD was inducible over a range of inducer concentrations (Fig. 4A and B). The rhaSR-PrhaBAD system, however, is much more sensitive to inducer than araC-ParaBAD. Maximal induced expression from araC-ParaBAD occurred with 0.8% arabinose, while maximal induced expression from rhaSR-PrhaBAD occurred with 0.01% rhamnose (80 times more sensitive than araC-ParaBAD). The maximal induced expression was maintained up to 0.8% rhamnose (data not shown). Most importantly, the noninduced expression from both lacIq-Ptac and rhaSR-PrhaBAD was significantly lower than that with araC-ParaBAD (about 4- and 11-fold lower, respectively) (Fig. 4C). As observed with araC-ParaBAD, the noninduced expression with both lacIq-Ptac and rhaSR-PrhaBAD was unaffected by the addition of glucose. Additionally, both of these inducible promoter systems maintained high maximal induced expression. The maximal induced expression from lacIq-Ptac was about the same as that of araC-ParaBAD, while it was slightly lower from rhaSR-PrhaBAD (about 30% lower). The induction ratio was 3.5-fold higher with lacIq-Ptac than with araC-ParaBAD, while it was 7.6-fold higher with rhaSR-PrhaBAD (Fig. 4D). Therefore, both lacIq-Ptac and rhaSR-PrhaBAD satisfy the two most important criteria of inducible promoter systems. They both drive low-level gene expression in the absence of inducer and high maximal induced expression in the presence of inducer, leading to a wide range within which a gene can be expressed. Of the three inducible promoter systems tested, the rhaSR-PrhaBAD system performed the best. It was the most tightly controlled, allowed expression over the widest range, and was the most sensitive to inducer.
FIG 4.
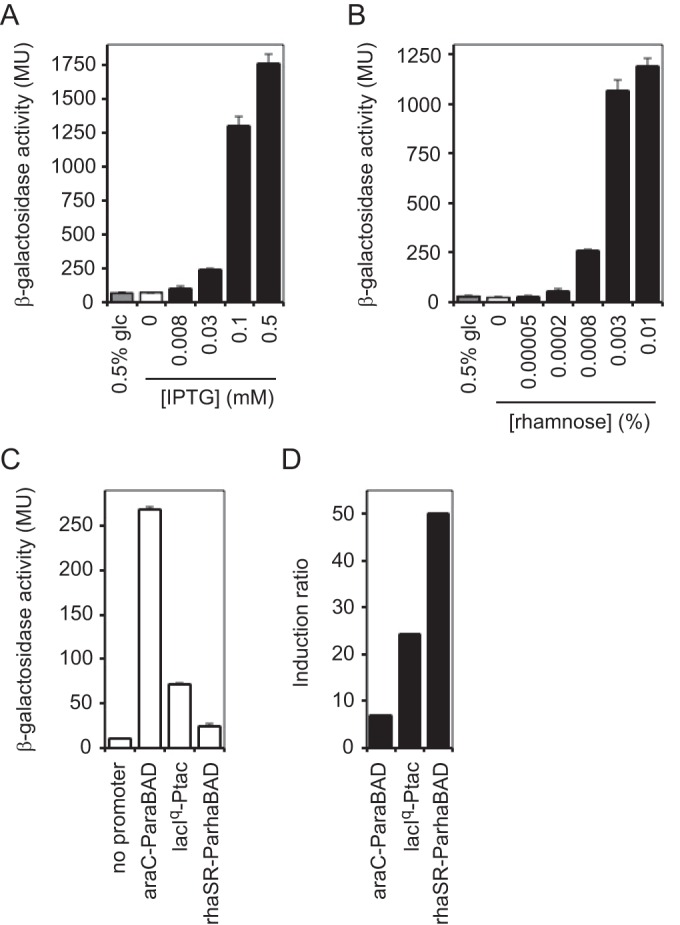
Lower noninduced expression from lacIq-Ptac and rhaSR-PrhaBAD. (A) PA103 attTn7::lacIq-Ptac-stRBS-lacZ frt strain was grown to mid-exponential growth phase in LB with or without glucose (glc) and with or without increasing concentrations of IPTG. β-Galactosidase activity was measured. (B) PA103 attTn7::rhaSR-PrhaBAD-stRBS-lacZ frt strain was grown to mid-exponential growth phase in LB with or without glucose and with or without increasing concentrations of rhamnose. β-Galactosidase activity was measured. (C) Comparison of noninduced activities of araC-ParaBAD, lacIq-Ptac, and rhaSR-PrhaBAD. (D) Comparison of induction ratios of araC-ParaBAD, lacIq-Ptac, and rhaSR-PrhaBAD. Induction ratio is maximum induced activity to noninduced activity.
Physiological consequences of noninduced gene expression from araC-ParaBAD but not rhaSR-PrhaBAD.
