

Abstract
Photoexcited ketones have diradical characteristics and are functionally similar to high-spin metal–oxo species that are frequently used to catalyze C–H oxidation. First discovered by Yang in 1958, photoexcited ketones can abstract a hydrogen atom from hydrocarbons inter- or intramolecularly. Coupling with atom-transfer, group-transfer, or radical addition, the Yang reaction can be used to achieve various types of C–H functionalization. We provide in this article an overview of triplet ketone-mediated or catalyzed C–H functionalization reactions.
Norrish–Yang cyclization is a well-known photochemical process that generates cyclobutanols from γ-hydrogen-bearing ketones. This intramolecular C–H functionalization reaction works by UV-irradiation of ketone to generate a diradical species that abstracts a γ-hydrogen to create a reactive site for cyclization (Fig. 1A). In contrast to Norrish type II cleavage wherein the diradical intermediate fragments into enol and olefin, combination of the radicals provides a cyclobutane ring. Shortly after N. C. Yang started his career at the University of Chicago, he reported in 1958 the discovery of this cyclization reaction with his wife and scientific collaborator D.-D. H. Yang.1 They showed that photoexcitation of ketone 1 in cyclohexane gave cyclobutanol 2 along with the Norrish type II cleavage products acetone (3) and 1-pentene (4) (Fig. 1B).
Fig. 1.
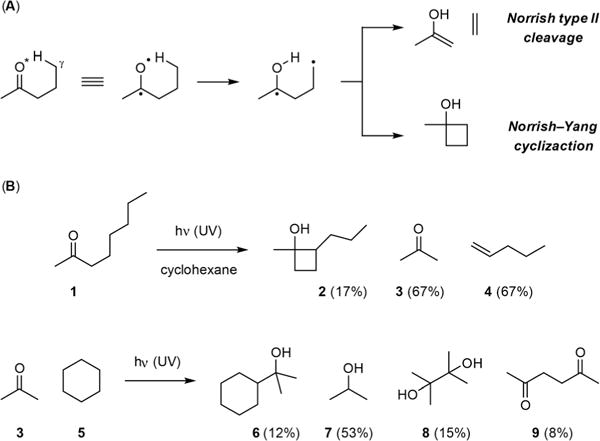
(A) The general reactions of Norrish type II cleavage and Norrish–Yang cyclization. (B) The Norrish–Yang cyclization and Yang C–H functionalization reactions reported by D.-D. H. Yang and N. C. Yang in 1958.
However, Norrish–Yang cyclization was an intramolecular variation of a more general reaction that the Yangs discovered. In the same report, they also described that UV-irradiation of a 1:8 mixture of acetone (3) and cyclohexane (5) afforded 2-cyclohexylpropan-2-ol (6), 2-propanol (7), pinacol (8), 2,5-hexanedione (9), and a small amount of methane and carbon monoxide, the Norrish type I cleavage products of acetone. They further proposed that photoexcited acetone (3*) abstracted a hydrogen atom from 5 to give the cyclohexyl radical and the acetone ketyl radical. Combination of these two radical species then yielded 6. It is somewhat surprising that the intermolecular Yang reaction was largely ignored despite the wide application of Norrish–Yang cyclization.
Photolytic C–H abstraction by ketones was generally regarded as photoreduction of triplet ketones.2 Both C–H and S–H are effective hydrogen donors.3–6 Through a series of careful kinetic studies, Walling concluded that triplet ketone has a reactivity similar to that of the tert-butoxy radical.4 He also found that UV-irradiation of a mixture of 5 and benzophenone (10) in carbon tetrachloride gave nearly quantitative yields of cyclohexyl chloride (11), benzopinacol (12), and hexachloroethane (13) based on consumed 5 (Fig. 2).7 Chlorination of 2,3-dimethylbutane (14) also proceeded with excellent regioselectivity to give chloride 15. However, there had been no further development of triplet ketone-mediated C–H halogenation reactions until recently.
Fig. 2.
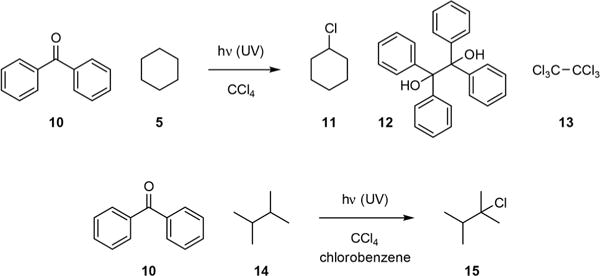
Performing Yang reaction in carbon tetrachloride led to the formation of C–H chlorination products.
The power of the Yang reaction has been recognized by Breslow.8 In a series of studies on remote C–H oxidation,9 he showed that fatty alcohols and steroids could be functionalized selectively at unactivated positions by a triplet benzophenone attached to a remote position of the molecule. For example, C–H abstraction of the C14-fatty alcohol derivative 16 occurred predominantly at the C12 position (Fig. 3).10 Oxidation of the steroid derivative 20 was also remarkably selective giving 21 as the only product.11 Here, the combination of radicals generated from the initial C–H abstraction at the C14 position was prohibited due to the rigid structure of the intermediate. Consequently, hydrogen transfer from C15 to the ketyl radical occurred to yield an olefin.
