

Abstract
Psychiatric diseases, notably major depression, are associated with imbalance of excitatory and inhibitory neurotransmission within the prefrontal cortex (PFC) and related limbic brain circuitry. In many cases these illnesses are precipitated or exacerbated by chronic stress, which also alters excitatory and inhibitory neurotransmitter systems. Notably, exposure to repeated uncontrollable stress causes persistent changes in the synaptic integrity and function of the principal glutamatergic excitatory neurons in the PFC, characterized by neuronal atrophy and loss of synaptic connections. This can lead to dysfunction of the PFC circuitry that is necessary for execution of adaptive behavioral responses. In addition, an emerging literature shows that chronic stress also causes extensive alteration of GABAergic inhibitory circuits in the PFC, leading to the hypothesis that inhibitory neurotransmitter deficits contribute to changes in PFC neuronal excitability and cognitive impairments. Here we review evidence in rodents and human, which point to the mechanisms underlying stress-induced alterations of GABA transmission in the PFC, and its relevance to circuit dysfunction in mood and stress related disorders. These findings suggest that alterations of GABA interneurons and inhibitory neurotransmission play a causal role in the development of stress-related neurobiological illness, and could identify a new line of GABA related therapeutic targets.
Introduction
The prefrontal cortex (PFC) plays a central role in stress adaptation (1, 2), and impaired circuitry and function of PFC subregions are pathological features of many psychiatric illnesses (3, 4). Clinical research has consistently reported that depression and other stress-related illnesses are associated with decreased volume, neuronal atrophy, and altered connectivity of PFC (5). These findings in humans are supported by rodent studies demonstrating that chronic stress exposure produces a number of alterations in the PFC, including dendritic atrophy and synapse loss (6–8), as well as loss of neurotrophic factor support. These core features of rodent stress studies have led to the hypothesis that reductions in neurotrophic factor expression result in neuronal and synaptic morphological deficits observed in human subjects (for review see (9)). A related hypothesis suggests that an imbalance in excitatory and inhibitory neurotransmission occurring directly through deficient GABAergic inhibitory signaling in the PFC could account for the outcomes observed in human subjects and rodent models.
In this paper, we review the literature and evidence demonstrating GABA dysfunction in human depression as well as in preclinical rodent stress studies. We then point to recent examples from studies in transgenic mice that shed light on how GABA interneuron subtypes can balance cortical transmission and ultimately shape top down control of depression- and anxiety-like behaviors. Finally, based on recent studies, we propose how intra-cortical GABA inhibition in the PFC can provide important therapeutic targets for the treatment of depression and other psychiatric illness.
GABAergic Neurotransmission in the PFC
The PFC is comprised of a heterogeneous population of neurons, including the glutamatergic principal excitatory neurons and GABAergic inhibitory interneurons. GABAergic interneurons comprise approximately 25% of the neurons within the neocortex (10), and are responsible for inhibitory control via activation of ionotropic GABAA receptors, which gate chloride entry into neurons (10). GABA transmission is also mediated through metabotropic GABAB receptors, which are generally thought to be active at higher GABA levels and can act as autoreceptors on presynaptic GABA terminals, as well as provide direct inhibition on principal neurons (11). GABAergic interneurons differ considerably in their morphology, electrophysiological properties, connectivity, and expression of neuropeptides and calcium-binding proteins (10). There are at least 20 different subtypes of cortical GABAergic interneurons that can be differentiated by expression of various molecular markers, such as, parvalbumin (PV), somastostatin (SST), cholecystokinin (CCK), calbindin, calretinin, and vasoactive intestinal peptide (10). Recent advances in genetic manipulation are allowing subtype specific modification of GABAergic interneurons, as well as specific GABAA receptor subunits that provide insight into the function of GABA mediated inhibition in stress associated disease states. In the following sections, we will discuss how chronic stress and depression cause diverse deficits in GABAergic inhibitory neurotransmission within the PFC, including reduced PFC GABA bioavailability, reduced levels of GABA receptors, and impaired function of specific subtypes of GABA interneurons which potentially contribute to pathological conditions.
PFC GABA Dysfunction in Stress and Depression
Until recently, much of the work on stress, depression, and PFC function has focused on alteration of the principle excitatory glutamatergic neurons. However, accumulating evidence suggests that loss of intra-cortical GABAergic transmission and consequent imbalances in excitatory and/or inhibitory neurotransmission in the PFC contribute to the etiology of stress-related psychiatric disorders (12). Clinical studies have reported reduced GABA levels in the frontal cortex of depressed individuals as compared to healthy humans. This includes a preliminary magnetic resonance spectroscopy study reporting that GABA levels are reduced in the occipital cortex of unmedicated depressed individuals (13). Further studies using the same approach demonstrated reduced GABA levels in PFC subregions, which are of greater relevance to depression and other psychiatric illnesses (14), and observed that remission was associated with normalization of GABA levels (15). Similarly, depressed patients that responded to repetitive transcranial magnetic stimulation also demonstrated an increase in GABA levels in the PFC that were absent in non-responders (16). Low plasma GABA levels were also reported to be predictive for the development of other psychiatric illness, such as, posttraumatic stress disorder (PTSD) (17), and maintenance of PTSD with comorbid depression (18).
Although, little is known about the regulation of GABA receptor subunits in psychiatric illness, accumulating evidence suggests that the GABA receptor expression is highly altered in depression. For example, reduced transcripts for GABAA subunits were observed in Brodmann areas 10 and 11, but not Brodmann 9 in depressed suicide victims (19). Subunit specific hypermethylation of GABAA promoters in suicide victims previously diagnosed with depression was also observed in Brodmann 10 providing support for transcriptional repression (20). GABAA subunit upregulation has also been observed most notably within the anterior cingulate region (Brodmann 24) (21, 22), which could represent a compensatory response to the reduced GABAergic tone. Reduced GABAA receptor binding has also been associated with PTSD (23). Similar studies in the frontal cortex of depressed patients are lacking but reduced GABAA receptor binding levels were observed in the parahippocampal and lateral temporal regions of depressed individuals (24).
