

Abstract
Axonal degeneration is a prominent feature of many forms of neurodegeneration, and also an early event in blast-mediated traumatic brain injury (TBI), the signature injury of soldiers in Iraq and Afghanistan. It is not known, however, whether this axonal degeneration is what drives development of subsequent neurologic deficits after the injury. The Wallerian degeneration slow strain (WldS) of mice is resistant to some forms of axonal degeneration because of a triplicated fusion gene encoding the first 70 amino acids of Ufd2a, a ubiquitin-chain assembly factor, that is linked to the complete coding sequence of nicotinamide mononucleotide adenylyltransferase 1 (NMAT1). Here, we demonstrate that resistance of WldS mice to axonal degeneration after blast-mediated TBI is associated with preserved function in hippocampal-dependent spatial memory, cerebellar-dependent motor balance, and retinal and optic nerve–dependent visual function. Thus, early axonal degeneration is likely a critical driver of subsequent neurobehavioral complications of blast-mediated TBI. Future therapeutic strategies targeted specifically at mitigating axonal degeneration may provide a uniquely beneficial approach to treating patients suffering from the effects of blast-mediated TBI.
Keywords: axonal degeneration, nicotinamide adenine dinucleotide, neurodegeneration, traumatic brain injury, WldS mouse
Significance Statement
Blast-mediated traumatic brain injury (TBI) is the signature injury of soldiers associated with chronic cognitive, motor, and visual dysfunction. An early event in blast-TBI is diffuse axonal damage, but it is not known whether this drives development of subsequent pathology. WldS mutant mice are resistant to axonal degeneration via a mutation that enables maintenance of neuronal nicotinamide adenine dinucleotide (NAD) levels after injury, and a number of different approaches have been proposed for augmenting NAD levels in the nervous system. We show that WldS mice are protected from axonal degeneration and deficits in cognition, movement, and vision after blast-TBI. Axonal degeneration is thus a critical early event in this prevalent injury, suggesting therapeutic potential of specifically mitigating early axonal degeneration after blast-TBI.
Introduction
Traumatic brain injury (TBI) is a leading cause of death and disability worldwide (Fleminger and Ponsford, 2005), with blast-mediated injury being the most common cause of TBI sustained by soldiers in the recent wars in Iraq (Operation Iraqi Freedom) and Afghanistan (Operation Enduring Freedom). Blast-mediated TBI places patients at risk for both acute and long-term neurologic complications, such as cognitive dysfunction, motor decline, psychiatric conditions, visual deficits, and neuropathologic features similar to Alzheimer’s disease (Hoge et al., 2008; Wolf et al., 2009; Goldstein et al., 2012; Shively et al., 2012). Sadly, there are currently no treatment options for patients beyond supportive and rehabilitative care.
Sheer forces associated with blast injury lead to widespread, diffuse, and progressive axonal injury, known to play a role in multiple forms of neurodegeneration (Raff et al., 2002; Nakagawa et al., 2011; Lingor et al., 2012; Magnuson et al., 2012; Yin et al., 2014). This form of injury and its associated behavioral deficits can be recapitulated in rodent models, which may therefore be useful for discovery and validation of new therapeutic approaches (Goldstein et al., 2012; Mohan et al., 2013; Yin et al., 2014). Pharmcologic agents shown to enhance flux of the nicotinamide adenine dinucleotide (NAD) salvage pathway in normal mammalian cells and facilitate NAD rebound following doxorubicin exposure (Pieper et al., 2010, 2014; MacMillan et al, 2011; Wang et al., 2014) confer protective efficacy on pathology and behavior in a rodent model of blast-mediated TBI (Yin et al., 2014), as well as other models of neurodegeneration in the central and peripheral nervous systems (De Jesús-Cortés et al., 2012, 2015,2016; Tesla et al., 2012; Blaya et al., 2014; Dutca et al., 2014; Naidoo et al., 2014; Kemp et al., 2015; Walker et al., 2015; Lee et al., 2016). In addition, treatment with NAD and NAD precursors, including nicotinamide, nicotonic acid mononucleotide, and nicotinamide mononucleotide (NMN), or overexpression of nicotinamide phosphoribosyltransferase protect axons in vitro (Araki et al., 2004; Wang et al., 2005; Sasaki et al., 2006).
To investigate whether NAD metabolism might be related to blast-mediated axonal degeneration in the brain, we applied the blast model of TBI to the Wallerian degeneration slow strain (WldS) of mice (Lunn et al., 1989). These mice were originally identified as being resistant to axonal degeneration after injury, and the wlds gene was subsequently shown to exist as a triplicated fusion gene encoding the first 70 amino acids of Ufd2a, a ubiquitin-chain assembly factor, that is linked directly to the complete coding sequence of nicotinamide mononucleotide adenylyl transferase 1 (Coleman et al., 1998; Conforti et al., 2000; Mack et al., 2001). WldS mice have shown resistance to neurodegeneration in multiple models, including Charcot–Marie–Tooth disease 1A (Meyer zu Horste et al., 2011), Parkinson’s disease (Sajadi et al., 2004), and retinal ganglion cell death after optic nerve crush injury (Lorber et al., 2012). These mice have also shown improved motor function, learning, and memory after concussive brain injury relative to wild-type littermates (Fox and Faden, 1998). Although multiple mechanisms have been proposed for how WldS mice are protected from axonal degeneration (Wang and Barres, 2012), it has recently been shown in vitro that NAD, the metabolite of WldS/nicotinamide mononucleotide adenylyltransferase enzymatic activity, is both sufficient and specific to recapitulate the axonal protection seen with the WldS mutation, thereby strongly suggesting that NAD is a likely molecular mediator of WldS axonal protection (Wang et al., 2015). Accordingly, we investigated whether WldS mice might be similarly protected from blast-induced TBI, using measures of both neurodegeneration and behavioral outcome.
Materials and Methods
Animals
All animal procedures were performed in accordance with the University of Iowa Carver College of Medicine animal care committee’s regulations. Animals were housed in temperature-controlled conditions, provided food and water ad libitum, and maintained on a 12-h light/dark cycle (6 a.m. to 6 p.m.). Heterozygous WldS mice (kindly provided by Dr. Karen O’Malley of Washington University, St. Louis, MO) were bred to generate WldS-positive mice and wild-type littermates. Genotyping was performed using genotyping primers: forward, CGTTGGCTCTAAGGACAGCAC, and reverse, CTGCAGCCCCCACCCCTT. Mice used were male and 8 weeks of age at the time of injury.
Blast-mediated TBI
Mice were anesthetized with 1 mg/kg ketamine and 0.1 mg/kg xylazine and placed in an enclosed blast chamber (50 cm long and 33 cm wide) constructed from an air tank partitioned into two sides. One side was pressurized with a 13-cm opening between the partitions and covered with a Mylar membrane. The unpressurized partition contained a restraint 10 cm from the Mylar membrane, into which the mouse was placed. The head was freely moving, whereas a metal tube shielded the body. Compressed air was forced into the pressurized partition until the Mylar membrane burst at 27 kPa. The blast wave impacted the test animal inside a foam-lined restraint to reduce blunt impact trauma of the head against the metal tube. The left side of the head was closest to the origin of the blast wave. Sham-injured animals were anesthetized in the same way and not subjected to the blast.