Fig. 3.
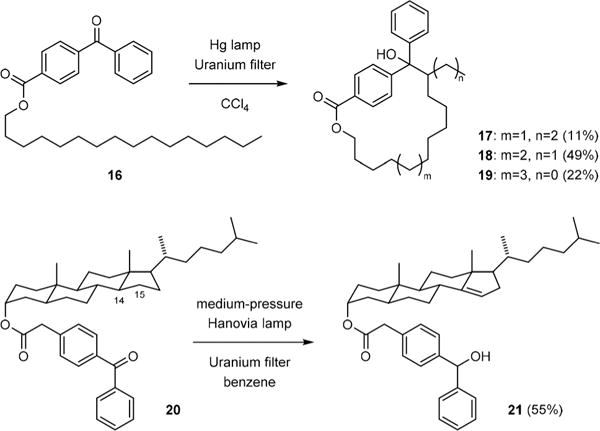
Breslow’s remote C–H oxidation reactions.
Ethereal C–H abstraction/radical addition is arguably the most explored Yang-type reaction. However, it was developed independent of Yang’s work and rooted in Ciamician’s discovery that exposure of benzophenone (10) in ethanol to sunlight (UV-visible light) gave benzopinacol (12).12 Schenck reported in 1957 that UV-irradiation of a mixture of 10, isopropanol (22), and maleic acid (23) gave tertiary alcohol 24 (Fig. 4), and offered a correct mechanistic interpretation for this coupling reaction.13 Unfortunately, Schenck’s work did not get much attention either. Following the cue of Pedersen,14 Elad studied ketone-initiated photoaddition of cyclic ethers to terminal olefins in 1964.15–17 He found that photoexcited acetophenone (25) could abstract an ethereal C–H from tetrahydrofuran (26). The resulting radical could then add to dimethyl maleate (27) to give adduct 28 after back-hydrogen transfer. Similarly, exposure of a mixture of acetone (3), 1,3-dioxolane (29) and 1-decene (30) to sunlight yielded protected undecanal 31.
Fig. 4.
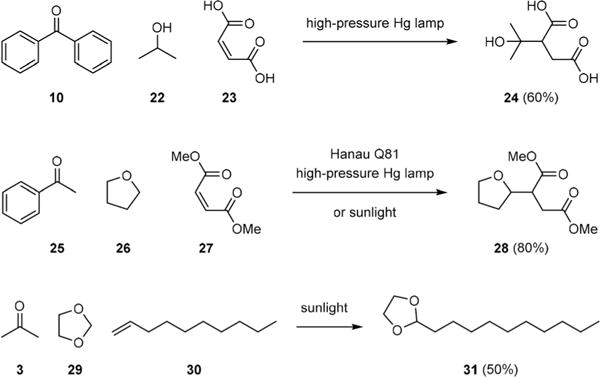
Schenck and Elad’s photochemical ethereal C–H abstraction/radical addition reactions.
It should be noted that ketones in these reactions were at times referred to as photosensitizers. However, as Elad noted, the term sensitizer should only be used when “a compound initiates a photochemical process through light absorption and does not undergo a chemical change itself”.16 In other words, sensitizers transfer triplet energy to the substrate without undergoing chemical transformations. Here, the photoexcited ketone should not be considered as a sensitizer because it abstracts a hydrogen atom from the substrate and is often consumed quickly due to facile Ciamician coupling. As a result, a substoichiometric to excess amount of ketone is typically required for this radical addition reaction.
Fraser-Reid demonstrated in 1972 that only a catalytic amount of benzophenone (10) was required to promote the coupling of alcohols and enones.18 He later achieved the synthesis of pillarose with this C–H functionalization reaction.19 UV-irradiation of enone 32 in the presence of 16 mol% of 10 and a large excess of ethylene glycol gave δ-lactone 33 (Fig. 5). Subsequent C6-deoxygenation and C7-oxidation afforded ethyl pillarose (34). Fraser-Reid has also provided evidence that 10 served as a C–H abstraction catalyst instead of a triplet energy-transfer photosensitizer in these coupling reactions.20
Fig. 5.
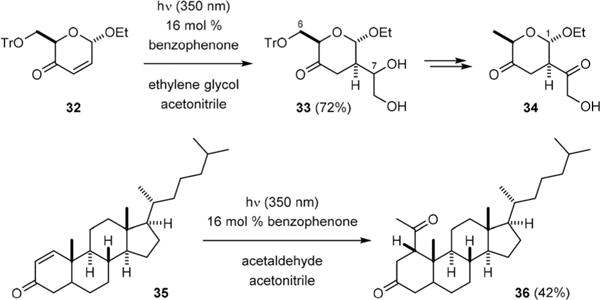
Fraser-Reid’s photochemical radical conjugate addition reactions.