Altered levels of other GABAergic markers have also been reported in depression and other psychiatric disorders. Postmortem brain samples of PFC subregions from unmedicated depressed individuals showed markedly decreased protein and mRNA levels of the GABA synthesizing enzyme glutamic acid decarboxylase (GAD) 67 that was not evident in subjects receiving treatment at the time of death (25). More recently, alterations in levels of specific GABAergic interneuron subtypes have been demonstrated. For example, reductions in the number and size of calbindin-positive neurons were reported in the dlPFC (26), as well as in occipital cortex (27) of depressed subjects. Transcript analyses of postmortem PFC from depressed patients also revealed a reduction in the expression of somastostatin (SST), a neuropeptide that is expressed in a subtype of GABA interneuron that makes up approximately 30% of all cortical GABA interneurons (28). Collectively, the literature summarized indicates that PFC GABAergic transmission is highly dysregulated in stress/depression and related disorders; however, the link between the diminished GABAergic transmission and affective disorders is still unknown.
For the purpose of this focused review, we have concentrated on the PFC and related subregions. However, multiple labs have contributed to the understanding GABAergic regulation in other brain regions in depression and other related disorders (12, 29).
GABA Deficits, PFC and Stress: Insights from Animal Models
Animal models of stress and depression are beginning to elucidate whether and how altered GABA transmission is causally linked to stress-related disorders. The strongest evidence lending support to this notion comes from studies using GABAA receptor mutant mice (mutant mice heterozygous for the γ2 subunit of the GABAA receptor, referred to as γ2+/−) (30, 31). These mice have reduced GABAA receptor binding, and the behavioral phenotype includes neophobia and behavioral inhibition (31), and anhedonia, which is a core symptom of depression (31). The γ2+/− mice also exhibit HPA axis hyperactivity (31), which is an endocrine hallmark of stress-related disorders. Increased anxiety- and depression-related behaviors have also been reported in mice lacking the α2 subunit of GABAA receptor that is highly expressed in the neocortex (12). Most recent evidence further indicates that γ2+/− mutant mice have significantly reduced cell surface expression of N-methyl-D-aspartate (NMDA)- and AMPA- type glutamate receptors along with deficits in density of functional glutamatergic markers in the PFC and hippocampus (32). Interestingly, these deficits in the PFC were normalized following a single dose of ketamine (a rapid-acting antidepressants) (32). These findings in GABAA mutant lines provide compelling evidence that loss of GABA function plays a casual role in the development of mood and cognitive disorders.
Experiments using chronic stress paradigms in rodents provide further evidence for GABA receptor dysregulation in mood disorders. For example, exposure to maternal separation stress during the first postnatal week decreased the expression of GABAA receptors in the frontal cortex and other stress-effected brain regions in adulthood (33). Other preclinical studies of chronic stress, such as, chronic cold (34), chronic foot shock (35), and social isolation (36), also report stress-induced decreases in GABAA receptor expression in cortical brain regions. However, it is important to note that the stress-induced changes in GABAA receptor expression are highly dependent on stressor modality, intensity, and brain regions analyzed. For example, chronic immobilization stress leads to increased GABAA receptor expression in the PFC (35). Collectively, the findings indicate that GABAergic system in the PFC is highly sensitive to stress.
Stress also reduces other markers of prefrontal GABA transmission. For instance, social isolation causes a 40% reduction in GABA transporter 1 immunolabeling in the PFC compared to socially housed littermate rats (37). A recent analysis of parvalbumin transcript levels and number of PV-positive cells in PFC following exposure to a chronic unpredictable mild stress paradigm revealed somewhat conflicting results (38). This is intriguing, and one possible explanation could be the sustained hypoactivity of SST interneurons. SST interneurons inhibit PV interneurons, and therefore, sustained SST interneuron loss of function could serve as a primary driver for the hyperactivity of PV interneurons. Further studies are required to test the link between SST and PV interneurons and output of excitatory neurons in the PFC. A similar phenomenon has been described in mouse motor cortex where it has been shown that SST interneurons provide an overall disinhibition signal to pyramidal neurons via inhibition of PV interneurons (39)
It has also been suggested that decreased GABA function could play a causative role in the reduction of excitatory synapses resulting from chronic unpredictable stress CUS (12). This is supported by evidence that partial (30%) ablation of SST neurons results in compensatory, long-lasting reductions in cortico-cortical excitatory drive (40). Decreased GABA function could contribute to reduction in 5-HT stimulated cortico-cortical drive that is caused by CUS (41). Together, the above evidence indicates that changes in PFC GABAergic transmission increase the vulnerability to stress-related illness.
GABA interneuron subtypes and microcircuit characteristics
The SST and the PV are the two major subtypes of cortical GABAergic interneurons that differ substantially with respect to morphology, electrophysiological properties, firing rate and most importantly targeting of specific cellular domains of pyramidal neurons and other interneurons (Figure 1). Approximately 25% of cortical GABA interneurons express SST and 40% express PV. Subtype specific targetting using CRE recombinase under the control of SST or PV promoters has provided important advances in our knowledge of the functions of interneuron subtypes. The SST and PV subtypes have received the most attention in studies of stress-related disease and are therefore the focus of the current review (Figure 1). However, additional GABA interneuron subtypes may also play an important role in mood disorders.
Figure 1. Impact of stress on local GABA interneuron activity and regulation of PFC microcircuitry.