Barnes maze
The Barnes maze test was conducted on a gray circular surface 91 cm in diameter, raised to a height of 90 cm, with 20 holes 5 cm in diameter equally spaced around the perimeter (Stoelting Co.). The surface was brightly lit and open to motivate the test animal to learn the location of a dark escape chamber recessed under one of the 20 holes, which was designated randomly. The maze was surrounded by a black circular curtain on which were hung four different and equally spaced visual cues (with different shapes and colors), for orientation to the designated location of the escape chamber. Each animal was subjected to 4 days of training comprising four trials per day. An area extending 4 cm from the escape hole in all directions was used as the target area for measurements (percent time in escape area, percent latency to escape, and nose pokes). A probe trial was conducted on the next day, during which time the escape chamber was removed and measurements were made to confirm the animal’s memory based on spatial cues. Measurements were acquired with Anymaze video tracking software (Stoelting Co.), and analysis was conducted blind to treatment group.
Foot slip assay
We used standard procedures described by Luong et al. (2011) to measure motor balance coordination. During the training period, mice were trained to cross the 80-cm beam to enter a black box with nesting material three times a day over two consecutive days. On test day, behavior was videotaped during the task, and foot slips were analyzed by an observer blind to condition and treatment group.
Provocative pattern-evoked electroretinography
Provocative pattern-evoked electroretinography (pPERG) was used to objectively measure the function of retinal ganglion cells by recording the amplitude of the PERG waveform 4 weeks after TBI. Mice were anesthetized with a combination of ketamine (0.03 mg/g, i.p.) and xylazine (0.005 mg/g, i.p.) and placed on a heated recording table to maintain body temperature. They were placed in a 60° head-down position using a custom-made PERG system, and responses were evoked using alternating, reversing, and black-and-white vertical stimuli delivered on a monitor (Jorvec, Miami, FL). A reference needle electrode was placed at the base of the head, and a ground electrode was placed at the base of the tail to complete the circuit. Each animal was placed at the same fixed position in front of the monitor to prevent recording variability caused by animal placement. Mice were positioned in a provocative head-down position for 15 min before initiation of the recording and remained in this position throughout the duration of the recording. Stimuli (18° radius visual angle subtended on full-field pattern, two reversals/s, 372 averaged signals with cutoff filter frequencies of 1–30 Hz, 98% contrast, 80 cd/m2 average monitor illumination intensity) were delivered under mesopic conditions without dark adaptation to exclude the possible effect of direct photoreceptor-derived evoked responses. A diffuser placed over the pattern on the monitor also did not elicit a measurable evoked potential, further ensuring that the electrical responses were elicited from retinal ganglion cells. The PERG response was evaluated by measuring the amplitude (peak to trough) of the waveform.
Immunohistochemistry
Mice were killed by transcardial perfusion with 4% paraformaldehyde at pH 7.4, and dissected brains were immersed in 4% paraformaldehyde overnight at 4°C and cryoprotected in sucrose for 72 h. Brains were then rapidly frozen in isopentane precooled to –70°C with dry ice. All brains were stored in a freezer at –80°C before sectioning. Serial sections (40 µm) were cut coronally through the cerebrum, approximately from bregma 3.20 mm to bregma –5.02 mm, and the brainstem and cerebellum, approximately from bregma –5.52 mm to bregma –6.96 mm (Paxinos and Franklin, 1997). Every section in a series of 12 sections (interval: 480 µm) was collected separately. All sections were stored free-floating in FD sections storage solution (FD Neurotechnologies, Columbia, MD) at –20ºC before further processing. For silver staining, sections were collected in 0.1 m phosphate buffer (pH 7.4) containing 4% paraformaldehyde and fixed for 5 days at 4°C. Sections were then processed for the detection of neurodegeneration with FD NeuroSilver Kit II (FD Neurotechnologies) according to the manufacturer’s instructions. Sections were subsequently mounted on slides, dehydrated in ethanol, cleared in xylene, and coverslipped with Permount (Fisher Scientific, Fair Lawn, NJ). All images were taken with an Aperio ScanScope (Leica Biosystems, Buffalo Grove, IL).
Immunohistochemistry quantification
Optical densitometry for quantification of immunohistochemical signal was modified from published methodology (Baldock and Poole et al. 1993). Images were captured with an upright microscope (Zeiss Axiolmager.M2) equipped with a monochromatic digital camera (Zeiss AxioCam MRm Rev.3) and processed with Zen imaging software (Zeiss 2012, Blue edition). The microscope light intensity and camera exposure were held constant. The operator outlined areas of interest around specific brain regions and recorded the intensity of light passing through the slide. Degenerating axons allowed less light to pass through the section owing to their uptake of silver stain, so lower light intensity correlated with increased degeneration. The operator performing quantification was blinded to condition and treatment.
Transmission electron microscopy
Mice were transcardially perfused with Karnovsky’s fixative solution (2% formaldehyde, 2.5% glutaraldehyde, 0.2 m sodium cacodylate buffer, 1 mm CaCl2, 2 mm MgCl2, and 42.8 mm NaCl, pH 7.4) 2 weeks after either sham or blast injury. Harvested brains were incubated in Karnovsky’s fixative solution overnight at 4°C. Whole brains were cut in the horizontal plane (100 µm) using a vibratome (Leica 1500). Sections that contained the hippocampus were selected, washed with 0.1 m sodium cacodylate buffer, and postfixed with 1% osmium fixative for 1 h. Sections were then dehydrated in a series of ethanol (50%, 75%, 95%, and 100%) followed by embedding in EPON resin overnight at 65°C. For transmission electron microscopy, ultrathin sections (60 nm) adjacent to semithin sections were cut with an ultramicrotome, loaded onto a Formvar 200-mesh Ni grid, and counterstained with uranyl acetate and lead citrate. Specimens were examined using a JEOL JEM 1230 electron microscope with a Gatan UltraScan 1000 2k x 2k charge-coupled device camera.
Statistical analysis
All data was compiled and analyzed using Graphpad Prism. Significance was performed with ANOVA and Tukey post hoc analysis.
Results
WldS mice are protected from learning and memory deficits after blast-mediated TBI
To evaluate the susceptibility of WldS mice to blast-mediated TBI, we used a model of blast injury in which a blast wave is intiated by rupture of a mylar membrane to expose anesthetized mice in an enclosed overpressure chamber composed of an air tank partitioned into two sides (Mohan et al., 2013; Yin et al., 2014)). A sealed mylar membrane covers a port between the two parts of the tank, and pressure is increased in the side without the mouse until the membrane ruptures at ∼27 kPa. This rupture generates a blast wave that travels through the mouse’s untethered head located in a padded holder, while the body is shielded by a metal tube. The intensity of the blast wave is 149.8 ± 2.09 kPa, and total duration of the pressue is ∼10–15 ms, composed of both blast wave and wind gust (Mohan et al., 2013; Yin et al., 2014).