Fraser-Reid further found that photoexcited benzophenone could catalyze conjugate addition of aldehydes to enones.21 For example, irradiation of 1-cholesten-3-one (35) with 16 mol% of 10 and a large excess of acetaldehyde gave ketone 36. However, this reaction is not mechanistically related to the Yang reaction because 10 worked as a photosensitizer to transfer its triplet energy to acetaldehyde. The triplet aldehyde then underwent Norrish type I cleavage to give a carbonyl radical that added to enone.22 Over the past few decades, the scope of photochemical C–H abstraction/radical addition has been expanded to include amines and alkanes as radical donors and alkynes as acceptors.23–27 In addition, other than the ester group, nitro, sulfone, amide, nitrile, and aldehyde groups can all be used as activating groups for the radical acceptors.28–32
Addition of radicals to substituted butenolides has also been shown to proceed with good diastereoselectivity.33–36 For example, Mann reported in 1994 that irradiation of butenolide 37 with 10 in methanol gave γ-lactone 38 as the only diastereomeric product (Fig. 6).34 Additionally, Hoffmann showed that 4,4′-dimethoxylbenzophenone is an effective catalyst for the coupling of N-methylpyrrolidine (39) and butenolide 40.35 The α-radical of 39 approached 40 from its less hindered face to yield 41 selectively.
Fig. 6.
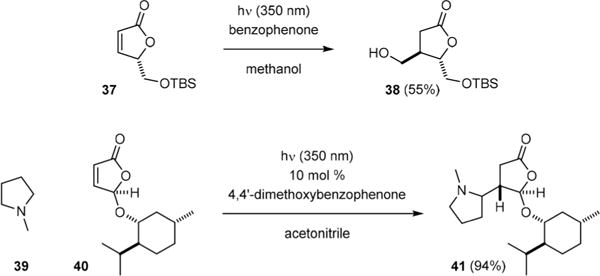
Diastereoselective radical addition to butenolides.
Various tandem processes has also been developed to elaborate the conjugate addition products. For example, Guzmán showed in 1981 that the coupling of isopropanol (22) and dihydropyranone 42 gave δ-lactone 43 that rearranged to γ-lactones 44 under the reaction conditions (Fig. 7).37 Gómez also found that photolysis of enone 45 with 10 in isopropanol (22) provided bicyclic lactone 46 through C–H abstraction of 22, conjugate addition to 45, 6-exo-trig cyclization, and back-hydrogen transfer.38
Fig. 7.
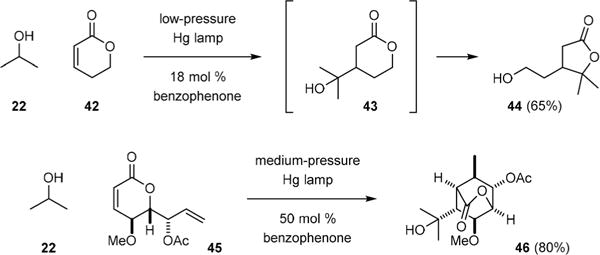
Tandem reactions involving photochemical radical addition to dihydropyranone.
A significant advancement in this field of research is the development of photochemical group-transfer reactions by Inoue.39–42 Inoue first showed in 2011 that C–H carbamoylation of tetrahydrofuran (26) could be achieved by irradiation of 26 with pentafluorophenyl isocyanate (47) in the presence of 10 (Fig. 8).39 The use of an electron-deficient isocyanate is important to the efficient formation of amide 48. He then showed that C–H cyanation of N-BOC-L-proline methyl ester (49) by tosyl cyanide (50) could be promoted by 10* to deliver the trans-cyanoproline derivative 51 with excellent regio- and diastereoselectivity.41
Fig. 8.
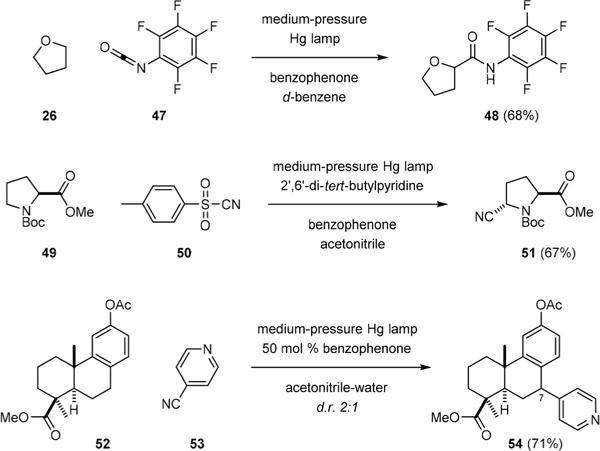
Inoue’s C–H carbamoylation, cyanation, and 4-pyridination reactions.
Inoue also demonstrated in 2013 that C–H pyridination of methyl O-acetylpodocarpate (52) could be realized by irradiation of 52 with 4-cyanopyridine (53) in the presence of 10.42 Unlike the Gif-type Minisci reaction,43 the addition of the benzylic radical of 52 to 53 was highly regioselective. The 4-pyridination product 54 was obtained as a 2:1 mixture of C7-diastereomers without 2-pyridination.
Although Walling has shown that the Yang reaction could be intercepted with chlorination, the development of a Yang-type C–H halogenation reaction did not occur until 2012.44 Serendipitously during the study of vanadium-catalyzed C–H fluorination, Chen found that irradiation of 4-ethylacetophenone (55) with light generated from a household compact fluorescent lamp (CFL) led to the formation of benzylic radical 56 that could be fluorinated to provide benzylic fluoride 57 (Fig. 9). This reaction proceeded through intermolecular hydrogen abstraction of 55 by 55* to give 56 and ketyl radical 58. After Selectfluor (59) donated a fluorine atom to 56, the resulting aminium radical cation 60 abstracted a hydrogen atom from 58 to turn over 55 and yield ammonium 61.