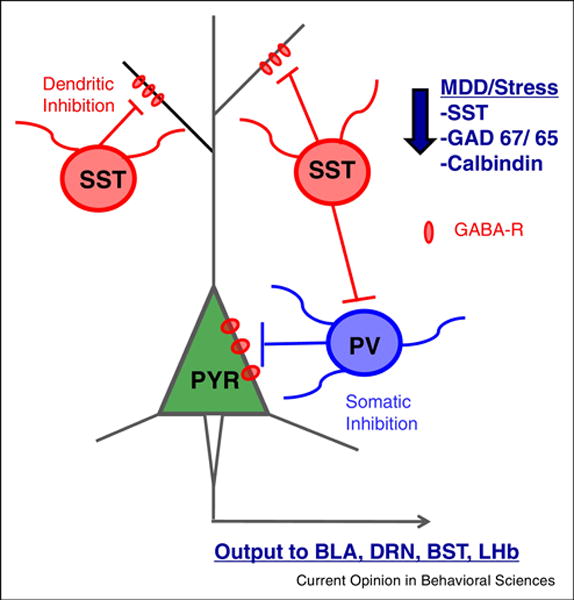
The PFC glutamatergic output neuron is under regulatory control of local GABA interneurons, predominantly somatostatin (SST) and parvalbumin (PV). SST provides inhibition of the dendritic compartment of excitatory pyramidal cells as well as to PV subtype GABA interneurons. PV interneurons provide peri-somatic inhibition to the pyramidal cells. The PFC projects to numerous subcortical and brainstem targets, such as the bed nucleus of the stria terminalis (BST), central nucleus of the amygdala (CeA), and dorsal raphe nucleus (DRn) that contribute to regulation of mood and emotion. Studies of MDD subjects and rodent stress models demonstrate reductions in levels of SST, GAD67/65, and calbindin, another marker of SST interneurons.
The SST GABAergic interneurons extend projections that target the dendritic compartment of PFC principal excitatory neurons and are therefore positioned to regulate the effects of incoming signals to principal neurons. Additionally, SST neurons play a role in establishing a balance of excitation and inhibition by directly inhibiting other classes of interneurons, notably PV cells. This has important behavioral implications as putative SST interneuron inhibition of PV cells in the PFC has been demonstrated to govern fear expression (42). Interestingly, investigation of post-mortem human tissue has demonstrated a reduction in SST content in the PFC of individuals with MDD (43–45). The functional consequences of decreased SST have been examined in an elegant study of SST deletion mutant mice. These mice display several depression related phenotypes, including increased basal corticosterone, reduced BDNF transcript levels, and increased anxiety- and depression-like behaviors (46). These findings support the hypothesis that decreased SST expression contributes to the depression related endocrine, neurotrophic, and behavioral symptoms.
Other studies extend this work by testing the influence of chemogenetic inhibition of SST interneurons in the dorsal PFC. The results of these studies show that acute inhibition produces changes in behavior similar to deletion of SST knock-out, an acute increase in depression-like behaviors (47). However, 3 weeks of repeated SST inhibition or ablation SST interneurons in adult mice produced the opposite effects, producing antidepressant-responses (47). One factor to consider, particularly with the acute studies is that analysis of behaviors during this period could be confounded by the acute inhibition of SST neurons and the resulting hyperexcitability of the principle neurons. Nevertheless, these findings demonstrate the complexity of modeling reduced GABAergic signaling in depression, but provide compelling evidence that SST interneurons may play a significant role in pathology of mood disorders.
PV interneurons are a large component of the basket cell population that targets the soma of PFC principal neurons, and exhibit a phasic, fast spiking electrophysiological profile (10). Soma targeting places PV interneurons in a position to gate the output of principal neurons, as opposed to the SST cells that gate input to these neurons. Consistent with this, cell type specific manipulation of PV interneurons has demonstrated their importance in maintaining balance between excitation and inhibition, and relevance to numerous emotional behaviors. Optogenetic studies demonstrate that inhibition of PV interneurons in the dorsal PFC is critical for inhibition of fear expression during extinction (42). In addition, reduced excitatory drive onto PV neurons in the dorsal PFC was observed in mice after exposure to extreme footshock that produces learned helplessness behavior (48); in addition, mice that were identified as resilient could be rendered helpless by chemogenetic inhibition of PV neurons. Based on recent advances and emerging evidence, we have provided a conceptual framework of how SST and PV interneurons gate PFC activity to regulate behavior, and how they interact within the PFC microcircuit prior to, and following, stress (Figure 1), however a clearer and more detailed understanding of the function of these interneuron subtypes will help inform our knowledge of the mechanisms underlying stress associated diseases.
Role of GABA Interneurons in the Actions of Antidepressants
Further support for aberrant regulation of GABAergic tone comes from studies demonstrating normalization of GABA transmission after antidepressant treatment (12). Patients exposed to treatment with SSRIs (49), electroconvulsant therapy (50), and transcranial magnetic stimulation (16) showed normalization of GABA levels. Consistent with human studies, preclinical data obtained from GABA receptor mutant mice (γ2+/−) that exhibit anxiety and depressive behaviors show partial normalization following fluoxetine treatment (31). Chronic fluoxetine treatment also increases extracellular GABA levels in brain (51), suggesting that increased GABA contributes to antidepressant behavioral responses. GABA neurotransmission, including synapse formation is controlled by BDNF-TrkB signaling (52) and it is possible that BDNF deficits caused by stress could contribute to deficits in GABA signaling. Conversely, up regulation of BDNF-TrkB signaling in response to antidepressant treatment could promote GABA synaptic activity (53).