The Barnes maze was used to evaluate hippocampal-dependent spatial learning in WldS mice after blast-induced TBI, with 25 animals per group. This task consists of a round table with equally spaced holes at its perimeter, one of which contains an escape cup. Mice are motivated to learn the location of the hole that houses the escape cup, so that they can enter the hole and hide in the cup to avoid exposure on the table. Testing was initiated 7 days after blast injury, beginning with 4 days of training in which mice were allowed to find and enter the escape hole and then rest in the protective cup. All mice, regardless of genotype or injury group, learned how to locate the platform more quickly over the course of the 4-day training period (Fig. 1A), indicating equal ability to learn in all four groups. On day 5, the probe test was conducted, in which the escape cup was removed and the ability of the mouse to remember the location of the cup was then assessed by measuring the amount of time the mouse spent in the area surrounding where the cup had been previously located. During the probe test, sham-injured wild-type animals spent ∼50% of their time in the escape area, defined as a 5-cm radius surrounding the escape hole (Fig. 1B). This indicates normal memory. There was no significant difference in performance in the probe test between sham-injured WldS mice and sham-injured wild-type littermate mice. In contrast, blast-injured wild-type mice spent only ∼20% of their time in the escape area (Fig. 1B; p < 0.0001 relative to sham-injury wild-type), indicating the expected degree of impaired memory after injury (Yin et al., 2014). No differences in this measure were seen between sham-injury and blast-injury WldS groups, in relation to each other or sham-injury wild-type mice. Importantly, both sham-injury and blast-injury WldS mice showed significantly greater time in the escape area than blast-injury wild-type mice, with ∼40% time in escape area for sham-injury WldS (p < 0.01 relative to sham-injury wild-type) and ∼43% time in escape area for blast-injury WldS (p < 0.0001 relative to sham-injury wild-type; Fig. 1B). This indicates that whereas the WldS mutation does not improve the animal’s memory under normal conditions, it does effectively block impairment in memory that is normally observed after blast injury. Importantly, none of the four groups differed in ability to physically participate in the task, as determined by comparable levels of average speed of locomotion and total distance traveled during the probe test (Fig 1C, D).
Figure 1.

WldS mice are protected from memory deficits after blast-mediated TBI. A, Latency to escape progressively decreases over the 4-day training period in all groups. B, Blast-mediated TBI wild-type (WT) mice spend less than half the time as sham-injury WTs in the escape area (5-cm radius around the escape hole) during the probe test of memory. Blast-mediated TBI WldS mice spend a amount of time in the escape area comparable to that of sham-injury WT and sham-injury WldS mice. C, The average locomotion speed during the probe trial was similar in all groups. D, The total distance traveled during the probe trial was similar in all groups. Each group consisted of 25 male congenic C57/Bl6 mice, aged 12–14 weeks. Data were collected and scored in an automated manner blind to treatment group. Data are represented as mean ± SEM. Significance was determined by two-way ANOVA with Bonferroni post hoc analysis. p-values labeled as **<0.01 and ****<0.0001 compared with blast-injured WT animals.
WldS mice are protected from motor coordination deficits after blast-mediated TBI
To assay balance and coordination, which are compromised after blast-mediated TBI in wild-type mice (Yin et al., 2014), we used the standard balance beam task (Luong et al., 2011). Mice were trained to cross an 80-cm-long balance beam over 2 days before the day of injury, and tested again 28 days after injury. Videorecording of all mice traversing the beam was analyzed for the number of foot slips by observers blind to genotype or injury group. Blast-injured wild-type mice displayed four times as many foot slips as sham-injured wild-type mice (p < 0.0001 relative to sham-injury wild-type), whereas sham- and blast-injury WldS mice showed no difference from sham-injury wild-type mice (Fig. 2). Thus, WldS mice are protected from the motor coordination deficits that are normally observed after blast-mediated TBI in mice.
Figure 2.
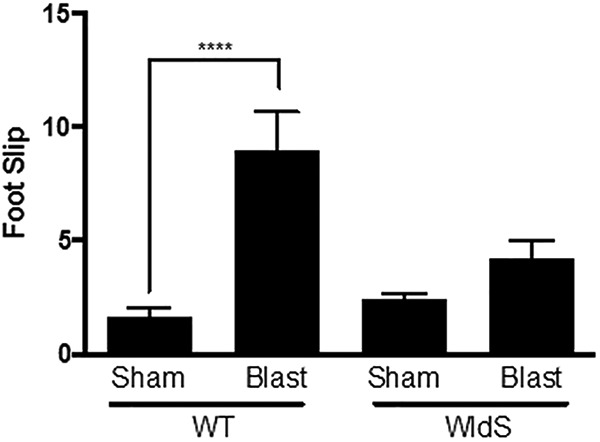
WldS mice are protected from motor coordination deficits after blast-mediated TBI. Blast-injured wild-type (WT) mice showed an increased number of foot slips relative to sham-injury WT mice 28 days after blast-mediated TBI. Blast-injured WldS mice show a similar number of foot slips as sham-injury WT and sham-injury WldS mice. Each group consisted of 25 male congenic C57/Bl6 mice, aged 12–14 weeks. Data was manually collected and scored blind to treatment group. Data are represented as mean ± SEM. Significance was determined by two-way ANOVA with Bonferroni post hoc analysis. Significance was determined by two-way ANOVA with Bonferroni post hoc analysis. p-values labeled as ****<0.0001 compared to sham-injured WT animals.
WldS mice are protected from axonal degeneration after blast-mediated TBI
Histologic examination of brain tissue for evidence of axonal degeneration was conducted via silver staining 12 days after injury, as previously established (Yin et al., 2014). In wild-type mice, blast injury was associated with prominent silver staining of degenerating axons in CA1 stratum radiatum of the hippocampus, cerebellum, cortex, corpus callosum, olfactory bulb, striatum, and thalamus, with no injury in the hypothalamus (Figs. 3 and 4), consistent with previous observations in this TBI model (Yin et al., 2014). Use of an established technique of automated optical densitometry to quantify the magnitude of silver staining, in which decreased signal indicates greater impedence of light through the tissue owing to silver staining of degenerating axons (Yin et al., 2014), showed that in all cases the extent of axonal degeneration in wild-type mice after TBI was statistically significant compared with the sham group (Fig. 5). There were no signficant differences between sham-injured WldS mice and sham-injured wild-type littermate mice. In addition, no significant differences were noted between sham- and blast-injury WldS mice, and both of these groups also showed statistically greater signal than blast-injury wild-type mice in CA1 stratum radiatum of the hippocampus, cerebellum, cortex, corpus callosum, and striatum (Fig. 5).