Fig. 9.
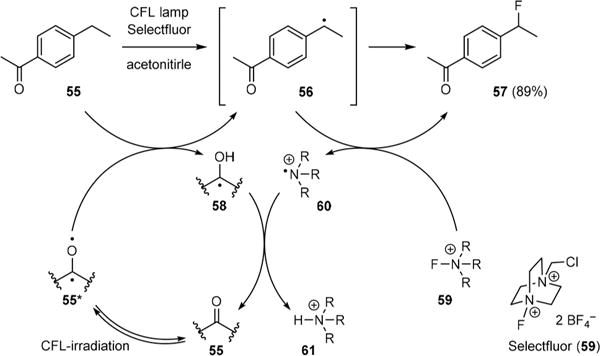
Chen’s discovery of a Yang-type C–H fluorination reaction.
Unlike most Yang reactions, fluorination of 55 by 59 was particularly efficient giving no Ciamician coupling product. Chen hypothesized that dimerization of 58 did not occur because of a facile hydrogen transfer from 58 to the highly energetic aminium radical cation 60, which quickly turned over 55. It was thus possible to use a low loading of exogenous ketone to catalyze C–H fluorination. Indeed, CFL-irradiation of ethylbenzene (57) in the presence of 5 mol% of acetophenone (25) yielded benzylic fluoride 58 (Fig. 10).45 The efficiency of this reaction was further improved when using 10 or 9-fluorenone (60) as the catalyst as both 10 and 60 absorb visible light much more efficiently than 55. C–H chlorination could also be achieved by replacing 59 with N-chlorosuccinimide (NCS) under otherwise the same reaction conditions.44
Fig. 10.
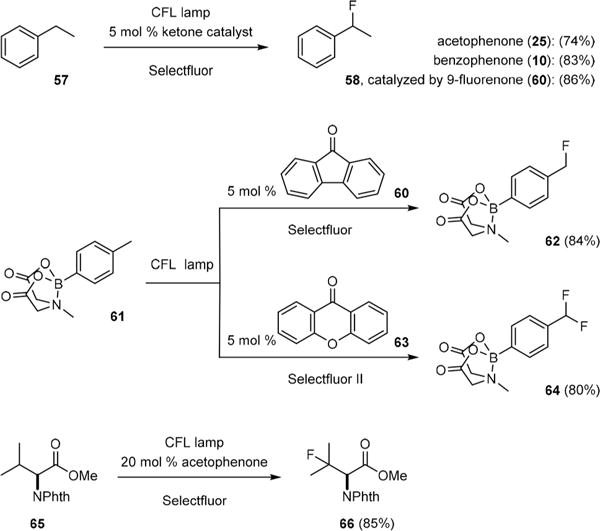
Chen’s triple ketone-catalyzed C–H fluorination reactions.
Because the n/π* energy gap can be tuned by changing the substituent groups of ketone, the reactivity and selectivity of triplet ketone can be modulated easily. In general, diarylketones are more effective benzylic C–H fluorination catalysts whereas monoarylketones are more reactive toward unactivated C–H. Selective formation of the mono- or difluorination products can also be achieved through catalyst-control. For example, 9-fluorenone (60) catalyzed benzylic C–H monofluorination of boronate 61 to give 62; xanthone (63) catalyzed benzylic C–H difluorination to yield 64. De Frutos and Kappe have further shown that the reaction efficiency could be significantly improved with flow chemistry and black-light irradiation.46
Intriguingly, acetophenone (25), with a larger n/π* gap, is a colorless oil and has only a trace amount of absorption in the visible light region. However, CFL-irradiation can activate 25 to abstract unactivated C(sp3)–H groups effectively presumably through residual absorption of light between 380 and 400 nm.47 Thus, C–H fluorination of N-phthaloyl-L-valine methyl ester 65 could be achieved under very mild conditions to give β-fluoro α-amino ester 66 with excellent regioselectivity. Chen has also demonstrated that CFL-irradiation could drive other photoreactions of monoarylketones and enones that have no apparent absorption above 380 nm.47 For example, CFL-irradiation induced Norrish type II cleavage and Norrish– Yang cyclization of valerophenone (67) to give acetophenone (25), propene (68), and cyclobutanols 69 and 70 (Fig. 11). The coupling of cyclopentenone (71) and 2-propanol (22) also proceeded smoothly to give adduct 72 under CFL-irradiation. Residual absorption has also been used by Melchiorre to induce aldehyde-catalyzed atom-transfer radical addition of haloalkanes to olefins.48
Fig. 11.
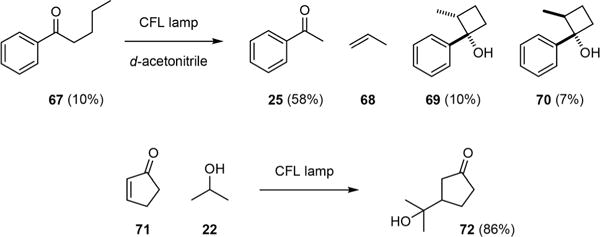
Chen’s demonstration of photoreactions driven by residual absorption.