Further evidence for GABA interneurons in the treatment of depression come from recent studies of rapid acting antidepressants, particularly ketamine. Clinical studies demonstrate that ketamine (NMDA receptor antagonist), produce rapid (within hours) antidepressant effects in treatment resistant patients (54, 55). This is particularly notable when compared with typical antidepressants that have a significant time lag (weeks to months) and modest efficacy. Interestingly, there is evidence that actions of ketamine include regulation of GABAergic signaling (29). Our previous studies reveal that ketamine rapidly increases synaptic connectivity, and reverses the neuronal atrophy and behavioral deficits caused by chronic stress (56, 57). These effects are activity dependent and are associated with a burst of glutamate in the PFC (58). NMDARs are expressed on interneurons as well as excitatory neurons and because interneurons are more active at resting state are more sensitive to the open channel blocker actions of ketamine. This is supported by evidence that ketamine administration leads to decreased spontaneous firing of GABA interneurons in the PFC and a delayed increase in the firing rate of pyramidal cells (29). Importantly, ketamine-induced disinhibition would be dependent on the presence of ongoing GABAergic suppression of neocortical activity. The GABA interneurons are divided into different classes with different firing responses (the low threshold spiking SST neurons, fast spiking PV cells, and delayed spiking non PV and non SST interneurons). Out if these, the SST interneurons exhibit high basal firing rates and a > 10 fold higher firing frequency (59), indicating that the spontaneous activity of these GABAergic interneurons mediates the maintenance of a highly suppressed state of cortical synaptic transmission. Together, These findings support a disinhibition hypothesis for the activity dependent actions of ketamine.
Similar effects have been observed for another rapid acting agent, scopolamine, an acetylcholine muscarinic receptor antagonist. Consistent with this hypothesis, our recent study demonstrated that M1-acetylcholine receptor (M1-AchR) expression in the SST interneurons is required for the rapid antidepressant-like effects of scopolamine (60). We found that M1-AChR activation on SST interneurons stimulates the firing of these inhibitory neurons, and that knockdown of M1-AChR on SST neurons blocks the antidepressant behavioral actions of scopolamine. This finding indicates that reduced SST activity within the dendritic field of PFC principal neurons is a necessary component of the rapid antidepressant response.
GABA hypofunction in response to stress and in MDD appears to contradict the ketamine disinhibition mechanism, but there are several key issues to consider. Importantly, the disinhibition hypothesis describes the initial actions (~1 hr) of ketamine, blockade of NMDA receptors on GABA interneurons that triggers a glutamate burst; this initiates, but is distinct from the long-lasting (1–7 d) synaptic (57) and therapeutic actions of ketamine (54, 61). Thus, we propose that ketamine-induced disinhibition causes an adaptive response that restores the excitatory/inhibitory balances (Figure 2). According to this hypothesis, acute, transient suppression of PFC GABAergic interneurons would produce antidepressant behavioral effects. Experiments to test this hypothesis require selective activation and/or inihibition of specific interneuron populations (SST or PV) using a combination of GABA interneuron specific Cre recombinase mice and light or chemically driven manipulation of cell activity.
Figure 2. Schematic representation of ketamine- and scopolamine- mediated disinhibition of mPFC pyramidal neurons via inhibition of local GABA interneurons.
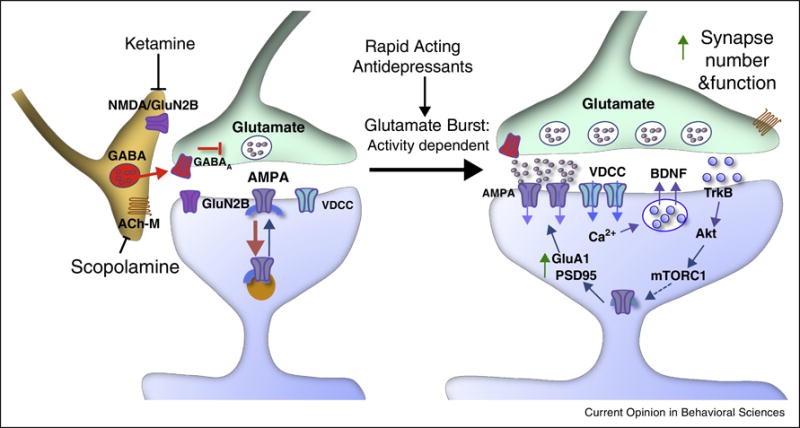
Ketamine triggers a burst of glutamate that is thought to occur via inhibition of GABA interneurons; the tonic firing of these GABA interneurons is driven by NMDA receptors, and the active, open-channel state allows ketamine to enter and block channel activity. The resulting glutamate burst stimulates AMPA receptors, which causes depolarization and activation of voltage-dependent Ca2+ channels (VDCC), leading to release of BDNF and stimulation of TrkB and Akt, which then activates mTORC1 signaling, leading to the increased synthesis of proteins that are required for synapse maturation and formation (i.e., GluA1 and PSD95). Scopolamine also causes a glutamate burst via blockade of acetylcholine muscarinic M1 (ACh-M1) receptors on GABA interneurons.
Conclusion
From early development to adulthood GABA interneurons play crucial role in assembling the microcircuitry and orchestrating the activity of the cerebral cortex. Impairments in the function of the cortical GABAergic transmission exert a strong influence on brain function, including cognitive, mood, learning and behavior. Here, we highlight recent findings that are beginning to delineate how changes in various components of the PFC GABAergic microcircuit are casually linked to stress and depression. Indeed, the studies of ketamine and scopolamine have generated considerable excitement, pointing to a key role of GABAergic transmission in the effects of rapid acting antidepressants, and in the development of next-generation therapeutics (Figure 2).
Despite intensive research, we are left with a number of significant gaps in our understanding of GABA/glutamate balance in the pathophysiology of depression and other stress related illnesses. One problem that has hindered the complete understanding of the underlying cause of depression is the lack of techniques to selectively manipulate each interneuron subtype. This is now being addressed with advances in optogenetics, chemogenetics, microendoscopy, and imaging technology; these approaches will allow studies to determine the influence of stress on the activity of GABA interneuron subtypes and the effects of activation or inhibition of specific GABA cell populations on neighboring GABA and principle neurons, as well as behavior. Moreover, analysis of sex specific differences in stress-induced effects on GABA interneuron populations are surprisingly incomplete, and could lead to improved treatments for women who experience higher rates of depression compared to men. Progress and new insights in these areas will help us to generate alternative and more efficacious therapeutic strategies and eventually prevention of stress related illnesses such as depression.
Highlights.
Chronic stress causes atrophy and decreased connectivity of excitatory neurons in PFC.