Figure 3.

WldS hippocampus and cerebellum are protected from axonal degeneration after blast-mediated TBI. High-power representative pictures with 40× objective from hippocampal CA1 stratum radiatum and cerebellum show prominent silver staining of degenerating axons (red arrows) in blast-injured wild-type mice 12 days after injury, with little to no axonal degeneration in WldS mice after the same injury. Images shown are representative of typical images from five animals in each group. Scale bar = 2.5 mm.
Figure 4.

WldS mice are broadly protected throughout the brain from axonal degeneration after blast-mediated TBI. As in hippocampus and cerebellum, protection was also noted in WldS cortex, corpus callosum, olfactory bulb, striatum, and thalamus. Noticeably, the hypothalamus is resistant to axonal degeneration in wild-type mice after blast-mediated TBI. Images shown are representative of typical images from five animals in each group. Scale bar = 2.5 mm.
Figure 5.
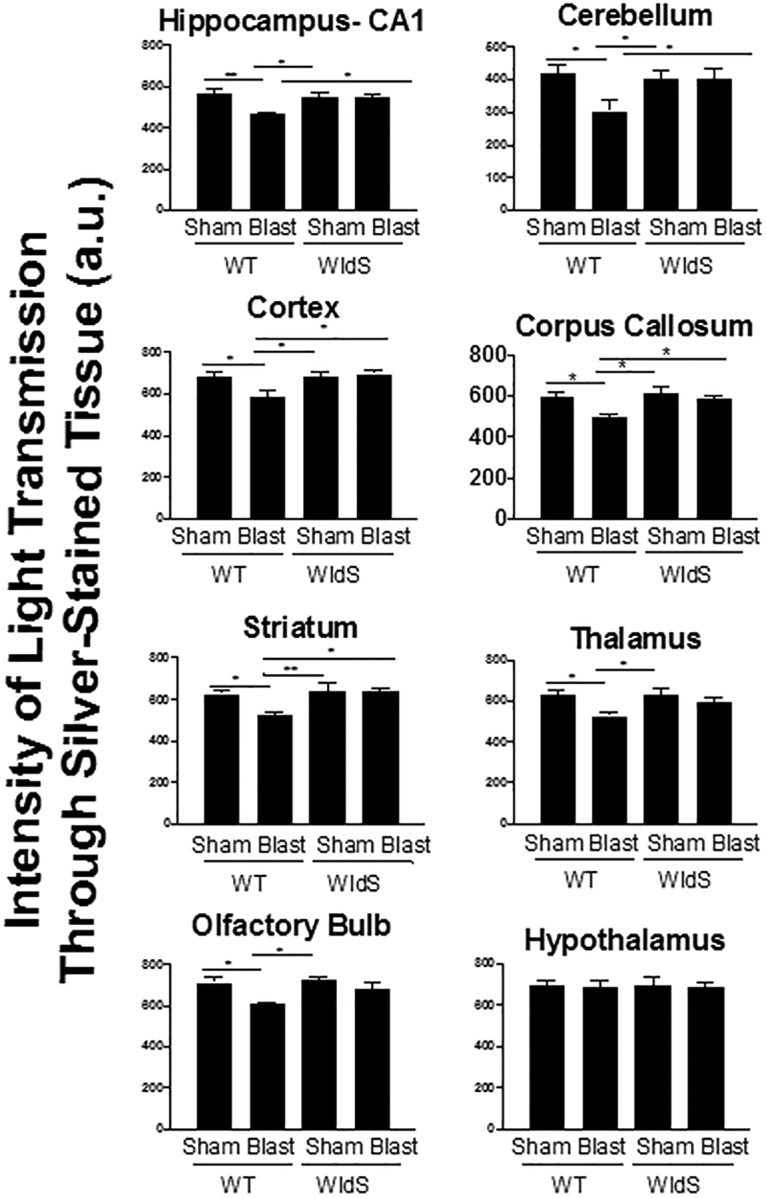
Optical densitometry of light transmitted through silver-stained brain regions from all animals in each group was used to quantify the protective effect. Signal was quantified for 18 sections for each of the five animals, spaced 480 mm apart. Here, a greater value indicates that more light was able to pass unimpeded through the section by virtue of less silver staining, which reflects less axonal degeneration. Data are represented as mean ± SEM. p-value *<0.05 and **<0.01 determined by two-way ANOVA with Bonferroni post hoc analysis compared with blast-injured WT animals.
Protective efficacy of the WldS mutation was confirmed by transmission electron microscopy of brain tissue 12 days after injury. This showed normal myelin and axonal mitochondrial structures in the CA1 stratum radiatum of sham-injury wild-type mice, as well as in sham- and blast-injury WldS mice (Fig. 6). Blast-injured wild-type mice, however, showed degeneration of the myelin sheath, as well as abnormal outer membrane and internal cristae structures within neuronal mitochondria.
Figure 6.
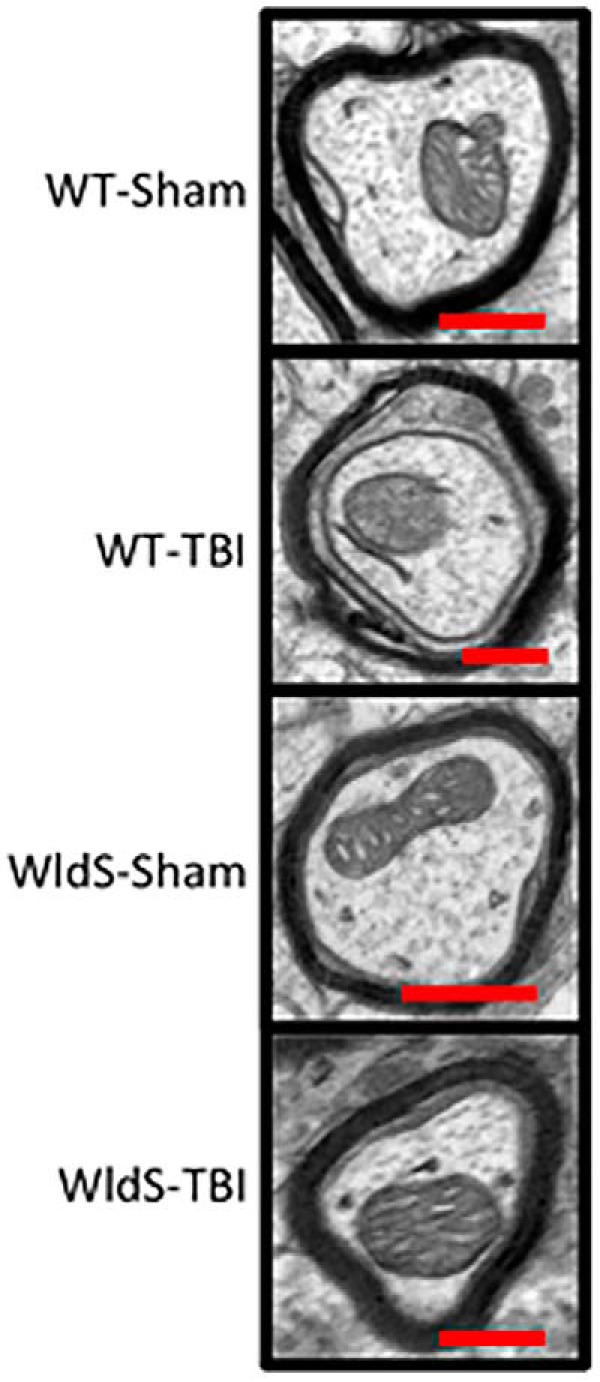
Transmission electron microscopy 12 days after injury shows normal myelin and axonal mitochondrial structures in the CA1 stratum radiatum of sham-injury wild-type mice, with no differences seen in sham- and blast-injury WldS mice. Blast-injured wild-type mice show degeneration of the myelin sheath along, as well as abnormal outer membrane and internal cristae structures within neuronal mitochondria.