To investigate the consequences of the different levels of noninduced expression from araC-ParaBAD and rhaSR-PrhaBAD on gene function, we examined a conditionally essential gene whose function resulted in a simple phenotype. We chose to use the aacC1 gene (from the miniTn7-gm delivery plasmid) (18) encoding a gentamicin acetyltransferase that inactivates gentamicin and leads to resistance to the antibiotic. In the absence of aacC1 expression, P. aeruginosa is sensitive to gentamicin. If the noninduced expression from araC-ParaBAD or rhaSR-PrhaBAD was sufficient for gentamicin acetyltransferase gene function, then the cells containing the aacC1 gene under the control of these promoters would become resistant to gentamicin in the absence of inducer. We cloned the aacC1 gene into our miniTn7-araC-ParaBAD and miniTn7-rhaSR-PrhaBAD plasmids, integrated them into the P. aeruginosa chromosome (and excised the FRT-flanked aacC1 from the miniTn7 backbone), and measured gentamicin resistance. P. aeruginosa with either empty miniTn7, miniTn7-araC-ParaBAD-aacC1, or miniTn7-rhaSR-PrhaBAD-aacC1 was grown to mid-exponential growth phase in LB and then diluted and grown in LB with or without increasing concentrations of gentamicin (from 0.08 to 20 μg/ml). The isogenic negative-control strain, the wild type with an empty miniTn7, grew in the presence of gentamicin until a MIC of 5 μg/ml (Fig. 5A, white triangles). The strain with miniTn7-araC-ParaBAD-aacC1 grew until a MIC of 20 μg/ml (Fig. 5A, white squares). This increased gentamicin resistance showed that the noninduced expression from araC-ParaBAD was sufficient for aacC1 gene function. The strain with miniTn7-rhaSR-PrhaBAD-aacC1, on the other hand, exhibited the same gentamicin sensitivity as the control strain, indicating that the noninduced expression from rhaSR-PrhaBAD was not sufficient for aacC1 gene function (Fig. 5A, white circles). This strain grew in the presence of 50 μg/ml gentamicin with 0.003125% rhamnose (Fig. 5A, black circles), indicating that the miniTn7-rhaSR-PrhaBAD-aacC1 construct was functional. To confirm these results in a different experimental context, each strain was grown to mid-exponential phase in LB, serially diluted 10-fold, and then spotted and grown on LB and LB with 10 μg/ml gentamicin. The strains bearing either an empty miniTn7 or miniTn7-rhaSR-PrhaBAD-aacC1 grew on LB but were unable to grow on LB with gentamicin, while the strain with miniTn7-araC-ParaBAD-aacC1 grew equally well on LB and LB with gentamicin (Fig. 5B). These results clearly show that the leakiness of the araC-ParaBAD system has physiological consequences, a problem not observed with the rhaSR-PrhaBAD system.
FIG 5.

Noninduced expression of aacC1 from araC-ParaBAD but not rhaSR-PrhaBAD is sufficient for gentamicin resistance at different chromosomal loci and in different strains. (A) PA103 attTn7::frt (no aacC1; triangles), PA103 attTn7::araC-ParaBAD-stRBS-aacC1 frt (ParaBAD; squares), and PA103 attTn7::rhaSR-PrhaBAD-stRBS-aacC1 frt (PrhaBAD; circles) strains were grown to mid-exponential growth phase in LB without inducer (empty shapes) or with inducer (arabinose or rhamnose; filled squares or circles, respectively). Cultures were diluted into LB with or without gentamicin and with or without inducer. Cultures were grown in 96-well plates at 37°C with shaking. Relative growth (OD600 of culture with gentamicin/without gentamicin) was calculated after 300 min (5 h). (B to E) Strains were grown to mid-exponential growth phase in LB. Cultures were serially diluted 10-fold and spotted onto LB and LB with 10 μg/ml gentamicin. Plates were grown at 37°C overnight. (B) PA103 attTn7::frt (no aacC1), PA103 attTn7::araC-ParaBAD-stRBS-aacC1 frt (ParaBAD), and PA103 attTn7::rhaSR-PrhaBAD-stRBS-aacC1 frt (PrhaBAD) strains. (C) PA103 attCTX::tet (no aacC1), PA103 attCTX::araC-ParaBAD-stRBS-aacC1 tet (ParaBAD), and PA103 attCTX::rhaSR-PrhaBAD-stRBS-aacC1 tet (PrhaBAD) strains. (D) PAO1 attCTX::tet (no aacC1), PAO1 attCTX::araC-ParaBAD-stRBS-aacC1 tet (ParaBAD), and PAO1 attCTX::rhaSR-PrhaBAD-stRBS-aacC1 tet (PrhaBAD) strains. (E) PA14 attCTX::tet (no aacC1), PA14 attCTX::araC-ParaBAD-stRBS-aacC1 tet (ParaBAD), and PA14 attCTX::rhaSR-PrhaBAD-stRBS-aacC1 tet (PrhaBAD) strains.