Stress causes disruption of GABAergic inhibitory neurons in the PFC.
GABA subtypes, somatostatin and parvalbumin, are dysregulated by stress.
Rapid acting antidepressants inhibit GABAergic interneurons.
Acknowledgments
This work was supported by NIMH R37MH45481, NIMH R01MH93897, Yale University School of Medicine and by the State of Connecticut.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Conflict of Interest
Dr. Duman has received consulting, speaking fees and/or grant support from Naurex, Taisho, Johnson & Johnson, Lilly, Lundbeck, Sunovion, Navitor, and Forest. Drs. Ghosal and Hare declare no competing interests.
References
* Indicates papers of particular interest
- 1.Duman RS. Neurobiology of stress, depression, and rapid acting antidepressants: remodeling synaptic connections. Depression and anxiety. 2014;31(4):291–6. doi: 10.1002/da.22227. Epub 2014/03/13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Popoli M, Yan Z, McEwen BS, Sanacora G. The stressed synapse: the impact of stress and glucocorticoids on glutamate transmission. Nature reviews Neuroscience. 2012;13(1):22–37. doi: 10.1038/nrn3138. Epub 2011/12/01. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Goto Y, Yang CR, Otani S. Functional and dysfunctional synaptic plasticity in prefrontal cortex: roles in psychiatric disorders. Biological psychiatry. 2010;67(3):199–207. doi: 10.1016/j.biopsych.2009.08.026. Epub 2009/10/17. [DOI] [PubMed] [Google Scholar]
- 4.Hains AB, Arnsten AF. Molecular mechanisms of stress-induced prefrontal cortical impairment: implications for mental illness. Learning & memory. 2008;15(8):551–64. doi: 10.1101/lm.921708. Epub 2008/08/08. [DOI] [PubMed] [Google Scholar]
- 5.Drevets WC, Price JL, Furey ML. Brain structural and functional abnormalities in mood disorders: implications for neurocircuitry models of depression. Brain structure & function. 2008;213(1–2):93–118. doi: 10.1007/s00429-008-0189-x. Epub 2008/08/16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Banasr M, Valentine GW, Li XY, Gourley SL, Taylor JR, Duman RS. Chronic unpredictable stress decreases cell proliferation in the cerebral cortex of the adult rat. Biological psychiatry. 2007;62(5):496–504. doi: 10.1016/j.biopsych.2007.02.006. Epub 2007/06/26. [DOI] [PubMed] [Google Scholar]
- 7*.Ota KTLR, Voleti B, Maldonado-Aviles JG, Duric V, Iwata M, Dutheil S, Duman C, Boikess S, Lewis DA, Stockmeier CA, DiLeone RJ, Rex C, Aghajanian GK, Duman RS. REDD1 is essential for stress-induced synaptic loss and depressive behavior. Nature medicine. 2014;20(5):531–5. doi: 10.1038/nm.3513. Epub 2014 Apr 13. This study provided evidence that REDD1 is a critical mediator of the atrophy of neurons and depressive behavior caused by chronic stress exposure in both human and preclinical model. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Radley JJ, Morrison JH. Repeated stress and structural plasticity in the brain. Ageing research reviews. 2005;4(2):271–87. doi: 10.1016/j.arr.2005.03.004. Epub 2005/07/05. [DOI] [PubMed] [Google Scholar]
- 9.Duman RS, Monteggia LM. A neurotrophic model for stress-related mood disorders. Biological psychiatry. 2006;59(12):1116–27. doi: 10.1016/j.biopsych.2006.02.013. Epub 2006/04/25. [DOI] [PubMed] [Google Scholar]
- 10.Markram H, Toledo-Rodriguez M, Wang Y, Gupta A, Silberberg G, Wu C. Interneurons of the neocortical inhibitory system. Nature reviews Neuroscience. 2004;5(10):793–807. doi: 10.1038/nrn1519. Epub 2004/09/21. [DOI] [PubMed] [Google Scholar]
- 11.Gassmann M, Bettler B. Regulation of neuronal GABA(B) receptor functions by subunit composition. Nature reviews Neuroscience. 2012;13(6):380–94. doi: 10.1038/nrn3249. Epub 2012/05/19. [DOI] [PubMed] [Google Scholar]
- 12.Luscher B, Fuchs T. GABAergic control of depression-related brain states. Advances in pharmacology. 2015;73:97–144. doi: 10.1016/bs.apha.2014.11.003. Epub 2015/02/01. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13*.Sanacora G, Mason GF, Rothman DL, Behar KL, Hyder F, Petroff OA, Berman RM, Charney DS, Krystal JH. Reduced cortical gamma-aminobutyric acid levels in depressed patients determined by proton magnetic resonance spectroscopy. Archives of general psychiatry. 1999;56(11):1043–7. doi: 10.1001/archpsyc.56.11.1043. Epub 1999/11/24. This study provided the first evidence that GABA levels are reduced in depression. [DOI] [PubMed] [Google Scholar]
- 14.Hasler G, van der Veen JW, Tumonis T, Meyers N, Shen J, WC D. Reduced prefrontal glutamate/glutamine and gamma-aminobutyric acid levels in major depression determined using proton magnetic resonance spectroscopy. Archives of general psychiatry. 2007;64(2):193–200. doi: 10.1001/archpsyc.64.2.193. Epub 2007/02/07. [DOI] [PubMed] [Google Scholar]
- 15.Hasler G, Neumeister A, van der Veen JW, Tumonis T, Bain EE, Shen J, Drevets WC, Charney DS. Normal prefrontal gamma-aminobutyric acid levels in remitted depressed subjects determined by proton magnetic resonance spectroscopy. Biological psychiatry. 2005;58(12):969–73. doi: 10.1016/j.biopsych.2005.05.017. Epub 2005/07/27. [DOI] [PubMed] [Google Scholar]