Tan has also reported the development of an interesting photochemical C–H fluorination in 2014.49 Although the reaction conditions were similar to that reported by Chen, a different mechanism of C–H abstraction was involved. He showed that fluorination of Boc-protected 1-adamantylamine (73) by 59 to give fluoride 74 could be achieved by CFL-irradiation of 73 in the presence of a catalytic amount of anthraquinone (75) (Fig. 12). This result is in sharp contrast to Chen’s fluorination wherein no reaction occurred in the presence of an N–H group. The selectivity of triplet 75 was also quite different from that of triplet 60. Kinetic and computational studies strongly support the hypothesis that photoexcited 75* transferred its triplet energy to 59 to generate 59* that donated its fluorine atom to 75. The resulting aminium radical dication 76 abstracted a hydrogen atom from 73 to yield a 3-adamantyl radical that incorporated a fluorine atom from [75–F]• to yield 74.
Fig. 12.
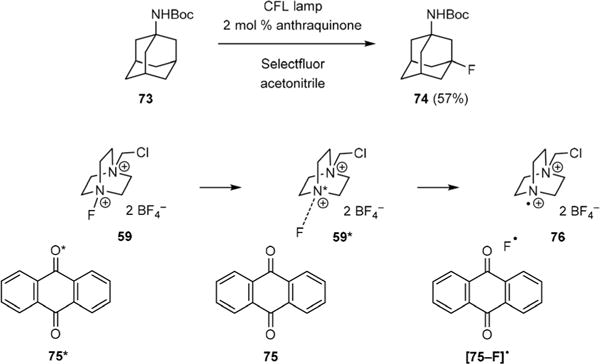
Tan’s photochemical C–H fluorination reaction.
Li has further extended the utility of Yang reaction from functionalization of C(sp3)–H to C(sp2)–H in 2016.50 He showed that the Yang C–H functionalization could be used to introduce a trifluoromethyl group to caffeine (77) via the radical adduct 78 to give the 8-substituted product 79 selectively (Fig. 13). This reaction started with single-electron oxidation of Langlois’ reagent (80) by triplet acetone (3*) to generate a trifluoromethanesulfinyl radical (81) and ketyl radical anion 82. Subsequent decomposition of 81 yielded a trifluoromethyl radical (83) and sulfur dioxide (84). Radical addition of 83 to 77 then provided 78. Abstraction of the ipso-hydrogen of 78 by 82 afforded 79 and alkoxide 85. Li also found that using diacetyl (86) as a co-solvent allowed for initiation of this photochemical process by visible light. Thus, trifluoromethylation of arene 87 could be achieved under very mild conditions to give 88.
Fig. 13.
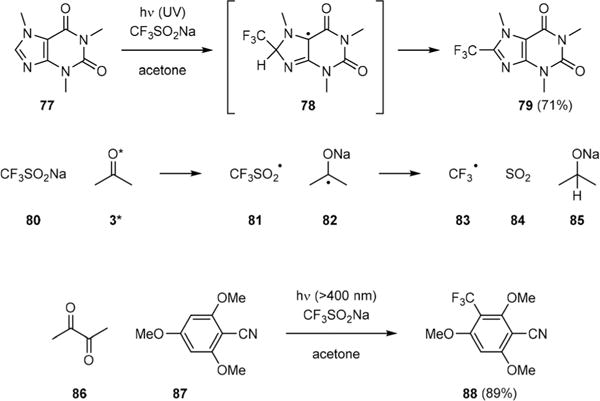
Li’s photochemical C–H trifluoromethylation reactions.
The past decade has witnessed rapid advancement in C–H functionalization and radical chemistry is an important branch in this research field.51–81 Using photochemical methods to induce radical reactions has also regained popularity recently. The diradical characteristics of triplet carbon–oxo (carbonyl) make photoexcited ketones functionally equivalent to high-spin metal–oxo species,82–90 the major C–H oxidation catalysts in nature. Similar to the ligands of metal–oxo complexes, the substituent groups of ketones can modulate the reactivity and selectivity of the catalyst. Since the discovery of the Yang C–H functionalization reaction more than fifty years ago, triplet ketones have been used to initiate a variety of atom-transfer, group-transfer, and radical addition reactions. The scope of Yang reaction has gone beyond enzymatic C–H functionalization; however, the selectivity and efficiency are still far from ideal. In particular, the regioselectivity of hydrogen abstraction largely depends on the stereoelectronic properties of the substrates, making it hard to achieve complete catalyst-controlled selectivity that overwrites the innate C–H reactivity. There is also no good strategy to establish a transient interaction between the catalyst and certain functional groups of the substrate to direct C–H abstraction. A list of ketones with different excitation wavelengths allowing for selective activation of the catalyst with narrow-band or laser excitation in the presence of multiple chromophores is also yet to be compiled. We hope that these challenges will inspire future endeavours that enable “precision chemical reactions”.
Acknowledgments
We thank NIH (NIGMS R01-GM079554), the Welch Foundation (I-1868), and UT Southwestern for financial support.