WldS mice are protected from damage to the visual system after blast-mediated TBI
In patients, TBI frequently leads to chronic visual dysfunction, including light sensitivity, ocular motility dysfunction, optic neuropathy, retinopathy, and visual field loss (Cockerham et al., 2009, 2011; Lemke et al., 2013). A measure of retinal ganglion cell and optic nerve damage is the pattern electroretinogram (PERG), a painless and noninvasive objective diagnostic measure of retinal function that requires no verbal communication between patient and clinicians. This latter feature is important, given the acute variance in mental functioning of patients after TBI. PERG is applicable to both human patients and mice, and measures stimulus-evoked electrical activity of retinal cells in response to contrast modulation of patterned visual stimuli, such as a checkerboard, at constant luminance (Porciatti, 2007). Because deficits in PERG typically do not arise until visual damage has been sustained, this procedure has been modified to the provocative PERG (pPERG), in which mice or people are tilted with their head down to increase intraocular pressure. This amplifies retinal sensitivity to damage and has recently been shown to provide an early, sensitive, and noninvasive indicator of future chronic visual dysfunction after TBI, including later development of retinal cell death (Dutca et al., 2014). When wild-type mice were exposed to blast injury, they exhibited significantly decreased pPERG amplitude 4 weeks later, relative to sham injury wild-type animals (p < 0.05; Fig. 7). Both sham- and blast-injury WldS animals, however, showed pPERG amplitude preserved to the wild-type sham-injury level (p < 0.01 relative to blast-injury wild-type animals; Fig. 7), indicating that the WldS mutation protects the visual system after TBI.
Figure 7.
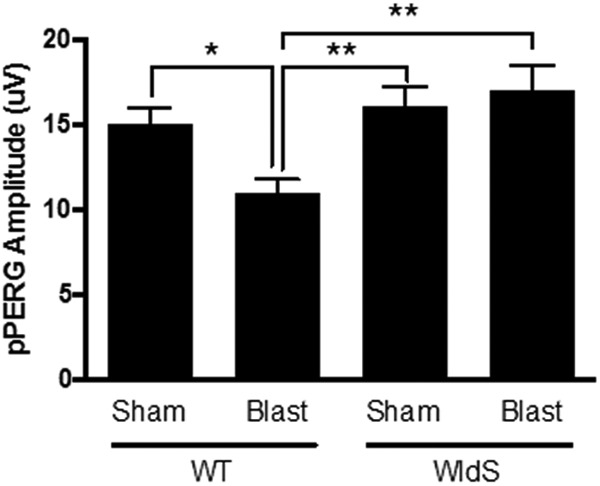
WldS mice are protected from pPERG deficits after blast-mediated TBI. pPERG serves as an early indicator of future chronic damage to the visual system, including retinal cell death. Wild-type (WT) mice exhibit ∼25% decrease in pPERG 4 weeks after blast injury, whereas both sham- and blast-injury WldS mice exhibit pPERG levels equivalent to sham-injury WT mice. Each group consisted of 25 male congenic C57/Bl6 mice, aged 12–14 weeks. Data were collected and scored in an automated manner blind to identification of the group and are represented as mean ± SEM. p-value *<0.05 and **<0.01 determined by-two way ANOVA with Bonferroni post hoc analysis, compared to blast-injured WT animals.
Discussion
An estimated 5 million people in the US currently experience long-term motor and cognitive disability related to TBI, at an annual cost exceeding $70 billion (Fleminger and Ponsford, 2005). Although axonal degeneration is a major aspect of many forms of neurodegeneration (Warner et al., 2010; Wang et al., 2012; Johnson et al., 2013; Smith et al., 2013), its contribution to the pathological and neurobehavioral deficits of blast-mediated TBI, the most common form of TBI sustained by soldiers in Iraq and Afghanistan, has not previously been investigated. Here, we show for the first time that early axonal degeneration is a critical driver of the development of neurologic deficits after blast-mediated TBI.
We addressed this issue by using WldS mice, which are are resistant to Wallerian degeneration, an active process of axon-autonomous self-destruction linked to neurodegeneration in both injury and disease (Conforti et al., 2014). These mice have been previously shown to be protected from Wallerian degeneration of axons after injury in different regions, including spinal cord (Fujiki et al., 1996; Zhang et al., 1996) and dentate gyrus (Schauwecker and Steward, 1997). Here, we report that these mice are broadly protected from axonal degeneration throughout the brain after blast injury. Importantly, this protection is also associated with complete preservation of normal cognitive, motor, and visual function after blast exposure. These preclinical studies were highly powered and rigorously executed, with data acquisition and analysis conducted blind to genotype and injury group. Given the current lack of efficacious treatment for patients with any form of TBI (Smith et al., 2013), including blast-mediated, these findings are highly clinically relevant. It is not known, however, whether the beneficial effect of WldS would also be effective post-injury, and future experiments will be needed to address this question. In addition, future work focused on specifically delineating the role of WldS in the distinct hippocampal, cerebellar, and visual neuronal circuitry underlying these disparate behavioral effects could add further insight into unique pathological processes involved in each domain after injury.
Although our results are highly suggestive that early thereapeutic intervention aimed at mitigating axonal degeneration is likely to benefit patients suffering from blast-mediated TBI, they do not provide definitive evidence that the functional improvement in WldS mice after blast-mediated TBI is exclusively the result of axonal protection. However, a complementary body of literature supports this notion. For example, it has recently been shown that genetic ablation of the Toll receptor adaptor sarm1 (sterile α/Armadillo/Toll-Interleukin receptor homology domain protein) gene, which is a key mediator of the active process of Wallerian degeneration, protects mice from multiple injury phenotypes after closed-head mild TBI (Henninger et al., 2016). These findings with a genetic loss-of-function model in TBI nicely complement our current findings with the gain-of-function WldS model.