- 16.Dubin MJ, Mao X, Banerjee S, Goodman Z, Lapidus KA, Kang G, Liston C, Shungu DC. Elevated prefrontal cortex GABA in patients with major depressive disorder after TMS treatment measured with proton magnetic resonance spectroscopy. Journal of psychiatry & neuroscience : JPN. 2016;41(3):E37–45. doi: 10.1503/jpn.150223. Epub 2016/02/24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Vaiva G, Thomas P, Ducrocq F, Fontaine M, Boss V, Devos P, Rascle C, Cottencin O, Brunet A, Laffargue P, M G. Low posttrauma GABA plasma levels as a predictive factor in the development of acute posttraumatic stress disorder. Biol Psychiatry. 2004;55(3):250–4. doi: 10.1016/j.biopsych.2003.08.009. Epub 2004/01/28. [DOI] [PubMed] [Google Scholar]
- 18.Vaiva G, Boss V, Ducrocq F, Fontaine M, Devos P, Brunet A, Laffargue P, Goudemand M, P T. Relationship between posttrauma GABA plasma levels and PTSD at 1-year follow-up. The American journal of psychiatry. 2006;163(8):1446–8. doi: 10.1176/ajp.2006.163.8.1446. Epub 2006/08/01. [DOI] [PubMed] [Google Scholar]
- 19.Merali Z, Du L, Hrdina P, Palkovits M, Faludi G, Poulter MO, Anisman H. Dysregulation in the suicide brain: mRNA expression of corticotropin-releasing hormone receptors and GABA(A) receptor subunits in frontal cortical brain region. The Journal of neuroscience : the official journal of the Society for Neuroscience. 2004;24(6):1478–85. doi: 10.1523/JNEUROSCI.4734-03.2004. Epub 2004/02/13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Poulter MO, Du L, Weaver IC, Palkovits M, Faludi G, Merali Z, Szyf M, Anisman H. GABAA receptor promoter hypermethylation in suicide brain: implications for the involvement of epigenetic processes. Biological psychiatry. 2008;64(8):645–52. doi: 10.1016/j.biopsych.2008.05.028. Epub 2008/07/22. [DOI] [PubMed] [Google Scholar]
- 21.Choudary PV, Molnar M, Evans SJ, Tomita H, Li JZ, Vawter MP, Myers RM, Bunney WE, Jr, Akil H, Watson SJ, Jones EG. Altered cortical glutamatergic and GABAergic signal transmission with glial involvement in depression. Proceedings of the National Academy of Sciences of the United States of America. 2005;102(43):15653–8. doi: 10.1073/pnas.0507901102. Epub 2005/10/19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Sequeira A, Klempan T, Canetti L, ffrench-Mullen J, Benkelfat C, Rouleau GA, T G. Patterns of gene expression in the limbic system of suicides with and without major depression. Molecular psychiatry. 2007;12(7):640–55. doi: 10.1038/sj.mp.4001969. Epub 2007/03/14. [DOI] [PubMed] [Google Scholar]
- 23.Geuze E, van Berckel BN, Lammertsma AA, Boellaard R, de Kloet CS, Vermetten E, Westenberg HG. Reduced GABAA benzodiazepine receptor binding in veterans with posttraumatic stress disorder. Molecular psychiatry. 2008;13(1):74–83, 3. doi: 10.1038/sj.mp.4002054. Epub 2007/08/02. [DOI] [PubMed] [Google Scholar]
- 24.Klumpers UM, Veltman DJ, Drent ML, Boellaard R, Comans EF, Meynen G, Lammertsma AA, Hoogendijk WJ. Reduced parahippocampal and lateral temporal GABAA-[11C]flumazenil binding in major depression: preliminary results. European journal of nuclear medicine and molecular imaging. 2010;37(3):565–74. doi: 10.1007/s00259-009-1292-9. Epub 2009/11/06. [DOI] [PubMed] [Google Scholar]
- 25.Karolewicz B, Maciag D, O’Dwyer G, Stockmeier CA, Feyissa AM, Rajkowska G. Reduced level of glutamic acid decarboxylase-67 kDa in the prefrontal cortex in major depression. The international journal of neuropsychopharmacology/official scientific journal of the Collegium Internationale Neuropsychopharmacologicum. 2010;13(4):411–20. doi: 10.1017/S1461145709990587. Epub 2010/03/20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Rajkowska G, O’Dwyer G, Teleki Z, Stockmeier CA, Miguel-Hidalgo JJ. GABAergic neurons immunoreactive for calcium binding proteins are reduced in the prefrontal cortex in major depression. Neuropsychopharmacology : official publication of the American College of Neuropsychopharmacology. 2007;32(2):471–82. doi: 10.1038/sj.npp.1301234. Epub 2006/10/26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Maciag D, Hughes J, O’Dwyer G, Pride Y, Stockmeier CA, Sanacora G, Rajkowska G. Reduced density of calbindin immunoreactive GABAergic neurons in the occipital cortex in major depression: relevance to neuroimaging studies. Biological psychiatry. 2010;67(5):465–70. doi: 10.1016/j.biopsych.2009.10.027. Epub 2009/12/17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lin LC, Sibille E. Reduced brain somatostatin in mood disorders: a common pathophysiological substrate and drug target? Frontiers in pharmacology. 2013;4:110. doi: 10.3389/fphar.2013.00110. Epub 2013/09/24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Duman RS, Aghajanian GK, Sanacora G, Krystal JH. Synaptic plasticity and depression: new insights from stress and rapid-acting antidepressants. Nature medicine. 2016;22(3):238–49. doi: 10.1038/nm.4050. Epub 2016/03/05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Smith KS, R U. Anxiety and depression: Mouse genetics and pharmacological approaches to the role of GABA(A) receptor subtypes. Neuropharmacology. 2012;62:54–62. doi: 10.1016/j.neuropharm.2011.07.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Shen Q, Lal R, Luellen BA, Earnheart JC, Andrews AM, Luscher B. gamma-Aminobutyric acid-type A receptor deficits cause hypothalamic-pituitary-adrenal axis hyperactivity and antidepressant drug sensitivity reminiscent of melancholic forms of depression. Biological psychiatry. 