In these experiments, the inducible promoter systems and gentamicin acetyltransferase gene were integrated at the Tn7 transposon insertion site (attTn7) near the chromosomal origin of replication (18, 35). To eliminate the possibility that this phenotypic consequence was unique to this particular chromosomal locus, we built strains with the inducible promoter systems and gentamicin acetyltransferase gene at the CTX bacteriophage integration site (attCTX) on the opposite side of the circular chromosome near the terminus of replication (22). We constructed miniCTX-araC-ParaBAD-aacC1 and miniCTX-rhaSR-PrhaBAD-aacC1 plasmids and integrated them into the P. aeruginosa chromosome. The strains were then grown to mid-exponential phase in LB, serially diluted, spotted, and grown on LB and LB with gentamicin. As observed at the Tn7 insertion site, the attCTX::araC-ParaBAD-aacC1 strain was gentamicin resistant, while the attCTX::rhaSR-PrhaBAD-aacC1 strain was gentamicin sensitive (Fig. 5C). We also wanted to rule out the possibility that this phenotype is exclusive to a certain P. aeruginosa strain. Each of the experiments described to this point were performed with the P. aeruginosa strain PA103. Thus, we also integrated miniCTX-araC-ParaBAD-aacC1 and miniCTX-rhaSR-PrhaBAD-aacC1 in the common laboratory strains PAO1 and PA14. Both the PAO1 and PA14 strains with attCTX::araC-ParaBAD-aacC1 were gentamicin resistant, while both strains with attCTX::rhaSR-PrhaBAD-aacC1 were gentamicin sensitive (Fig. 5D and E). Therefore, the physiological consequences of the noninduced gene expression from araC-ParaBAD were observed at two distinct chromosomal loci and in three different P. aeruginosa strains. Furthermore, the tightly controlled rhaSR-PrhaBAD system prevented the antibiotic resistance phenotype at both chromosomal loci and in each of the three strains tested.
Modulation of growth rate through inducible gene expression from rhaSR-PrhaBAD.
With the knowledge that the noninduced expression from araC-ParaBAD was sufficient for the function of a conditionally essential nonchromosomal gene, we examined whether this noninduced expression could also be problematic with conditionally essential chromosomal genes. When provided with a suitable carbon and nitrogen source, wild-type P. aeruginosa can synthesize all of the compounds it needs to grow, such as nucleic acids, amino acids, lipids, and sugars. The genes responsible for the synthesis of amino acids, for example, are conditionally essential. When an amino acid is available in the growth medium, the genes necessary for its synthesis are dispensable for growth. When the amino acid is absent, the bacterium must make it and the biosynthesis genes become essential for growth. We took advantage of the conditional essentiality of the tryptophan biosynthesis genes to examine the effects of noninduced expression from araC-ParaBAD and rhaSR-PrhaBAD inducible promoter systems on chromosomal P. aeruginosa genes.
To determine if the noninduced expression from araC-ParaBAD was sufficient for the tryptophan biosynthesis gene function, we cloned trpF, trpC, and trpA from P. aeruginosa strain PA14 into our miniCTX-araC-ParaBAD and miniCTX-rhaSR-PrhaBAD plasmids. We choose to work with these trp genes because each gene is either alone (not part of an operon) or is located at the 3′ end of an operon (see Fig. S1 in the supplemental material) (36–38), meaning that transposon insertions would only disrupt the target genes and not affect the expression of downstream genes. Importantly, each of the trp genes have different translation initiation sequences (RBS and start codons). To ensure that we were testing the outcome of gene expression with a variety of different native translation initiation sequences, we preserved these sequences (trpF, CAACAGGAGTCGAAAGCATG, 8-nucleotide spacing between RBS and start codon; trpC, AGAGAGGAGAACGCACAGTG, 8-nucleotide spacing between RBS and start codon; and trpA, TCGAGGATTCTCCGTCGTTG, 11-nucleotide spacing between RBS and start codon) in each miniCTX1 construct. After the miniCTX constructs were integrated into the chromosome of P. aeruginosa strain PA14 trpF, trpC, and trpA transposon insertion mutants (39), we grew the auxotrophic mutants and their complemented derivatives in M9 minimal medium supplemented with succinate or M9 succinate with tryptophan. We began these experiments by comparing the wild-type PA14 and the PA14 trpF mutant. The wild-type strain grew equally well in M9 succinate with and without tryptophan, while the trpF mutant only grew in M9 succinate with tryptophan (Fig. 6A). The trpF mutant complemented with attCTX::araC-ParaBAD-trpF grew as well in M9 succinate as the wild-type strain. This showed that the noninduced expression from araC-ParaBAD was sufficient for trpF function. The trpF mutant complemented with attCTX::rhaSR-PrhaBAD-trpF did not grow in M9 succinate, indicating that the noninduced expression from rhaSR-PrhaBAD was insufficient for trpF function. The strain grew well in M9 succinate with 0.003% rhamnose, confirming that the attCTX::rhaSR-PrhaBAD-trpF construct was functional. We observed the same results with the complemented trpC and trpA mutants (see Fig. S2), despite the fact that the genes have different translational initiation sequences and encode different enzymes. Together these results show that the noninduced expression from araC-ParaBAD can be problematic with conditionally essential chromosomal genes with different translation initiation sequences. Importantly, this problem can be resolved by utilizing the more tightly controlled rhaSR-PrhaBAD system.
FIG 6.