In addition, recent pharmacologic agents shown to enhance flux of the NAD salvage pathway in normal mammalian cells have also demonstrated axonal protection associated with similar behavioral protection when administered after blast-mediated TBI (Yin et al., 2014), as well as behavioral and histological protection in concussive TBI models (Blaya et al., 2014). Furthermore, other avenues of augmenting neuronal NAD levels, such as administration of nicotinamide (Hoane et al., 2006, 2008; Goffus et al., 2010), poly(ADP-ribose) polymerase inhibition (Clark et al., 2007; Stoica et al., 2014), or intranasal delivery of NAD (Won et al., 2012) also show protective efficacy in multiple histologic and behavioral outcome measures after concussive TBI.
Thus, taken together, our findings support the notion that Wallerian degeneration is an important underlying pathological feature of blast-mediated TBI and its behavioral consequences. This illustrates the translational potential of NAD-augmenting therapies known to promote axonal survival, or future alternative approaches for promoting axonal survival, as a fruitful avenue for clinical treatment of patients suffering from the effects of blast-mediated TBI.
Acknowledgments
Acknowledgments: We thank Dr. Karen O’Malley of Washington University (St. Louis, MO) for providing breeding pairs of WldS mice.
Synthesis
The decision was a result of the Reviewing Editor Michael Sofroniew and the peer reviewers coming together and discussing their recommendations until a consensus was reached. A fact-based synthesis statement explaining their decision and outlining what is needed to prepare a revision is listed below. The following reviewers agreed to reveal their identity: Rajiv Ratan, John Greer
The paper is now acceptable.
References
- Araki T, Sasaki Y, Milbrandt J (2004) Increased nuclear NAD biosynthesis and SIRT1 activation prevent axonal degeneration. Science 305:1010–1013. 10.1126/science.1098014 [DOI] [PubMed] [Google Scholar]
- Blaya MO, Bramlett HM, Naidoo J, Pieper AA, Dietrich WD (2014) Neuroprotective efficacy of a proneurogenic compound after traumatic brain injury. J Neurotrauma 31:476–486. 10.1089/neu.2013.3135 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baldock RA, Poole I (1993) Video camera calibration for optical densitometry. J Microsc Oct. 172(Pt1):49–54. [DOI] [PubMed] [Google Scholar]
- Clark RS, Vagni VA, Nathaniel PD, Jenkins LW, Dixon CE, Szabó C (2007) Local administration of the poly(ADP-ribose) polymerase inhibitor INO-1001 prevents NAD+ depletion and improves water maze performance after traumatic brain injury in mice. J Neurotrauma 24:1399–1405. 10.1089/neu.2007.0305 [DOI] [PubMed] [Google Scholar]
- Cockerham GC, Goodrich GL, Weichel ED, Orcutt JC, Rizzo JF, Bower KSA, Schuchard RA (2009) Eye and visual function in traumatic brain injury. J Rehabil Res Dev 46:811–818. [DOI] [PubMed] [Google Scholar]
- Cockerham GC, Rice TA, Hewes EH, Cockerham KP, Lemke S, Wang G, Lin RC, Glynn-Milley C, Zumhagen L (2011) Closed-eye ocular injuries in the Iraq and Afghanistan wars. N Engl J Med 364:2172–2173. 10.1056/NEJMc1010683 [DOI] [PubMed] [Google Scholar]
- Coleman MP, Conforti L, Buckmaster EA, Tarlton A, Ewing RM, Brown MC, Lyon MF, Perry VH (1998) An 85-kb tandem triplication in the slow Wallerian degeneration (Wlds) mouse. Proc Natl Acad Sci U S A 95:9985–9990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conforti L, Gilley J, Coleman MP (2014) Wallerian degeneration: an emerging axon death pathway linking injury and disease. Nat Rev Neurosci 15:394–409. 10.1038/nrn3680 [DOI] [PubMed] [Google Scholar]
- Conforti L, Tarlton A, Mack TG, Mi W, Buckmaster EA, Wagner D, Perry VH, Coleman MP (2000) a Ufd2/D4Cole1e chimeric protein and overexpression of Rbp7 in the slow Wallerian degeneration (Wlds) mouse. Proc Natl Acad Sci U S A 97:11377–11382. 10.1073/pnas.97.21.11377 [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Jesús-Cortés H, Miller AD, Britt JK, DeMarco AJ, De Jesús-Cortés M, Stuebing E, Naidoo J, Vázquez-Rosa E, Morlock L, Williams NS, Ready JM, Narayanan NS, Pieper AA (2015) Protective efficacy of P7C3-S243 in the 6-hydroxydopamine model of Parkinson’s disease. NPJ Parkinsons Dis 1:pii:15010. 10.1038/npjparkd.2015.10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Jesús-Cortés H, Rajadhyaksha AM, Pieper AA (2016) Cacna1c: protecting young hippocampal neurons in the adult brain. Neurogenesis 10.1080/23262133.2016.1231160 [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Jesús-Cortés H, Xu P, Drawbridge J, Estill SJ, Huntington P, Tran S, Britt J, Tesla R, Morlock L, Naidoo J, Melito LM, Wang G, Williams NS, Ready JM, McKnight SL, Pieper AA (2012) Neuroprotective efficacy of aminopropyl carbazoles in a mouse model of Parkinson disease. Proc Natl Acad Sci U S A 109:17010–17015. 10.1073/pnas.1213956109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dutca LM, Stasheff SF, Hedberg-Buenz A, Rudd DS, Batra N, Blodi FR, Yorek MS, Yin T, Shankar M, Herlein JA, Naidoo J, Morlock L, Williams NS, Kardon RH, Anderson MG, Pieper AA, Harper MM (2014) Early detection of subclinical visual damage after blast-mediated TBI enables prevention of chronic visual deficit by treatment with P7C3-S243. Invest Ophthalmol Vis Sci 55:8330–8341. 10.1167/ioversus14-15468 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fleminger S, Ponsford J (2005) Long term outcome after traumatic brain injury. bmj 331:1419–1420. 10.1136/bmj.331.7530.1419 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fox G, Faden AI (1998) Traumatic brain injury causes delayed motor and cognitive impairment in a mutant mouse strain known to exhibit delayed Wallerian degeneration. J Neurosci Res 53:718–727. 10.1002/(SICI)1097-4547(19980915)53:6<718::AID-JNR9>3.0.CO;2-8 [DOI] [PubMed] [Google Scholar]
- Fujiki M, Zhang Z, Guth L, Steward O (1996) Genetic influences on cellular reactions to spinal cord injury: activation of macrophages/microglia and astrocytes is delayed in mice carrying a mutation (WldS) that causes delayed Wallerian degeneration. J Comp Neur 317:469–484. 10.1002/(SICI)1096-9861(19960729)371:3<469::AID-CNE9>3.3.CO;2-2 [DOI] [PubMed] [Google Scholar]