2010;68(6):512–20. doi: 10.1016/j.biopsych.2010.04.024. Epub 2010/06/29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32*.Ren Z, Pribiag H, Jefferson SJ, Shorey M, Fuchs T, Stellwagen D, Luscher B. Bidirectional Homeostatic Regulation of a Depression-Related Brain State by Gamma-Aminobutyric Acidergic Deficits and Ketamine Treatment. Biological psychiatry. 2016 doi: 10.1016/j.biopsych.2016.02.009. Epub 2016/04/12. This study examined the consequences of diminished GABA receptor function in regulating synaptic integrity within the PFC and shows that ketamine reverses these GABA deficits. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Caldji C, Francis D, Sharma S, Plotsky PM, Meaney MJ. The effects of early rearing environment on the development of GABAA and central benzodiazepine receptor levels and novelty-induced fearfulness in the rat. Neuropsychopharmacology : official publication of the American College of Neuropsychopharmacology. 2000;22(3):219–29. doi: 10.1016/S0893-133X(99)00110-4. Epub 2000/02/29. [DOI] [PubMed] [Google Scholar]
- 34.Acosta GB, Otero Losada ME, Rubio MC. Area-dependent changes in GABAergic function after acute and chronic cold stress. Neuroscience letters. 1993;154(1–2):175–8. doi: 10.1016/0304-3940(93)90200-5. Epub 1993/05/14. [DOI] [PubMed] [Google Scholar]
- 35.Braestrup C, Nielsen M, Nielsen EB, L M. Benzodiazepine receptors in the brain as affected by different experimental stresses: the changes are small and not undirectional. Psychopharmacology (Berl) 1979;65(3):273–7. doi: 10.1007/BF00492215. [DOI] [PubMed] [Google Scholar]
- 36.Matsumoto K, Puia G, Dong E, P G. GABA(A) receptor neurotransmission dysfunction in a mouse model of social isolation-induced stress: possible insights into a non-serotonergic mechanism of action of SSRIs in mood and anxiety disorders. Stress. 2007;10(1):3–12. doi: 10.1080/10253890701200997. [DOI] [PubMed] [Google Scholar]
- 37.Bloomfield C, French SJ, Jones DN, Reavill C, Southam E, Cilia J, Totterdell S. Chandelier cartridges in the prefrontal cortex are reduced in isolation reared rats. Synapse. 2008;62(8):628–31. doi: 10.1002/syn.20521. Epub 2008/05/31. [DOI] [PubMed] [Google Scholar]
- 38.Shepard R, Page CE, C L. Sensitivity of the prefrontal GABAergic system to chronic stress in male and female mice: Relevance for sex differences in stress-related disorders. Neuroscience. 2016;332:1–12. doi: 10.1016/j.neuroscience.2016.06.038. [DOI] [PubMed] [Google Scholar]
- 39*.Zhang W, Zhang L, Liang B, Schroeder D, Zhang ZW, Cox GA, Li Y, Lin DT. Hyperactive somatostatin interneurons contribute to excitotoxicity in neurodegenerative disorders. Nature neuroscience. 2016;19(4):557–9. doi: 10.1038/nn.4257. Epub 2016/02/24. This study used optogentics and cell selective approaches to show that SST-mediated disinhibition of pyramidal neurons occurs via inhibition of PV interneurons. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Seybold BA, Stanco A, Cho KK, Potter GB, Kim C, Sohal VS, Rubenstein JL, Schreiner CE. Chronic reduction in inhibition reduces receptive field size in mouse auditory cortex. Proceedings of the National Academy of Sciences of the United States of America. 2012;109(34):13829–34. doi: 10.1073/pnas.1205909109. Epub 2012/07/04. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Li N, Liu RJ, Dwyer JM, Banasr M, Lee B, Son H, Li XY, Aghajanian G, Duman RS. Glutamate N-methyl-D-aspartate receptor antagonists rapidly reverse behavioral and synaptic deficits caused by chronic stress exposure. Biological psychiatry. 2011;69(8):754–61. doi: 10.1016/j.biopsych.2010.12.015. Epub 2011/02/05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Courtin J, Chaudun F, Rozeske RR, Karalis N, Gonzalez-Campo C, Wurtz H, Abdi A, Baufreton J, Bienvenu TC, Herry C. Prefrontal parvalbumin interneurons shape neuronal activity to drive fear expression. Nature. 2014;505(7481):92–6. doi: 10.1038/nature12755. Epub 2013/11/22. [DOI] [PubMed] [Google Scholar]
- 43*.Sibille E, Morris HM, Kota RS, Lewis DA. GABA-related transcripts in the dorsolateral prefrontal cortex in mood disorders. The international journal of neuropsychopharmacology/official scientific journal of the Collegium Internationale Neuropsychopharmacologicum. 2011;14(6):721–34. doi: 10.1017/S1461145710001616. Epub 2011/01/14. This study provided evidence that supports the idea that PFC GABAergic deficits are highly associated with mood disorders. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Tripp A, Kota RS, Lewis DA, Sibille E. Reduced somatostatin in subgenual anterior cingulate cortex in major depression. Neurobiology of disease. 2011;42(1):116–24. doi: 10.1016/j.nbd.2011.01.014. Epub 2011/01/15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Seney ML, Tripp A, McCune S, Lewis DA, Sibille E. Laminar and cellular analyses of reduced somatostatin gene expression in the subgenual anterior cingulate cortex in major depression. Neurobiology of disease. 2015;73:213–9. doi: 10.1016/j.nbd.2014.10.005. Epub 2014/10/16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Lin LC, Sibille E. Somatostatin, neuronal vulnerability and behavioral emotionality. Mol Psychiatry. 