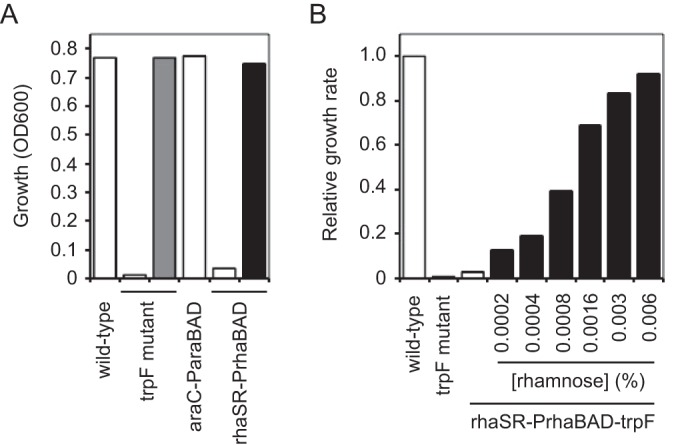
Complementation of tryptophan auxotrophy and modulation of growth rate with inducible expression of tryptophan biosynthesis gene from rhaSR-PrhaBAD. (A) PA14 (wild-type), PA14 trpF::TnMar (trpF mutant), PA14 trpF::TnMar attCTX::araC-ParaBAD-trpF tet (araC-ParaBAD), and PA14 trpF::TnMar attCTX::rhaSR-PrhaBAD-trpF tet (rhaSR-PrhaBAD) strains were grown overnight (about 15 h) in M9 succinate (white bars), M9 succinate with tryptophan (gray bar), or M9 succinate with rhamnose (black bar). The measured OD600 of each overnight culture is shown. (B) PA14 (wild-type), PA14 trpF::TnMar (trpF mutant), and PA14 trpF::TnMar attCTX::rhaSR-PrhaBAD-trpF tet (rhaSR-PrhaBAD-trpF) cultures were grown in M9 succinate (white bars) or M9 succinate with increasing concentrations of rhamnose (black bars) in 96-well plates at 37°C with shaking for 8 h. Relative growth rates were calculated with respect to the wild-type PA14 culture.
Using our miniTn7-lacZ reporter in P. aeruginosa strain PA103, we showed that the rhaSR-PrhaBAD inducible promoter system allowed gene expression over a wide range (Fig. 4B). With the assumption that gene expression from rhaSR-PrhaBAD can be similarly tuned in P. aeruginosa strain PA14, we expected to be able to modulate tryptophan biosynthesis by inducing different levels of trpF gene expression. To test this assumption, we grew wild-type PA14, the trpF mutant, and the trpF mutant complemented with miniCTX-rhaSR-PrhaBAD in M9 succinate or M9 succinate with increasing concentrations of rhamnose (Fig. 6B; also see Fig. S3 in the supplemental material). In minimal medium, the rate of cell growth should be proportional to cellular tryptophan synthesis. If trpF gene expression from rhaSR-PrhaBAD is tunable, then the growth rate of the attCTX-rhaSR-PrhaBAD-trpF strain should increase with increasing rhamnose concentration. As shown in Fig. 6B, we observed an increase in growth rate of the trpF mutant complemented with miniCTX-rhaSR-PrhaBAD in the presence of increasing concentrations of rhamnose from 0.002 to 0.006%. When grown in M9 succinate with 0.0002% rhamnose, the growth rate of the complemented mutant was nearly 10 times slower than that of the wild type. As the concentration of rhamnose was increased, we observed a progressive increase in growth rate until the wild-type growth rate was reached with 0.006% rhamnose. These results not only demonstrate the utility of the rhaSR-PrhaBAD inducible promoter system but also provide an experimental platform to precisely tune P. aeruginosa metabolism and growth.
DISCUSSION
The aims of the work described here were to describe the shortcomings of the araC-ParaBAD inducible promoter system and reengineer it to improve its functionality in P. aeruginosa, as well as to identify a system that is tightly controlled in the absence of inducer and inducible over a wide range of expression levels. These criteria are important because a failure to achieve tight control of gene expression can obscure the study of gene function, while a narrow range of inducible gene expression can make it difficult to match an induced expression level with the native expression level. Our data show that the araC-ParaBAD inducible promoter system is not tightly controlled in the absence of arabinose in P. aeruginosa, as it is in E. coli, and the leakiness cannot be reduced through carbon catabolite repression. Although araC-ParaBAD exhibits high maximally induced expression, the high noninduced expression narrows the range within which expression can be modulated. Both the lacIq-Ptac and rhaSR-PrhaBAD inducible promoter systems display significantly lower noninduced expression than araC-ParaBAD, and they both maintain high maximal induced expression. Consequently, expression from lacIq-Ptac and rhaSR-PrhaBAD is inducible over a broader range than that of araC-ParaBAD. Of these three inducible promoter systems, rhaSR-PrhaBAD is the most tightly controlled and allows expression over the widest range.