Biography

Chuo Chen was born in Taipei, Taiwan, ROC in 1973. He worked in the laboratory of Professor Tien-Yau Luh as an undergraduate student at National Taiwan University where he obtained his B.S. degree in 1995. He then studied natural product synthesis under the direction of Professor Matthew D. Shair at Harvard University. After receiving his Ph.D. degree in 2001, he worked as a Howard Hughes Medical Institute (HHMI) postdoctoral fellow in Professor Stuart L. Schreiber’s laboratory at Harvard University before joining the faculty of the University of Texas Southwestern Medical Center in 2004. He is now a Professor in Biochemistry and a Southwestern Medical Foundation Scholar in Biomedical Research. His research interests include natural product synthesis, synthetic methodology, and chemical biology.
Footnotes
Dedicated to Professor Tien-Yau Luh on the occasion of his 70th birthday.
Notes and references
- 1.Yang NC, Yang DDH. J Am Chem Soc. 1958;80:2913–2914. [Google Scholar]
- 2.Hammond GS, Turro NJ. Science. 1963;142:1541–1553. doi: 10.1126/science.142.3599.1541. [DOI] [PubMed] [Google Scholar]
- 3.Pitts JN, Jr, Letsinger RL, Taylor RP, Patterson JM, Recktenwald G, Martin RB. J Am Chem Soc. 1959;81:1068–1077. [Google Scholar]
- 4.Walling C, Gibian MJ. J Am Chem Soc. 1965;87:3361–3364. [Google Scholar]
- 5.Cohen SG, Rose AW, Stone PG, Ehret A. J Am Chem Soc. 1979;101:1827–1832. [Google Scholar]
- 6.Stone PG, Cohen SG. J Am Chem Soc. 1982;104:3435–3440. [Google Scholar]
- 7.The formation of 11 was first noted when Freon 112 (CFCl2CFCI2) was used as an inert internal standard for GLC analysis (ref. 4, footnote 10).
- 8.The name “Yang reaction” was coined by Breslow (ref. 10).
- 9.Breslow R. Chem Rec. 2001;1:3–11. doi: 10.1002/1528-0691(2001)1:1<3::AID-TCR3>3.0.CO;2-B. [DOI] [PubMed] [Google Scholar]
- 10.Breslow R, Winnik MA. J Am Chem Soc. 1969;91:3083–3084. [Google Scholar]
- 11.Breslow R, Baldwin S, Flechtner T, Kalicky P, Liu S, Washburn W. J Am Chem Soc. 1973;95:3251–3262. doi: 10.1021/ja00791a031. [DOI] [PubMed] [Google Scholar]
- 12.Ciamician G, Silber P. Ber Dtsch Chem Ges. 1900;33:2911–2913. [Google Scholar]
- 13.Schenck GO, Koltzenburg G, Grossmann H. Angew Chem. 1957;69:177–178. [Google Scholar]
- 14.Diekmann J, Pedersen CJ. J Org Chem. 1963;28:2879–2880. [Google Scholar]
- 15.Elad D, Youssefyeh RD. J Org Chem. 1964;29:2031–2032. [Google Scholar]
- 16.Rosenthal I, Elad D. Tetrahedron. 1967;23:3193–3204. [Google Scholar]
- 17.Rosenthal I, Elad D. J Org Chem. 1968;33:805–811. [Google Scholar]
- 18.Fraser-Reid B, Holder NL, Yunker MB. J Chem Soc, Chem Commun. 1972:1286–1287. [Google Scholar]
- 19.Walker DL, Fraser-Reid B. J Am Chem Soc. 1975;97:6251–6253. doi: 10.1021/ja00854a055. [DOI] [PubMed] [Google Scholar]
- 20.Benko Z, Fraser-Reid B. J Org Chem. 1988;53:2066–2072. [Google Scholar]
- 21.Fraser-Reid B, Anderson RC, Hicks DR, Walker DL. Can J Chem. 1977;55:3986–3995. [Google Scholar]
- 22.Kharasch MS, Urry WH, Kuderna BM. J Org Chem. 1949;14:248–253. doi: 10.1021/jo01154a009. [DOI] [PubMed] [Google Scholar]
- 23.de Alvarenga ES, Mann J. J Chem Soc, Perkin Trans 1. 1993:2141–2142. [Google Scholar]
- 24.Mella M, Fagnoni M, Albini A. Eur J Org Chem. 1999:2137–2142. [Google Scholar]
- 25.Geraghty NWA, Hannan JJ. Tetrahedron Lett. 2001;42:3211–3213. [Google Scholar]
- 26.Bach T, Aechtner T, Neumüller B. Chem – Eur J. 2002;8:2464–2475. doi: 10.1002/1521-3765(20020603)8:11<2464::AID-CHEM2464>3.0.CO;2-S. [DOI] [PubMed] [Google Scholar]
- 27.Fernández-González M, Alonso R. J Org Chem. 2006;71:6767–6775. doi: 10.1021/jo060883y. [DOI] [PubMed] [Google Scholar]