- Goffus AM, Anderson GD, Hoane MR (2010) Sustained delivery of nicotinamide limits cortical injury and improves functional recovery following traumatic brain injury. Oxid Med Cell Longev 3:145–152. 10.4161/oxim.3.2.11315 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldstein LE, Fisher AM, Tagge CA, Zhang XL, Velisek L, Sullivan JA, Upreti C, Kracht JM, Ericsson M, Wojnarowicz MW, Goletiani CJ, Maglakelidze GM, Casey N, Moncaster JA, Minaeva O, Moir RD, Nowinski CJ, Stern RA, Cantu RC, Geiling J, Blusztajn JK, Wolozin B, Ikezu T, Stein TF, Budson AE, Kowall NW, Chargin D, Sharon A, Saman S, Hall GF, Moss WC, Cleveland RO, Tanzi RE, Stanton PK, McKee AC (2012) Chronic traumatic encephalopathy in blast-exposed military veterans and a blast neurotrauma mouse model. Sci Transl Med 4:34ra60 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henninger N, Bouley J, Sikoglu EM, An J, Moore CM, King JA, Bowser R, Freeman MR, Brown JRF (2016) Attenuated traumatic axonal injury and improved functional outcome after traumatic brain injury in mice lacking Sarm1. Brain 139:1094–1105. 10.1093/brain/aww001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoane MR, Pierce JL, Holland MA, Anderson GD (2008) Nicotinamide treatment induces behavioral recovery when administered up to 4 hours following cortical contusion injury in the rat. Neuroscience 154:861–868. 10.1016/j.neuroscience.2008.04.044 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoane MR, Tan AA, Pierce JL, Anderson GD, Smith DC (2006) Nicotinamide treatment reduces behavioral impairments and provides cortical protection after fluid percussion injury in the rat. J Neurotrauma 23:1535–1548. 10.1089/neu.2006.23.1535 [DOI] [PubMed] [Google Scholar]
- Hoge CW, McGurk D, Thomas JL, Cox AL, Engel CC, Castro CA (2008) Mild traumatic brain injury in US soldiers returning from Iraq. N Engl J Med 358:453–463. 10.1056/NEJMoa072972 [DOI] [PubMed] [Google Scholar]
- Johnson VE, Stewart W, Smith DH (2013) Axonal pathology in traumatic brain injury. Exp Neurol 246:35–43. 10.1016/j.expneurol.2012.01.013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kemp SW, Szynkaruk M, Stanoulis KN, Wood MD, Liu EH, Willand MP, Morlock L, Naidoo J, Williams NS, Ready JM, Mangano TJ, Beggs S, Salter MW, Gordon T, Pieper AA, Borschel GH (2015) Pharmacologic rescue of motor and sensory function by the neuroprotective compound P7C3 following neonatal nerve injury. Neuroscience 284:202–216. 10.1016/j.neuroscience.2014.10.005 [DOI] [PubMed] [Google Scholar]
- Lee AS, De Jesús-Cortés H, Kabir ZD, Knobbe W, Orr M, Burgdorf C, Huntington P, McDaniel L, Britt JK, Hoffman F, Brat DJ, Rajadhyaksha AM, Pieper AA (2016) The neuropsychiatric disease-associated gene cacna1c mediates survival of young hippocampal neurons. eNeuro 3(2):1–11. 10.1523/ENEURO.0006-16.2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lemke S, Cockerham CG, Glynn-Milley C, Cockerham KP (2013) Visual quality of life in veterans with blast-induced traumatic brain injury. JAMA Ophthalmology 131:1602–1609. 10.1001/jamaophthalmol.2013.5028 [DOI] [PubMed] [Google Scholar]
- Lingor P, Koch JC, Tönges L, Bähr M (2012) Axonal degeneration as a therapeutic target in the CNS. Cell Tissue Res 349:289–311. 10.1007/s00441-012-1362-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lorber B, Tassoni A, Bull ND, Moschos MM, Martin KR (2012) Retinal ganglion cell survival and axon regeneration in Wlds transgenic rats after optic nerve crush and lens injury. BMC Neuroscience 13:56. 10.1186/1471-2202-13-56 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lunn ER, Perry VH, Brown MC, Rosen H, Gordon S (1989) Absence of Wallerian degeneration does not hinder regeneration in peripheral nerve. Eur J Neurosci 1:27–33. [DOI] [PubMed] [Google Scholar]
- Luong TN, Carlisle HJ, Southwell A, Patterson PH (2011) Assessment of motor balance and coordination in mice using the balance beam. J Vis Exp 10:2376 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mack TG, Reiner M, Beirowski B, Mi W, Emanuelli M, Wagner D, Thomson D, Gillingwater T, Court F, Conforti L, Fernando FS, Tarlton A, Andressen C, Addicks K, Magni G, Ribchester RR, Perry VH, Coleman MP (2001) Wallerian degeneration of injured axons and synapses is delayed by a Ube4b/Nmnat chimeric gene. Nat Neurosci 4:1199–1206. 10.1038/nn770 [DOI] [PubMed] [Google Scholar]
- MacMillan KS, Naidoo J, Liang J, Melito L, Williams NS, Morlock L, Huntington PJ, Estill SJ, Longgood J, Becker GL, McKnight SL, Pieper AA, De Brabander JK, Ready JM (2011) Development of proneurogenic, neuroprotective small molecules. J Am Chem Soc 133:1428–1437. 10.1021/ja108211m [DOI] [PMC free article] [PubMed] [Google Scholar]
- Magnuson J, Leonessa F, Ling GS (2012) Neuropathology of explosive blast traumatic brain injury. Curr Neurol Neurosci Rep 12:570–579. 10.1007/s11910-012-0303-6 [DOI] [PubMed] [Google Scholar]
- Meyer zu Horste G, Miesbach TA, Muller JI, Fledrich R, Stassart RM, Kieseier BC, Coleman MP, Sereda MW (2011) The Wlds transgene reduces axon loss in a Charcot-Marie-Tooth disease 1A rat model and nicotinamide delays post-traumatic axonal degeneration. Neurobiol Dis 42:1–8. 10.1016/j.nbd.2010.12.006 [DOI] [PubMed] [Google Scholar]
- Mohan K, Kecova H, Hernandez-Merino E, Kardon RH, Harper MM (2013) Retinal ganglion cell damage in an experimental rodent model of blast-mediated traumatic brain injury. Invest Ophthalmol Vis Sci 54:3440–3450. 10.1167/ioversus12-11522 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naidoo J, De Jesús-Cortés H, Huntington P, Estill S, Morlock LK, Starwalt R, Mangano TJ, Williams NS, Pieper AA, Ready JM (2014) Discovery of a neuroprotective chemical, (S)-N-(3-3,6-dibromo-9H-carbazol-9-yl)-2-fluoropropyl)-6-methoxypyridin-2-amine [(-)-P7C3-S243], with improved druglike properties. J Med Chem 57:3746–3754. 10.1021/jm401919s [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakagawa A, Manley GT, Gean AD, Ohtani K, Armonda R, Tsukamoto A, Yamamoto H, Takayama K, Tominaga T (2011) Mechanisms of primary blast-induced traumatic brain injury: insights from shock-wave research. J Neurotrauma 28:1101–1119. 10.1089/neu.2010.1442 [DOI] [PubMed] [Google Scholar]