Although our data describe the different behaviors of these inducible promoter systems in P. aeruginosa, they do not necessarily explain why the differences exist. In E. coli, the promoters of many catabolic genes (including the ara, lac, and rha operons) tend to be weak and need to be activated by transcription factors to allow the formation of the RNA polymerase-promoter complex and transcriptional initiation (40). Alterations that strengthen these promoters by making the sequence closer to the consensus sequence recognized by RNA polymerase (TTGACA-17-TATAAT in E. coli) can eliminate the need for activation. (DNA sequences shown in boldface indicate the consensus promoter sequence recognized by σ70-RNA polymerase in E. coli.) The lacUV5 promoter (TTTACA-18-TATAAT), for example, is a derivative of the lac promoter (TTTACA-18-TATGTT) with mutations that strengthen the promoter and suppress the need for cAMP-CRP activation (41). The tac promoter (TTGACA-16-TATAAT) used in this study is a trp-lacUV5 hybrid that strengthens the promoter even further (33). If promoter strength was the only factor that contributes to noninduced expression, then the tac promoter may be expected to be the least tightly controlled. Presumably, the architecture of the regulatory system and the cellular levels of the regulatory proteins are also important factors. The regulatory system that controls the tac promoter is comprised of a single regulatory protein, the LacI transcriptional repressor. LacI binds to an operator site that overlaps the promoter and prevents the formation of the RNA polymerase-promoter complex. The lacIq gene has a mutation that strengthens the promoter that controls expression of lacI, leading to an increase in the amount of LacI produced, thereby decreasing noninduced expression from the tac promoter (42, 43). The rhaBAD promoter (AGGTCG-17-TAGACT) is weak in E. coli. Activation of the rhaBAD promoter occurs through a regulatory cascade involving RhaR and RhaS (26). RhaR is a transcription factor that binds rhamnose and activates the transcription of the rhaS gene, as well as its own gene. When RhaS, also a transcription factor, accumulates to sufficient amounts, it activates transcription from the rhaBAD promoter. Perhaps this architecture creates a regulatory buffer that maintains low noninduced expression (27). The araBAD promoter (CTGACG-18-TACTGT) is also quite weak in E. coli. It is repressed by AraC (in the absence of arabinose) and then activated by the arabinose-AraC complex. One possible explanation for its leakiness is that the cellular levels of AraC are lower and the promoter is stronger in P. aeruginosa than E. coli. In this scenario, AraC repression may be less complete and the need for activation by arabinose-AraC could be diminished in P. aeruginosa compared to that in E. coli.
Despite its leakiness in P. aeruginosa, the araC-ParaBAD system has been used to induce the expression of several genes (13–17). For some applications of inducible promoter systems, tightly controlled gene expression may not be particularly important. In cases where the amount of gene product necessary for gene function is large, the leaky gene expression from araC-ParaBAD may be insufficient for gene function. On the other hand, there are presumably many other cases where only a small amount of gene product is needed for gene function. In this study, we presented two distinct examples that demonstrate the problems associated with the araC-ParaBAD system in P. aeruginosa. Noninduced expression of a gentamicin acetyltransferase gene (aacC1) from araC-ParaBAD was sufficient to confer gentamicin resistance. Similarly, noninduced expression of three different tryptophan biosynthesis genes (trpF, trpC, and trpA) from araC-ParaBAD was sufficient to support tryptophan biosynthesis and cell growth in minimal medium. The leakiness of the araC-ParaBAD system was already encountered in a study of essential cell envelope biosynthesis genes (14). To overcome this problem, the authors constructed a suicide plasmid (pBEM10) for integration of araC-ParaBAD upstream of target genes at their native chromosomal loci. The distinguishing feature of this integration plasmid was the inclusion of a weak RBS (TTGGGCTAACCTTCTGAAAAGCTTATG; ΔG of −3.09 kcal/mol; 19-nucleotide RBS start codon spacing). This weak RBS would offset the effects of araC-ParaBAD leakiness by decreasing translational initiation and accumulation of the resulting protein gene product. Subsequently, miniCTX1 derivatives that preserve this weak RBS were constructed and used to study other essential envelope biosynthesis genes (16, 44). As we demonstrated in our study, decreasing RBS strength can reduce the problem of promoter leakiness, but it also reduces the maximal induced amount of gene product and narrows the range within which the gene product can be modulated. Therefore, the existing method for overcoming the problems with the araC-ParaBAD system incompletely achieves the goals of tightly controlled noninduced expression and high maximum induced expression, allowing a broad range of inducible gene expression. Furthermore, this approach does not resolve the problem when the gene product in not a protein, such as noncoding RNAs that are not translated into proteins.
In addition to modulating RBS strength, we also explored an alternative approach to mitigate the effects of araC-ParaBAD leakiness. We introduced a portion of the 5′ UTR from the amiE gene downstream of araC-ParaBAD in an attempt to subject target genes to carbon catabolite repression (translational inhibition by Hfq) in P. aeruginosa. The expectation was that the addition of the 5′ UTR would allow us to decrease translation of target mRNA when the cells were grown with a preferred carbon source (such as amino acids, which elicit strong catabolite repression). Indeed, the 5′ UTR of amiE improved the functionality of the araC-ParaBAD systems by decreasing the noninduced activity of the lacZ gene about 6-fold and increasing the induction ratio about 5-fold in rich medium (LB). The reduction was more modest than we expected, however, when cells were grown in minimal medium supplemented with Casamino Acids compared to minimal medium with glucose. The 5′ UTR of the amiE gene is 134 bp in length and contains a transcriptional terminator (amiL) that prevents read-through transcription of the ami operon in the absence of aliphatic amides, followed by a 40-bp region with the CA motif recognized by Hfq (11, 12). Using a translational lacZ fusion, the 134-bp 5′ UTR resulted in an approximately 2-fold decrease in activity in minimal medium supplemented with succinate compared to glucose and about a 10-fold decreased compared to activity in minimal medium with mannitol (12). We observed only about a 10% decrease in activity with our amiE-lacZ translational fusion in minimal medium with succinate compared to glucose. These results suggest that the CA motif is not sufficient for catabolite repression of amiE and indicate that additional upstream sequences also are involved. A more complete understanding of catabolite repression in P. aeruginosa should allow the design and implementation of a posttranscriptional regulatory module that could augment the usefulness of araC-ParaBAD as well as other inducible and constitutive promoter systems.