- 28.Sakakibara T, Nakagawa T. Carbohydr Res. 1987;163:239–246. [Google Scholar]
- 29.Ogura K, Kayano A, Sumitani N, Akazome M, Fujita M. J Org Chem. 1995;60:1106–1107. [Google Scholar]
- 30.Drew MGB, Harrison RJ, Mann J, Tench AJ, Young RJ. Tetrahedron. 1999;55:1163–1172. [Google Scholar]
- 31.Cardarelli AM, Fagnoni M, Mella M, Albini A. J Org Chem. 2001;66:7320–7327. doi: 10.1021/jo010400k. [DOI] [PubMed] [Google Scholar]
- 32.Dondi D, Caprioli I, Fagnoni M, Mella M, Albini A. Tetrahedron. 2003;59:947–957. [Google Scholar]
- 33.Mann J, Weymouth-Wilson A. Carbohydr Res. 1992;216:511–515. [Google Scholar]
- 34.Mann J, Weymouth-Wilson AC. J Chem Soc, Perkin Trans 1. 1994:3141–3148. [Google Scholar]
- 35.Bertrand S, Glapski C, Hoffmann N, Pete JP. Tetrahedron Lett. 1999;40:3169–3172. [Google Scholar]
- 36.Mosca R, Fagnoni M, Mella M, Albini A. Tetrahedron. 2001;57:10319–10328. [Google Scholar]
- 37.Guzmán A, Mendoza S, Diaz E. Synthesis. 1981:989–991. [Google Scholar]
- 38.Gómez AM, Mantecón S, Valverde S, López JC. J Org Chem. 1997;62:6612–6614. [Google Scholar]
- 39.Kamijo S, Hoshikawa T, Inoue M. Tetrahedron Lett. 2011;52:2885–2888. [Google Scholar]
- 40.Kamijo S, Hoshikawa T, Inoue M. Org Lett. 2011;13:5928–5931. doi: 10.1021/ol202659e. [DOI] [PubMed] [Google Scholar]
- 41.Hoshikawa T, Yoshioka S, Kamijo S, Inoue M. Synthesis. 2013:874–887. [Google Scholar]
- 42.Hoshikawa T, Inoue M. Chem Sci. 2013;4:3118–3123. [Google Scholar]
- 43.Minisci F, Fontana F. Tetrahedron Lett. 1994;35:1427–1430. [Google Scholar]
- 44.J. Xia and C. Chen, unpublished results.
- 45.Xia JB, Zhu C, Chen C. J Am Chem Soc. 2013;135:17494–17500. doi: 10.1021/ja410815u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Cantillo D, de Frutos O, Rincón JA, Mateos C, Kappe CO. J Org Chem. 2014;79:8486–8490. doi: 10.1021/jo5016757. [DOI] [PubMed] [Google Scholar]
- 47.Xia JB, Zhu C, Chen C. Chem Commun. 2014;50:11701–11704. doi: 10.1039/c4cc05650g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Arceo E, Montroni E, Melchiorre P. Angew Chem, Int Ed. 2014;53:12064–12068. doi: 10.1002/anie.201406450. [DOI] [PubMed] [Google Scholar]
- 49.Kee CW, Chin KF, Wong MW, Tan CH. Chem Commun. 2014;50:8211–8214. doi: 10.1039/c4cc01848f. [DOI] [PubMed] [Google Scholar]
- 50.Li L, Mu X, Liu W, Wang Y, Mi Z, Li CJ. J Am Chem Soc. 2016;138:5809–5812. doi: 10.1021/jacs.6b02782. [DOI] [PubMed] [Google Scholar]
- 51.Seregina IV, Gevorgyan V. Chem Soc Rev. 2007;36:1173–1193. doi: 10.1039/b606984n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Li CJ. Acc Chem Res. 2008;42:335–344. doi: 10.1021/ar800164n. [DOI] [PubMed] [Google Scholar]
- 53.Davies HML, Morton D. Chem Soc Rev. 2011;40:1857–1869. doi: 10.1039/c0cs00217h. [DOI] [PubMed] [Google Scholar]
- 54.Zhou M, Crabtree RH. Chem Soc Rev. 2011;40:1875–1884. doi: 10.1039/c0cs00099j. [DOI] [PubMed] [Google Scholar]
- 55.McMurray L, O’Hara F, Gaunt MJ. Chem Soc Rev. 2011;40:1885–1898. doi: 10.1039/c1cs15013h. [DOI] [PubMed] [Google Scholar]
- 56.Boorman TC, Larrosa I. Chem Soc Rev. 2011;40:1910–1925. doi: 10.1039/c0cs00098a. [DOI] [PubMed] [Google Scholar]
- 57.Collet F, Lescot C, Dauban P. Chem Soc Rev. 2011;40:1926–1936. doi: 10.1039/c0cs00095g. [DOI] [PubMed] [Google Scholar]
- 58.Zhang SY, Zhang FM, Tu YQ. Chem Soc Rev. 2011;40:1937–1949. doi: 10.1039/c0cs00063a. [DOI] [PubMed] [Google Scholar]
- 59.Gutekunst WR, Baran PS. Chem Soc Rev. 2011;40:1976–1991. doi: 10.1039/c0cs00182a. [DOI] [PubMed] [Google Scholar]
- 60.Hartwig JF. Chem Soc Rev. 2011;40:1992–2002. doi: 10.1039/c0cs00156b. [DOI] [PubMed] [Google Scholar]
- 61.Herrmann P, Bach T. Chem Soc Rev. 2011;40:2022–2038. doi: 10.1039/c0cs00027b. [DOI] [PubMed] [Google Scholar]