- Paxinos G, Franklin K (1997) Mouse brain in stereotaxic coordinates. Los Angeles: Academic. [Google Scholar]
- Pieper AA, McKnight SL, Ready JM (2014) P7C3 and an unbiased approach to drug discovery for neurodegenerative disease. Chem Soc Rev 43:6716–6726. 10.1039/c3cs60448a [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pieper AA, Xie S, Capota E, Estill SJ, Zhong J, Long JM, Becker GL, Huntington P, Goldman SE, Shen CH, Capota M, Britt JK, Kotti T, Ure K, Brat DJ, Williams NS, MacMillan KS, Naidoo J, Melito L, Hsieh J, De Brabander J, Ready JM, McKnight SL (2010) Discovery of a proneurogenic, neuroprotective chemical. Cell 142:39–51. 10.1016/j.cell.2010.06.018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Porciatti V (2007) The mouse pattern electroretinogram. Doc Ophthalmol 115:145–153. 10.1007/s10633-007-9059-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raff MC, Whitmore AV, Finn JT (2002) Axonal self-destruction and neurodegeneration. Science 296:868–871. 10.1126/science.1068613 [DOI] [PubMed] [Google Scholar]
- Sajadi A, Schneider BL, Aebischer P (2004) Wlds-mediated protection of dopaminergic fibers in an animal model of Parkinson disease. Curr Biol 14:326–330. 10.1016/j.cub.2004.01.053 [DOI] [PubMed] [Google Scholar]
- Sasaki Y, Araki T, Milbrandt J (2006) Stimulation of nicotinamide adenine dinucleotide biosynthetic pathways delays axonal degeneration after axotomy. J Neurosci 26:8484–8491. 10.1523/JNEUROSCI.2320-06.2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schauwecker PE, Steward O (1997) Genetic influences on cellular reactions to brain injury: activation of microglia in denervated neuropil in mice carrying a mutation (Wld(S)) that causes delayed Wallerian degeneration. J Comp Neur 380:82–94. [DOI] [PubMed] [Google Scholar]
- Shively S, Scher AI, Perl DP, Diaz-Arrastia R (2012) Dementia resulting from traumatic brain injury: what is the pathology? Arch Neurol 69:1245–1251. 10.1001/archneurol.2011.3747 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith DH, Hicks R, Povlishock JT (2013) Therapy development for diffuse axonal injury. J Neurotrauma 30:307–323. 10.1089/neu.2012.2825 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stoica BA, Loane DJ, Zhao Z, Kabadi SV, Hanscom M, Byrnes KR, Faden AI (2014) PARP-1 inhibition attenuates neuronal loss, microglia activation and neurological deficits after traumatic brain injury. J Neurotrauma 31:758–772. 10.1089/neu.2013.3194 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tesla R, Wolf HP, Xu P, Drawbridge J, Estill SJ, Huntington P, McDaniel L, Knobbe W, Burket A, Tran S, Starwalt R, Morlock L, Naidoo J, Williams NS, Ready JM, McKnight SL, Pieper AA (2012) Neuroprotective efficacy of aminopropyl carbazoles in a mouse model of amyotrophic lateral sclerosis. Proc Natl Acad Sci U S A 109:17016–17021. 10.1073/pnas.1213960109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walker AK, Rivera PD, Wang W, Chuang JC, Tran S, Osborne-Lawrence S, Estill SJ, Starwalt R, Huntington P, Morlock L, Naidoo J, Williams NS, Ready JM, Eisch AJ, Pieper AA, Zigman JM (2015) The P7C3 class of neuroprotective compounds exerts antidepressant efficacy in mice by increasing hippocampal neurogenesis. Mol Psychiatry 20:500–508. 10.1038/mp.2014.34 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang G, Han T, Nijhawan D, Theodoropoulos P, Naidoo J, Yadavalli S, Mirzaei H, Pieper AA, Ready JM, McKnight SL (2014) P7C3 neuroprotective chemicals function by activating the rate-limiting enzyme in NAD salvage. Cell 158:1324–1334. 10.1016/j.cell.2014.07.040 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang J, Zhai Q, Chen Y, Lin E, Gu W, McBurney MW, He Z (2005) A local mechanism mediates NAD-dependent protection of axon degeneration. J Cell Biol 170:349–355. 10.1083/jcb.200504028 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang JT, Barres BA (2012) Axon degeneration: where the WldS things are. Curr Biol 22:R221–R223. 10.1016/j.cub.2012.02.056 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang JT, Medress ZA, Barres BA (2012) Axon degeneration: molecular mechanisms of a self-destruction pathway. J Cell Biol 196:7–18. 10.1083/jcb.201108111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang JT, Medress ZA, Vargas ME, Barres BA (2015) Local axonal protection by WldS as revealed by conditional regulation of protein stability. Proc. Natl. Acad. Sci USA 112:10092–10100. 10.1073/pnas.1508337112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warner MA, Marquez de la Plata C, Spence J, Wang JY, Harper C, Moore C, Devous M, Diaz-Arrastia R (2010) Assessing spatial relationships between axonal integrity, regional brain volumes, and neuropsychological outcomes after traumatic axonal injury. J Neurotrauma 27:2121–2130. 10.1089/neu.2010.1429 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolf SJ, Bebarta VS, Bonnet CJ, Pons PT, Cantrill SV (2009) Blast injuries. Lancet 374:405–415. 10.1016/S0140-6736(09)60257-9 [DOI] [PubMed] [Google Scholar]
- Won SJ, Choi BY, Yoo BH, Sohn M, Ying W, Swanson RA, Suh SW (2012) Prevention of traumatic brain injury-induced neuron death by intranasal delivery of nicotinamide adenine dinucleotide. J Neurotrauma 29:1401–1409. 10.1089/neu.2011.2228 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yin TC, Britt JK, De Jesús-Cortés H, Lu Y, Genova RM, Khan MZ, Voorhees JR, Shao J, Katzman AC, Huntington PJ, Wassink C, McDaniel L, Newell EA, Dutca LM, Naidoo J, Cui H, Bassuk AG, Harper MM, McKnight SL, Ready JM, Pieper AA (2014) P7C3 neuroprotective chemicals block axonal degeneration and preserve function after traumatic brain injury. Cell Reports 8:10–11. 10.1016/j.celrep.2014.08.030 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Z, Fujiki M, Guth L, Steward O (1996) Genetic influences on cellular reactions to spinal cord injury: a wound-healing response present in normal mice is impaired in mice carrying a mutation (WldS) that causes delayed Wallerian degeneration. J Comp Neur 371:485–495. 10.1002/(SICI)1096-9861(19960729)371:3&lt;485::AID-CNE10&gt;3.0.CO;2-I [DOI] [PubMed] [Google Scholar]