Although our primary motivation to identify an inducible promoter system that is tightly controlled in P. aeruginosa was the analysis of essential gene function with the aim of developing new antibiotics to treat P. aeruginosa infections, the utility of tightly controlled inducible promoters is not restricted to the study of essential genes. They can also be applied to understanding essential cellular functions maintained through parallel gene pathways. Such parallel pathways are employed by cells to promote phenotypic stability despite cellular and environmental variability. Genes in these pathways are often synthetically lethal, meaning that mutants in individual gene pathways are viable and mutant combinations are lethal. The study of synthetic lethal genes often involves conditional expression of a gene with an inducible promoter that allows inactivation of its synthetic lethal gene pairs. Tightly controlled inducible promoters can also be used to modify genetic circuits to understand the design principles of natural circuits, as well as to replace natural genetic circuits with controllable synthetic ones that have altered functionality. Such genetic engineering can also be applied to precisely alter cell metabolism and improve a targeted cellular function (45). In this context, tightly controlled inducible promoters are used to shift metabolic flow toward a desired product by amplifying the desired pathway or limiting an alternative pathway. The improved and expanded repertoire of inducible promoter systems provided in this work, in particular rhaSR-PrhaBAD, should help progress toward an understanding of gene function and the engineering of metabolic capabilities in P. aeruginosa.
Supplementary Material
ACKNOWLEDGMENTS
We thank Dominique Limoli, Alexander Meeske, Brian Meehan, and Charles Moran for helpful comments and suggestions during the preparation of the manuscript.
Footnotes
Supplemental material for this article may be found at http://dx.doi.org/10.1128/AEM.02041-16.
REFERENCES
- 1.Strateva T, Yordanov D. 2009. Pseudomonas aeruginosa–a phenomenon of bacterial resistance. J Med Microbiol 58:1133–1148. doi: 10.1099/jmm.0.009142-0. [DOI] [PubMed] [Google Scholar]
- 2.Lee N, Francklyn C, Hamilton EP. 1987. Arabinose-induced binding of AraC protein to araI2 activates the araBAD operon promoter. Proc Natl Acad Sci U S A 84:8814–8818. doi: 10.1073/pnas.84.24.8814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Guzman LM, Belin D, Carson MJ, Beckwith J. 1995. Tight regulation, modulation, and high-level expression by vectors containing the arabinose PBAD promoter. J Bacteriol 177:4121–4130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Schleif R. 2010. AraC protein, regulation of the l-arabinose operon in Escherichia coli, and the light switch mechanism of AraC action. FEMS Microbiol Rev 34:779–796. doi: 10.1111/j.1574-6976.2010.00226.x. [DOI] [PubMed] [Google Scholar]
- 5.Miyada CG, Stoltzfus L, Wilcox G. 1984. Regulation of the araC gene of Escherichia coli: catabolite repression, autoregulation, and effect on araBAD expression. Proc Natl Acad Sci U S A 81:4120–4124. doi: 10.1073/pnas.81.13.4120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Görke B, Stülke J. 2008. Carbon catabolite repression in bacteria: many ways to make the most out of nutrients. Nat Rev Microbiol 6:613–624. doi: 10.1038/nrmicro1932. [DOI] [PubMed] [Google Scholar]
- 7.Rojo F. 2010. Carbon catabolite repression in Pseudomonas: optimizing metabolic versatility and interactions with the environment. FEMS Microbiol Rev 34:658–684. doi: 10.1111/j.1574-6976.2010.00218.x. [DOI] [PubMed] [Google Scholar]
- 8.Collier DN, Hager PW, Phibbs PV. 1996. Catabolite repression control in the Pseudomonads. Res Microbiol 147:551–561. doi: 10.1016/0923-2508(96)84011-3. [DOI] [PubMed] [Google Scholar]
- 9.Yuste L, Canosa I, Rojo F. 1998. Carbon-source-dependent expression of the PalkB promoter from the Pseudomonas oleovorans alkane degradation pathway. J Bacteriol 180:5218–5226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hester KL, Lehman J, Najar F, Song L, Roe BA, MacGregor CH, Hager PW, Phibbs PV, Sokatch JR. 2000. Crc is involved in catabolite repression control of the bkd operons of Pseudomonas putida and Pseudomonas aeruginosa. J Bacteriol 182:1144–1149. doi: 10.1128/JB.182.4.1144-1149.