- 62.Bras JL, Muzart J. Chem Rev. 2011;111:1170–1214. doi: 10.1021/cr100209d. [DOI] [PubMed] [Google Scholar]
- 63.Yeung CS, Dong VM. Chem Rev. 2011;111:1215–1292. doi: 10.1021/cr100280d. [DOI] [PubMed] [Google Scholar]
- 64.Sun CL, Li BJ, Shi ZJ. Chem Rev. 2011;111:1293–1314. doi: 10.1021/cr100198w. [DOI] [PubMed] [Google Scholar]
- 65.Ackermann L. Chem Rev. 2011;111:1315–1345. doi: 10.1021/cr100412j. [DOI] [PubMed] [Google Scholar]
- 66.Liu C, Zhang H, Shi W, Lei A. Chem Rev. 2011;111:1780–1824. doi: 10.1021/cr100379j. [DOI] [PubMed] [Google Scholar]
- 67.Engle KM, Mei TS, Wasa M, Yu JQ. Acc Chem Res. 2012;45:788–802. doi: 10.1021/ar200185g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Vedernikov AN. Acc Chem Res. 2012;45:803–813. doi: 10.1021/ar200191k. [DOI] [PubMed] [Google Scholar]
- 69.Colby DA, Tsai AS, Bergman RG, Ellman JA. Acc Chem Res. 2012;45:814–825. doi: 10.1021/ar200190g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Brückl T, Baxter RD, Ishihara Y, Baran PS. Acc Chem Res. 2012;45:826–839. doi: 10.1021/ar200194b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Powers DC, Ritter T. Acc Chem Res. 2012;45:840–850. doi: 10.1021/ar2001974. [DOI] [PubMed] [Google Scholar]
- 72.Campbell AN, Stahl SS. Acc Chem Res. 2012;45:851–863. doi: 10.1021/ar2002045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Hartwig JF. Acc Chem Res. 2012;45:864–873. doi: 10.1021/ar200206a. [DOI] [PubMed] [Google Scholar]
- 74.Sigman MS, Werner EW. Acc Chem Res. 2012;45:874–884. doi: 10.1021/ar200236v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Hashiguchi BG, Bischof SM, Konnick MM, Periana RA. Acc Chem Res. 2012;45:885–898. doi: 10.1021/ar200250r. [DOI] [PubMed] [Google Scholar]
- 76.Boisvert L, Goldberg KI. Acc Chem Res. 2012;45:899–910. doi: 10.1021/ar2003072. [DOI] [PubMed] [Google Scholar]
- 77.Roizen JL, Harvey ME, Du Bois J. Acc Chem Res. 2012;45:911–922. doi: 10.1021/ar200318q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Davies HML, Lian Y. Acc Chem Res. 2012;45:923–935. doi: 10.1021/ar300013t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Neufeldt SR, Sanford MS. Acc Chem Res. 2012;45:936–946. doi: 10.1021/ar300014f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Haibach MC, Kundu S, Brookhart M, Goldman AS. Acc Chem Res. 2012;45:947–958. doi: 10.1021/ar3000713. [DOI] [PubMed] [Google Scholar]
- 81.Daugulis O, Roane J, Tran LD. Acc Chem Res. 2015;48:1053–1064. doi: 10.1021/ar5004626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Vaillancourt FH, Yeh E, Vosburg DA, Garneau-Tsodikova S, Walsh CT. Chem Rev. 2006;106:3364–3378. doi: 10.1021/cr050313i. [DOI] [PubMed] [Google Scholar]
- 83.Neumann CS, Fujimori DG, Walsh CT. Chem Biol. 2008;15:99–109. doi: 10.1016/j.chembiol.2008.01.006. [DOI] [PubMed] [Google Scholar]
- 84.Que L, Jr, Tolman WB. Nature. 2008;455:333–340. doi: 10.1038/nature07371. [DOI] [PubMed] [Google Scholar]
- 85.Punniyamurthy T, Rout L. Coord Chem Rev. 2008;252:134–154. [Google Scholar]
- 86.Friedle S, Reisner E, Lippard SJ. Chem Soc Rev. 2010;39:2768–2779. doi: 10.1039/c003079c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Borovik AS. Chem Soc Rev. 2011;40:1870–1874. doi: 10.1039/c0cs00165a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Lu H, Zhang XP. Chem Soc Rev. 2011;40:1899–1909. doi: 10.1039/c0cs00070a. [DOI] [PubMed] [Google Scholar]
- 89.Che CM, Lo VKY, Zhou CY, Huang JS. Chem Soc Rev. 2011;40:1950–1975. doi: 10.1039/c0cs00142b. [DOI] [PubMed] [Google Scholar]
- 90.Lewis JC, Coelho PS, Arnold FH. Chem Soc Rev. 2011;40:2003–2021. doi: 10.1039/c0cs00067a. [DOI] [PMC free article] [PubMed] [Google Scholar]