2015;20(3):377–87. doi: 10.1038/mp.2014.184. Epub 2015/01/21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Soumier A, Sibille E. Opposing effects of acute versus chronic blockade of frontal cortex somatostatin-positive inhibitory neurons on behavioral emotionality in mice. Neuropsychopharmacology. 2014;39(9):2252–62. doi: 10.1038/npp.2014.76. Epub 2014/04/03. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Perova Z, Delevich K, Li B. Depression of excitatory synapses onto parvalbumin interneurons in the medial prefrontal cortex in susceptibility to stress. The Journal of neuroscience : the official journal of the Society for Neuroscience. 2015;35(7):3201–6. doi: 10.1523/jneurosci.2670-14.2015. Epub 2015/02/24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Bhagwagar Z, Wylezinska M, Taylor M, Jezzard P, Matthews PM, Cowen PJ. Increased brain GABA concentrations following acute administration of a selective serotonin reuptake inhibitor. The American journal of psychiatry. 2004;161(2):368–70. doi: 10.1176/appi.ajp.161.2.368. Epub 2004/02/03. [DOI] [PubMed] [Google Scholar]
- 50.Sanacora G, Mason GF, Rothman DL, Hyder F, Ciarcia JJ, Ostroff RB, Berman RM, Krystal JH. Increased cortical GABA concentrations in depressed patients receiving ECT. The American journal of psychiatry. 2003;160(3):577–9. doi: 10.1176/appi.ajp.160.3.577. Epub 2003/03/04. [DOI] [PubMed] [Google Scholar]
- 51.Goren MZ, Kucukibrahimoglu E, Berkman K, Terzioglu B. Fluoxetine partly exerts its actions through GABA: A neurochemical evidence. Neurochem Res. 2007;32(9):1559–65. doi: 10.1007/s11064-007-9357-2. [DOI] [PubMed] [Google Scholar]
- 52.Chen AI, Nguyen CN, Copenhagen DR, Badurek S, Minichiello L, Ranscht B, Reichardt LF. TrkB (tropomyosin-related kinase B) controls the assembly and maintenance of GABAergic synapses in the cerebellar cortex. The Journal of neuroscience : the official journal of the Society for Neuroscience. 2011;31(8):2769–80. doi: 10.1523/JNEUROSCI.4991-10.2011. Epub 2011/03/19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Vithlani MHR, Zhong P, Terunuma M, Hines DJ, Revilla-Sanchez R, Jurd R, Haydon P, Rios M, Brandon N, Yan Z, Moss SJ. The ability of BDNF to modify neurogenesis and depressive-like behaviors is dependent upon phosphorylation of tyrosine residues 365/367 in the GABA(A)-receptor γ2 subunit. The Journal of neuroscience : the official journal of the Society for Neuroscience. 2013;33(39):155567–77. doi: 10.1523/JNEUROSCI.1845-13.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Berman RM, Cappiello A, Anand A, Oren DA, Heninger GR, Charney DS, Krystal JH. Antidepressant effects of ketamine in depressed patients. Biological psychiatry. 2000;47(4):351–4. doi: 10.1016/s0006-3223(99)00230-9. Epub 2000/02/25. [DOI] [PubMed] [Google Scholar]
- 55.Zarate CA, Jr, Singh JB, Carlson PJ, Brutsche NE, Ameli R, Luckenbaugh DA, Charney DS, Manji HK. A randomized trial of an N-methyl-D-aspartate antagonist in treatment-resistant major depression. Archives of general psychiatry. 2006;63(8):856–64. doi: 10.1001/archpsyc.63.8.856. Epub 2006/08/09. [DOI] [PubMed] [Google Scholar]
- 56.Voleti B, Navarria A, Liu RJ, Banasr M, Li N, Terwilliger R, Sanacora G, Eid T, Aghajanian G, Duman RS. Scopolamine rapidly increases mammalian target of rapamycin complex 1 signaling, synaptogenesis, and antidepressant behavioral responses. Biological psychiatry. 2013;74(10):742–9. doi: 10.1016/j.biopsych.2013.04.025. Epub 2013/06/12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57*.Li N, Lee B, Liu RJ, Banasr M, Dwyer JM, Iwata M, Li XY, Aghajanian G, Duman RS. mTOR-dependent synapse formation underlies the rapid antidepressant effects of NMDA antagonists. Science. 2010;329(5994):959–64. doi: 10.1126/science.1190287. Epub 2010/08/21. This is the first direct demonstatration of the signaling pathways involved in the antidepressant activity of ketamine. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58*.Moghaddam B, Adams B, Verma A, Daly D. Activation of glutamatergic neurotransmission by ketamine: a novel step in the pathway from NMDA receptor blockade to dopaminergic and cognitive disruptions associated with the prefrontal cortex. The Journal of neuroscience : the official journal of the Society for Neuroscience. 1997;17(8):2921–7. doi: 10.1523/JNEUROSCI.17-08-02921.1997. Epub 1997/04/15. This is the first direct demonstatration of ketamine-mediated activation of gluatamatrigic neurotransmission in the PFC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Urban-Ciecko J, Fanselow EE, Barth AL. Neocortical somatostatin neurons reversibly silence excitatory transmission via GABAb receptors. Current biology : CB. 2015;25(6):722–31. doi: 10.1016/j.cub.2015.01.035. Epub 2015/03/03. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Wohleb ES, Wu M, Gerhard DM, Taylor SR, Picciotto MR, Alreja M, Duman RS. GABA interneurons mediate the rapid antidepressant-like effects of scopolamine. The Journal of clinical investigation. 2016;126(7):2482–94. doi: 10.1172/JCI85033. Epub 2016/06/09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Newport DJ, Carpenter LL, McDonald WM, Potash JB, Tohen M, Nemeroff CB, Biomarkers APACoRTFoN, Treatments Ketamine and Other NMDA Antagonists: Early Clinical Trials and Possible Mechanisms in Depression. The American journal of psychiatry. 2015;172(10):950–66. doi: 10.1176/appi.ajp.2015.15040465. Epub 2015/10/02. [DOI] [PubMed] [Google Scholar]