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sonnleitner E, Bläsi U. 2014. Regulation of Hfq by the RNA CrcZ in Pseudomonas aeruginosa carbon catabolite repression. PLoS Genet 10:e1004440. doi: 10.1371/journal.pgen.1004440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Sonnleitner E, Abdou L, Haas D. 2009. Small RNA as global regulator of carbon catabolite repression in Pseudomonas aeruginosa. Proc Natl Acad Sci U S A 106:21866–21871. doi: 10.1073/pnas.0910308106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Baynham PJ, Ramsey DM, Gvozdyev BV, Cordonnier EM, Wozniak DJ. 2006. The Pseudomonas aeruginosa ribbon-helix-helix DNA-binding protein AlgZ (AmrZ) controls twitching motility and biogenesis of type IV pili. J Bacteriol 188:132–140. doi: 10.1128/JB.188.1.132-140.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Mdluli KE, Witte PR, Kline T, Barb AW, Erwin AL, Mansfield BE, McClerren AL, Pirrung MC, Tumey LN, Warrener P, Raetz CRH, Stover CK. 2006. Molecular validation of LpxC as an antibacterial drug target in Pseudomonas aeruginosa. Antimicrob Agents Chemother 50:2178–2184. doi: 10.1128/AAC.00140-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Qiu D, Damron FH, Mima T, Schweizer HP, Yu HD. 2008. PBAD-based shuttle vectors for functional analysis of toxic and highly regulated genes in Pseudomonas and Burkholderia spp. and other bacteria. Appl Environ Microbiol 74:7422–7426. doi: 10.1128/AEM.01369-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.DeLucia AM, Six DA, Caughlan RE, Gee P, Hunt I, Lam JS, Dean CR. 2011. Lipopolysaccharide (LPS) inner-core phosphates are required for complete LPS synthesis and transport to the outer membrane in Pseudomonas aeruginosa PAO1. mBio 2:e00142-11. doi: 10.1128/mBio.00142-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Damron FH, McKenney ES, Schweizer HP, Goldberg JB. 2013. Construction of a broad-host-range Tn7-based vector for single-copy P(BAD)-controlled gene expression in gram-negative bacteria. Appl Environ Microbiol 79:718–721. doi: 10.1128/AEM.02926-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Choi K-H, Gaynor JB, White KG, Lopez C, Bosio CM, Karkhoff-Schweizer RR, Schweizer HP. 2005. A Tn7-based broad-range bacterial cloning and expression system. Nat Methods 2:443–448. doi: 10.1038/nmeth765. [DOI] [PubMed] [Google Scholar]
- 19.Fürste JP, Pansegrau W, Frank R, Blöcker H, Scholz P, Bagdasarian M, Lanka E. 1986. Molecular cloning of the plasmid RP4 primase region in a multi-host-range tacP expression vector. Gene 48:119–131. doi: 10.1016/0378-1119(86)90358-6. [DOI] [PubMed] [Google Scholar]
- 20.Becher A, Schweizer HP. 2000. Integration-proficient Pseudomonas aeruginosa vectors for isolation of single-copy chromosomal lacZ and lux gene fusions. Biotechniques 29:948–952. [DOI] [PubMed] [Google Scholar]
- 21.Gibson DG, Young L, Chuang R-Y, Venter JC, Hutchison CA, Smith HO. 2009. Enzymatic assembly of DNA molecules up to several hundred kilobases. Nat Methods 6:343–345. doi: 10.1038/nmeth.1318. [DOI] [PubMed] [Google Scholar]
- 22.Hoang TT, Kutchma AJ, Becher A, Schweizer HP. 2000. Integration-proficient plasmids for Pseudomonas aeruginosa: site-specific integration and use for engineering of reporter and expression strains. Plasmid 43:59–72. doi: 10.1006/plas.1999.1441. [DOI] [PubMed] [Google Scholar]
- 23.Choi K-H, Kumar A, Schweizer HP. 2006. A 10-min method for preparation of highly electrocompetent Pseudomonas aeruginosa cells: application for DNA fragment transfer between chromosomes and plasmid transformation. J Microbiol Methods 64:391–397. doi: 10.1016/j.mimet.2005.06.001. [DOI] [PubMed] [Google Scholar]
- 24.Hoang TT, Karkhoff-Schweizer RR, Kutchma AJ, Schweizer HP. 1998. A broad-host-range Flp-FRT recombination system for site-specific excision of chromosomally-located DNA sequences: application for isolation of unmarked Pseudomonas aeruginosa mutants. Gene 212:77–86. doi: 10.1016/S0378-1119(98)00130-9. [DOI] [PubMed] [Google Scholar]
- 25.Kelly M, Clarke PH. 1962. An inducible amidase produced by a strain of Pseudomonas aeruginosa. J Gen Microbiol 27:305–316. doi: 10.1099/00221287-27-2-305. [DOI] [PubMed] [Google Scholar]
- 26.Egan SM, Schleif RF. 1993. A regulatory cascade in the induction of rhaBAD. J Mol Biol 234:87–98. doi: 10.1006/jmbi.1993.1565. [DOI] [PubMed] [Google Scholar]
- 27.Haldimann A, Daniels LL, Wanner BL. 1998. Use of new methods for construction of tightly regulated arabinose and rhamnose promoter fusions in studies of the Escherichia coli phosphate regulon. J Bacteriol 180:1277–1286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Cardona ST, Valvano MA. 2005. An expression vector containing a rhamnose-inducible promoter provides tightly regulated gene expression in Burkholderia cenocepacia. Plasmid 54:219–228. doi: 10.1016/j.plasmid.2005.03.004. [DOI] [PubMed] [Google Scholar]
- 29.Cardona ST, Mueller CL, Valvano MA. 2006. Identification of essential operons with a rhamnose-inducible promoter in Burkholderia cenocepacia. Appl Environ Microbiol 72:2547–2555. doi: 10.1128/AEM.72.4.2547-2555.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ortega XP, Cardona ST, Brown AR, Loutet SA, Flannagan RS, Campopiano DJ, Govan JRW, Valvano MA. 2007. A putative gene cluster for aminoarabinose biosynthesis is essential for Burkholderia cenocepacia viability. J Bacteriol 189:3639–3644. doi: 10.1128/JB.00153-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Juhas M, Stark M, Mering von C, Lumjiaktase P, Crook DW, Valvano MA, Eberl L. 2012. High confidence prediction of essential genes in Burkholderia cenocepacia. PLoS One 7:e40064. doi: 10.1371/journal.pone.0040064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Mohamed YF, Valvano MA. 2014. A Burkholderia cenocepacia MurJ (MviN) homolog is essential for cell wall peptidoglycan synthesis and bacterial viability. Glycobiology 24:564–576. doi: 10.1093/glycob/cwu025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.de Boer HA, Comstock LJ, Vasser M. 1983. The tac promoter: a functional hybrid derived from the trp and lac promoters. Proc Natl Acad Sci U S A 80:21–25. doi: 10.1073/pnas.80.1.21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Bagdasarian MM, Amann E, Lurz R, Rückert B. 1983. Activity of the hybrid trp-lac (tac) promoter of Escherichia coli in Pseudomonas putida. Construction of broad-host-range, controlled-expression vectors. Gene 26:273–282. [DOI] [PubMed] [Google Scholar]
- 35.Peters JE, Craig NL. 2001. Tn7: smarter than we thought. Nat Rev Mol Cell Biol 2:806–814. doi: 10.1038/35099006. [DOI] [PubMed] [Google Scholar]
- 36.Hadero A, Crawford IP. 1986. Nucleotide sequence of the genes for tryptophan synthase in Pseudomonas aeruginosa. Mol Biol Evol 3:191–204. [DOI] [PubMed] [Google Scholar]
- 37.Essar DW, Eberly L, Han CY, Crawford IP. 1990. DNA sequences and characterization of four early genes of the tryptophan pathway in Pseudomonas aeruginosa. J Bacteriol 172:853–866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Murata T. 1996. The trpF nucleotide sequence and its promoter analysis in Pseudomonas aeruginosa. Microbiol Immunol 40:107–114. doi: 10.1111/j.1348-0421.1996.tb03324.x. [DOI] [PubMed] [Google Scholar]
- 39.Liberati NT, Urbach JM, Miyata S, Lee DG, Drenkard E, Wu G, Villanueva J, Wei T, Ausubel FM. 2006. An ordered, nonredundant library of Pseudomonas aeruginosa strain PA14 transposon insertion mutants. Proc Natl Acad Sci U S A 103:2833–2838. doi: 10.1073/pnas.0511100103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Busby S, Ebright RH. 1999. Transcription activation by catabolite activator protein (CAP). J Mol Biol 293:199–213. doi: 10.1006/jmbi.1999.3161. [DOI] [PubMed] [Google Scholar]
- 41.Silverstone AE, Arditti RR, Magasanik B. 1970. Catabolite-insensitive revertants of lac promoter mutants. Proc Natl Acad Sci U S A 66:773–779. doi: 10.1073/pnas.66.3.773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Müller-Hill B, Crapo L, Gilbert W. 1968. Mutants that make more lac repressor. Proc Natl Acad Sci USA 59:1259–1264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Calos MP. 1978. DNA sequence for a low-level promoter of the lac repressor gene and an “up” promoter mutation. Nature 274:762–765. doi: 10.1038/274762a0. [DOI] [PubMed] [Google Scholar]
- 44.Lo Sciuto A, Fernández-Piñar R, Bertuccini L, Iosi F, Superti F, Imperi F. 2014. The periplasmic protein TolB as a potential drug target in Pseudomonas aeruginosa. PLoS One 9:e103784. doi: 10.1371/journal.pone.0103784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Bailey JE. 1991. Toward a science of metabolic engineering. Science 252:1668–1675. doi: 10.1126/science.2047876. [DOI] [PubMed] [Google Scholar]
- 46.Choi K-H, Mima T, Casart Y, Rholl D, Kumar A, Beacham IR, Schweizer HP. 2008. Genetic tools for select-agent-compliant manipulation of Burkholderia pseudomallei. Appl Environ Microbiol 74:1064–1075. doi: 10.1128/AEM.02430-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Figurski DH, Helinski DR. 1979. Replication of an origin-containing derivative of plasmid RK2 dependent on a plasmid function provided in trans. Proc Natl Acad Sci U S A 76:1648–1652. doi: 10.1073/pnas.76.4.1648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Choi K-H, Schweizer HP. 2006. mini-Tn7 insertion in bacteria with single attTn7 sites: example Pseudomonas aeruginosa. Nat Protoc 1:153–161. doi: 10.1038/nprot.2006.24. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.